
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Structural aspects of activation pathways of aspartic protease zymogens and viral 3C protease precursors
AMIR R. KHAN*, NINA KHAZANOVICH-BERNSTEIN, ERNST M. BERGMANN, AND MICHAEL N. G. JAMES†
Medical Research Council Group in Protein Structure and Function, Department of Biochemistry, University of Alberta, Edmonton, Alberta T6G 2H7, Canada
ABSTRACT The three-dimensional structures of the inactive protein precursors (zymogens) of the serine, cysteine, aspartic, and metalloprotease classes of proteolytic enzymes are known. Comparisons of these structures with those of the mature, active proteases reveal that, in general, the preformed, active conformations of the residues involved in catalysis are rendered sterically inaccessible to substrates by the residues of the zymogens’ N-terminal extensions or prosegments. The prosegments interact in nonsubstrate-like fashions with the residues of the active sites in most of the cases. The gastric aspartic proteases have a well-characterized zymogen conversion pathway. Structures of human progastricsin, the inactive intermediate 2, and active human pepsin are known and have been used to define the conversion pathway. The structure of the zymogen precursor of plasmepsin II, the malarial aspartic protease, shows a new twist on the mode of inactivation used by the gastric zymogens. The prosegment of proplasmepsin disrupts the active conformation of the two catalytic aspartic acid residues by inducing a major reorientation of the two domains of the mature protease. The picornaviral 2A and 3C proteases have a chymotrypsin-like tertiary structure but with a cysteine nucleophile. These enzymes cleave themselves from the viral polyprotein in cis (intramolecular cleavage) and carry out trans cleavages of other scissile peptides important for the virus life cycle. Although the structure of the precursor viral polyprotein is unknown, it probably resembles the organization of the proenzymes of the bacterial serine proteases, subtilisin, and α-lytic protease. Cleavage of the prosegment is known to occur in cis for these precursor molecules.
Zymogens of proteolytic enzymes consist of the intact protease with an N-terminal extension. Conversion of the inactive zymogen to the mature, active protease requires limited proteolysis usually of a single peptide bond (1). Molecular rearrangements accompany the proteolytic removal of the prosegment of the zymogen, eventually leading to the mature protease. The prosegments of the zymogens range in size from two residues for some of the granzymes to more than 150 residues for a-lytic protease, a bacterial serine protease (2).
The conversion of zymogens to the respective active enzymes is achieved by several different mechanisms (3). The active serine proteases of the chymotrypsin family result from limited proteolysis of the zymogens by convertases. For example, the cascade of the blood-clotting enzymes (4) involves the conversion of inactive forms (e.g., prothrombin) to active forms of the enzyme (thrombin) by a highly specific catalytic cleavage by another of the clotting enzymes (factor Xa). On the other hand, simply changing the pH of the solution in which the gastric aspartic protease zymogens are dissolved from ≈6.5 to 3.0 (an increase in [H+] of ≈3,100-fold) is sufficient to bring about the conversion (5). In a similar fashion, the zymogens of the papain-like cysteine proteases are converted to the active enzymes in a pH-regulated fashion. The in vitro activation of propapain is consistent with an initial intramolecular cleavage event (6). The conversion of procarboxypeptidase is initiated by trypsin cleavage of the Arg-99p-Ala-1 bond at the prosegment to mature enzyme junction (7). Prostromelysin-1 can be converted to the active form by other proteolytic enzymes, heat, or the presence of organomercurial agents (8).
There are some generalities regarding zymogen conversion that one can make in light of the three-dimensional structures of both the zymogens and the respective active enzymes (3). First, the residues that constitute the active sites of the protease portions of the zymogens have virtually identical conformations to those of the mature, active proteases. The major exceptions are the serine proteases of the chymotrypsin family. The activation process involves the formation of an ion pair between the newly formed N-terminal residue Ile-16 NH3+ and the β-carboxylate of Asp-194 (9), which triggers the conformational changes that form the oxyanion binding pocket and the active conformation of the S1 specificity pocket [the nomenclature of Schechter and Berger (10) is used throughout this manuscript].
Second, the preformed active sites of the protease portions of zymogens are generally not accessible to substrates because residues of the prosegments sterically block the approach to the active sites. This statement does not hold for the chymotrypsin-like serine proteases as the active sites of these zymogens are able to bind protein inhibitors that induce conformational changes that form the oxyanion hole in spite of the ion pair involving Asp-194 and Ile-16 being absent (11).
Proteolysis of the portion of the prosegments that interact with the active site residues is prevented in several different ways. In prostromelysin the prosegment passes through the active site in the reverse polypeptide direction (N→C) relative to substrates or transition state mimics (12). A reverse orientation of the prosegment blocking the active site in the cysteine protease zymogens also has been observed in the structures of rat procathepsin B (13) and human procathepsin L (14). The region of the prosegment of the gastric aspartic proteases interacting with the catalytic residues Asp-32 and Asp-215 (porcine pepsin numbering) most intimately, includes a highly conserved lysine at position 36p (the residue numbers of the prosegment are followed by p). The εNH3+ group of Lys-36p forms an ion pair with each of the two active site carboxylate groups (15).
Conversion of Gastric Aspartic Protease Zymogens
The molecular structures of human progastricsin (16), activation intermediate 2 of human gastricsin (17), and a structural
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviation: HPV, human polio virus. |
|
* |
Present address: Department of Molecular and Cellular Biology, Harvard University, 7 Divinity Avenue, Cambridge, MA 02138. |
|
† |
To whom reprint requests should be addressed. E-mail: michael.james@ualberta.ca. |
homolog to mature, human gastricsin, human pepsin 3 (18) allow one to construct a reasonably detailed view of the pathway followed in the conversion of the inactive zymogen to the active protease. Fig. 1 shows stereo ribbon diagrams of each of these three molecular structures. Fig. 2 shows a diagrammatic view of the conversion pathway. This pathway is a general pathway for the gastric aspartic proteases but the individual enzymes differ in detail.
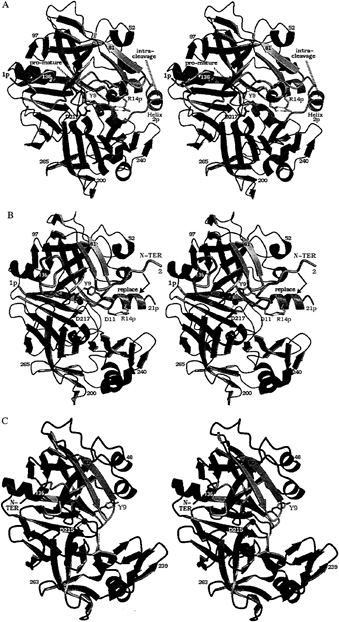
FIG. 1. Structures on the conversion pathway of the aspartic protease zymogen progastricsin. The structure of human gastricsin is not known; the human pepsin structure therefore has been used as a model for gastricsin. This figure, as well as Figs. 3–6 have been prepared with BOBSCRIPT (19) and RASTER 3D (20). (A) The structure of human progastricsin (16) represented in stereo. The residues of the prosegment (Ala-1p to Leu-43p) are in green, those of the gastricsin portion of the zymogen are in blue except for those regions that undergo large conformational changes, Ser-1 to Ala-13, Phe-71 to Thr-81 and Tyr-125 to Ala-136, which are represented in mauve. The promature junction is Leu-43p-Ser-1, the peptide bond cleaved intramolecularly is Phe-26p to Leu-27p. The side chains of Asp-32 and Asp-217 are represented in red. (B) Stereo view of the molecular structure of intermediate 2 on the activation pathway of human gastricsin (17). The color scheme used is the same as in A. The residues missing on this figure, Leu-22p to Phe-26p and Ser-1, are disordered in the structure, and there is no interpret able electron density for them on the maps. The water molecule bound between the two carboxyl groups of Asp-32 and Asp-217 is shown as a red sphere. The final step in the conversion involves the dissociation of the peptide Ala-1p to Phe-26p from gastricsin with the N-terminal residues of gastricsin, Ser-1 (N-ter) to Ala-13, replacing the N-terminal ß-strand of the prosegment. (C) The structure of human pepsin (18) shown as a model of human gastricsin. The regions of gastricsin that undergo large conformational changes from their positions in progastricsin are shown in pink, and the active site aspartates with the bound catalytic H2O molecule are colored red. Reproduced with permission from ref. 3.
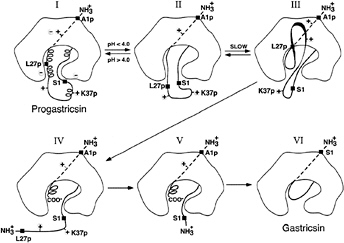
FIG. 2. A diagrammatic representation of the conversion pathway of progastricsin to gastricsin. The prosegment of progastricsin (I), A1p to L43p, has three helical segments and a net positive charge forming several ion pair interactions and electrostatic stabilization with the mature portion of the zymogen. The highly conserved K37p interacts directly with the catalytic aspartates Asp-32 and Asp-217. Lowering the pH of the solution below 4.0 converts progastricsin into intermediate 1 (26) represented by II. Refolding of the prosegment in the vicinity of the active gastricsin brings the first scissile bond Phe-26p-Leu-27p to the exposed active site aspartates (III). Cleavage at the premature junction, Leu-43p-Ser-1, (IV) is likely an intermolecular cleavage (28) and results in intermediate 2 (V), a molecular species that consists of Ala-1p to Phe-26p noncovalently associated with mature gastricsin (17). The final step in the conversion results from the dissociation of the N-terminal peptide 1–26 of the prosegment and refolding the residues Ser-1 to Ala-13 to replace the region of the prosegment in the six-stranded ß-sheet of gastricsin (VI).
Progastricsin consists of a single polypeptide chain of 372 aa (21). The N-terminal extension or prosegment is 43 aa in length and comprises residues Ala-1p to Leu-43p. The prosegment is folded into a compact domain having an initial extended ß-strand (Val-3p to Lys-8p) followed by three helical segments: Ile-13p to Lys-20p, Leu-22p to Arg-28p, Pro-34p to Arg-39p (16). The third helical segment (a 310 helix) packs against the active site residues and the eNH3+ group of a conserved lysine residue (Lys-37p in progastricsin) forms ion pair interactions with the carboxyl groups of the two catalytic aspartates, Asp-32 and Asp-217. Two tyrosine side chains Tyr-38p and Tyr-9 form symmetric H-bonded interactions with the carboxylates of Asp-217 and Asp-32, respectively, further restricting access to the active site. The phenolic side chain of Tyr-9 occupies the S1 binding pocket; Tyr-38p is in the S1' binding pocket. The tertiary structure of the prosegment (Leu-1p to Tyr-37p) in porcine pepsinogen (15) is virtually identical to that described above for progastricsin (16).
The polypeptide chain from Tyr-38p to Tyr-9 in progastricsin adopts a conformation that is different from the equivalent segment of chain in the pepsinogens (15, 22). As well, a portion of the polypeptide chain in gastricsin Tyr-125 to Ala-136 (Fig. 1A) is displaced from the position that this chain segment occupies in all other aspartic protease zymogens and active enzymes (16).
The trigger for initiating the conversion of the gastric aspartic protease zymogens is a drop in pH (5). At neutral pH, the structures of the zymogens are stabilized by the electrostatic interactions of the ion pairs and the inactive conformation is maintained (16). However, when the zymogens reach the acid pH (˜2.0) of the lumen of the stomach, the carboxylate groups become protonated and the repulsive interactions among the net positive charges of the prosegment destabilize its interactions in the active site of the protease. Kinetic studies in the late 1930s showed that the conversion of porcine pepsinogen into pepsin was an autocatalytic process (5, 23). In addition, the fact that the loss of pepsinogen was not accompanied by an equivalent increase in the appearance of pepsin implied the presence of intermediate species on the pathway (5). Spectroscopic studies of this conversion process established that there are conformational changes (24) in the 5-ms to 2-s time scale (25). Rapidly changing the pH back to neutrality can reverse these conformational changes.
Biochemical studies of the conversion of human progastricsin to gastricsin showed the presence of at least two intermediates (26). Intermediate 1 is the species formed rapidly after the pH was dropped below 4.0. The prosegment is unfolded in intermediate 1 and the active site of gastricsin is exposed and accessible to substrates. The first hydrolytic event detected during the activation of progastricsin is the intramolecular cleavage of the Phe-26p to Leu-27p peptide bond (26). Subsequently, an intermolecular cleavage at the Leu-43p-Ser-1 peptide bond (the promature junction) results in the formation of transient intermediate 2 that can be stabilized by transferring the pH to neutrality (>6.5). The resulting molecular species has been characterized biochemically (26) and comprises residues Ala-1p to Phe-26p noncovalently associated with mature gastricsin (Ser-1 to Ala-329).
Intermediate 2 recently has been characterized structurally (17), and its structure is depicted in Fig. 1B. The ß-strand (Val-3p to Lys-8p) is in the same position as observed in the structure of progastricsin. In addition, the first helix (Ile-13p to Lys-20p) is intact and is very similarly oriented as it is in the zymogen structure. The two catalytic aspartates, Asp-32 and Asp-217, have a water molecule bound between them in the same position as the nucleophilic water observed in the native structures of all mature aspartic proteases (27). The S1 binding site is occluded, however, as the side chain of Tyr-9 still forms a hydrogen bond with the carboxylate of Asp-32. The segment Tyr-125 to Ala-136 has moved from its position in progastricsin
(Fig. 1A) to the position and conformation common among the mature enzymes whose structures have been solved (Fig. 1 B and C).
The ion pair Arg-14p to Asp-11 in intermediate 2 at pH ˜6.5 stabilizes the N-terminal peptide, Ala-1p to Phe-26p, in its original location in the zymogen preventing the N-terminal residues of gastricsin (Ser-1 to Ala-12) from adopting their final position in the mature enzyme. On the other hand, intermediate 2 would be relatively short-lived at acid pH values <3.0. At these low pH values the carboxylate of Asp-11 would be protonated, and therefore the ion pair with Arg-14p would be severely weakened. Prolonged exposure of intermediate 2 to an acid environment favors dissociation of the ß-strand of the zymogen and its replacement by the N terminus of mature gastricsin (Fig. 1C).
Progastricsin (Fig. 2I) has stabilizing electrostatic interactions between the positively charged residues of the prosegment and the negatively charged groups of the gastricsin portion of the zymogen (16). These electrostatic interactions are weakened by the drop in pH that results in the protonation of the carboxylate groups of aspartic and glutamic residues (29–31). In particular, protonation of the catalytic aspartate groups, Asp-32 and Asp-217, weakens the interactions with Lys-37p, allowing the uncoiling of the 310 helix and permitting the diffusion of this segment away from the active site. The residues surrounding Lys-37p in progastricsin (Thr-29p to Asp-33p and Phe-40p to Leu-43p) all have substantially higher than average B factors, indicating that they are highly mobile and would easily undergo conformational changes that would expose the preformed active site (Fig. 2II).
In contrast, the ß-strand at the N terminus of the prosegment (Val-3p to Lys-8p) associates with the mature gastricsin through hydrogen bonding and hydrophobic interactions. These forces are not pH dependent. With the helical regions of the prosegment uncoiled (Fig. 2II) and the polypeptide from roughly Lys-11p to Ala-13 in a dynamic state of flux, eventually a sensitive peptide (e.g., Phe-26p-Leu-27p) would diffuse to the preformed active site and intramolecular cleavage would occur (Fig. 2III). The resulting cleaved form of the zymogen (Fig. 2IV) is enzymatically active and also would be free to catalyze intermolecular cleavages that have been detected kinetically with pepsinogen (32). This is the likely fate of the bond at the prosegment to mature junction (Leu-43p-Ser-1); it is cleaved intermolecularly and the peptide Leu-27p to Leu-43p dissociates from the complex. The noncovalent complex of Ala-1p to Phe-26p bound to gastricsin (intermediate 2 or Fig. 2V) can be stabilized by returning the pH to neutrality. The final step (Fig. 2 V to VI) in the conversion process involves a dissociation of the ß-strand and helical regions (Ala-1p to Phe-26p) of the prosegment from gastricsin and its replacement by the N-terminal residues of gastricsin.
The prosegments of the pepsinogens and the progastricsins have very similar sequences and three-dimensional structures. The sequences of prosegments of other aspartic proteases are also similar to those of the gastric enzymes, suggesting that the general features of the conversion process are shared among the chymosins and cathepsins D. Differences in the sites of internal cleavage and the kinetics of the activation process (33) are explained partly by the positions of the cleavage sites in the different prosegments (34).
Conversion of Proplasmepsin II
The plasmepsin system presents a different view of aspartic protease activation than do the gastric proteases. Plasmepsin is the aspartic protease used by the malaria parasite Plasmodium to degrade hemoglobin in red blood cells. The plasmepsins are synthesized as inactive zymogens, the proplasmepsins, having N-terminal prosegments that differ both in sequence and in size from other known aspartic protease zymogens. Proplasmepsin prosegments contain approximately 125 aa and lack sequence similarity with the archetypal gastric zymogen prosegments, which are typically about 45 residues long (35–37). Proplasmepsin prosegments also contain a transmembrane helix that anchors these zymogens to the membrane during delivery from the endoplasmic reticulum to the digestive vacuole where activation and hemoglobin digestion occur (38). Activation of proplasmepsin in vivo is carried out by a maturase at acidic pH (38). Additionally, proplasmepsin II and P. vivax proplasmepsin can be activated autocatalytically at low pH, with the cleavage occurring upstream of the wild-type mature N terminus (39).
The crystal structure of proplasmepsin II from P. falciparum revealed some surprising contrasts with the gastric aspartic protease zymogens (40). Instead of blocking a preformed active site, as in the gastric zymogens, the prosegment in proplasmepsin causes a major distortion of the molecule, preventing the formation of a functional active site.
The recombinant proplasmepsin II used in the crystallographic studies had the prosegment truncated by the first 76 residues to facilitate expression (39). Almost the entire length of this shortened prosegment interacts with the mature portion of proplasmepsin II. The prosegment has a well-defined secondary structure, consisting of an initial ß-strand, followed by two a-helices and a coil connection to the mature N terminus (Fig. 3). As in the gastric zymogens (15, 16, 22), the prosegment ß-strand participates in the six-stranded ß-sheet, the central motif of aspartic proteases, and becomes replaced by the mature N terminus upon activation (Fig. 3). Although the position of the prosegment ß-strand is similar to that seen in gastric zymogens, the remainder of the prosegment adopts a very different disposition. Instead of running through the substrate-binding cleft, the two helices interact exclusively with the C domain of the molecule. The promature junction is located in a tight loop comprised of residues Tyr-122p to Asp-4, the Tyr-Asp loop, where Asp-4 plays a key role in maintaining the structure of the loop (with hydrogen bonds to Tyr-122p and Ser-1) and anchoring it to the C domain (with hydrogen bonds to Lys-238 and Phe-241) (Fig. 4a).
The N terminus of the mature plasmepsin sequence differs in conformation between proplasmepsin II and plasmepsin II (40, 41). Upon activation, residues 1–14 undergo a large conformational change, placing Asp-4 to Phe-11 into the central ß-sheet. Residues 15–29 make a more subtle rearrangement that alters their interactions with the active site Psi loops (Fig. 4 b and c). When the central ß-sheet motif and C domain (residues 138–329) of plasmepsin II and proplasmepsin II are superimposed, their N domains (residues 30–129) are related by a rotation of 14° about an axis running roughly in the plane of the central ß-sheet and perpendicular to the strands. This domain movement observed in proplasmepsin II is novel in terms of its division into rigid bodies, magnitude, direction, and effect on activity (42). It renders the active site cleft more open in the zymogen than in the enzyme, severely distorting the geometry from that of the active site in plasmepsin II. In proplasmepsin II, the active site Psi loops are farther apart relative to plasmepsin II (Fig. 4b). Asp-34 and Asp-214 are too far apart in the zymogen to carry out the general base activation of a nucleophilic water molecule. The so-called immature active site is therefore catalytically inactive, and upon activation must collapse to the fireman’s grip configuration that defines the active site in all aspartic proteases of known structure (43) (Fig. 4c).
The method of inactivation in proplasmepsin II is different from that observed in the gastric aspartic protease zymogens. In proplasmepsin there is no positively charged moiety (such as Lys-36p in pepsinogen) to neutralize the charge repulsion between the catalytic Asps at neutral pH (44). Instead, the two Asp residues are kept apart from each other and are engaged in a network of hydrogen bonds both within and between the
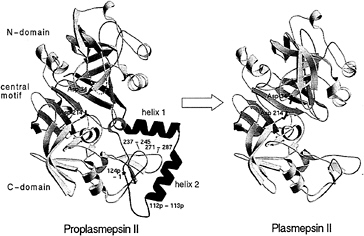
FIG. 3. A structural view of the conversion of proplasmepsin II (Left) (40) to plasmepsin II (Right) (41). The N and C domains are colored yellow, the central motif is green, the prosegment (with its helices labeled) is magenta, and the N-terminal 30 aa of the mature sequence are cyan. The catalytic aspartic acid residues (34 and 214) are colored red. The tip of the flap in proplasmepsin II, which is disordered in the crystal structure, is shown as a dotted line. The peptide bonds cleaved in autoactivation (112p–113p) and in the maturase-assisted activation (124p–1) are marked by asterisks in proplasmepsin II.
Psi loops (Fig. 4b). The function of the prosegment, together with the rearranged mature N terminus, is to maintain the molecule in the open conformation, leaving the active site accessible but greatly distorted.
The structure of proplasmepsin II suggests that disruption of three salt bridges (Glu-87p with Arg-92p, Asp-91p with His-164, and Glu-108p with Lys-107p) at low pH plays a key role in autoactivation. Dissociation of these interactions at low pH should destabilize the prosegment structure and weaken the association between the prosegment and the C domain. The most dramatic effect of acidification, however, should occur at Asp-4. This residue keeps the promature junction locked in the compact Tyr-Asp loop and tethers this loop to the C domain (Fig. 4a). Protonation of the Asp-4 side chain should disrupt the interactions of its carboxylate oxygens (with Tyr-122p, Lys-238, and Phe-241), opening up the Tyr-Asp loop and introducing a slack of five residues into the prosegment harness. With this region of the prosegment loosened, the molecule may adopt the domain-closed form with a functional active site. It should be noted that the bond cleaved in autoactivation of proplasmepsin II, Phe-112p to Leu-113p, is located at the C terminus of the prosegment helix 2, which must be one of the early locations to lose its secondary structure upon acidification. Once the active site is formed, the scissile bond, now in an extended conformation, then can be presented for cleavage either in cis or in trans.
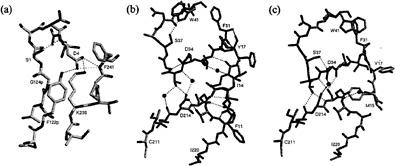
FIG. 4.(a) The promature junction in proplasmepsin II. The hydrogen bonding network of Asp-4, within the Tyr-Asp loop (Tyr-122p-Asp-4) and to the C-domain residues Lys-238 and Phe-241, is shown, (b) The immature active site in proplasmepsin II. The Psi loops (31–41 and 211–220) interact with each other through direct and water-mediated hydrogen bonds. In addition, both Psi loops form hydrogen bonds with the N-terminal residues 11–17. (c) The active site of plasmepsin II, showing the symmetrical arrangement of hydrogen bonds around the catalytic aspartates known as the fireman’s grip. The sphere between Asp-34 and Asp-214 is the oxygen atom from pepstatin that was present in the crystal structure (41). The N-terminal residues 15–18 and their interactions with the Psi loops also are shown.
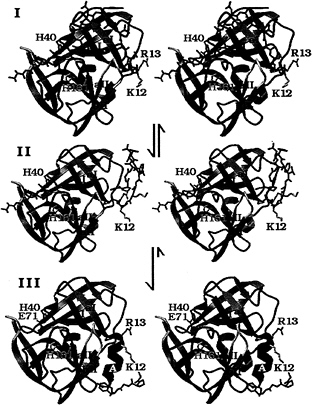
FIG. 5. A structural model for the N-terminal, autocatalytic excision of the enteroviral 3C proteases. The model is based on the crystal structure of the poliovirus 3C protease (HPV 3C) (63). The secondary structure of HPV 3C is shown in a ribbon representation. The N-terminal ß-barrel domain is blue and the C-terminal ß-barrel domain is mauve. (I) Model of the precursor of the 3C protease with the 3B|3C cleavage site sequence bound in the active site in the conformation of a cognate substrate of the 3C protease. The model was derived from the P5 to p2' residues of OMTKY3 bound in the active site of Streptomyces griseus protease B (SGPB) (65) after the optimal superposition of HPV 3C and SGPB (63). Included in this figure are the residues starting at the P5 (Thr) position of the 3B protein. The P5 to P1 residues and residues 1–5 of HPV 3C (P1' to P5') are colored gray; residues 6–13 are dark gray. The side chains of three active site residues, the nucleophile Cys-147 (yellow), the general acid-base catalyst His-40 (blue), and the S1 specificity determinant His 161 (light blue), are included. Residues 1–11 of HPV 3C reach into the active site of the protease and are in a mostly extended conformation. After the intramolecular cleavage the new N terminus Gly-1 dissociates from the active site while the P5 to P1 residues are still bound (II). Subsequently residues 6–13 of HPV 3C fold into a stable a-helix (colored black), which prevents the new N terminus from binding again to the active site and renders the conformational change irreversible. Arg-13 of the conserved sequence motif K/RR/KNL/I, which forms the last turn of the N-terminal helix in HPV 3C, anchors the N terminus to the structure. (III) The crystal structure of HPV 3C is shown with the N-terminal a-helix in black. The rearrangement of the N terminus in this model is accompanied by small conformational changes of ß-strands aI and bI of the N-terminal domain and the loop (yellow) that connects ß-strands aII and bII of the C-terminal domain.
Autoactivation of proplasmepsin II takes place readily between pH 3.8 and 4.7 (45). The lower pH range covers the pKas of Asp and Glu carboxylates in proteins (46), even taking into account some pKa depression that may be expected because of these residues’ participation in salt bridges and hydrogen bonds. For instance, the involvement of Asp-4 in a number of hydrogen bonds and a salt bridge (Fig. 4a) is likely to lower its side-chain pKa relative to that of a solvent-exposed carboxylate.
The requirement for low pH for activation by a maturase is less conclusive based on the proplasmepsin II structure. The promature junction is located at the surface of the molecule and therefore should be accessible to the external maturase. Acidification may be necessary to induce the Tyr-Asp loop opening for the Gly-124p to Ser-1 scissile bond to assume an extended conformation suitable for proteolytic cleavage. Alternatively, low pH may be required if the maturase itself has an acidic pH optimum. Further studies of the maturase will be needed to resolve this issue.
Autocatalytic Excision of Picornaviral 3C Proteases
Picornaviruses constitute a large family of positive-sense, single-stranded RNA viruses (47). An early and important step in the picornaviral lifecycle is the translation of the single-stranded viral RNA genome into a single large polyprotein (48,
49). The viral polyprotein is processed into the individual viral gene products by the viral 3C protease, itself a part of the polyprotein (50). The picornaviral 3C protease is the prototype of the new class of chymotrypsin-like cysteine proteases (49, 51, 52). It can cleave itself out of the viral polyprotein in cis and in trans, when it is expressed as part of the polyprotein or separately (53, 54).
The autocatalytic excision is not correlated with concomitant development of the proteolytic activity. The precursors of the 3C gene product already have proteolytic activity (55–58). In the viruses of the genus enterovirus the precursor 3CD (the 3D gene product constitutes the RNA-dependent RNA polymerase) shows a proteolytic activity that is distinct from that of 3C. In poliovirus, and presumably in most other enteroviruses, the proteolytic activity of the precursor 3CD is required for the efficient processing of the capsid precursor proteins (58). In hepatitis A virus the 3ABC gene product appears to be an important, proteolytically active intermediate of the polyprotein processing (57).
Palmenberg and Rueckert (59) examined the kinetics of the polyprotein processing in the picornavirus, encephalomyocarditis virus. Their data suggest that the autocatalytic excision of the 3C gene product can be a truly intramolecular event. Further evidence for an intramolecular excision of the 3C protease from poliovirus was provided by Hanecak et al. (60). Taken together, these data suggest that the autocatalytic excision of 3C from the polyprotein at both the N and C termini can be either intramolecular or intermolecular.
Once atomic resolution structures of 3C proteases became available (61–63), it was possible to develop structural models for the autocatalytic excision of the picornaviral 3C proteases. The crystal structures confirmed that the picornaviral 3C proteases are structurally related to the chymotrypsin family of serine proteases. Based on some of the unique structural details, the authors of the crystal structure papers (61–63) proposed similar models for an autocatalytic, intramolecular cleavage at the N terminus of 3C. They also agreed that it is much less obvious how an intramolecular cleavage could occur at the C terminus of 3C.
The N-terminal residues of the picornaviral 3C proteases form a stable a-helix that precedes the first strand of the N-terminal ß-barrel domain. This helix packs against the surface of the C-terminal domain of 3C. The last turn of this a-helix is formed by the residues of a highly conserved sequence motif K/RR/KNI/L (48).
Another unusual feature of the picornaviral 3C proteases is an antiparallel ß-ribbon that extends from the C-terminal ß-barrel (49). It forms an extension of the second and third ß-strands of the C-terminal domain and corresponds topologically to the methionine loop of the chymotrypsin-like serine proteases. This feature is also present in the bacterial serine proteases such as a-lytic protease and Streptomyces griseus protease B (64, 65). The recent crystal structure of a-lytic protease complexed with its prosegment (2) revealed that this feature plays an important role in the folding of the protease and in the autocatalytic, intramolecular processing of the precursor of a-lytic protease.
The structural model of an intramolecular cleavage at the N terminus of the picornaviral 3C proteases (Fig. 5) predicts that the N-terminal a-helix folds to its final conformation only after the 3C protease has cleaved its own N terminus (61–63). Before the intramolecular cleavage at the 3B|3C site, the corresponding residues [Gly-1 to Lys-12 in human polio virus (HPV) 3C] must be in an extended conformation (Fig. 5I) to reach into the active site through the cleft between ß-strand bI from the N-terminal domain and the loop connecting ß-strands aII and bII from the C-terminal domain. The loop that connects ß-strands aII and bII had to be moved in the model of the precursor molecule (Fig. 5I), with respect to its position in the native HPV 3C protease structure (Fig. 5III) to accommodate this. Several residues from ß-strand aI also are slightly moved away from their positions in the structure of the native 3C protease to widen the cleft between the N- and C-terminal domains through which the N terminus passes.
After the autocatalytic cleavage at Gly-1 of HPV 3C the new N terminus dissociates out of the active site (Fig. 5II). The folding of residues 5–13 into a stable helix, which packs tightly onto the surface of the molecule, subsequently would render this conformational change irreversible. It is necessary to remove the new N terminus from the protease active site to prevent intramolecular, competitive product inhibition of the protease.
The conserved sequence motif K/RR/KNI/L that eventually forms the last turn of the N-terminal helix anchors the residues of the N-terminal helix to the core structure of the protease. The side chains of Arg-13 and Asn-14 interact with the highly conserved sequence motif KFRDI of the RNA-binding site of the 3C protease. The residues that will become the N-terminal helix are in an extended conformation in the precursor (Fig. 5I). The up-down side-chain pattern in this extended conformation places the small side chains of Ala-7, Ala-9, and Ala-11 (P7', P9', and P11') into the cleft between the two domains of the proteases and the larger side chains of residues Tyr-6, Val-8, and Met-10 point to the surface. Larger side chains than alanine in positions 7, 9, and 11 would not have fitted easily into the surface of the cleft. We suggest therefore that the three alanine residues are important for the conformation of the N terminus in the precursor as well as for the formation of the N-terminal helix.
It is much more difficult to envision an intramolecular cleavage of the picornaviral 3C protease at its own C terminus. The crystal structure of the core proteins from Sindbis and Semlicki forest viruses (66) show how an additional ß-strand can reach from the C terminus to the active site of a chymotrypsin-like protease; however, the unique antiparallel ß-ribbon of the picornaviral 3C proteases that extends from ß-strands bII and cII and interacts with the N-terminal domain would prevent this (Fig. 5III).
We thank Perry d’Obrennan for help in making Fig. 2. Mae Wylie has been very helpful in getting the manuscript into its final polished form. A.R.K. was supported by a Medical Research Council of Canada Studentship; N.K.-B. was the holder of an Alberta Heritage Foundation for Medical Research Studentship. This work has been supported by the Medical Research Council of Canada and by Grant UO1AI38249 from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.
1. Neurath, H. (1957) in Advances in Protein Chemistry XII, eds. Anfinsen, C.B., Jr., Anson, M.L., Bailey, K. & Edsall, J.T. (Academic, New York), pp. 319–386.
2. Sauter, N.K., Mau, T., Rader, S.D. & Agard, D.A. (1998) Nat. Struct. Biol 5, 945–950.
3. Khan, A.R. & James, M.N.G. (1998) Protein Sci. 7, 815–836.
4. Davie, E.W., Fujikawa, K. & Kisiel, W. (1991) Biochemistry 30, 10363–10370.
5. Herriott, R.M. (1939) J. Gen. Physiol. 22, 65–78.
6. Vernet, T., Khouri, H.E., Laflamme, P., Tessier, D.C., GourSalin, B., Storer, A.C. & Thomas, D.Y. (1991) J. Biol. Chem. 266, 21451–21457.
7. Aviles, F.X., Vendrell, J., Guasch, A., Coll, M. & Huber, R. (1993) Eur. J. Biochem. 211, 381–389.
8. Nagase, H., Enghild, J.J., Suzuki, K. & Salvesen, G. (1990) Biochemistry 29, 5783–5789.
9. Huber, R. & Bode, W. (1978) Acc. Chem. Res. 11, 114–122.
10. Schechter, I. & Berger, A. (1967) Biochem. Biophys. Res. Commun. 27, 157–162.
11. Bode, W., Schwager, P. & Huber, R. (1978) J. Mol. Biol. 118, 99–112.
12. Becker, J.W., Marcy, A.I., Rokosz, L.L., Axel, M.G., Burbaum, J.J., Fitzgerald, P.M.D., Cameron, P.M., Esser, C.K., Hagmann, W.K., Hermies, J.D. & Springer, J.P. (1995) Protein Sci. 4, 1966–1976.
13. Turk, D., Podobnik, M., Kuhelj, R., Dolinar, M. & Turk, V. (1996) FEBS Lett. 384, 211–214.
14. Coulombe, R., Grochulski, P., Sivaraman, J., Menard, R., Mort, J.S. & Cygler, M. (1996) EMBO J. 15, 5492–5503.
15. James, M.N.G. & Sielecki, A.R. (1986) Nature (London) 319, 33–38.
16. Moore, S.A., Sielecki, A.R., Chernaia, M.M., Tarasova, N.I.& James, M.N.G. (1995) J. Mol. Biol 247, 466–485.
17. Khan, A.R., Cherney, M.M., Tarasova, N.I. & James, M.N.G. (1997) Nat. Struct. Biol 4, 1010–1015.
18. Fujinaga, M., Chernaia, M.M., Tarasova, N.I., Mosimann, S.C. & James, M.N.G. (1995) Protein Sci. 4, 960–972.
19. Esnouf, R.M. (1997) J. Mol Graphics 15, 133–138.
20. Merritt, E.A. & Murphy, M.E.P. (1994) Acta Crystallogr. D 50, 869–873.
21. Taggart, R.T., Cass, L.G., Mohandas, T.K, Derby, P., Barr, P.J., Pals, G. & Bell, G.I. (1989) J. Biol Chem. 264, 375–379.
22. Bateman, K.S., Cherney, M.M., Tarasova, N.I. & James, M.N.G. (1998) in The Aspartic Proteases: Retroviral and Cellular Enzymes, ed. James, M.N.G. (Plenum, New York), pp. 259–263.
23. Herriott, R.M. (1938) J. Gen. Physiol. 21, 501–540.
24. McPhie, P. (1972) J. Biol Chem. 247, 4277–4281.
25. Auer, H.E. & Glick, D.M. (1984) Biochemistry 23, 2735–2739.
26. Foltmann, B. & Jensen, A.L. (1982) Eur. J. Biochem. 128, 63–70.
27. Davies, D.R. (1990) Anna. Rev. Biophys. Chem. 19, 189–215.
28. Al-Janabi, J., Hartsuck, J. & Tang, J. (1971) J. Biol Chem. 247, 4628–4632.
29. Foltmann, B. (1981) Essays Biochem. 17, 52–84.
30. Perlmann, G.E. (1963) J. Mol Biol 6, 452–464.
31. Glick, D.M., Shalitin, Y. & Hitt, C.R. (1989) Biochemistry 28, 2626–2630.
32. Marciniszyn, J., Huang, J.S., Hartsuck, J.A. & Tang, J. (1976) J. Biol Chem. 251, 7095–7102.
33. Kageyama, T., Ichinose, M., Miki, K, Athauda, S.B., Tanji, M. & Takahashi, K. (1989) J. Biochem. (Tokyo) 105, 15–22.
34. Dunn, B. (1997) Nat. Struct. Biol 4, 969–972.
35. Dame, J.B., Reddy, R.G., Yowell, C.A., Dunn, B.M., Kay, J. & Berry, C. (1994) Mol Biochem. Parasitol. 64, 177–190.
36. Berry, C., Dame, J.B., Dunn, B.M. & Kay, J. (1995) in Aspartic Proteases: Structure, Function, Biology, and Biomedical Implications, ed. Takahashi, K. (Plenum, New York), pp. 511–518.
37. Francis, S.E., Gluzman, I.Y., Oksman, A., Knickerbocker, A., Mueller, R., Bryant, M.L., Sherman, D.R., Russell, D.G. & Goldberg, D.E. (1994) EMBO J. 13, 306–317.
38. Francis, S.E., Banerjee, R. & Goldberg, D.E. (1997) J. Biol Chem. 272, 14961–14968.
39. Hill, J., Tyas, L., Phylip, L., Kay, J., Dunn, B.M. & Berry, C. (1994) FEBS Lett. 352, 155–158.
40. Khazanovich Bernstein, N., Cherney, M.M., Loetscher, H., Ridley, R.G. & James, M.N.G. (1999) Nat. Struct. Biol 6, 32–37.
41. Silva, A.M., Lee, A.Y., Gulnik, S.V., Maier, P., Collins, J., Bhat, T.N., Collins, P.J, Cachau, R.E., Luker, K.E., Gluzman, I.Y., et al. (1996) Proc. Natl. Acad. Sci. USA 93, 10034–10039.
42. Sali, A., Veerapandian, B., Cooper, J.B., Moss, D.D., Hofmann, T. & Blundell, T.L. (1992) Proteins 12, 158–170.
43. Fusek, M. & Vetvicka, V. (1995) Aspartic Proteases: Physiology and Pathology (CRC, New York), pp. 22–24.
44. Sielecki, A.R., Fujinaga, M., Read, R.J. & James, M.N.G. (1991) J. Mol Biol 219, 671–692.
45. Moon, R.P., Bur, D., Loetscher, H., D’Arcy, A., Tyas, L., Oefner, C., Grueninger-Leitch, F., Mona, D., Rupp, K, Dorn, A., et al. (1997) Eur. J. Biochem. 244, 552–560.
46. Tanford, C. (1962) Adv. Protein Chem. 17, 69–165.
47. Rueckert, R.R. (1996) in Fields Virology, eds. Fields, B.N., Knipe, D.M., Howley, P.M., Channock, R.M., Melnick, J.L., Monath, T.P., Roizmann, B. & Straus, S.E. (Lippincott-Raven, Philadelphia), pp. 609–654.
48. Bergmann, E.M. & James, M.N.G. (1999) in Proteases of Infectious Agents, ed. Dunn, B. (Academic, San Diego), pp. 139–163.
49. Bergmann, E.M. & James, M.N.G. (1999) in Handbook of Experimental Pharmacology, eds. von der Helm, K. & Korant, B. (Springer, Heidelberg), in press.
50. Palmenberg, A.C. (1990) Annu. Rev. Microbiol. 44, 602–623.
51. Gorbalenya, A.E. & Snijder, E.J. (1996) Perspect. Drug Discovery Des. 6, 64–86.
52. Ryan, M.D. & Flint, M. (1997) J. Gen. Virol. 78, 699–723.
53. Harmon, S.A., Updike, W., Jia, X.-Y., Summers, D.F. & Ehrenfeld, E. (1992) J. Virol. 66, 5242–5247.
54. Richards, O.C., Ivanoff, L.A., Bienkowska-Szewczyk, K., Butt, B., Petteway, S.R., Jr., Rothstein, M.A. & Ehrenfeld, E. (1987) Virology 161, 348–356.
55. Davis, G.J., Wang, Q.M., Cox, G.A., Johnson, R.B., Wakulchik, M., Datson, C.A. & Villarreal, E.C. (1997) Arch. Biochem. Biophys. 346, 125–130.
56. Jürgensen, D., Kusov, Y.Y., Facke, M., Kräusslich, H.G. & Gauss-Müller, V. (1993) J. Gen. Virol. 74, 677–683.
57. Probst, C., Jecht, M. & Gauss-Müller, V. (1998) J. Virol. 72, 8013–8020.
58. Ypma-Wong, M.F., Dewalt, P.G., Johnson, V.H., Lamb, J.G. & Semler, B.L. (1988) Virology 166, 265–270.
59. Palmenberg, A.C. & Rueckert, R.R. (1982) J. Virol. 41, 244–249.
60. Hanecak, R., Semler, B.L., Ariga, H., Anderson, C.W. & Wimmer, E. (1984) Cell 37, 1063–1073.
61. Bergmann, E.M., Mosimann, S.C., Chernaia, M.M., Malcolm, B.A. & James, M.N.G. (1997) J. Virol. 71, 2436–2448.
62. Matthews, D.A., Smith, W.W., Ferre, R.A., Condon, B., Budahazi, G., Sisson, W., Villafranca, J.E., Janson, C.A., McElroy, H.E., Gribskov, C.L. & Worland, S. (1994) Cell 77, 761–771.
63. Mosimann, S.C., Chernaia, M.M., Sia, S., Plotch, S. & James, M.N.G. (1997) J. Mol Biol 273, 1032–1047.
64. Fujinaga, M., Delbaere, L.T.J., Brayer, G.D. & James, M.N.G. (1985) J. Mol Biol 184, 479–502.
65. Huang, K, Lu, W., Anderson, S., Laskowski, M., Jr. & James, M.N.G. (1995) Protein Sci. 4, 1985–1997.
66. Tong, L., Wengler, G. & Rossmann, M.G. (1993) J. Mol Biol 230, 228–247.








