
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
The structure of the human ßII-tryptase tetramer: Fo(u)r better or worse
CHRISTIAN P. SOMMERHOFF*†, WOLFRAM BODE‡, PEDRO J. B. PEREIRA‡, MILTON T. STUBBS§, JÖRG STURZEBECHER¶, GERD P. PIECHOTTKA*, GABRIELE MATSCHINER*, AND ANDREAS BERGNER‡
*Abteilung Klinische Chemie und Klinische Biochemie in der Chirurgischen Klinik und Poliklinik, Klinikum Innenstadt der Ludwig-Maximilians-Universität, Nußbaumstrasse 20, D-80336 Munich, Germany; ‡Abteilung für Strukturforschung, Max-Planck-Institut für Biochemie, Am Klopferspitz 18a, D-82152 Martinsried, Germany; §Institut für Pharmazeutische Chemie der Philipps-Universität Marburg, Marbacher Weg 6, D-35032 Marburg, Germany; and ¶Klinikum der Universität Jena, Zentrum für Vaskuläre Biologie und Medizin, Nordhäuserstrasse 78, D-99089 Erfurt, Germany
ABSTRACT Tryptases, the predominant serine proteinases of human mast cells, have recently been implicated as mediators in the pathogenesis of allergic and inflammatory conditions, most notably asthma. Their distinguishing features, their activity as a heparin-stabilized tetramer and resistance to most proteinaceous inhibitors, are perfectly explained by the 3-Å crystal structure of human ßII-tryptase in complex with 4-amidinophenylpyruvic acid. The tetramer consists of four quasiequivalent monomers arranged in a flat frame-like structure. The active centers are directed toward a central pore whose narrow openings of approximately 40 A× 15 Å govern the interaction with macromolecular substrates and inhibitors. The tryptase monomer exhibits the overall fold of trypsin-like serine proteinases but differs considerably in the conformation of six surface loops arranged around the active site. These loops border and shape the active site cleft to a large extent and formallcontacts with neighboring monomers via two distinct interfaces. The smaller of these interfaces, which is exclusively hydrophobic, can be stabilized by the binding of heparin chains to elongated patches of positively charged residues on adjacent monomers or, alternatively, by high salt concentrations in vitro. On tetramer dissociation, the monomers are likely to undergo transformation into a zymogen-like conformation that is favored and stabilized by intramonomer interactions. The structure thus provides an improved understanding of the unique properties of the biologically active tryptase tetramer in solution and will be an incentive for the rational design of mono- and multi-functional tryptase inhibitors.
Human mast cell tryptases (EC 3.4.21.59) comprise a family of trypsin-like serine proteinases closely related in sequence that are derived from =3 nonallelic genes (1, 2). Tryptases (at least isoenzymes aI, ßI, ßII, and ßIII) are highly and selectively expressed in mast cells and to a lesser extent in basophils (3, 4). Only ß-tryptases, however, appear to be activated intracellularly and stored in secretory granules (5, 6), accumulating to much larger amounts than any other of the granule-associated serine proteinases of leukocytes and lymphocytes. On mast cell activation, ß-tryptases are secreted bound to heparin in diverse allergic and inflammatory conditions ranging from asthma and rhinitis to psoriasis and multiple sclerosis. Various studies performed in animals and humans have provided considerable evidence that tryptases are directly involved in the pathogenesis of asthma (7–9), a hypothesis also supported by apparent genetic links of tryptases to airway reactivity (10, 11).
Several unique properties distinguish tryptases from other trypsin-like proteinases (reviewed in refs. 12 and 13). Most notably, tryptases are enzymatically active in the form of a noncovalently linked tetramer. The tetramer is stabilized by association with negatively charged aminoglycans such as heparin or high ionic strength conditions in vitro. On dissociation, reversible only under certain conditions, the monomers lose activity, apparently because of transition into a zymogen-like state (14, 15). This mechanism is thought to govern tryptase activity in vivo. With the exception of the “atypical” Kazal-type inhibitor leech-derived tryptase inhibitor (LDTI) (16, 17), human tryptases are resistant to inhibition by proteinaceous inhibitors. In accordance with their trypsin-like activity, tryptases efficiently hydrolyze a number of peptide substrates including the neuropeptides “vasoactive intestinal peptide” and “peptide histidine methionine” (18). Few macromolecular substrates are cleaved, however, leading to the activation of prostromelysin, prourokinase, and the protein-ase-activated receptor-2 (19–21) and the inactivation of fibronectin and of the procoagulant functions of high molecularmass kininogen and fibrinogen (22–24).
These distinguishing features are well explained by the crystal structure of the human lung ßII-tryptase tetramer, whose overall architecture has been summarized recently (25). Here, we describe the identification of the tetramer within the crystal packing, the detailed structure of the monomers, and their interactions in the tetramer. In addition, structural features likely to favor a zymogen-like conformation of isolated monomers and models of the interaction with stabilizing heparin proteoglycans and inhibitors are presented.
Identification of the Relevant Tryptase Tetramer. In the x-y plane of the tryptase crystals, the tryptase-monomers are arranged in flat rectangular tetrameric aggregates that form extended protein layers (Fig. 1a). Within these layers, each tetramer is rotated about the crystallographic a- and b-axes by ˜7°, in agreement with the self-rotation function. The tetramers appear well separated from their neighbors in one direction (x-direction in Fig. 1a) but are in somewhat closer contact in the perpendicular direction (y in Fig. 1a). In the z-direction, the tetramers are stacked along the crystallographic 41 screw axis. Because of the 7° tilt of each tetramer from the x–y plane, their projections (Fig. 1b) alternate between leaning to the left, being horizontal, and leaning to the right, respectively, giving rise to a 7° precession motion of the
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviations: APPA, 4-amidinophenylpyruvic acid; LDTI, leech-derived tryptase inhibitor. |
|
|
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1A0L). |
|
† |
To whom reprint requests should be addressed. E-mail: sommerhoff@clinbio.med.uni-muenchen.de. |
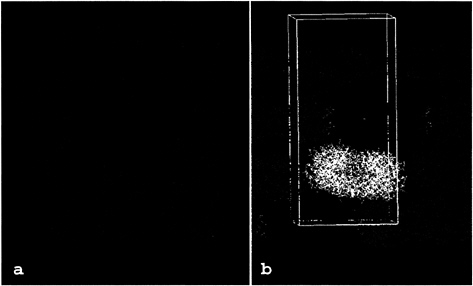
FIG. 1. Packing of the human ßII tryptase crystal, (a) View along thez-axis showing one layer of tryptase molecules in the x-y plane. The tryptase monomers are grouped into tetrameric aggregates that form extended sheets. Each of these tryptase tetramers is clearly delimited from its neighbors in both directions. A “reference” tetramer is shown in red for simplicity, (b) View across the z-axis. In the z direction, layers of tetramers are stacked on each other along the 41 screw axis. The local 2-fold symmetry axis is tilted from the z direction by ˜7°, causing increased crystal-stabilizing contacts between layers stacked in the z-direction. One unit cell (82.9×82.9×172.9Å), occupied by four tryptase tetramers, is indicated by a white bordered box.
local (2-fold; see below) rotation axis along the crystallographic 41 screw axis. The largely complementary interaction surfaces between the monomers of the tetramer are typical for intersubunit contacts, whereas neighboring tetramers interact with one another via much more usual crystal contacts. Thus, within a tetramer, monomer A (Fig. 2) interacts with monomers B and D via interfaces of sizes 540 Å2 and 1,075 Å2, respectively (solvent inaccessible surface probed by using a sphere of 1.4-Å radius; Collaborative Computational Project No. 4 suite). In contrast, the four monomers of one given tetramer interact with monomers from neighboring tetramers via interfaces of less than 280 Å2 (in the x-y plane) and 265 Å2 (along the z-axis), respectively. The contacts between tetramers include a number of hydrogen bonds and six unique salt bridges and thus are qualitatively similar to those usually observed in typical crystal contacts.
These packing considerations suggest that the tetramer emphasized in Fig. 1 represents the enzymatically active tetramer of human ß-tryptase. This tetramer selection is supported by the finding that the six loops that deviate most from the structures of other trypsin-like proteinases are aII involved in forming monomer-monomer contacts within a tetramer. More important, this unique tetramer perfectly explains the distinguishing properties of tryptase in solution, e.g., the resistance to proteinaceous inhibitors other than LDTI, the unusual substrate specificity, and the stabilization by the binding of heparin-like glycosaminoglycans (see below).
Overall Tetramer Structure. In the tryptase tetramer, monomers (arbitrarily assigned A, B, C, and D in Fig. 2) are positioned at the corners of a flat rectangular frame leaving a continuous central pore. The tetramer displays almost perfect 222 symmetry that, however, is not exact because of the crystallographically asymmetric environment and an imperfect internal packing (see below). The horizontal and the vertical 2-fold axes, which cross each other in the center of the tetramer, relate monomers A to B and C to D, or A to D and B to C, respectively. The third 2-fold symmetry axis relating monomers A to C and B to D is arranged virtually perpendicular to the other 2-fold axes and runs almost through their point of intersection in the central pore.
The active centers of the four monomers are directed toward the central pore (Fig. 2). This pore exhibits a rectangular cross section and is twisted by ˜30° about the tetramer axis. It possesses two narrow openings of dimension 40 Å×15 Å, and widens in its central part to a cross section of 50 Å×25 Å, just large enough for elongated peptides of the diameter of an a-helix to thread though the exits and to interact with the active sites. Both pore entrances are partially obscured by the 147-loops (see below), which project from each of the monomers but on alternative entrance sides, so that only two diagonally arranged active centers can be viewed directly (Fig. 2). With 33 basic (including 12 His residues) and 24 acidic residues per monomer, human tryptase exhibits an average percentage of charged residues comparable to related serine proteinases, but is only slightly positively charged at neutral pH. These charges are not evenly distributed along the molecular surface, however. Rather, negatively charged residues cluster preferentially on the inner pore-facing surface, conferring the pore with a quite negative electrostatic potential, and along the peripheral A–D (and B–C) edges. In contrast, the A–B (and C–D) peripheries and one front side of the monomer surface are positively charged and probably are involved in heparin binding (see below and Fig. 6).
Monomer Structure. The tryptase monomer exhibits the typical ß-strand-dominated fold seen in other trypsin-like serine proteinases. The core is made by two six-stranded
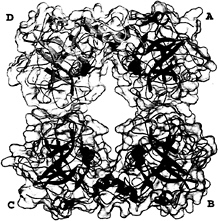
FIG. 2. Overall structure of the tryptase tetramer. The four monomers A, B, C, and D (clockwise) are shown as blue, red, green, and yellow ribbons, each surrounded by a semitransparent surface. The inhibitor molecules APPA are given as orange CPK models, each binding into one of the four S1 specificity pockets.
β-barrels that are packed together and further clamped by three transdomain segments (Fig. 3). This core structure is covered by a number of polypeptide loops, a short α-helical turn (Ala-55-Gly-66, not shown in Fig. 3a), and two regular α-helices, the so-called “intermediate helix” (Glu-164-Leu-173A) and the C-terminal helix (Arg-230-Val-242). The catalytic residues Ser-195, His-57, and Asp-102 (chymotrypsinogen numbering) are located in the junction between both barrels. The active-site cleft runs perpendicular to this barrel junction. In the “standard orientation” shown in Fig. 3, this cleft runs approximately horizontally across the molecular surface facing the viewer and is ready to accommodate and bind extended peptide substrates extending from left to right. One hundred sixty-two and 168 residues of the tryptase monomer are topologically equivalent to the archetypal proteinases chymotrypsin (26) and trypsin (27), respectively, with an rms deviation of their α-carbon atoms of 0.65 Å for both comparisons. The numbering of the tryptase residues given in this article is predominantly based on the equivalence with chymotrypsinogen (28) and at only a few trypsin-characteristic sites on that with trypsin (27).
In detail, however, the topology of the tryptase monomers deviates significantly from these reference proteinases (Fig. 3b), probably more than any other trypsin-like serine proteinase. In particular, six surface loops that border and shape the active-site cleft are unique (Fig. 3a). These loops comprise the 147-loop (including the 152-“spur”), the 70- to 80-loop, the 37-loop, the 60-loop, the 97-loop, and the 173-loop (Fig. 3a). The 147-loop, which together with Gln-192 forms the rather acidic southern wall of the active-site cleft, is shortened by one residue in its initial part, but contains a two-residue insertion (Pro-152-Pro-152A-cisPro-152B-Phe-153-Pro-154) in its proline-rich and hydrophobic 152-spur. The neighboring 70- to 80-loop to the east, which in the calcium-binding serine proteinases winds around a stabilizing calcium ion (27), is three residues shorter and more compact in tryptase. It is probably not designed for calcium binding, in spite of topologically similar liganding groups; Glu-70 and Asp-80, involved in a partially buried salt bridge cluster with Arg-34, are
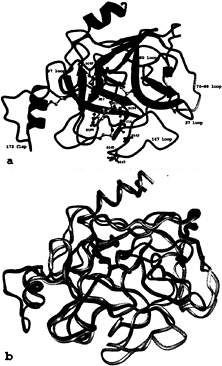
FIG. 3. The tryptase monomer in standard orientation, i.e., as seen approximately from the middle of the central pore of the tetramer toward the active site of monomer A (represented by Ser-195, His-57, and Asp-102), (a) Ribbon representation of a tryptase monomer. The amidino group of the APPA molecule interacts with Asp-189 in the S1 pocket. Ser-195 O-γ is bound covalently to the APPA carbonyl group forming a hemiketal. The six unique surface loops of tryptase that surround the active site and are engaged in intermonomer contacts are shown in special colors, namely (anticlockwise) the 147-loop (light blue), the 70- to 80-loop (yellow), the 37-loop (orange), the 60-loop (magenta), the 97-loop (green), and the 173-flap (red). All other tryptase segments are given in dark blue. The side chains of the catalytic triad residues as well as Asp-143, Asp-145, and Asp-147 in the acidic 147-loop are shown as a ball-and-stick model. (b) Overlay of the structures of the tryptase monomer and bovine trypsin, both given as ropes. The color-coding of tryptase is as in a, whereas trypsin is shown in gray. The most relevant deviations from the trypsin backbone appear in the colored loop regions of tryptase.
oppositely arranged to the two calcium-binding Glu residues in trypsin. The 37-loop, above the 70- to 80-loop, possesses two additional residues (Pro-37A and Tyr-37B), which bulge away from the loop axis. The adjacent 60-loop, with five inserted residues, turns away from the cleft abruptly to the north, where it kinks at cisPro-60A to approach the general main chain course of other serine proteinases. At position 69, a buried Arg replaces the Gly residue that is strictly conserved in most other homologous proteinases, allowing for a special conformation. Although the 97-loop, at the northern rim of the cleft, contains the same number of residues as other serine proteinases, it differs considerably in conformation. The N-terminal part is shortened by two residues between positions 96 and 97, thus placing Ala-97 in the position normally occupied by residue 99,
whereas its C-terminal part makes an unusual extra helical turn before arriving at Asp-102. By far the largest insertion, with nine residues, occurs in the 173-loop. After the unusually long three-turn intermediate helix, the 10 residues from His-173 to Val-173I form an exposed flap centered around the imidazole side chain of His-173.
With 245 amino acid residues, the tryptase monomer possesses 15 and 22 residues more than the B-chains of chymotrypsin and trypsin, respectively. Compared with chymotrypsinogen, most of these extra residues present in all tryptases known so far are inserted in the 37-loop (two residues), the 60-loop (+5), the 147-loop (+1), the 173-loop (+9), at position 221A (+1) and at the C terminus (+1), whereas the 70- to 80-loop (–3) and the 214- to 220-loop (–1, as inalltrypsin-like serine proteinases) are shorter. On the reverse side, the largely hydrophobic cluster of four Trp residues (Trp-27, -29, -207, and -137) is noteworthy. Only the indole moieties of the latter two Trp are significantly exposed to the surface. At the C terminus, only the main chain atoms of the two penultimate residues Lys-244 and Lys-245 are well defined by electron density, while the C-terminal Pro-246 could not be located. The side chain of the single N-linked sugar attachment site in human ßII-tryptase, Asn-204, extends away from the molecular surface opposite to the active site. Some residual electron density exists distal to its carboxamide group, which is not large enough to account for a covalently linked sugar residue.
As found in almost all trypsin-like serine proteinases [except, e.g., single-chain tissue type plasminogen activator (29)], the N-terminal Ile-16-Val-17 segment is inserted in the Ile-16 pocket, forming a solvent inaccessible salt bridge between its free Ile-16 a-amino group and the carboxylate group of Asp-194. The formation of this salt bridge after activation cleavage creates a functional substrate recognition site by reorienting the Asp-194 side chain from an external position in the zymogen, where it might hydrogen bond to a surface located His-40··· Ser-32 pair forming the so-called “zymogen triad,” to an internal position in the active molecule (30, 31). This reorientation restructures the surrounding “activation domain,” which in trypsin(ogen) mainly includes the linings of the Ile-16 pocket and the S1 specificity pocket (i.e., segments Ile-16-Gly-19, Tyr-184-Asp-194, Gly-216-Asn-223, and Gly-142-Tyr-151), and the “oxyanion hole” formed by the amide groups of Gly-193 and Ser-195 (28, 32, 33). The single-chain zymogen and the activated monomer are adequately described by a two-state model, in which an inactive conformation is in equilibrium with an active form possessing a structured activation domain (31). The partition between both forms depends on environmental conditions such as the endogenous free Ile-16-Val-17 N-terminal segment (34), free Ile-Val dipeptide (31), ligands in the substrate binding site (30, 36), or other effectors such as fibrin with respect to tissue plasminogen activator or tissue factor in the case of Factor VIIa (29, 37). This conformational partition can be influenced by internal molecular groups that stabilize or destabilize one or the other state. Tryptase possesses the zymogen triad residues His-40 and Ser-32, which would stabilize the zymogen state. In addition, the acidic residues Asp-143, Asp-145, and Asp-147 arranged around the Ile-16 cleft could form a negatively charged anchoring site that could compete with the Ile-16 pocket for the Ile-16 a-amino group, thus destabilizing the structured active state of the tryptase monomer. Furthermore, some of the loops in contact with the activation domain of tryptase, such as the long 173-loop or the 70- to 80-loop, which has been shown to be strongly correlated with the equilibrium state in bovine elastase “subunit III” (38), could influence the structured state. The conformation of the tryptase 173-loop, probably held in place in the tetramer by contacts with monomer D, certainly has an effect on the stability of the integrated monomer. Interestingly, tissue factor, thought to support insertion of the N-terminal Ile-16 a-amino terminus of activated Factor VIIa B-chain on complex formation (37), likewise binds to the 173-loop at the intermediate helix flank (39).
Interfaces. All monomer-monomer contacts within the tetramer are realized via six loops arranged around the active center. These loops, emphasized by special colors in Figs. 3–5, differ fundamentally in their conformation and partly in size from those of other trypsin-like serine proteinases. Monomers A and B interact with one another through the 147-loop, the 70- to 80-loop, and the 37-loop (Fig. 4d). Each 152-spur slots into a cleft formed by the 37- and the 70- to 80-loop of its own monomer and the 152-spur of the opposing neighbor. At the center of the interface, the side chains of Phe-153 and Tyr-75 from each subunit form an approximate tetrahedron (Fig. 5a). The side chain of Tyr-75 from monomer B (D) would clash with the equivalent A (C) side chain if they were arranged in a symmetrical manner. Instead, the phenolic group of Tyr-75 of monomer A turns in the opposite direction, breaking the 2-fold symmetry (see the partial electron density in Fig. 5a). This A–B (C–D) interface is exclusively hydrophobic, with a remarkable number of Tyr and Pro side chains involved, and lacks any intermonomer hydrogen bonds. Toward the pore, the side chains of the two Arg-150 residues oppose one another. The charges of their guanidyl groups presumably make unfavorable energy contributions to the A–B interaction.
Monomer A interacts with monomer D through the entire northern rim consisting of the 173-flap, the 97-loop, and the 60-loop (Figs. 4a and 5b), again via equivalent loops. Both 97-loops rest with their 95–99 segments on one another (Fig. 4a), with both Ile-99 side chains in direct contact. Further toward both peripheries, segment Pro-60A-Asp-60B and the opposing segment Gly-173B-Tyr-173D run antiparallel to one another, forming two-rung antiparallel ladders between Gly-173B-Tyr-173D and Pro-60A-Val-60C (Fig. 5b). Each Tyr-95 aromatic side chain nestles into the bend of the opposing 173-flap, and each Tyr-173D phenolic side chain slots into a hydrophobic cleft made by the 60-loop and the 97-loop of the opposing monomer. In addition, both monomers are cross-connected by salt bridges between Asp-60B and Arg-224 and

FIG. 4. Loop arrangements in the tetramer. The six special loops engaged in monomer-monomer interactions are shown in the color coding introduced in Fig. 3. (a) The D–A dimer as seen from outside of the tetramer along the local 2-fold axis, (b) The monomer viewed in standard orientation, (c) Front view of the tetramer. (d) The A–B dimer seen from outside of the tetramer along the local 2-fold axis.
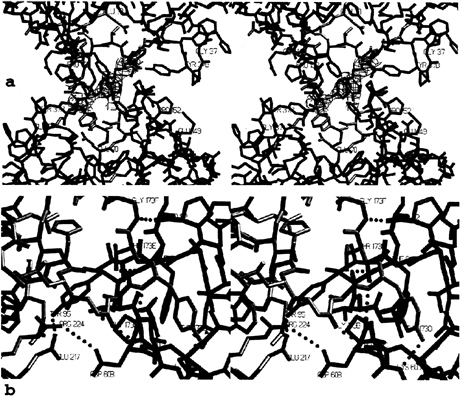
FIG. 5. Stick representation of the contact interfaces between monomers, (a) The AB-interface seen from inside the tetramer along the local 2-fold axis, shown together with the final 2F0-Fc electron density map for both Tyr-75 side chains contoured at 1 σ level. The monomers and loops are given in the color coding introduced in Figs. 3 and 4. (b) The AD-interface (half side) observed approximately perpendicular to the local 2-fold axis, shown together withallintermonomer hydrogen bonds and salt bridges (green dots). Segments of monomers A and D are given in blue and yellow, respectively.
by four hydrogen bonds involving both main and side chains (Fig. 5b). Thus, the A–D (and the corresponding B–C) interface comprises a number of polar/charged interactions in addition to several hydrophobic contacts.
The A–B homodimer carries a number of positively charged residues at the periphery, which cluster and form an obliquely oriented two-lobed patch of positive charges that extends toward one of the front sides of each monomer, giving rise to the blue-colored electrostatic potential surfaces in Fig. 6. With an overall length of almost 100 Å, this patch would allow tight electrostatic binding of an extended heparin chain of ≈20 sugars running obliquely along the A–B edge as shown in Fig. 6. The length of such heparin chains is in good agreement with the experimentally observed stabilization of the tetramer by heparin fractions of molecular mass 5,500 Da and above (40). On the peripheral surface of the A–D (and the corresponding B–C) homodimer, in contrast, positive charges are counter-balanced by adjacent negative ones.
Interaction with Substrates and Inhibitors. The immediate vicinity of the tryptase active site is quite similar in structure to that of trypsin. The specificity S1 pocket, which opens to the west of the reactive Ser-195 (Fig. 3a), is virtually identical to that of trypsin and well suited to accommodate P1-Lys and Arg side chains. The 4-amidinophenylpyruvic acid (APPA) molecule inserts into this pocket in the same manner as in the complex with trypsin (41). Thus, its amidino group hydrogen is bonded to both Asp-189 carboxylate oxygens, Gly-219 O and Ser-190 Oγ, and its phenyl ring is sandwiched between peptide planes 215–216 and 190–192. Ser-195 Oγ bonds to the carbonyl group of the tetrahedral pyruvate part of APPA (Fig. 3a), and hydrogen bonds to His-57 Nε. As indicated by the relatively low equilibrium dissociation constant of the APPA-tryptase complex [Ki 0.71 μM; (42)], APPA fits well to the tryptase active site. Toward the south of the active site of tryptase, the side chains of Asp-143, Asp-145, and Asp-147 protrude from the relatively flat and hydrophobic southern embankment (Fig. 3a). The resulting negative charge cluster provides a second anchoring point for dibasic synthetic tryptase inhibitors such as bis-benzamidines (17, 42, 43), allowing favorable interactions with a distal basic group such as in pentamidine. The structural basis of the unexpected high affinity of bifunctional inhibitors containing suitably arranged adjacent imidazole moieties such as present in the inhibitor BABIM and closely related analogues (43, 44) has recently been revealed: two nitrogen atoms
FIG. 6. Model of the binding of a 20-mer heparin-like glycosaminoglycan chain along the A–B edge of the tryptase-tetramer. The solid-surface representation of tryptase indicates positive (blue) and negative (red) electrostatic potential contoured from –4 kT/e to 4 kT/e. The heparin chain (green/yellow/red stick model) is long enough to bind to clusters of positively charged residues on both sides of the monomer-monomer interface, thereby bridging and stabilizing the interface which is exclusively hydrophobic in nature (see Fig. 5a).
of the two methylene-connected benzimidazoles coordinate a zinc zinc that also binds to the active-site located Ser-195 Oγ and His-57 Nε (44). The zinc-mediated binding enhancement of BABIM-like inhibitors is particularly large but not restricted to tryptase.
Toward the east, the substrate-binding site of tryptase is not only bounded by the side chains of Tyr-37B and Tyr-74 of monomer A, but also by the Phe-153 benzyl group and the 152-spur of the neighboring monomer B. Thus, binding of extended substrate chains is limited to about P5′ (Fig. 7).

FIG. 7. View from the LDTI inhibitor (represented only by its reactive site loop P7 to P3′) toward the active-site cleft. The P1 Lys residue is buried.
Toward the north, the 97-loop of monomer A borders the substrate binding region in a manner different from most other serine proteinases, and together with the side chains of Phe-94, Ala-97, and Gln-98 of monomer D forms a projecting “canopy.” The S2 subsite underneath is open and larger than that of trypsin. The S3/S4 subsite above the Trp-215 indole moiety is fully blocked by the side chain of Gln-98 and the phenolic group of Tyr-95 provided by monomer D. Toward the west, however, the substrate-binding site is bordered exclusively by segments of the D-monomer, in particular the His-57 imidazole ring and segment 57–60. Thus, the active centers of monomers A and D (B and C) are spatially close (distance ≈23 Å for the A–D pair) to each other in the tryptase tetramer, rendering the tryptase tetramer suitable for the specific binding of bifunctional inhibitors with relatively short spacers.
The central pore of tryptase restricts the size of accessible substrates and inhibitors considerably. For larger proteins such as fibronectin and the zymogens of stromelysin-1 and urokinase-type plasminogen activator, the cleavage sites must be extended into the active sites. Docking experiments with C-terminally truncated prostromelysin-1 (45) and with single-chain tissue plasminogen activator (29) as a model for prourokinase show that the activation cleavage loops of these proproteinases must be extracted from their crystal structures to allow binding in the tryptase active center. More flexible peptides, in contrast, could easily thread through the pore of the tetramer to be processed or destroyed. Flexible polypeptide chains with two distant basic residues, as in “vasoactive intestinal peptide” (18), might even dock to adjacent active sites simultaneously to produce fragments of distinct length.
The active centers of the tryptase monomers are also largely inaccessible for macromolecular inhibitors. The only exception known is LDTI, an “atypical” Kazal-type inhibitor that is smaller than the classical members of this family (16). LDTI has been shown to bind to trypsin through its reactive-site loop (residues P4 to P4′) in a canonical manner (17, 46). In the model of the complex with tryptase monomer A, the four N-terminal residues preceding this binding segment could bend toward the south (with respect to Figs. 3 and 7), leading to the juxtaposition of the basic Lys-I1-Lys-I2 amino terminus (with the suffix I identifying inhibitor residues) with the carboxylate groups of Asp-143 and Asp-147 of monomer A. Alternatively, the two Lys residues could interact with Asp-60B of molecule D. The involvement of such electrostatic interactions is supported by the deleterious effect of deletions and substitutions of these basic residues on the affinity of LDTI toward tryptase but not trypsin (17). The LDTI reactivesite loop, running from Cys-I14 (P5) to Pro-I22 (P4′; ovomucoid numbering), is relatively small compared with classical Kazal-type inhibitors, allowing good overall fit to the restricted substrate binding groove (Figs. 7 and 8a). Furthermore, its central helix is one turn shorter, so that it just fits into the central pore of the tetramer on canonical binding to the active site of monomer A with only a few narrow contacts of its molecular antipole, opposite to its reactive-site loop, with the 147-loop of monomer D. Docking of a second LDTI molecule is possible at the opposite active centers of either monomer B or monomer C (Fig. 8a). A slight collision between Cys-I56 and Gly-I28 of two bound LDTI molecules could be relieved by minor torsion in the proteinase-inhibitor interfaces, as observed for other canonically binding inhibitors such as eglin c (46). Any such torsion in the LDTI molecule bound to monomer A would impose an opposing torsion in the LDTI molecule bound to monomer B, facilitating such a relaxation. The simultaneous binding of two LDTI molecules to the tetramer is in good agreement with experimental results showing ≈50% inhibition of the cleavage activity toward small chromogenic substrates by nanomolar LDTI concentrations (16). Modeling experiments with more elongated classical Kazal-type inhibitors or with the prototypical bovine pancre-

FIG. 8. Models of the interaction of the human tryptase tetramer with proteinaceous inhibitors. The tryptase tetramers are shown as green ribbons. An inhibitor molecule (blue) is modeled into the active site of monomer A by superposition of the proteinase moiety of known proteinase-inhibitor complexes to a tryptase monomer. For LDTI and BPTI the target proteinase was trypsin (17, 49), for MPI chymotrypsin (47). The active sites of the other tryptase monomers are occupied by APPA molecules (orange). Parts of the inhibitors clashing with the structure of tryptase (i.e., a distance smaller than 1.5 Å between the Ca-atoms of the respective molecules) are highlighted in red. (a) In addition to one molecule of the “atypical” Kazal-type inhibitor LDTI bound to the tryptase monomer A a second molecule (shown in pink and yellow) can bind to the active site of either monomer B or C. (b) Bovine pancreatic trypsin inhibitor (aprotinin). (c) Human mucous proteinase inhibitor bound to tryptase with its inhibitorily active second domain.
atic trypsin inhibitor indicate strong collisions of their distal pole segments with the neighboring monomers D and B, in particular with the 147-loops, explaining the observed inactivity of these inhibitors toward tryptase (Fig. 8b). The central portion of the two-domain mucous proteinase inhibitor (MPI=SLPI=HUSI-I) would clash with the A–D interface region of the tryptase tetramer if bound to the active site of monomer A (Fig. 8c) via its inhibitorily active second domain (47). Similarly, elafin (=SKALP), an inhibitor corresponding to the MPI second domain (48), should not be able to inhibit tryptase. The much larger plasma proteinase inhibitors are clearly far too bulky to fit into the narrow pore of the tryptase tetramer and gain access to one of the active centers.
CONCLUSION
In summary, the structure of the ßII-tryptase tetramer has been identified based on the four crystallographically independent quasiidentical monomers and the analysis of their arrangement within the crystal packing. With its frame-like architecture and its active centers facing a narrow central pore, the resulting tryptase tetramer structure explains most of the distinct properties of the biologically active tryptase tetramer in solution. The unusual substrate specificity, with a preference for peptidergic substrates, and the resistance to proteinaceous inhibitors other than LDTI are both caused by the limited accessibility of the active sites within the narrow central pore. The tetramer can be stabilized by heparin glycosaminoglycan chains larger than ˜20 sugar residues, a length required to bridge the weaker of the two distinct monomer-monomer interfaces. The loss of enzymatic activity on dissociation of the tetramer is caused by stabilization by internal molecular groups of a zymogen-like rather than the active state. Finally, the knowledge of the structure of the active center of the monomer as well as of the distances between neighboring active sites allows the rational design of multifunctional inhibitors. Such inhibitors that bind to more than one active center will ideally have potentiated affinity, conferring selectivity for the tryptase tetramer. Such inhibitors will be valuable as pharmacological tools to probe the pathophysiological function(s) of tryptases in vivo and may have therapeutic potential against asthma and other mast-cell related disorders.
We are grateful to R.Huber and H.Fritz for their generous support. We thank D.Grosse and R.Mentele for their excellent help in crystallization and amino acid sequence analysis. This work was supported by Sonderforschungsbereich 469 of the University of Munich, the Deutsche Forschungsgemeinschaft (STU 161, BO 1279), the Fonds der Chemischen Industrie, and programs BIO4-CT98–0418 and TMR ERBFXCT 98–0193 of the European Union.
1. Miller, J.S., Westin, E.H. & Schwartz, L.B. (1989) J. Clin. Invest. 84, 1188–1195.
2. Pallaoro, M., Fejzo, M.S., Shayesteh, L., Blount, J.L. & Caughey, G.H. (1999) J. Biol Chem. 274, 3355–3362.
3. Schwartz, L.B., Irani, A.M., Roller, K., Castells, M.C. & Schechter, N.M. (1987) J. Immunol. 138, 2611–2615.
4. Xia, H.Z., Kepley, C.L., Sakai, K., Chelliah, J., Irani, A.M. & Schwartz, L.B. (1995) J. Immunol 154, 5472–5480.
5. Schwartz, L.B., Sakai, K., Bradford, T.R., Ren, S., Zweiman, B., Worobec, A.S. & Metcalfe, D.D. (1995) J. Clin. Invest. 96, 2702–2710.
6. Sakai, K., Ren, S. & Schwartz, L.B. (1996) J. Clin. Invest. 97, 988–995.
7. Caughey, G.H. (1997) Am.J.Respir. Cell Mol. Biol. 16, 621–628.
8. Johnson, P.R. A., Ammit, A.J., Carlin, S.M., Armour, C.L., Caughey, G.H. & Black, J.L. (1997) Eur. Respir. J. 10, 38–43.
9. Rice, K.D., Tanaka, R.D., Katz, B.A., Numerof, R.P. & Moore, W.R. (1998) Curr. Pharm. Des. 4, 381–396.
10. De Sanctis, G.T., Merchant, M., Beier, D.R., Dredge, R.D., Grobholz, J.K., Martin, T.R., Lander, E.S. & Drazen, J.M. (1995) Nat. Genet. 11, 150–154.
11. Hunt, J.E., Stevens, R.L., Austen, K.F., Zhang, J., Xia, Z. & Ghildyal, N. (1996) J. Biol. Chem. 271, 2851–2855.
12. Schwartz, L.B. (1994) Methods Enzymol. 244, 88–100.
13. Caughey, G.H. (1995) Mast Cell Proteases in Immunology and Biology (Dekker, New York).
14. Ren, S., Sakai, K. & Schwartz, L.B. (1998) J. Immunol. 160, 4561–4569.
15. Selwood, T., McCaslin, D.R. & Schechter, N.M. (1998) Biochemistry 37, 13174–13183.
16. Sommerhoff, C.P., Söllner, C., Mentele, R., Piechottka, G.P., Auerswald, E.A. & Fritz, H. (1994) Biol. Chem. Hoppe-Seyler 375, 685–694.
17. Stubbs, M.T., Morenweiser, R., Stürzebecher, J., Bauer, M., Bode, W., Huber, R., Piechottka, G.P., Matschiner, G., Sommerhoff, C.P., Fritz, H., et al. (1997) J. Biol. Chem. 272, 19931–19937.
18. Tam, E.K. & Caughey, G.H. (1990) Am.J.Respir. Cell Mol Biol 3, 27–32.
19. Gruber, B.L., Marchese, M.J., Suzuki, K., Schwartz, L.B., Okada, Y., Nagase, H. & Ramamurthy, N.S. (1989) J. Clin. Invest. 84, 1657–1662.
20. Stack, M.S. & Johnson, D.A. (1994) J. Biol. Chem. 269, 9416–9419.
21. Molino, M., Barnathan, E.S., Numerof, R., Clark, J., Dreyer, M., Cumashi, A., Hoxie, J.A., Schechter, N., Woolkalis, M. & Brass, L.F. (1997) J. Biol. Chem. 272, 4043–4049.
22. Lohi, J., Harvima, I. & Keski-Oja, J. (1992) J. Cell. Biochem. 50, 337–349.
23. Little, S.S. & Johnson, D.A. (1995) Biochem. J. 307, 341–346.
24. Schwartz, L.B., Bradford, T.R., Littman, B.H. & Wintroub, B.U. (1985) J. Immunol. 135, 2762–2767.
25. Pereira, P.J., Bergner, A., Macedo-Ribeiro, S., Huber, R., Matschiner, G., Fritz, H., Sommerhoff, C.P. & Bode, W. (1998) Nature (London) 392, 306–311.
26. Blevins, R.A. & Tulinsky, A. (1985) J. Biol. Chem. 260, 4264– 4275.
27. Bode, W. & Schwager, P. (1975) J. Mol. Biol. 98, 693–717.
28. Wang, D., Bode, W. & Huber, R. (1985) J. Mol. Biol. 185, 595–624.
29. Renatus, M., Engh, R.A., Stubbs, M.T., Huber, R., Fischer, S., Kohnert, U. & Bode, W. (1997) EMBO J. 16, 4797–4805.
30. Huber, R. & Bode, W. (1978) Acc. Chem. Res. 11, 114–122.
31. Bode, W. (1979) J. Mol. Biol. 127, 357–374.
32. Freer, S.T., Kraut, J., Robertus, J.D., Wright, H.A.T. & Xuong, N.H. (1970) Biochemistry 9, 1997–2009.
33. Bode, W., Fehlhammer, H. & Huber, R. (1976) J. Mol. Biol. 106, 325–335.
34. Hedstrom, L., Lin, T.Y. & Fast, W. (1996) Biochemistry 35, 4515–4523.
35. Bode, W., Schwager, P. & Huber, R. (1978) J. Mol. Biol. 118, 99–112.
36. Bolognesi, M., Gatti, G., Menagatti, E., Guarneri, M., Marquart, M., Papamokos, E. & Huber, R. (1982) J. Mol. Biol. 162, 839–868.
37. Higashi, S. & Iwanaga, S. (1998) Int.J.Hematol. 67, 229–241.
38. Pignol, D., Gaboriaud, C., Michon, T., Kerfelec, B., Chapus, C. & Fontecilla Camps, J.C. (1994) EMBO J. 13, 1763–1771.
39. Banner, D.W., D’Arcy, A., Chene, C., Winkler, F.W., Guha, A., Konigsberg, W.H., Nemerson, Y. & Kirchhofer, D. (1996) Nature (London) 380, 41–46.
40. Alter, S.C., Metcalfe, D.D., Bradford, T.R. & Schwartz, L.B. (1987) Biochem. J. 248, 821–827.
41. Walter, J. & Bode, W. (1983) Hoppe-Seylers Z. Physiol. Chem. 364, 949–959.
42. Stürzebecher, J., Prasa, D. & Sommerhoff, C.P. (1992) Biol Chem. Hoppe-Seyler 373, 1025–1030.
43. Caughey, G.H., Raymond, W.W., Bacci, E., Lombardy, R.J. & Tidwell, R.R. (1993) J. Pharmacol. Exp. Ther. 264, 676–682.
44. Katz, B.A., Clark, J.M., Finer Moore, J.S., Jenkins, T.E., Johnson, C.R., Ross, M.J., Luong, C., Moore, W.R. & Stroud, R.M. (1998) Nature (London) 391, 608–612.
45. Becker, J.W., Marcy, A. L, Rokosz, L.L., Axel, M.G., Burbaum, J.J., Fitzgerald, P.M., Cameron, P.M., Esser, C.K., Hagmann, W.K., Hermes, J.D., et al. (1995) Protein Sci. 4, 1966–1976.
46. Bode, W. & Huber, R. (1992) Eur. J. Biochem. 204, 433–451.
47. Grütter, M.G., Fendrich, G., Huber, R. & Bode, W. (1988) EMBO J. 7, 345–351.
48. Tsunemi, M., Matsuura, Y., Sakakibara, S. & Katsube, Y. (1996) Biochemistry 35, 11570–11576.
49. Huber, R., Kukla, D., Bode, W., Schwager, P., Bartels, K., Deisenhofer, J. & Steigemann, W. (1974) J. Mol. Biol. 89, 73–101.








