
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Sonic hedgehog protein signals not as a hydrolytic enzyme but as an apparent ligand for Patched
NAOYUKI FUSE*†, TAPAN MAITI*†, BAOLIN WANG*, JEFFERY A. PORTER*‡, TRACI M. TANAKA HALL§¶, DANIEL J. LEAHY§, AND PHILIP A. BEACHY*||
* Department of Molecular Biology and Genetics and §Department of Biophysics and Biophysical Chemistry, Howard Hughes Medical Institute, Johns Hopkins University School of Medicine, Baltimore, MD 21205
ABSTRACT The amino-terminal signaling domain of the Sonic hedgehog secreted protein (Shh-N), which derives from the Shh precursor through an autoprocessing reaction mediated by the carboxyl-terminal domain, executes multiple functions in embryonic tissue patterning, including induction of ventral and suppression of dorsal cell types in the developing neural tube. An apparent catalytic site within Shh-N is suggested by structural homology to a bacterial carboxypeptidase. We demonstrate here that alteration of residues presumed to be critical for a hydrolytic activity does not cause a loss of inductive activity, thus ruling out catalysis by Shh-N as a requirement for signaling. We favor the alternative, that Shh-N functions primarily as a ligand for the putative receptor Patched (Ptc). This possibility is supported by new evidence for direct binding of Shh-N to Ptc and by a strong correlation between the affinity of Ptc-binding and the signaling potency of Shh-N protein variants carrying alterations of conserved residues in a particular region of the protein surface. These results together suggest that direct Shh-N binding to Ptc is a critical event in transduction of the Shh-N signal.
Hedgehog (Hh) proteins constitute a family of secreted signaling molecules that govern patterns of cellular differentiation during embryogenesis (reviewed in refs. 1–3). The hedgehog (hh) gene was first identified and isolated in Drosophila, where its multiple roles include patterning of larval segments and adult appendages. Vertebrate hh homologues also are involved in many aspects of developmental patterning. The Sonic hedgehog (Shh) member of this family, for example, is required for patterning of the neural tube and other tissues (4).
Hedgehog protein biogenesis (reviewed in ref. 5) has been best studied for the Drosophila protein but very likely is similar for Hedgehog proteins fromallspecies. After cleavage of an amino-terminal signal sequence on entry into the secretory pathway, the Hh protein undergoes an intramolecular autoprocessing reaction that involves internal cleavage between the Gly-Gly residues of an absolutely conserved GCF tripeptide (6, 7). The amino-terminal product of this cleavage, which is the species active in signaling (7), also receives a covalent cholesteryl adduct (8). Autoprocessing at this site and covalent linkage to cholesterol have been experimentally confirmed for the Shh protein (7–9). In Drosophila, a Hedgehog protein from a construct truncated at the internal site of cleavage is active in signaling, but this protein is not spatially restricted in its signaling activity and therefore causes gross mispatterning and lethality in embryos (10). The autoprocessing reaction thus is required not only to release the active signal from the precursor but also to specify the appropriate spatial distribution of this signal within developing tissues, presumably through insertion of the cholesteryl moiety into the lipid bilayer of the plasma membrane. Recent studies also have revealed palmi-toylation of the amino-terminal cysteine of the amino-terminal signaling domain of the Shh secreted protein (Shh-N); the occurrence of this second lipid modification is regulated by autoprocessing and may also influence the activity and distribution of Shh-N (9).
The patterning of the ventral neural tube is thought to require an inductive signal from the underlying mesodermal cells of the notochord (11). Shh protein is synthesized in the notochord and can induce differentiation of ventral cell types such as floor plate cells and motor neurons from neural plate explants in vitro (12); a similar role for Shh in vivo is confirmed by a loss of these cell types in mice lacking Shh gene function (4). Shh protein thus appears to constitute the inductive patterning signal from the notochord, and in vitro explant experiments have demonstrated a concentration-dependent response, with low concentrations of Shh-N protein inducing motor neuron differentiation and higher concentrations inducing increasing numbers of floor plate cells, ultimately to the exclusion of motor neurons (12, 13). Shh-N protein at concentrations below those required to induce differentiation of motor neurons or floor plate cells can repress expression of cell markers of dorsal neural tube, such as Pax-7 and Pax-3, in neural plate explants (14, 15). This repression of dorsal cell markers is presumed to mediate the transition of naive neural plate cells into ventral progenitor cells, which then differentiate into motor neurons or ventral interneurons at later stages of embryogenesis. Thus, the concentration-dependent activity of Shh-N has been proposed to regulate the dorso-ventral patterning of the developing neural tube.
Several components have been identified as candidates for receptor function in transduction of the Hh protein signal (reviewed in ref. 3). The patched (ptc) gene, originally identified in Drosophila, encodes a multipass transmembrane protein (Ptc). ptc mutations in Drosophila embryos cause inappropriate activation of wingless gene expression, a phenotype opposite that of hh mutations, thus suggesting that ptc functions as a negative effector in hh signaling (16, 17). The observations that hh ptc double mutant embryos resemble ptc mutants and
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviations: Shh-N, the amino-terminal signaling domain of the Sonic hedgehog secreted protein; HNF-3ß, hepatocyte nuclear factor 3ß; Ptc-CTD, Ptc with a truncation resulting in a 140-residue carboxyl-terminal deletion. |
|
† |
N.F. and T.M. contributed equally in this work. |
|
‡ |
Present address: Ontogeny, Inc., Cambridge, MA 02138. |
|
¶ |
Present address: National Institute on Environmental and Health Sciences, Research Triangle Park, NC 27709. |
|
|| |
To whom reprint requests should be addressed at: Department of Molecular Biology and Genetics, Johns Hopkins University School of Medicine, 725 North Wolfe Street PCTB-714, Baltimore, MD 21205. E-mail: pbeachy@jhmi.edu. |
that, in a ptc mutant background, ectopic Hh expression produces no further phenotypic effects, together suggest that the Ptc gene product acts downstream of Hh to regulate its signaling activity (16, 18, 19). Genetic epistasis studies further suggest that the smoothened gene (smo), which encodes another transmembrane protein (Smo), functions downstream of ptc in the hh signaling cascade (reviewed in ref. 3). Because smo is required for hh signaling, it has been proposed that Smo activates the Hh pathway and that Ptc inhibits Smo activity. Genetic mosaic analysis in the Drosophila wing imaginal disc showed that Ptc has, in addition to a cell-autonomous negative effect on Hh signaling, an ability to sequester the Hh protein and prevent its movement to adjacent cells (20).
Vertebrate homologues of both ptc and smo genes have been identified (reviewed in ref. 3). Shh-N was found to bind to cells expressing Ptc or both Ptc and Smo, but not to cells expressing Smo alone (21, 22). Moreover, Ptc interacted with Smo independently of the presence of Shh-N, suggesting that the two transmembrane proteins form a complex. An integrated view of Drosophila genetic analyses and biochemical studies of vertebrate homologues suggests a model in which the Ptc-Smo complex might function as Hh receptor, with direct binding of Hh to Ptc releasing Smo activity from inhibition by Ptc. It must be noted, however, that these biochemical studies did not examine the role of a physical interaction between Shh-N and Ptc in activation of the Shh pathway. In addition, these biochemical studies did not exclude the possibility that Shh-N interacts not directly with Ptc but with another component of a complex that includes Ptc, because the crosslinked binding complexes were extremely large and were not analyzed with regard to their composition.
The model just described assumes a role for Shh-N as a ligand for a receptor. The crystal structure of the Shh-N protein, however, suggested an alternative possibility. This structure revealed a zinc ion coordinated in an arrangement remarkably similar to that of thermolysin, carboxypeptidase A, and other zinc hydrolases (23). Even more striking is the remarkable similarity in folded structure of a portion of Shh-N to the catalytic domain of D,D-carboxypeptidase from Streptomyces albus, a cell wall enzyme closely related in structure and activity to other bacterial enzymes involved in conferring vancomycin resistance (Fig. 1 B and D) (24, 25). Although the functional role of this putative hydrolase in Shh-N is not known, one possibility is that signaling requires Shh-N hydrolytic activity on as yet unknown substrates. Thus, several fundamental questions about the mechanisms of Shh-N signaling remain unanswered. Does Shh-N function as a ligand or as an enzyme? Is Ptc interaction with Shh-N direct and is this a critical event in transduction of the Shh-N signal? To illuminate these issues, we used the structure as the basis for design of mutations expected to abolish zinc hydrolase activity within Shh-N. We also used structure-based systematic mutagenesis to produce Shh-N proteins with alterations in evolutionarily conserved surface residues and then compared the signaling activity of these altered proteins in neural plate explants to their capacity for binding to Ptc-expressing cultured cells. We found that Shh-N signaling does not require catalytic activity and instead correlates critically with the ability of Shh-N to bind directly to Ptc.
MATERIALS AND METHODS
Preparation of Recombinant Shh-N Mutant Proteins. Constructs for altered Shh-N were made by standard methods (26). Recombinant proteins were expressed in Escherichia coli and purified as described previously (12). To prepare the 32P-labeled Shh-N protein, a protein kinase A site tag (RRASV) was introduced at the carboxy terminus of Shh-N, and the tagged Shh-N was phosphorylated in a reaction containing
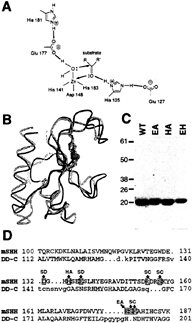
FIG. 1. A possible catalytic site in Shh-N. (A) Model for an apparent zinc hydrolase catalytic site derived from the crystal structure of Shh-N (23). Glu-177 and His-135 residues are presumed to be essential for catalysis (see text), and His-141, Asp-148, and His-183 coordinate the Zn2+ ion. (B) Superimposed alpha-carbon traces of Shh-N (yellow) and D,D-carboxypeptidase from Streptomyces albus (green). The portion of these proteins displaying structural homology is drawn, with the Zn2+ ions shown as blue spheres. Residues within the structurally homologous portion of Shh-N that are altered in SC (four of six) and SD (two of three) (see text) are located in structurally diverged loops and are highlighted in red. (C) Coomassie blue staining of purified recombinant wild type (WT) and E177A (EA), H135A (HA) and double mutant (EH) Shh-N proteins resolved in SDS/PAGE (15%); molecular mass markers are indicated at left (kDa). (D) Structure-based alignment of amino acid sequence from the portions of mouse Shh (mSHH) and Streptomyces albus D,D-carboxypeptidase (DD-C) shown in B. The residues involved in zinc coordination or hydrogen bonding of the water molecule are shown in dark blue, and other conserved residues are in light blue. Target sites for mutagenesis are indicated in green (for zinc hydrolase mutants) or red (for SC and SD mutants, see below).
[γ-32P]ATP. Cy2-labeled recombinant Shh-N was prepared by using CyDye FluoroLink Reactive Dye (Amersham).
Chicken Neural Plate Explant Culture. Chicken intermediate neural plate explant culture methods have been described previously (12, 27). Neural plate explants were stained with either mouse anti-Pax-7 [PAX7, Developmental Studies Hybridoma Bank (DSHB)], rabbit anti-hepatocyte nuclear factor (HNF)-3β (K2, a gift from T.M.Jessell, Columbia University), or mouse anti-Islet-1(40.2D6, DSHB) antibodies.
Cell Culture for Ptc Expression. Fragments encoding full-length mouse Ptc and carboxyl-terminal Myc-tagged Ptc-CTD (Ptc with a truncation resulting in a 140-residue carboxyl-terminal deletion; amino acids nos. 1–1,291, a gift from M.P. Scott, Stanford University) were inserted into pIND(Sp) vector (Invitrogen). To make stable cell lines, EcR-293 cells
(Invitrogen) were transfected with recombinant constructs or empty vector, and several independent clones for each construct were isolated.
Shh-N-Ptc-Binding Assay. Ptc expression was induced in cloned stable derivatives of the cell line EcR-293 by addition of ponasterone A (Invitrogen). After induction, 2.5×105 cells were mixed with increasing concentrations (0.1 nM-50 nM) of 32P-Shh-N (for Scatchard analyses) or with a fixed concentration (0.9 nM) of 32P-Shh-N and various concentrations of competitors (for competitive binding assays). After incubation at 4°C, cells were collected, and the bound 32P-Shh-N was determined. For the qualitative Ptc-binding assay, QT6 cells transiently transfected with pRK5-Ptc-CTD were incubated with 2 nM Cy2-labeled Shh-N protein and 160 nM unlabeled competitor. The ability of the unlabeled protein to compete for binding of Cy2-Shh-N protein to cells was directly observed by fluorescence microscopy.
Crosslinking of 32P-Shh-N to Ptc. Induced EcR-293 cells were incubated with the 32P-labeled Shh-N at a final concentration of 2 nM at 4°C. Unlabeled Shh-N was added as competitor to 200 nM. After the cells were washed once with PBS, labeled Shh-N was crosslinked to cells by adding freshly prepared disuccinimidyl suberate (Pierce) to 5 mM in PBS and incubating for 50 min. at 4°C. Crosslinked cells were washed with cold PBS and lysed in 0.15 mM NaCl/0.05 mM Tris-HCl, pH 7.2/1% Triton X-100/1% sodium deoxycholate/0.1% SDS (RIPA) buffer containing proteinase inhibitors. Lysate proteins were fractionated by SDS/PAGE (6%) and visualized by staining with Coomassie blue. After the gel was dried, crosslinked products were visualized by autoradiography.
RESULTS
Zinc Hydrolase Activity Is Not Required for Shh-N Signaling. To determine whether Shh-N acts as an enzyme, glutamate-177 (E177) and histidine-135 (H135) were substituted by alanine. E177 forms a hydrogen bond to a zinc-bound water molecule, and H135 is positioned to stabilize a potential tetrahedral intermediate (Fig. 1A; ref. 23). By analogy with other zinc hydrolases, both residues are likely to be essential for catalytic activity (28). Furthermore, the VanX protein, a structural homologue of Shh-N, displays a reduction in activity
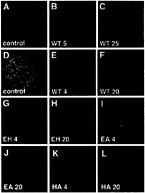
FIG. 2. Signaling activities of Shh-N zinc hydrolase mutants. (A–C) Chicken intermediate neural plate explants double stained for expression of the motor neuron marker Islet-1 (blue) and the floor plate marker HNF-3ß (red). No Islet-1- or HNF-3ß-positive cells were observed in control explants (A), whereas 5 nM (B) and 25 nM (C) concentrations of wild-type Shh-N induced expression of Islet-1 and HNF-3ß, respectively. (D–L) Neural plate explants double stained with antibodies against a dorsal marker Pax-7 (green) and the floor plate marker HNF-3ß (red). Explants cultured with medium only (D) express Pax-7 but not HNF-3ß. Wild-type Shh-N protein fully repressed expression of Pax-7 at 4 nM (E) and uniformly induced HNF-3ß inallcells at 20 nM (F). The EH and EA mutant proteins repressed Pax-7 at 4 nM (G, I, respectively), albeit somewhat less efficiently, and were able to uniformly induce HNF-3ß expression at 20 nM (H and J, respectively). The H135A (HA) mutant protein was indistinguishable from wild type (K, at 4 nM and L, at 20 nM). Images were captured using a×40 objective.
of more than six orders of magnitude on alteration of the R71 residue (29), which corresponds to H135 in Shh-N (25). These substitutions (E177A, H135A) were introduced individually and in combination into an E.coli expression vector, and purified altered proteins were prepared (Fig. 1C).
Table 1. Properties of altered Shh-N proteins
|
Protein |
Mutation sites |
Pax-7 repression, nM |
HNF-3ß induction, nM |
Ptc-CTD affinity, nM |
5E1 IP |
Heparin binding |
|
WT |
Wild type (a a 25–198) |
~4 |
=20 |
0.48 |
++ |
+ |
|
HA |
H135A |
~4 |
=20 |
0.63 |
ND |
+ |
|
EA |
E177A |
~10 |
=20 |
1.7 |
ND |
+ |
|
EH |
E177A, H135A |
~10 |
=20 |
1.7 |
ND |
+ |
|
SA |
K75A, E76A, Y81A, D105A, N116A, E189A, K195A |
~4 |
=20 |
0.66 |
++ |
+ |
|
SB |
N51A, V52A, T56A, E168A |
~4 |
=20 |
0.48 |
++ |
+ |
|
SC |
P42A, K46A, R154A, S157A, S178A, K179A |
»1,000 |
»1,000 |
»36 |
– |
– |
|
SD |
E90A, D132A, E138A |
~10 |
=20 |
0.84 |
++ |
+ |
|
SE |
P42A, K46A |
~20 |
~100 |
2.4 |
– |
+ |
|
SF |
R154A, S157A |
~70 |
=100 |
9.1 |
+ |
+ |
|
SG |
S178A, K179A |
~30 |
~100 |
4.3 |
+ |
+ |
|
Protein signaling was tested at initial concentrations of 4, 20,100, 500, and 1,000 nM and subsequently at 10 nM concentration intervals for EA, HA, EH, SD, SE, SF, and SG. The minimum concentration required for complete repression of Pax-7 and for uniform induction of HNF-3ß is shown for each protein. As an indication of affinity for Ptc-CTD, binding coefficients (KI) for binding of mutant Shh-N proteins to Ptc-CTD were derived from competitive binding experiments in Fig. 6 A and B by using the equation KI=[IC50]/(l+[L]/KL), where [IC50] is the concentration of unlabelled mutant proteins required for 50% competition. [L] is the concentration of unbound wild-type protein (32P-Shh-N) and KL is the dissociation constant for wild-type Shh-N. Immunoprecipitation by 5E1 monoclonal antibody (Fig. 7) and binding to heparin-agarose are indicated. ND, not determined. |
||||||
We used a chicken intermediate neural plate explant culture system to test the signaling activity of recombinant proteins (12). Wild-type Shh-N protein applied to these explants induced motor neurons at 5 nM and predominantly floor plate cells at 25 nM, as monitored by expression of Islet-1 and HNF-3β, respectively (Fig. 2 A–C). Shh-N protein at 4 nM sufficed for suppression of the dorsal marker Pax-7 (Fig. 2 D and E). The concentrations of Shh-N required for these inductive events, although slightly higher than previously reported (12, 15, 27), were reproducible in the assay protocol used in this study.
All three of the zinc hydrolase mutant Shh-N proteins tested, E177A (EA), H135A (HA), and the double mutant (EH), retained the capacity to repress Pax-7 expression and to induce floor plate cells in the explants (Fig. 2 G–L). Whereas the EA and EH mutant proteins displayed slightly reduced signaling
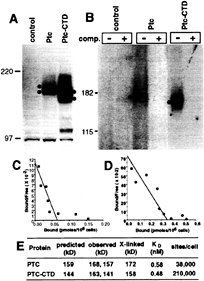
FIG. 3. Direct binding of Shh-N to Ptc. (A) Ptc and Ptc-CTD expression in stably transfected cloned cell lines. Cell lysates were prepared from stable EcR-293 cell lines carrying pIND(Sp) (empty vector control), pIND(Sp)-Ptc, or pIND(Sp)-Ptc-CTD, and proteins were fractionated by SDS/PAGE (6%) followed by blotting and detection with anti-Ptc antibody (Santa Cruz Biotechnology). Two bands (dots) were detected in lysates from Ptc or from Ptc-CTD cells, but not from control cells. (B) Crosslinking of 32P-labeled Shh-N protein to Ptc and Ptc-CTD. EcR-293 cells expressing Ptc and Ptc-CTD were incubated with 32P-Shh-N protein in absence (–) or presence (+) of a 100-fold excess of unlabeled Shh-N protein and then crosslinked. Cell lysates were subjected to SDS/PAGE (6%) and crosslinked products detected by autoradiography. Autoradiographic images for control and Ptc and for Ptc-CTD are presented at distinct contrast settings to highlight the crosslinked species. Migration of marker proteins (in kDa) is shown at left. (C, D) Scatchard analysis of the high-affinity component of 32P-Shh-N binding to EcR-293 cells expressing Ptc (C) or Ptc-CTD (D). (E) Summary of predicted molecular masses of Ptc and Ptc-CTD, experimental values estimated from Western blotting (A), and apparent masses of crosslinked products (B). Experimental values are the average of several independent determinations. Also shown are estimates of the binding coefficients of Shh-N for Ptc and for Ptc-CTD, and estimates of the number of binding sites per cell.
activity, the HA protein was indistinguishable from wild type (Fig. 2 G–L; Table 1). Because the altered residues are absolutely critical for catalytic activity in other zinc hydrolases (28, 29), retention of signaling activity by Shh-N hydrolase mutant proteins indicates that catalytic activity is not required for signaling. The reduced potency for EH and EA in signaling may reflect a destabilization of folded protein structure, as might be expected from substitution of Ala for the largely buried side chains of the Glu-177 and His-135 residues. Indeed, the EA and EH altered proteins displayed a somewhat reduced affinity for Ptc-CTD protein, which may account for their reduced potency, whereas HA was essentially indistinguishable from wild type (Table 1; see below).
Direct Binding of Shh-N Protein to Ptc. Because the analyses above suggested a noncatalytic function of Shh-N protein, we next focused on Shh-N interaction with Ptc (21, 22). To determine whether Shh-N protein directly interacts with Ptc, we generated stable cloned EcR-293 cell lines for ecdysone-inducible expression of full length Ptc and Ptc-CTD (see Materials and Methods). Such stable cell lines, but not a control line carrying the empty vector, expressed Ptc and Ptc-CTD proteins when induced with the ecdysone analog, ponasterone A (Fig. 3A). On protein blots probed with anti-Ptc antibody, two broad bands were detected for Ptc (dots, 168 kDa and 157 kDa) or for Ptc-CTD (dots, 163 kDa and 141 kDa). The estimated masses of the faster-migrating species were close to the molecular masses predicted from primary sequence (159 kDa for Ptc and 144 kDa for Ptc-CTD) (Fig. 3E).
For sensitive detection of Shh-N binding to Ptc, a 32P-labeled Shh-N protein was prepared by introducing a protein kinase A (PKA) site at the carboxy terminus of Shh-N followed by labeling of the purified recombinant protein with PKA and [γ-32P]ATP (see Materials and Methods). Addition of this kinase site at the carboxy terminus did not affect signaling activity of Shh-N (data not shown). We performed crosslinking of 32P-labeled Shh-N protein to EcR-293 cells expressing Ptc or Ptc-CTD in the presence of a bivalent crosslinker, disuccinimidyl suberate. As shown in Fig. 3B, crosslinked products were detected in lysates of Ptc and Ptc-CTD cells, but not in those of control cells. These crosslinked species were abolished by competition with unlabeled Shh-N protein (+ lanes), demonstrating a specific interaction. The crosslinked species form a single band, not two as detected in Western blotting, suggesting that a particular form of Ptc or Ptc-CTD might bind to Shh-N. The estimated molecular masses of the crosslinked products (172 kDa for Ptc and 158 kDa for Ptc-CTD) differ by 14 kDa, which corresponds closely to the differences in mass between Ptc and Ptc-CTD and definitively indicates the participation of Ptc and Ptc-CTD in these complexes. The apparent masses of these complexes furthermore are close to the sums of the masses of Shh-N plus Ptc or of Shh-N plus Ptc-CTD (178 kDa and 163 kDa, respectively) (Fig. 3E), suggesting a 1:1 stoichiometry of Ptc and Shh-N in these complexes. These results strongly suggest that Shh-N interacts directly with Ptc protein.
Quantitative analysis of 32P-Shh-N binding to these cells revealed a high-affinity Ptc-dependent component of binding that could be competed by nanomolar concentrations of unlabeled Shh-N and a low-affinity component that was not dependent on Ptc expression and that could not be competed by Shh-N. Scatchard analysis of the Ptc- and Shh-N-specific high affinity component (Fig. 3 C and D) indicated that the binding coefficients of Ptc and Ptc-CTD for 32P-Shh-N protein are similar (0.58 nM and 0.48 nM respectively; Fig. 3E) (21). Assuming, as argued above, that one Shh-N ligand binds to one Ptc molecule, the number of binding sites per cell for Ptc-CTD (210,000) is about 5.5 times higher than that for Ptc (38,000) (Fig. 3E). The temperature utilized in these binding studies (4°C) is not permissive of endocytosis, indicating that Shh-N binding initially occurs on the cell surface, even though immunofluorescence studies clearly demonstrate that Ptc and

FIG. 4. Alteration of Shh-N surface residues. (A) Ribbon diagram and (B, C) surface representations of Shh-N. B is shown in the same orientation asy A, but C is rotated 180° about a vertical axis relative to A and B. Surface-exposed evolutionarily conserved residues that were selected for alteration cluster into four major regions: SA (blue), SB (green), SC (red), and SD (yellow). Residues mutagenized are indicated in Table 1. (D) Coomassie blue stain of an SDS/PAGE separation of purified mutant Shh-N proteins. SE, SF, and SG denote proteins with distinct subsets of the altered residues in SC (see Table 1). Migration of molecular mass markers indicated at left (in kDa). Figs. 1B and 4A made with MOLSCRIPT (36); Fig. 4 B and C were made with GRASP (37).
Ptc-CTD proteins are predominantly localized inside cells (data not shown). The difference in number of binding sites for these two proteins thus could be caused either by a higher degree of surface localization for Ptc-CTD or, alternatively, by a higher level of Ptc-CTD expression as compared with Ptc (Fig. 3A), a phenomenon also consistently observed in transiently transfected cells (data not shown). Thus we cannot at present distinguish whether the 140 residues absent from Ptc-CTD influence the subcellular localization of the Ptc protein or its steady-state levels within the cell.
The Role of Shh-N Surface Residues in Signaling and in Ptc Binding. Having demonstrated a direct and high-affinity interaction between Ptc and Shh-N, we set out to determine the significance of this interaction by examining the correlation between Ptc binding and signaling potency of altered Shh-N proteins. The Shh-N protein was subjected to systematic mutagenesis to identify surface residues involved in signaling and potential ligand/receptor interactions. Because Hh pro
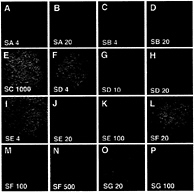
FIG. 5. Signaling activities of Shh-N proteins with altered surface residues. Neural plate explants stained for expression of Pax-7 (green) and HNF-3ß (red). Explants were cultured in the presence of the indicated proteins at the indicated concentrations (nM). SA and SB proteins are as active as wild-type Shh-N, because they repress expression of Pax-7 at 4 nM and induce expression of HNF-3ß at 20 nM. The SD protein is slightly less active than wild-type protein, and the SC mutant protein is completely inactive. The SE and SG proteins display reduced activity, and the SF protein is even less active. Results are summarized in Table 1. Images were captured using a×40 objective.
teins can act similarly across species and in distinct biological settings [Shh, for example, is active in Drosophila (30, 31) and distinct vertebrate proteins can act in common pathways (32)], it seems likely that surface residues potentially important in inductive activities and ligand/receptor interactions would be conserved. The Shh-N structure was used to identify surface residues based on degree of side chain exposure to solvent (23). Among these surface residues, those that are evolutionarily conserved were geographically divided into four major regions named SA, SB, SC, and SD (Fig. 4 A–C) and subjected to mutagenesis. We initially generated four mutant proteins, each containing multiple alanine substitutions at the conserved surface residues within each region (see Table 1). Because the side chains of the residues selected are solvent exposed, we expected that the folded structures of these proteins would not be affected.
The altered proteins were purified (Fig. 4D) and applied to chicken neural plate explant cultures. The SA and SB altered proteins repressed Pax-7 expression and induced floor plate cells in the explants as well as the wild-type protein (Fig. 5 A–D), and the SD altered protein displayed an approximate 2.5-fold reduction in activity (Fig. 5 F–H). In striking contrast, no signaling activity of the SC mutant could be detected even at 1 µM, a concentration 250-fold higher than that required for Pax-7 repression by wild-type protein (Fig. 5E; results summarized in Table 1). We next examined Ptc binding for these altered proteins using a competition binding assay. The SA, SB, and SD mutant proteins competed with the 32P-labeled wild-type Shh-N protein for binding to Ptc-CTD expressing cells as well or nearly as well as the wild-type protein (Fig. 6A), yielding similar binding coefficients (Table 1). Ptc-binding activity of the SC mutant, however, was not detectable (Fig. 6A; Table 1), suggesting a possible correlation between Ptc binding and signaling activity for the Shh-N protein.
To explore this correlation further, we tested three additional proteins (SE, SF, and SG), each with alterations in two amino acid residues that comprise distinct subsets of the six residues altered in SC (see Table 1). All three of these mutant proteins displayed signaling activity in the explant culture assay, but only at significantly reduced levels. At 4 nM none of these three proteins repressed Pax-7 (Fig. 5I; data not shown); at 20 nM the SE and SG proteins repressed Pax-7 almost completely or partially, respectively, but SF did not (Fig. 5 J, L, and O). At 100 nM, the SE and SG proteins induced HNF-3ß expression in most cells of the explant, but SF did so only in a small number of cells (Fig. 5 K, M, and P). Further assays at concentration intervals of 10 nM pinpointed the minimal concentrations required for Pax-7 repression, with values of ˜20, ˜30, and ˜70 nM for SE, SG, and SF, respectively (results in Table 1). Competition binding assays
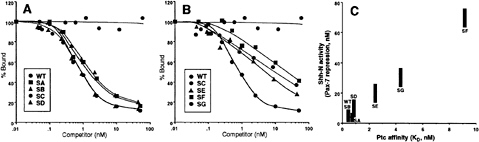
FIG. 6. Binding of altered Shh-N proteins to Ptc. (A and B) Competition by altered proteins for binding of 32P-Shh-N to EcR-293 cells expressing Ptc-CTD. Binding of 32P-Shh-N in the presence of each altered protein at various concentrations is normalized to the total value of 32P-Shh-N bound (approximately 35% of input) in the absence of competitor. The SC mutant, inactive in signaling, also fails to compete for binding to Ptc-CTD. The SE, SF, and SG proteins with intermediate levels of signaling activity, displayed intermediate levels of competition for binding to Ptc-CTD. Data are summarized in Table 1. (C) Signaling activity as a function of Ptc affinity. On the basis of neural plate signaling assays (Figs. 2, 5; Table 1), protein concentrations required for Pax-7 repression are plotted as a function of Ptc-binding affinity. The protein concentrations are plotted as ranges centered about the concentrations presented in Table 1. Note that there is an excellent correlation between Ptc binding and activity in Pax-7 repression. The zinc hydrolase mutants EA, HA, and EH (Table 1) also corroborate this correlation but are omitted for clarity.
also revealed a significantly reduced affinity of the SE and SG proteins for Ptc-CTD, and an even lower affinity for the SF protein (Fig. 6B; Table 1). These results indicate that normal Ptc binding and neural plate signaling activities require distinct contributions from multiple individual residues in the SC surface region. Furthermore, among proteins with alterations in distinct subsets of the SC mutant residues, Ptc-binding affinity correlated extremely well with neural plate signaling activity (Fig. 6C).
We also purified Shh-N proteins with deletions of amino- or carboxyl-terminal residues and examined their activities qualitatively in signaling and in Ptc binding (Table 2). An altered protein lacking nine amino-terminal residues (∆N34) displayed signaling and Ptc-binding activities indistinguishable from wild type. In contrast, ∆N50, which lacks 25 amino-terminal residues, completely lost both activities. We note that the residues deleted in ∆N50 include P42 and K46, which were altered in the SC and SE mutant proteins, and that the mutant ∆N45 (lacking 20 amino-terminal residues), which does not contain P42, also lost signaling activity. The activity defects in these proteins are more severe than those of the SE protein, suggesting that loss of these amino-terminal residues may have some effect on the overall structure or stability of the Shh-N protein. A deletion mutant lacking residues from 166 to the carboxy terminus, ∆C165, had neither signaling nor Ptc-binding activities, and ∆C101 also lost signaling activity (Table 2). These deletions also remove residues that are altered in the SC protein (R154, S157, S178, and K179 in ∆C101; S178 and
Table 2. Properties of Shh-N deletion derivatives
|
Protein |
Residues present |
HNF-3β induction |
Ptc-CTD binding |
5E1 IP |
Heparin binding |
|
WT |
wild type (a a 25–198) |
+ |
+ |
+ |
+ |
|
∆N34 |
a a 34–198 |
+ |
+ |
+ |
+ |
|
∆N45 |
a a 45–198 |
– |
ND |
ND |
– |
|
∆N50 |
a a 50–198 |
– |
– |
– |
– |
|
∆C165 |
a a 25–165 |
– |
– |
– |
+ |
|
∆C101 |
a a 25–101 |
– |
ND |
ND |
+ |
|
The properties of these mutant proteins were either indistinguishable or completely different from wild type in a qualitative neural plate assay for induction of HNF-3β or in a qualitative Ptc-binding competition assay using QT6 cells transiently transfected with Ptc-CTD and Cy2-labeled Shh-N (see Material and Methods). Immunoprecipitation by 5E1 monoclonal antibody and binding to heparin agarose are indicated. ND, not determined. |
|||||
K179 in ∆C165), but again, the deleted regions are sufficiently extensive that they would be expected to affect protein structure.
We note that inallof the altered proteins tested we failed to find a single example of a protein that retained signaling activity while losing the ability to bind Ptc. As seen in Fig. 6C, there is an excellent correlation between Ptc binding and signaling activity inallaltered proteins for which these properties can be measured, and these results strongly suggest that Ptc binding may be a critical requirement for signaling.
Antibody Recognition and Heparin Binding of Altered Proteins. The monoclonal antibody 5E1, directed against Shh-N, blocks signaling in neural plate explants (14) (data not shown) and also blocks binding of the Shh-N protein to Ptc-expressing cells (data not shown). The reactivity of the 5E1 antibody with altered Shh-N proteins was examined by immunoprecipitation. We found thatallproteins that retain signaling and Ptc-binding activities, including wild type, SA, SB, SD, and ∆N34, also retain full reactivity with 5E1 (Fig. 7A; Tables 1, 2; data not shown). In contrast, the altered proteins SC, ∆N50, and ∆C165, which lost both signaling and Ptc-binding activities, were not immunoprecipitated by 5E1 (Fig. 7A; Tables 1, 2; data not shown). Altered proteins with intermediate signaling and Ptc-binding properties, such as SE, SF, and SG, displayed intermediate reactivities with 5E1 (Fig. 7B; Table 1). Reactivity of 5E1 with Shh-N proteins thus correlates well with Ptc binding and neural plate signaling activities.
Because 5E1 works well for immunoprecipitation and for immunocytochemistry but very poorly in Western analysis (data not shown), it appears to recognize an epitope present on the native Shh-N protein but not in denatured protein. The strong correlation between 5E1 binding, Ptc binding, and neural plate signaling furthermore suggests that the 5E1 epitope coincides with determinants required for these activities. One possible explanation for the coordinate loss of signaling, 5E1 binding, and Ptc binding in the SC protein is that the folded structure of this protein might be disrupted. Circular dichroism analysis, however, indicates that the secondary structure profile of SC is similar to that of wild-type Shh-N (data not shown), suggesting that any disruption in folded structure must be highly local in nature. In addition, mutations in distinct subsets of the residues altered in SC display intermediate phenotypes, suggesting multiple independent contributions of individual residues in formation of the Ptc-interacting region of the protein surface.
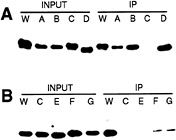
FIG. 7. Reactivity of 5E1 antibody with altered Shh-N proteins. After immunoprecipitation (IP) with the 5E1 monoclonal antibody, altered proteins were detected by Western blotting using a polyclonal antibody. The starting (input) and precipitated (IP) material are shown for each protein. (A) The wild-type, SA, SB, and SD altered proteins were precipitated well by the 5E1 antibody, but the SC protein was not. (B) SE, SF, and SG displayed intermediate reactivity with the 5E1 antibody.
The Shh-N protein also binds to heparin [(12); data not shown], and the crystal structure contains a sulfate anion at a location near the SC region (23). In addition, recent evidence suggests that tout velu, a Drosophila gene whose mammalian homologues function in the polymerization of glycosamines for synthesis of heparan sulfate proteoglycans (33, 34), plays a role in the reception and transport of the Hh signal (35). We therefore tested whether the alterations in our proteins affect their ability to bind to heparin agarose. As seen in Tables 1 and 2, only three of the proteins tested, SC, ∆N45, and ∆N50, lost the ability to bind heparin agarose, and these three proteins are completely inactive in Ptc binding and signaling. Some of the proteins that lose signaling and Ptc-binding activity retained the ability to bind heparin, indicating that heparin binding is not sufficient for Ptc binding and for signaling. Our data, however, would be consistent with the idea that heparin binding may be necessary for Ptc binding and for signaling.
DISCUSSION
The Putative Zinc Hydrolase in Shh-N. Alterations in residues that should be critical for the putative zinc hydrolase activity of Shh-N did not disrupt its ability to induce ventral neural cell types or to suppress dorsal markers, suggesting that catalytic activity is not required for Shh signaling in the neural plate. Although residues constituting the putative zinc hydrolase active site are widely conserved among Hh family members, they are not fully conserved in Drosophila (23), suggesting that hydrolase function is not required for signaling in this organism. We also introduced and ectopically expressed the E177A and EH mutant Shh constructs into Drosophila and compared their ability to mispattern the embryonic cuticle with that of wild-type Shh (30) and could detect no significant difference between them (H.E.F.Takahashi and P.A.B., unpublished data), further substantiating dispensability of catalytic activity for Shh-N signaling function in the context of developing Drosophila embryos. Furthermore, experiments with mutant proteins expressed in cultured cells suggest that the putative hydrolase activity is not required for the normal biogenesis and processing of Shh, nor for its normal state of modification (data not shown). We also note that we failed to detect any hydrolase activity of Shh-N in biochemical assays with a variety of substrates, including some like those for D,D-carboxypeptidase, which contained D-amino acid residues.
The putative zinc hydrolase of Shh-N has thus resisted our attempts to reveal an activity, either in biochemical or in in vitro or in vivo signaling assays, raising the possibility that the putative catalytic site represents an evolutionary vestige of its common ancestry with the D,D-carboxypeptidase family of proteins. In this view, the zinc atom may have lost its ancestral role in catalysis but could have retained a role in stabilizing protein structure through interactions with the side chains of coordinating residues. The lack of conservation of coordinating residues in the Drosophila protein may indicate a replacement of these interactions by other stabilizing interactions. General dispensability of hydrolase activity in Hh signaling is consistent with the importance of surface residues conserved among Hh proteins for binding to Ptc and for signaling (see below). Alternatively, it is possible that Shh-N hydrolase retains a role not detected by our biochemical or in vitro and in vivo signaling assays. Such a role likely would be modulatory in nature, given the essentially normal signaling activity of hydrolase mutant proteins, and its discovery may require targeted recombination to mutagenize the endogenous mouse Shh gene.
Direct Binding of Shh-N to Ptc and Activation of the Shh Pathway. Previous genetic and biochemical studies are consistent with the idea that Ptc may function as a Hh receptor. The biochemical analyses demonstrated that Shh-N protein binds to Ptc-expressing cells, that Ptc is coimmunoprecipitated with Shh-N and vice versa, and that Ptc can be crosslinked in a complex containing Shh-N (21, 22). Because the composition of the crosslinked complexes was not characterized, however, these studies could not exclude the possibility that instead of binding directly to Ptc, Shh-N may bind to another component of a complex that includes Ptc. These biochemical studies also did not examine the role of such an interaction in the activation of the Shh pathway. The latter is a particularly significant issue given the genetically demonstrated role of Ptc in sequestration of Hh protein to restrict its movement within Drosophila tissues (20).
In addressing these questions, we have identified a crosslinked product containing radiolabeled Shh-N that is specifically competed by unlabeled Shh-N but not by the unlabeled mutant SC protein. This crosslinked complex also contains Ptc because its formation depends on Ptc expression and because it displays an apparent molecular mass difference that corresponds closely to the differences between full-length Ptc or Ptc-CTD. Finally, for both Ptc molecules, the apparent mass of the complex is close to the sum of Shh-N plus Ptc. Thus, although direct binding ideally would be demonstrated by studies with purified components, the properties of our crosslinked complexes strongly suggest a direct association between Shh-N and Ptc with a probable stoichiometry of 1:1. Given the possible anomalies in migration of such crosslinked species, we cannot rule out the possibility that more than one Shh-N molecule is present in these complexes, nor can we distinguish between the participation of the slower- or faster-migrating Ptc forms, which probably differ in their glycosylation (22). Although it has been reported that Ptc interacts with Smo independently of Shh-N (21), the apparent masses of our crosslinked products would appear to exclude Smo, which has a predicted mass of 87 kDa (21). It is possible that the cells we utilized do not express Smo endogenously or that high-level expression of Smo is required for formation of a complex with Ptc/Shh. Alternatively, our experimental conditions for crosslinking might disrupt Ptc-Smo interaction or fail to capture Smo protein.
We also have identified, using altered Shh-N proteins, the region of the Shh-N protein surface that is involved in Ptc binding and have used these altered proteins to show that neural plate signaling activity is retained in proportion to the binding affinity for Ptc. Thus, the extensive alterations in surface residues of the SA, SB, and SD proteins do not affect
or only mildly affect Ptc binding, and these proteins retain normal or nearly normal signaling activity. At the other extreme, the SC altered protein displays a complete loss of Ptc binding, and this is reflected in a complete loss of neural plate signaling activity. Even more telling, proteins carrying distinct subsets of the residues altered in SC result in intermediate levels of Ptc-binding activity and corresponding intermediate levels of neural plate signaling activity (Fig. 6C). Multiple individual residues within the SC surface region that contribute independently to Ptc binding thus also similarly contribute to signaling potency. Although our results cannot exclude a role for interactions with other proteins in reception of the Shh signal, they do strongly suggest that direct binding to Ptc is a critical step, and this information will serve as the basis for further elucidation of downstream events.
We thank Drs. M.Scott and T.Jessell for various cDNAs and antibodies. We also thank J.Wrable for help with circular dichroism analysis, Dr. J.Taipale for suggestions on the crosslinking experiment, Dr. M.Cooper and K.Young for preparation of some recombinant proteins, Dr. O.Sundin for help with chicken embryo experiments, Dr. R.Mann for comments on the manuscript, and members of the Beachy laboratory for discussions and suggestions. N.F. was a postdoctoral fellow of the Human Frontier Science Program. D.J.L. and P.A.B. are investigators of Howard Hughes Medical Institute. This work was supported in part by grants from the American Paralysis Association and the Ara Parseghian Medical Research Foundation.
Under a licensing agreement between Ontogeny, Inc. and the Johns Hopkins University, Dr. Beachy and the University hold equity in Ontogeny and are entitled to a share of royalties from sales of products related to the research described in this article. Dr. Beachy serves on Ontogeny’s Scientific Advisory Board and as a consultant to the company. All financial aspects of these arrangements are managed by the University in accordance with its policies.
1. Perrimon, N. (1995) Cell 80, 517–520.
2. Hammerschmidt, M., Brook, A. & McMahon, A.P. (1997) Trends Genet. 13, 14–21.
3. Goodrich, L.V. & Scott, M.P. (1998) Neuron 21, 1243–1257.
4. Chiang, C., Litingtung, Y., Lee, E., Young, K., Corden, J.L., Westphal, H. & Beachy, P. (1996) Nature (London) 383, 407–413.
5. Beachy, P.A., Cooper, M.K., Young, K.E., von Kessler, D.P., Park, W.J., Hall, T.M., Leahy, D.J. & Porter, J.A. (1997) Cold Spring Harbor Symp. Quant. Biol. 62, 191–204.
6. Lee, J.J., Ekker, S.C., von Kessler, D.P., Porter, J. A, Sun, B.I. & Beachy, P.A. (1994) Science 266, 1528–1537.
7. Porter, J. A, von Kessler, D.P., Ekker, S.C., Young, K.E., Lee, J.J., Moses, K. & Beachy, P.A. (1995) Nature (London) 374, 363–366.
8. Porter, J.A., Young, K.E. & Beachy, P.A. (1996) Science 274, 255–259.
9. Pepinsky, R.B., Zeng, C., Wen, D., Rayhorn, P., Baker, D.P., Williams, K.P., Bixler, S.A., Ambrose, C.M., Garber, E.A., Miatkowski, K., et al (1998) J. Biol. Chem. 237, 14037–14045.
10. Porter, J.A., Ekker, S.C., Park, W.-J., von Kessler, D.P., Young, K.E., Chen, C.-H., Ma, Y., Woods, A.S., Cotter, R.J., Koonin, E.V., et al (1996) Cell 86, 21–34.
11. Tanabe, Y. & Jessell, T.M. (1996) Science 274, 1115–1123.
12. Roelink, H., Porter, J.A., Chiang, C., Tanabe, Y., Chang, D.T., Beachy, P.A. & Jessell, T.M. (1995) Cell 81, 445–455.
13. Tanabe, Y., Roelink, H. & Jessell, T. (1995) Curr. Biol. 5, 651–658.
14. Ericson, J., Morton, S., Kawakami, A, Roelink, H. & Jessell, T.M. (1996) Cell 87, 661–673.
15. Ericson, J., Rashbass, P., Schedl, A., Brenner-Morton, S., Kawakami, A., van Heyningen, V., Jessell, T.M. & Briscoe, J. (1997) Cell 90, 169–180.
16. Ingham, P.W., Taylor, A.M. & Nakano, Y. (1991) Nature (London) 353, 184–187.
17. Ingham, P.W. & Hidalgo, A. (1993) Development (Cambridge, U.K.) 117, 283–291.
18. Tabata, T. & Kornberg, T.B. (1994) Cell 76, 89–102.
19. Ingham, P.W. (1993) Nature (London) 366, 560–562.
20. Chen, Y. & Struhl, G. (1996) Cell 87, 553–563.
21. Stone, D.M., Hynes, M., Armanini, M., Swanson, T.A., Gu, Q., L, J.R., Scott, M.P., Pennica, D., Goddard, A., Phillips, H., Noll, M.,, et al (1996) Nature (London) 384, 129–134.
22. Marigo, V., Davey, R. A, Zuo, Y., Cunningham, J.M. & Tabin, C.J. (1996) Nature (London) 384, 176–179.
23. Hall, T.M.T., Porter, J.A., Beachy, P.B. & Leahy, D.J. (1995) Nature (London) 378, 212–216.
24. Dideberg, O., Charlier, P., Dive, G., Joris, B., Frere, J.M. & Ghuysen, J.M. (1982) Nature (London) 299, 469–470.
25. Bussiere, D.E., Pratt, S.D., Katz, L., Severin, J.M., Holzman, T. & Park, C.H. (1998) Mol. Cell 2, 75–84.
26. Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A. & Struhl, K. (1994) Current Protocols in Molecular Biology (Wiley, New York).
27. Cooper, M.K., Porter, J.A., Young, K.E. & Beachy, P.A. (1998) Science 280, 1603–1607.
28. Christianson, D.W. (1991) Adv. Protein Chem. 42, 281–355.
29. Lessard, I.A. & Walsh, C.T. (1999) Chem. Biol. 6, 177–187.
30. Chang, D.T., Lopez, A, von Kessler, D.P., Chiang, C., Simandl, B.K., Zhao, R., Seldin, M.F., Fallen, J.F. & Beachy, P.A. (1994) Development (Cambridge, U.K.) 120, 3339–3353.
31. Krauss, S., Concordet, J.-P. & Ingham, P.W. (1993) Cell 75, 1431–1444.
32. Ekker, S.C., McGrew, L.L., Lai, C.-J., Lee, J.J., von Kessler, D.P., Moon, R.T. & Beachy, P.A. (1995) Development (Cambridge, U.K.) 121, 2337–2347.
33. McCormick, C., Leduc, Y., Martindale, D., Mattison, K., Esford, L.E., Dyer, A.P. & Tufaro, F. (1998) Nat. Genet. 19, 158–161.
34. Lind, T., Tufaro, F., McCormick, C., Lindahl, U. & Lidholt, K. (1998) J. Biol. Chem. 273, 26265–8.
35. Bellaiche, Y., The, I. & Perrimon, N. (1998) Nature (London) 394, 85–88.
36. Kraulis, P.J. (1991) J. Appl. Crystallogr. 24, 946–950.
37. Nicholls, A., Sharp, K.A. & Honing, B. (1991) Proteins 11, 281–296.








