
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
The catalytic sites of 20S proteasomes and their role in subunit maturation: A mutational and crystallographic study
MICHAEL GROLL*, WOLFGANG HEINEMEYERI†, SIBYLLE JÄGER†, TOBIAS ULLRICH*, MATTHIAS BOCHTLER*, DIETER H. WOLF†, AND ROBERT HUBER*‡
*Max-Planck-Institut für Biochemie, D-82152 Martinsried, Germany; and †Institut für Biochemie, Universität Stuttgart, D-70569 Stuttgart, Germany
ABSTRACT We present a biochemical and crystallographic characterization of active site mutants of the yeast 20S proteasome with the aim to characterize substrate cleavage specificity, subunit intermediate processing, and maturation. β1(Pre3), β2(Pupl), and β5(Pre2) are responsible for the postacidic, tryptic, and chymotryptic activity, respectively. The maturation of active subunits is independent of the presence of other active subunits and occurs by intrasubunit autolysis. The propeptides of β6(Pre7) and β7(Pre4) are intermediately processed to their final forms by β2(Pup1) in the wild-type enzyme and by β5(Pre2) and β1(Pre3) in the β2(Pup1) inactive mutants. A role of the propeptide of β1(Pre3) is to prevent acetylation and thereby inactivation. A gallery of proteasome mutants that contain active site residues in the context of the inactive subunits β3(Pup3), β6(Pre7), and β7(Pre4) show that the presence of Gly-1, Thr1, Asp17, Lys33, Ser129, Asp166, and Ser169 is not sufficient to generate activity.
Proteasomes are essential, ubiquitous intracellular proteases that degrade a broad variety of cytoplasmic, nuclear, and membrane proteins that have been marked for degradation by the attachment of polyubiquitin chains (1–3). Eukaryotic proteasomes are large protein complexes with a molecular mass around 2,000 kDa, with a modular architecture (4, 5). The catalytic core of the molecule is the 20S proteasome, a cylindrical particle that consists of four heptameric rings made from seven different subunits each, which are present in two copies and in unique locations so that the particle has overall 2-fold symmetry (1, 4–7). The yeast 20S proteasome subunits fall into two different classes phylogenetically related to the two subunits α and β of the archaebacterial proteasome (8) and have been named accordingly (7). The α-subunits are not catalytically active and form antechambers to the central cavity of the 20S complex that is built from the β-subunits. In Thermoplasma acidophilum proteasomes,all β-subunits are transcribed and translated from one gene only and are expressed as precursors. In the process of particle maturation, aII copies of the β-subunit become active, so that two rings of seven catalytic sites each are formed on the inner walls of the central chamber. The N-terminal threonine residue is exposed by this processing activity as the nucleophile in peptide bond hydrolysis (9, 10). It will subsequently be referred to as Thr1, thus assigning negative integers to residues of the propeptide. Based on the crystal structure of the T.acidophilum 20S proteasome, the distance between active site threonines was suggested as the molecular ruler that determines the length distribution of proteasome generated peptides (9).
A more complex picture for the mechanism of oligopeptide product generation was suggested by the crystal structure of the yeast 20S proteasome (7). It contains seven different α- and β-type subunits arranged in unique locations (Fig. 1). Four β-type subunits are inactive because they contain either unprocessed [β3(Pup3) and β4(Pre1)] or intermediately processed propeptides [β6(Pre7) and β7(Pre4)]. The remaining three subunits β1(Pre3), β2(Pupl), and β5(Pre2) have N-terminal threonine residues, are active, and have specificities determined largely by the nature of their S1 pockets (7). Specific mutants of the active β-type subunits have been isolated (11). They allowed the identification of different substrate specificities (11, 12) of the proteasome and led to a hypothesis for an intermolecular processing mechanism of inactive β-subunits.
Functional and structural analysis of the mutant proteasomes allows us to investigate substrate specificities, catalytic and autolytic mechanisms, and intermediate processing of propeptides. They also provide hints to the role of propeptides in proteasome maturation and enzymatic activity and help to clarify the mechanism by which peptide product length is controlled. They provide critical tests of possible allosteric interactions in the proteasome. A number of mutants of inactive subunits was generated to define the roles of individual residues for inactivity with the ultimate goal to activate those subunits.
MATERIALS AND METHODS
Protein Preparation and Analysis. Yeast strains that express mutant proteasomes were generated as described (11). Cells were grown on a 51 scale, and the modified enzymes were purified as reported for the wild-type (7). 20S proteasomes were separated into subunits by reversed phase HPLC. One-hundred-microgram samples were loaded on a RP60 Supersphere column (Merck). The column was washed with a gradient from 0 to 30% acetonitrile in 0.1% trifluoroacetic acid. Single subunits were eluted in a gradient from 30 to 60% acetonitrile in 0.1% trifluoroacetic acid at a flow rate of 0.3 ml/min and at a back pressure of 140 bar (1 bar=100 kPa). Peaks were identified and propeptides characterized by N-terminal sequence analysis and mass spectrometry.
Crystals of 20S proteasome mutants from Saccharomyces cerevisiae were grown in hanging drops at 24°C as described (7). The crystals were frozen in a stream of cold nitrogen gas (90 K). Data were collected by using synchrotron radiation with λ=1.1 Å on the BW6 beamline at the Deutschen Elektronen-synchrotron Centre (Hamburg, Germany) (Table 3). The anisotropy of diffraction was corrected by an overall temper-
|
|
PNAS is available online at www.pnas.org. |
|
|
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1RYP). |
|
‡ |
To whom reprint requests should be addressed. E-mail: huber@biochem.mpg.de. |
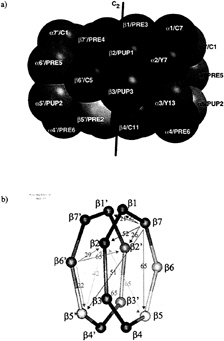
FIG. 1. (a) Topology of the yeast 20S proteasome. The active site threonine 1 residues are located at the inner wall of the cylindrical particle. (b) Scheme of the ß-rings with given distances between the active site threonines.
ature factor by comparing observed and calculated structure amplitudes by using X-PLOR (13). Electron density was averaged 10 times over the 2-fold noncrystallographic symmetry axis by using MAIN (14). Model building was carried out with FRODO (15).
RESULTS AND DISCUSSION
Subunit Processing. The topology of the yeast 20S proteasome is shown in Fig. 1a with the relevant distances between the active sites given in Fig. 1b. We have purified mutant yeast 20S proteasomes with a reduced number of active subunits carrying exchanges of Thr1 for Ala in ß1 and ß2. In ß5, Lys33 was exchanged for Ala or Arg because ß5T1A is not viable. Double mutants of ß1 and ß2 can be made. Some of these mutants show reduced growth (11), but 20S proteasomes can be isolated. We have characterized the ß-subunits chemically by Edman degradation and, in some cases, by mass spectrometry after separation of the individual subunits by HPLC (Tables 1 and 2).
The active subunits ß1, ß2, and ß5 are processed autocatalytically and independently of each other. Inactivating ß1 does not affect processing of ß2 and vice versa. Similarly, the mutation of ß5K33A and ß5K33R leads to inactivity of ß5 but has no effect on maturation of ß1 and ß2. This is consistent with earlier findings (16, 17), including pulse-chase experiments, which demonstrate that subunit maturation occurs late in proteasome assembly (11, 16, 18–20) after the formation of 15S–16S proteasome precursor particles. These particles are believed to be half proteasomes. As the active sites in 20S proteasomes are nearly 30 Å apart from each other, it appeared not possible that the Gly-1Thr1 cleavage occurs by a neighboring subunit.
The data on ß5 maturation are less straightforward to interpret. ß5K33R has very low enzymatic activity but is autoprocessed. ß5K33A is also inactive, but partially processed. We find clear electron density for the propeptide to residue Cys-8 in this mutant, but we can isolate by HPLC and mass spectrometry also the autoprocessed species (Table 2). An explanation might be an exceptional lability of the Gly-1Thr1 bond under the strongly acidic conditions of sample preparation for mass spectrometry.
The ß1 and ß2 T1A exchange in both the single and the double mutants leads to a failure in autoprocessing and to the presence of intact or intermediately processed propeptides of these subunits. In the ß1T1A ß2T1A double mutant, ß1 has its full length propeptide attached, and ß2 is -intermediately processed after Leu-15. ß7 is cleaved after Ile-19. In ß6, cleavage after Ala-17 and Thr-14 is found. Cleavage occurs after nonpolar residues, consistent with cleavage by ß5. Cleavage sites are at a sufficient distance from residue 1 to reach the remaining active centers of ß5 in the same ring for ß6 and ß7 and in the opposite ring for ß2 (Fig. 1b).
In the single ß1TlA-mutant, processing of ß6 and ß7 is as in the wild type, but ß1 is cleaved after Arg-10, obviously by ß2, whereas in ß2T1A the ß6 and ß7 propeptides are longer than in the wild type. Here, ß2 itself could not be characterized.
The inactive subunits ß6 and ß7 are intermediately processed by one of the active subunits. ß6 is adjacent to ß5 on the same ring and to ß2 on the opposite ring but further away from ß1 on both rings of the 20S proteasome (Fig. 1). The nine amino acid propeptide in the mature wild-type protein is too short to span the distance to either of the ß1 subunits. Experimentally, we find that inactivating ß1 in the ß1T1A-mutant, ß5 in the ß5K33A-mutant, and ß1 and ß5 in the ß1TIA ß5K33R mutant has no effect on the propeptide processing of ß6. In contrast, a significantly longer propeptide remains attached to ß6 in the ß2T1A-mutant. We conclude that ß6 is processed by ß2. Cleavage occurs after His-10 (Table 1), consistent with the trypsin-like activity of ß2. Because ß2 in the same ring is too far away to be reached by a nonapeptide Gln-9 to Gly-1, ß2 of the opposite ring must be the subunit that processes ß6.
In the case of ß7, the situation is similar, but the subunits ß5 and ß1 swap roles. ß7 is close to ß2 on the opposite ring and to the subunits ß1 on both rings. ß5 is too far away to be involved in the final maturation step. Experimentally, the wild-type propeptide of ß7 is found in the ß1T1A, in the ß5K33A mutant, and in the ß1T1Aß5K33R double mutant. In the ß2T1A mutant, the cleavage that occurs in the wild type is suppressed, identifying ß2 as the responsible subunit in the wild type. The cut occurs after Asn-9, a residue for which ß2 has some specificity (12). These data substantiate previous biochemical findings on ß7 maturation in the ß2T1A single and ß1T1A ß2T1A double mutant, which led to the hypothesis that inactive ß-subunits are processed by the closest active neighbor subunit (11).
The intermediately processed propeptides of ß6 and ß7 had been found in well defined locations in the molecular structure of the wild-type protein such that their N termini lie at the inner annulus of the ß-subunit rings far removed from the sites of proteolytic cleavage defined here (7). The same holds for the intermediately processed ß1 propeptide in ß1TIA and in ß1T1Aß5K33R. It has defined electron density to Leu-9, which also lies at the inner annulus, not far (16 Å) from ß6Gln-9 and ß7Thr-8. These observations indicate a major rearrangement of the propeptides after intermediate process-
Table 1. Yeast 20S proteasome mutants prepared and analyzed by N-terminal sequencing
|
|
ß1(Pre3) |
ß2(Pup1) |
ß3(Pup3) |
ß4(Pre1) |
ß5(Pre2) |
ß6(Pre7) |
ß7(Pre4) |
|
Wild type |
Gly-1 |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
|
Thr1 |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 ß2(Pup1)' |
Thr-8 ß2(Pup1)' |
|
ß1(Pre3) |
Acetyl |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
without propeptide |
(mass*) |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 ß2(Pup1)' |
Thr-8 ß2(Pup1)' |
|
ß1(Pre3) |
Arg-10 |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
T1A |
Leu-9 ß2(Pup1) |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 ß2(Pup1)' |
Thr-8 ß2(Pup1)' |
|
ß2(Pup1) |
Gly-1 |
|
Gly-1 |
Ala-17 |
Val-10 |
||
|
T1A |
Thr1 |
XXX |
Met-9 |
Met-1 |
Thr1 |
Ser-16 ß5(Pre2)' |
Asn-9 ß1(Pre3)' |
|
ß5(Pre2) |
Not viable |
Not viable |
Not viable |
Not viable |
Not viable |
Not viable |
Not viable |
|
T1A |
|
||||||
|
ß5(Pre2) |
Gly-1 |
Gly-1 |
|
His-10 |
Asn-9 |
||
|
K33A |
Thr1 |
Thr1 |
Met-9 |
Met-1 |
XXX (mass*) |
Gln-9 ß2(Pup1)' |
Thr-8 ß2(Pup1)' |
|
ß1(Pre3) |
|
Leu-15 |
|
Gly-1 |
Ala-17 |
Ile-19 |
|
|
T1A |
Met-19 |
Ala-14 |
Met-9 |
Met-1 |
Thr1 |
Ser-16 |
Ala-18 |
|
ß2(Pup1) |
|
ß5(Pre2); |
|
(mass*) |
ß5(Pre2); |
||
|
T1A |
ß5(Pre2)' |
ß5(Pre2); |
ß5(Pre2)' |
||||
|
ß1(Pre3) |
Arg-10 |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
T1A |
Leu-9 |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 |
Thr-8 |
|
ß5(Pre2) |
ß2(Pup1) |
|
ß2(Pup1)' |
ß2(Pup1)' |
|||
|
K33R |
|
||||||
|
ß2(Pup1) |
|||||||
|
T1A |
Not viable |
Not viable |
Not viable |
Not viable |
Not viable |
Not viable |
Not viable |
|
ß5(Pre2) |
|
||||||
|
K33R |
|||||||
|
ß3(Pup3) |
Gly-1 |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
G1T |
Thr1 |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 ß2(Pup1)' |
Thr-8 ß2(Pup1)' |
|
ß6(Pre7) |
Gly-1 |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
G1T/ |
Thr1 |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 |
Thr-8 |
|
A129S/ |
|
ß2(Pup1)' |
ß2(Pup1)' |
||||
|
A130G/ |
(mass*) |
|
|||||
|
H166D/ |
|
||||||
|
V169S |
|||||||
|
ß7(Pre4) |
Gly-1 |
Gly-1 |
|
Gly-1 |
His-10 |
Asn-9 |
|
|
R33K/ |
Thr1 |
Thr1 |
Met-9 |
Met-1 |
Thr1 |
Gln-9 |
Thr-8 |
|
F129S |
|
ß2(Pup1)' |
ß2(Pup1)' |
||||
|
P1 and P1' cleavage sites of the processed subunits are given, and responsible active subunits are indicated, (mass*), a hint for comparison with analysis by mass spectroscopy in Table 2. |
|||||||
ing and fixation at the final sites seen in the crystal structure. In ß1T1A ß2T1A, the full length propeptide of ß1 and the intermediately processed propeptides of ß2, ß6, and ß7 have well defined electron density up to residues Met-19 (ß1), Ala-14(ß2), Gln-9(ß6), and Thr-8(ß7), respectively.
Implications for Cleavage Specificity. Two ß subunits, ß3 and ß4, have propeptides of eight and one amino acids, respectively, which are too short to reach any catalytic site in the mature particle and are, indeed, not cleaved. The propeptides ofallother subunits are longer, and processing intermediates are observed. The discussed mutants are defective in some of the final maturation steps and show changes in the processing pattern. As shown above, most of the subunits responsible for these cleavages are defined and can be related to cleavage specificities.
In the ß1T1A-mutant, a nine-residue propeptide cleaved after Arg-10 is found, consistent with cleavage by ß2 in the same ring, according to its tryptic specificity and distance. Processing is completely suppressed in the ß1T1Aß2T1A double mutant, and the ß1Met-19 N terminus is observed. In the ß2TlA-mutant, autoactivation is suppressed, but the subunit could no longer be separated by HPLC. We were able to characterize the cleavage site of the propeptide of ß2 in the double mutant ß1T1A ß2T1A between Leu-15 and Ala-14. As the only active subunit left, ß5 must be responsible for this cut, assigning to it branched chain amino acid preferring (BrAAP) specificity, consistent with previous studies (12).
In the case of ß5, we have mutated Lys 33 to Ala and to Arg, abolishing activity. In the ß5K33A mutant, the resulting propeptide of ß5 is heterogeneous and could not be analyzed by Edman degradation. A fraction is found that is autolysed and has a Thr1 N terminus. In the x-ray structure, however, there is defined density to Cys-8, indicating, that the major proportion is not autolysed. However, ß5K33R is fully autolysed.
Table 2. Results of mass spectrometry of the subunits in the different yeast 20S proteasome mutants
ß6 and ß7 are processed to their final forms by ß2 of the opposite ring. Therefore, we have analyzed the ß2T1A-mutant for changes in the cleavage pattern of ß6 and ß7 propeptide. In ß6, the cut occurs between Ala-17 and Ser-16, as analyzed by Edman degradation of an HPLC fraction. In the ß1T1A ß2T1A double mutant, a component with cleavage between Thr-14 and Pro-13 is found by mass spectrometry. This bond must be hydrolyzed by ß5, assigning small neutral amino acid preferring (SNAAP) specificity to ß5.
In the ß2T1A-mutant, ß7 has one extra amino acid at the N terminus compared with the wild type. Inactivating ß1 in addition to ß2 shifts the cleavage further upstream to Ile-19 Ala-18. We conclude that ß1 and ß5 cleave after Val-10 and Ile-19, respectively, demonstrating BrAAP activity for both subunits, consistent with the apolar character of the P1 pocket of ß5. In the case of ß1, we assume that the positive charge of Arg 45 at the base of its P1 pocket is compensated by a bound bicarbonate anion to allow binding of neutral ligands, as had been observed before in the Leu-Leu-Norleucinal complex of the wild-type protein (7, 21).
The Role of the ß1 Propeptide. The propeptide of ß5 has been shown to be essential for cell viability but is functional when expressed in trans, suggesting a chaperone-like role in proteasome biogenesis (20). To investigate the role of the propeptide of ß1, we have replaced its propeptide with ubiquitin. As in other linear ubiquitin fusions (22, 23), ubiquitin is cleaved by ubiquitin C-terminal hydrolases (24) to liberate the N-terminal threonine. The mutant proteasomes were inactive when assayed for postacidic cleavage (PGPH) activity. Their ß1 subunit could be isolated by HPLC but was blocked for N-terminal sequencing. Structural analysis of the mutant proteasomes showed no significant differences to the wild-type structure except for extra density at the amino group of Thr1 that was interpreted as an acetyl group (Fig. 2) and confirmed by mass spectroscopy (Table 2). We conclude that the propeptide of ß1 has a role in preventing co- or posttranslational
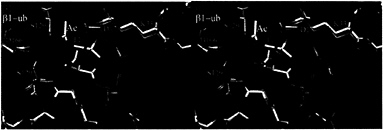
FIG. 2. Stereodiagram of the superposition of ß1 (green) and N-acetyl-ß1 (yellow) around the Thr1 site. The structures match closely.
Table 3. Crystallographic data of data collection and refinement
|
|
β2(Pup1) |
β5(Pre2) |
β1(Pre3) |
β1(Pre3) |
β3(Pup3) |
β6(Pre7) –5* |
β1(Pre3) without propeptide |
|
Space group |
P21 |
P21 |
P21 |
P21 |
P21 |
P21 |
P21 |
|
Cell |
a=136.7 |
α=135.6 |
α=135.4 |
α=135.5 |
α=135.5 |
α=135.7 |
α=135.9 |
|
constants (Å/°) |
b=300.6 |
β=300.3 |
β=302.5 |
β=300.7 |
β=301.2 |
β=300.3 |
β=301.6 |
|
|
c=145.2 |
γ=144.0 |
γ=145.5 |
γ=144.4 |
γ=146.5 |
γ=144.6 |
γ=144.5 |
|
β=113.1 |
β=113.0 |
β=112.6 |
β=112.9 |
β=112.8 |
β=113.2 |
β=112.7 |
|
|
Resolution, Å |
50–2.5 |
50–2.5 |
50–2.7 |
50–1.9 |
50–2.9 |
50–1.95 |
50–2.9 |
|
Observation, 2σ |
606959 |
951542 |
702094 |
2181093 |
631736 |
1755406 |
600449 |
|
Uniques |
289028 |
343517 |
270036 |
752101 |
225640 |
731544 |
218.345 |
|
Completeness |
88.3 |
93.3 |
93.8 |
92.9 |
94.5 |
91.1 |
94.2 |
|
Rmerge, % |
14.4 |
12.9 |
12.8 |
11.9 |
12.2 |
12.1 |
13.6 |
|
R/Rfree, % |
30.3/36.5 |
26.0/31.2 |
27.5/36.4 |
26.8/33.0 |
21.1/27.3 |
28.5/32.1 |
22.7/297 |
|
rms bonds, Å |
0.012 |
0.012 |
0.012 |
0.011 |
0.011 |
0.011 |
0.012 |
|
rms angles, ° |
2.0 |
1.9 |
1.84 |
1.8 |
1.85 |
1.933 |
1.89 |
acetylation and inactivation of this subunit. The lack of enzymatic activity of the N-acetyl-β1 mutant supports the proposed mechanism of catalysis (9) assigning to the amino group of site Thr1 the role of the proton acceptor, but steric hindrance of substrate docking by the acetyl group also may contribute to inactivity. It is noted that the acetyl group is not cleaved via of the conserved Lys33 is in maintaining the appropriate structure and electrostatic potential in the vicinity of the active autolysis, probably for steric and electronic reasons. The role (7, 21).
The Role of Lys33 in the Enzymatic Mechanism. The conservative exchange of Lys33 to arginine abolishes both autolysis and proteolysis in T.acidophilum proteasomes (25). We were particularly interested in this mutation because
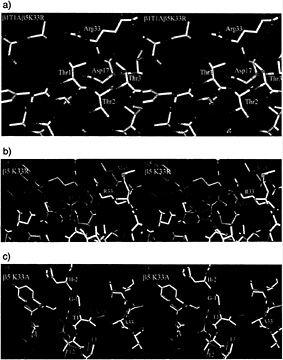
FIG. 3. (a) Stereodiagram of the β1T1A β5K33R double mutant in the vicinity of residue Thr1 in β5. The electron density is calculated with phases from the wild-type β5 model, (b) Stereodiagram of wild-type (green) and β5K33R (white) mutant around Thr1. They superimpose closely except for the site of mutation. β5K33R autolyses and has a free Thr1. (c) Comparison of the wild-type (green) and β5K33A (white) mutant. Loss of the Lys33 side chain leads to a large movement of the backbone of Thr1. The mutant is unable to autolyse and has the propeptide attached.

FIG. 4. A gallery of superposition of main chain traces around Thr 1. a and b show the three active subunits ß1, ß2 and ß5. In c and d, ß1 is compared with wild-type ß3 and ß3G1T, respectively, in e and f, ß1 is superimposed with wild-type ß6 and the 5-fold ß6 mutant (ß6*), and, in g and h, ß1 is compared with ß7 and ß4.
arginine in position 33 occurs naturally in the subunit ß7 of the yeast proteasome, where it displaces the Thr1 side chain, leading to incompetence in autolysis and to enzymatic inactivity (7). We exchanged Lys33 in ß5 of the yeast proteasome with arginine. Crystals that diffract to 1.9-Å resolution (Table 3) could be obtained with a double mutant, which additionally has Ala exchanged for Thr1 in subunit ß1. In contrast to wild-type ß7 and the quintuple mutant of ß6 (see below), aII residues in the vicinity of the active site of ß5, including Thr1, remain in their wild-type positions. The arginine residue has its side chain in the same orientation as the lysine residue, but its guanidino group is tilted with respect to the position of the amino group in the lysine residue to avoid a clash with Thr1 (Fig. 3 a and b). As in T.acidophilum proteasomes, the chymotryptic activity of this mutant against chromogenic substrates is abolished. However, in contrast to results obtained for the T.acidophilum proteasome, the propeptide in the yeast mutant is cleaved. We attribute this observation to a weak residual activity that suffices for autolysis during particle maturation. The mutant grows slowly at 30°C but not at 37°C (11), and it overexpresses 20S proteasomes. The phenotype could be attributable either to the lack of chymotryptic activity or to delayed or impaired proteasome maturation. Genetic studies favor the latter explanation (11, 20). Because autolysis still occurs in the ß5K33R mutant, we analyzed the ß5 mutant carrying the Lys33Ala mutation. The mutant strain was viable, although again it grew slowly and contained unusually high amounts of 20S proteasome. As expected, both autolysis and proteolysis did no longer occur. The 2.5-Å crystal structure of this mutant shows defined density for the propeptide and a major rearrangement of the position of Thr1 that fills the cavity created by the loss of the lysine residue and displaces Met45 (Fig. 3c). Mass spectrometry of a fraction separated by HPLC, however, showed also the presence of some correctly processed species (Table 2).
Some Structural and Functional Comparisons. A comparison of the refined molecular models of the mutants ß1T1A, ß2T1A, and ß1TIA ß2T1A showed no significant variation of subunit positions or backbone structures. The activity against chromogenic substrates of a particular subunit is insignificantly altered by the presence or absence of intact sites of other subunits. We had previously shown that the covalent binding of a specific bound irreversible inhibitor of ß2 has no significant influence on the PGPH and chymotryptic activity associated with ß1 and ß5 and does not show noticeable structural changes (26). Similarly, there is no measurable change in the activity and structure of ß1 and ß5 by strong binding of bifunctional reversible inhibitors to ß2 (27). Also, yeast 20S

FIG. 5. Stereo diagram of the 5-fold ß6 mutant in the vicinity of residue 1. The electron density is calculated with phases from the wild-type ß6 model (black bonds). The mutations A129S, G1T, and the rearrangement of K33 are clearly visible (red bonds).
proteasome with lactacystin bound to ß5 shows no structural change compared with the unligated species (7). These results do not support the existence of allosteric interactions between the active sites in general and argue against interactions mediated by conformational equilibria in particular. We are aware, however, that crystal lattice forces may oppose ligand-induced conformational changes occurring in solution.
Reactivation Studies. All proteasomal ß-subunits are members of a family of proteins having diverged from a single ancestor possibly similar to the archaebacterial ß subunit. Nevertheless, only three subunits, ß1, ß2, and ß5, are proteolytically active in yeast and higher eukaryotes The other ß-subunits, ß3, ß4, ß6, and ß7, are inactive and unable to autolyse. ß3, ß4, and ß6 lack the nucleophilic threonine in position 1, and ß7 has Arg33 and Phe 129 instead of Lys33 and Ser129, respectively, as the most conspicuous changes.
The conservation of backbone geometry and of the majority of residues making up the active site also in inactive proteasome ß-subunits has prompted us to investigate the possibility of reactivating inactive subunits. We first chose the inactive subunits ß3 and ß6 as promising targets for subunit activation experiments because of the close similarity of their backbone fold with the active subunits ß1, ß2, and ß5 (Fig. 4 a-c and e).
ß3. Gly1 replaces the canonical threonine in ß3 as the most conspicuous exchange. It was mutated to threonine. The resultant yeast strain is viable and does not show a growth phenotype. Purified proteasomes from this strain show a blocked N terminus as the wild type. An antibody was raised against ß3, and the migration of the mutant and of the wild-type subunit on denaturing SDS gels was compared. No difference could be observed, implying that the propeptide was not cleaved and the subunit remains inactive. Mass spectrometry confirms these results (Table 2). Additionally, we determined the crystal structure of this mutant, which, when compared with the wild-type ß3-subunit, does not show major rearrangements and confirms that the propeptide is attached (Fig. 4 c and d).
ß6. We repeated the experiment in an analogous manner with ß6. Although this subunit has a severely impaired catalytic machinery with Gly1, Ala129, His166, and Val169 instead of the cannonical Thr1, Ser129, Asp166, and Ser169, its backbone superimposes well with those of the active subunits, and the position of Lys33 is identical (Fig. 4e). Gly1 is shifted slightly toward Lys33 compared with the active subunits. We have replaced Gly1, Ala129, His166, and Val169 by their equivalents in active subunits. In addition, we exchanged Ala130 with glycine because this residue is conserved inallthree active subunits, although its role in catalysis is not obvious. The 5-fold mutant is again viable, but it has a severe growth defect. In comparison with the wild type, cells are up to 10×larger and express severalfold more proteasome, which could be purified and crystallized. The crystal structure analysis at 1.95-Å resolution shows defined electron density at ß6 for all nine residues of the partially processed propeptide, but it is substantially lower than in the wild type and particularly blurred at residues Asn-2 and Gly-1. Temperature factors of the propeptide are very high. Also, the mass spectrum of the corresponding HPLC fraction showed the molecular weight of the Gln-9 species, but a component with the molecular weight, corresponding to the autolysed species, also occurs. We conclude that the mutant protein is partially autolysed. Residues Asp17, Ser129, Asp166, and Ser169 of the mutant subunit ß6 are positioned as the corresponding residues in active wildtype subunit, but Thr1 remains where Gly1 in the wild-type ß6 subunit is. A close contact between the Thr1 and Lys33 side chains displaces the lysine side chain into an outwardly oriented position, where it is stabilized by hydrogen bonds to Glu31 and Asp53 (Figs. 4f and 5). The distortion of Thr1 with respect to its position in active subunits prevents the binding of a water molecule in the vicinity of Thr1, as seen in active subunits (7). As the phenotype of the mutant is unlikely be accounted for by an extra proteasomal activity, we have looked for other explanations. The major activities of wild-type proteasomes are present but somewhat reduced. Therefore, we suspect a decreased stability of the quintuple mutant. ß6 is in contact with ß5 and ß7 in the same ring and ß2 and ß3 of the opposite ring. His166 and Val169 contact ß5, ß2, and ß3 whereas Ala129 and Ala130 contact ß7. As seen from the lack of a phenotype of the triple mutant, ß6G1T A129S A130G, which presumably has a displaced lysine residue and impaired contacts with ß7, and from the lack of a phenotype of the quadruple mutant ß6A129S A130G H166D V169S, individual
residue effects count to be weak. Only in the quintuple mutant, where contacts of ß6 with all neighboring ß-subunits are disturbed, is a notable phenotype seen.
ß7. Based on our observation that the displacement of the mutationally introduced Thr1 with respect to its position in active subunits could explain the failure to activate ß3 and ß6, and based on the perfect match of the polypeptide backbone around Thr1 of ß7 and of the active subunits (Fig. 4g), we then attempted to activate ß7. Two residues have to be replaced, Arg33 and Phe129. The resulting yeast strain was viable and indistinguishable from the wild-type. N-terminal sequencing of ß7 revealed the presence of the wild-type propeptide. In the absence of a crystal structure, we can only suspect that the distortion in the backbone of wild-type ß7 in the region around Phe129, which we attribute mainly to unfavorable interactions with Asp 166 (Fig. 4g), is still present in the mutant and responsible for the inactivity and inability to autolyse. We did not try to activate ß4 because major differences between the Ca-traces of this subunit and of the active subunits exist (Fig. 4h).
Note Added in Proof. While this paper was in press, a publication by Arendt and Hochstrasser (28) appeared suggesting acetylation of ß1, ß2, and ß5 subunits by genetic methods in mutants lacking the respective propeptides. These results are in agreement with our findings in ß1 by analytical methods.
We thank Silvia Körner and Frank Siedler (Max-Planck-Institut für Biochemie, Martinsried, Germany) for help with mass spectrometry, Karlheinz Mann (Max-Planck-Institut für Biochemie, Martinsried, Germany) for help with N-terminal sequence analysis, and G.B. Bourenkow and H.Bartunik (DESY, Hamburg, Germany) for assistance with the x-ray experiments. The Sonderforschungsbereich 469 provided financial support. The work was furthermore supported by a grant from the Deutsche Forschungsgemeinschaft (Bonn) and the Fonds der Chemischen Industrie (Frankfurt).
1. Hilt, W. & Wolf, D.H. (1996) Trends Biochem. Sci. 21, 96–102.
2. Hershko, A. & Ciechanover, A. (1998) Annu. Rev. Biochem. 67, 425–479.
3. Hochstrasser, M. (1996) Annu. Rev. Genet. 30, 405–409.
4. Baumeister, W., Walz, J., Zühl, F. & Seemüller, E. (1998) Cell. 92, 367–380.
5. Peters, J.M., Cejka, Z., Harris, R.J., Kleinschmidt, J.A. & Baumeister, W. (1993) J. Mol Biol. 234, 932–937.
6. Coux, O., Tanaka, K. & Goldberg, A.L. (1996) Annu. Rev. Biochem. 65, 801–847.
7. Groll, M., Ditzel, L., Löwe, J., Stock, D., Bochtler, M., Bartunik, H.D. & Huber, R. (1997) Nature (London) 386, 463–471.
8. Dahlmann, B., Kopp, F., Kuehn, L., Niedel, B., Pfeifer, G., Hegerl, R. & Baumeister, W. (1989) FEBS Lett. 251, 125–131.
9. Löwe, J., Stock, D., Jap, B., Zwickl, P., Baumeister, W. & Huber, R. (1995) Science 268, 533–539.
10. Seemüller, E., Lupas, A, Stock, D., Löwe, J., Huber, R. & Baumeister, W. (1995) Science 268, 579–581.
11. Heinemeyer, W., Fischer, M., Krimmer, T., Stachon, U. & Wolf, D.H. (1997) J. Biol. Chem. 272, 25200–25209.
12. Nussbaum, A.K., Dick, T.P., Keilholz, W., Schirle, M., Stevanovic, S., Dietz, K., Heinemeyer, W., Groll, M., Wolf, D.H., Huber, R., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 12504– 12509.
13. Brunger, A. (1992) X-PLOR Version 3.1; A System for X-Ray Crystallography and NMR (Yale Univ. Press, New Haven, CT).
14. Turk, D. (1992) Ph.D. thesis (Technical Univ.; Munich).
15. Jones, T.A. (1978) J. Appl. Crystallogr. 15, 24–31.
16. Schmidtke, G., Kraft, R., Kostka, S., Henklein, P., Frömmel, C, Löwe, J., Huber, R., Kloetzel, P.M. & Schmidt, M. (1996) EMBO J. 15, 6887–6898.
17. Ditzel, L., Stock, D. & Löwe, J. (1997) Biol. Chem. 378, 239–247.
18. Frentzel, S., Pesold-Hurt, B., Seelig, A. & Kloetzel, P.M. (1994) J. Mol. Biol. 236, 975–981.
19. Nandi, D., Woodward, E., Ginsburg, D.B. & Monaco, J.J. (1997) EMBO J. 16, 5363–5375.
20. Chen, P. & Hochstrasser, M. (1996) Cell 86, 961–972.
21. Ditzel, L., Huber, R., Mann, K., Heinemeyer, W., Wolf, D.H. & Groll, M. (1998) J. Mol. Biol. 279, 1187–1191.
22. Bachmair, A, Finley, D. & Varshavsky, A. (1986) Science 234, 179–186.
23. Arfin, S.M. & Bradshaw, R.A. (1988) Biochemistry 27, 7979– 7984.
24. Wilkinson, K.D. (1997) FASEB J. 11, 1245–1256.
25. Seemüller, E., Lupas, A. & Baumeister, W. (1996) Nature (London) 382, 468–470.
26. Loidl, G., Groll, M., Musiol, H.-J., Ditzel, L., Huber, R. & Moroder, L. (1999) Chem. Biol. 6, 197–204.
27. Loidl, G., Groll, M., Musiol, H.-J., Huber, R. & Moroder, L. (1999) Proc. Natl. Acad. Sci. USA 96, 5418–5422.
28. Arendt, C.S. & Hochstrasser, M. (1999) EMBO J. 18, 3575–3585.








