
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes
D. A. MATTHEWS*, P. S. DRAGOVICH, S. E. WEBBER, S. A. FUHRMAN, A. K. PATICK, L. S. ZALMAN, T. F. HENDRICKSON, R. A. LOVE, T. J. PRINS, J. T. MARAKOVITS, R. ZHOU, J. TIKHE, C. E. FORD, J. W. MEADOR, R. A. FERRE, E. L. BROWN, S. L. BINFORD, M. A. BROTHERS, D. M. DELISLE, AND S. T. WORLAND
Agouron Pharmaceuticals, Inc., 3565 General Atomics Court, San Diego, CA 92121
ABSTRACT Human rhinoviruses, the most important etiologic agents of the common cold, are messenger-active single-stranded monocistronic RNA viruses that have evolved a highly complex cascade of proteolytic processing events to control viral gene expression and replication. Most maturation cleavages within the precursor polyprotein are mediated by rhinovirus 3C protease (or its immediate precursor, 3CD), a cysteine protease with a trypsin-like polypeptide fold. High-resolution crystal structures of the enzyme from three viral serotypes have been used for the design and elaboration of 3C protease inhibitors representing different structural and chemical classes. Inhibitors having a,ß-unsaturated carbonyl groups combined with peptidyl-binding elements specific for 3C protease undergo a Michael reaction mediated by nucleophilic addition of the enzyme’s catalytic Cys-147, resulting in covalent-bond formation and irreversible inactivation of the viral protease. Direct inhibition of 3C proteolytic activity in virally infected cells treated with these compounds can be inferred from dose-dependent accumulations of viral precursor polyproteins as determined by SDS/PAGE analysis of radiolabeled proteins. Cocrystal-structure-assisted optimization of 3C-protease-directed Michael acceptors has yielded molecules having extremely rapid in vitro inactivation of the viral protease, potent antiviral activity against multiple rhinovirus serotypes and low cellular toxicity. Recently, one compound in this series, AG7088, has entered clinical trials.
Picornaviruses are small nonenveloped RNA viruses with a single strand of messenger-active genomic RNA 7,500–8,000 nucleotides in length, which is replicated in the cytoplasm of infected cells. The family currently is divided into six genera with similar genetic organization and translational strategies. Among its members are several important human and veter-inary pathogens, including poliovirus and coxsackievirus (Enterovirus), foot-and-mouth disease virus (Aphthovirus), encephalomyocarditis virus (Cardiovirus), hepatitis A virus (Hepatovirus), and human rhinoviruses (Rhinovirus). As a consequence of limitations imposed by a small monocistronic RNA viral genome, picornaviruses depend on a strategy for temporal gene expression that includes highly controlled cotranslational and posttranslational processing of a precursor polyprotein by virally encoded proteases to generate the individual structural and nonstructural proteins needed for viral replication. While still in the process of synthesis, the polyprotein is cleaved proteolytically by the virally encoded 2A protease to release P1, the precursor to capsid proteins, from P2–P3. Subsequent processing of P1 to 1AB, 1C, and 1D and all P2 and P3 processing to release proteins needed for RNA replication depend on viral 3C protease activity (1–3).
In addition to its role in polyprotein processing, picornavirus 3C sequences are involved in proteolytic degradation of specific cellular proteins associated with host-cell transcription and in direct binding to viral RNA as part of a replication complex required for synthesis of plus-strand viral RNA (4–7).
Rhinoviruses are primary causative agents of the common cold. Whereas these infections are usually mild and self-limiting, consequences can be more severe for the elderly, for immune-compromised individuals, and for those predisposed to respiratory illness such as asthma (8). In the case of picornaviruses with limited serotypic diversity, such as poliovirus, foot-and-mouth disease virus, and hepatitis A virus, highly protective vaccines have been developed that are in use worldwide. On the other hand, developing effective immunizations against rhinovirus infections or against the pathogenic nonpolio enteroviruses is anticipated to be more challenging, owing to the large number of existing serotypes: at least 100 rhinoviruses and 65 enteroviruses. In an attempt to address this need, we have undertaken a program directed at discovering rhinovirus 3C protease inhibitors with antiviral activity against the spectrum of known rhinovirus serotypes. The results of these efforts and the identification of an antirhinoviral compound now entering clinical trials are described below.
Picornaviral 3C Proteases
Picornaviral 3C proteases are small monomeric proteins with molecular masses around 20 kDa. Crystal structures exist for 3C proteases from type 14 human rhinovirus (9), hepatitis A (10), and poliovirus (11). Viral 3C proteases fold into two topologically equivalent six-stranded ß-barrels with an extended shallow groove for substrate binding located between the two domains. In rhinovirus 3C protease, the catalytically important residues Cys-147, His-40, and Glu-71 form a linked cluster of amino acids with an overall geometry similar to the Ser-His-Asp catalytic triad found in the trypsin-like family of serine proteases. The highly conserved sequence Gly-X-Cys-Gly-Gly in viral 3C proteases serves to position Cys-147 for nucleophilic attack on the substrate’s carbonyl carbon and to orient backbone NH groups of Gly-145 and Cys-147 to form an “oxyanion hole” for stabilization of a tetrahedral transition state (9). Thus, the catalytic machinery for activation of the
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviation: CBZ, benzyloxycarbonyl. |
|
|
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB code 1CQQ). |
|
* |
To whom reprint requests should be addressed. E-mail: David.Matthews@Agouron.com. |
attacking nucleophile and stabilization of a tetrahedral intermediate-transition state in 3C proteases closely resembles that of trypsin-like serine proteases, suggesting that the viral 3C proteases are related mechanistically to serine proteases rather than to the papain-like cysteine proteases. Picornaviral 3C proteases process a limited number of cleavage sites in the virally encoded polyprotein. Most cleavages occur between Gln-Gly peptide bonds with distinct differences in the efficiency of cleavage at various junction sites. Recombinant rhinovirus 3C protease has an absolute requirement for Gln-Gly cleavage junctions in peptide substrates ranging from 7 to 11 aa in length (12).
Inhibitors of 3C Protease and the Issue of Serotypic Diversity Among Rhinoviruses
Picornaviral 3C proteases represent a unique class of enzymes that integrate characteristics of both serine and cysteine proteases with an unusual specificity for Gln-Gly cleavage junctions. The absence of known cellular homologues contributes to interest in 3C protease as a potentially important target for antiviral drug design. However, the vast serotypic diversity among rhinoviruses raises the question of whether or not a single agent can effectively target 3C proteases from the 100 or so rhinovirus serotypes capable of infecting humans. Primary sequence data are available for 3C proteases from 10 different rhinovirus serotypes, including the type 2 and type 14 enzymes that have less than 50% amino acid identity.
To address these diversity concerns before initiating a concerted drug-discovery effort, we undertook a program to obtain structural information on peptide-based inhibitors bound to 3C proteases from multiple rhinovirus serotypes. We wanted to identify the geometric and electronic factors that modulate protein/substrate (inhibitor) recognition, the extent to which specific residues that form the substrate (inhibitor) binding site of 3C protease are conserved across rhinovirus serotypes, and whether or not these binding-site residues are arranged similarly in 3C proteases from different virus serotypes.
Peptide Aldehydes Bound to Serotype 2 Rhinovirus 3C Protease. Peptide aldehydes have been used extensively as inhibitors of serine and cysteine proteases, although they typically have not proven effective as drug candidates because of their poor pharmacological properties. They bind as reversible adducts in which the nucleophilic cysteine or serine makes a covalent bond with the carbonyl carbon of the aldehyde, forming a stable tetrahedral species. Short peptidic aldehydes having sequences similar to canonical 3C protease cleavage sites have been reported as inhibitors of both rhinovirus and hepatitis A viral proteases (13–15). The combination of glutamine at P1 with aldehyde functionality causes cyclization on the aldehyde. (16). To circumvent this problem, replacements for the γ-carboxamide were sought that prevent internal cyclization but retain high affinity for the 3C protease S1 specificity pocket (15). Compound I (Fig. 1) is an N-terminal protected tripeptide aldehyde in which the -CH2C(O)NH2 of Gln is replaced with an N-acetyl isostere. Compound I is a 6-nM inhibitor of type 14 human rhinovirus 3C protease. Whereas the original x-ray structural studies of rhinovirus 3C protease were performed by using the serotype 14 enzyme (9), subsequent analysis of inhibitor binding was carried out mainly with type 2 3C protease, both because of the relative ease in obtaining cocrystals and their generally superior diffraction properties. Fig. 2 shows the 2.2-Å x-ray structure of compound I complexed with serotype 2 rhinovirus 3C protease (15).
The peptide aldehyde I binds to rhinovirus 3C protease in a partially extended conformation with inhibitor backbone atoms aligned for antiparallel β-sheet-type hydrogen bonding with an exposed β-strand (βE2) of the protein comprising
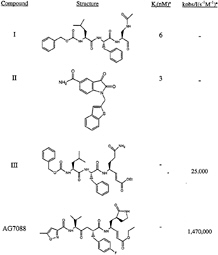
FIG. 1. Rhinovirus 3C protease inhibitors. Ki, inhibition constant; kobs, observed rate of inactivation; I, inhibitor concentration.
residues 162–165. The inhibitor’s P1 side chain lies in a shallow pocket bounded by βE2, by residues 142–144, and by His-161, the last of which donates a hydrogen bond to the N-acetyl oxygen. This oxygen accepts a second hydrogen bond from the side-chain hydroxyl of Thr-142. The inhibitor’s acetyl methyl group is close to the backbone carbonyl of Thr-142 (3.3 Å), suggesting that substrates or inhibitors having a similarly positioned P1 glutamine-like side chain could form a third hydrogen bond to enhance specific recognition of a γ-carboxamide group.
The P1 backbone amide makes a weak (3.2-Å) hydrogen bond with the carbonyl oxygen of Val-162. The deep S2 pocket easily accommodates the inhibitor’s bulky P2 Phe side chain, which is bounded on one side by the side chain of His-40 and on the other side by residues 127–130. Two ordered water molecules reside at the back of the S2 pocket. The side-chain hydroxyl and backbone NH of Ser-128 form hydrogen bonds with the inhibitor’s P2 NH and the carbonyl oxygen of the terminal benzyloxycarbonyl (CBZ) group, respectively. Two main-chain hydrogen bonds tether the inhibitor’s P3 Leu to backbone atoms of Gly-164, whereas the isobutyl side chain is mostly solvent-exposed. The benzyl portion of the CBZ group packs into a shallow hydrophobic pocket that probably accommodates a substrate’s P4 side chain. The side chain of Asn-165 is positioned directly above the benzene of CBZ, with its carboxamide NH pointing into the face of the aromatic ring, suggesting that some additional binding energy probably derives from this favorable amino-aromatic interaction (17).
The affinity of peptide aldehyde inhibitors for trypsin-like serine proteases has been attributed to their ability to form, with the active-site serine, hemiacetals that resemble the transition state in amide hydrolysis, with the oxyanion stabilized in a structurally conserved oxyanion hole. Considering the structural homology between 3C protease and trypsin-like serine proteases, we anticipated that the tetrahedral hemithioacetal oxygen of compound I when bound to 3C would be positioned similarly within the oxyanion hole. Indeed, we showed previously that 2,3-dioxindole inhibitors (see Fig. 1, compound II) form stable tetrahedral adducts with 3C pro-
FIG. 2. Compound I bound to serotype 2 human rhinovirus 3C protease. The protein is rendered as a semitransparent solvent-accessible surface with associated protein backbone and side-chain atoms colored pink. Catalytic triad residues are blue. Red spheres represent ordered solvent molecules. Inhibitor atoms are colored green for carbon, blue for nitrogen, and red for oxygen. The inhibitor carbon covalently bonded to Cys-147 is highlighted in light green.
tease in which the O3 oxygen is stabilized in just this manner (18). However, compound I binds in a non-transition-state conformation with the oxygen of the hemithioacetal stabilized by hydrogen bonding to Ne2 of His-40.
Compared with 3C protease complexes with 2,3-dioxindole inhibitors, the complex with compound I also differs in the main-chain conformation for protein residues 144–145. The peptide linkage joining Ser-144 and Gly-145 flips around so that NH (residue 145), instead of pointing into the oxyanion hole, is directed out toward solvent where it hydrogen bonds with an ordered water molecule. This structure suggests that, for 3C, optimum alignment of NH dipoles to form a classically configured oxyanion hole analogous to that seen in trypsin-like serine proteases may not occur in the native protein but rather requires a conformational change induced by substrate (or inhibitor) binding.
The Extended Substrate (Inhibitor) Binding Site for Rhinovirus 3C Protease Is Highly Conserved Among Different Viral Serotypes. High-resolution x-ray crystal structures for serotype 2 and serotype 16 3C proteases (overall amino acid sequence identity of 80%) bound to various peptide-based aldehyde inhibitors reveal that the two respective active sites are nearly identical (D.A.M., unpublished results). Not only do protein backbone atoms superpose within experimental error (<0.3 Å), but amino acid side chains interacting with peptide aldehyde inhibitors are identically conserved and oriented similarly in the complexes, except at position 130. Even for the more distantly related rhinovirus serotypes, there is a high level of amino acid identity for 3C protease residues that modulate binding of peptide aldehyde inhibitors such as compound I. There are 21 residues in serotype 2 3C protease that interact directly with the bound inhibitor. Of these, 17 are identically conserved in the 10 3C proteases of known sequence from different rhinovirus serotypes. For three of the nonconserved residues (residues 126, 144, and 146), interactions with the bound inhibitor are modulated by peptide backbone atoms only, suggesting that side-chain variation at these positions may not affect inhibitor binding significantly. Only in the case of residue 130 is there a nonconserved amino acid with a side chain directly contacting compound I. Residue 130 is either Asn or Thr in the 10 known rhinovirus 3C protease sequences. In the type 2 enzyme, Asn-130 is positioned at the back of the S2 specificity pocket where its side chain is in van der Waals contact with the inhibitor’s P2 benzyl group (Fig. 2). Nearby, but not directly contacting the P2 Phe, is a second nonconserved residue at position 69 (Lys or Asn, depending on serotype) that hydrogen bonds to ordered water molecules at the back of the S2 pocket. In summary, the available crystallographic and amino acid sequence data suggest that inhibitors of rhinovirus 3C protease could be expected to show efficacy against the enzyme from multiple viral serotypes provided they do not depend on binding determinants at the back of the S2 specificity pocket where structural variability between serotypes may be most pronounced.
Strategies for Rhinovirus 3C Protease Inhibitor Design. Several considerations come into play when developing strategies for design of therapeutically efficacious serine and cysteine protease inhibitors. For many of. these proteins, specificity pockets for substrate (or inhibitor) recognition are shallow, and binding determinants are widely dispersed over
large surface areas. Difficulties inherent in discovering small molecules with high affinity for such binding sites are in many respects analogous to those encountered in attempting to disrupt protein-protein interactions with small effector molecules. Serine proteases such as factor Xa and thrombin, proteins involved in the blood-coagulation pathway with deep well defined S1 specificity pockets, have been targeted effectively with structurally diverse, small, noncovalent inhibitors and thus are exceptions to this generalization (19). However, for virally encoded serine and cysteine proteases of known structure, such as the herpes family of serine proteases, hepatitis C NS3 protease, and picornavirus 3C proteases, the fact that substrate recognition is modulated by extensive protein-protein interactions represents a significant impediment for design of specific inhibitors. We know that inhibitor potency can be enhanced by taking advantage of the possibility for covalent adduct formation afforded by the presence of a reactive serine or cysteine at the active sites of these proteases. In the case of 3C, these effects are dramatic. Whereas compound I has a Ki of 6 nM against the serotype 14 enzyme, reduction of the aldehyde functionality to the corresponding alcohol yields a molecule with no measurable inhibition at concentrations as high as 100 µM (15). An optimized 9-aa substrate for 3C has a Km of only 400 µM, showing weak binding to this protease even for relatively large peptide substrates (12).
Not surprisingly, in light of these results, we have had little success identifying small noncovalent inhibitors of 3C protease. The alternative approach of incorporating specific noncovalent recognition plus an electrophile that can react covalently with the active site nucleophile is conceptually attractive. However, potency and the inherent chemical reactivity of the electrophilic center are usually correlated. Highly reactive electrophiles are likely to target nonselectively other cellular proteins and nonenzymatic biological nucleophiles, such as glutathione, rendering such agents unacceptable as drug candidates. In earlier work, we reported on the design of potent reversible 3C protease inhibitors based on a 2,3-dioxindole (isatin) core (18). When elaborated with substituents providing recognition in the S1 and S2 specificity pockets of 3C protease, inhibitors with low nanomolar Ki were obtained. An x-ray cocrystal structure of compound II revealed covalent attachment of Cys-147 to the electrophilic center (C2) with the carboxamide and benzothiophene groups positioned as expected in the S1 and S2 pockets (18). Unfortunately, all isatin inhibitors tested were devoid of antiviral activity and/or were toxic, properties most probably attributable to their high electrophilic reactivity. These findings led us to consider other types of covalent inhibitors where the chemical reactivity of the electrophilic center can be more effectively modulated in the context of molecules having high specificity for 3C protease.
Irreversible Michael Acceptors as Inhibitors of 3C Protease
Peptidic substrates in which the scissile amide carbonyl is replaced by a Michael acceptor were first introduced as specific irreversible inhibitors of the cysteine protease papain by Hanzlik and coworkers (20, 21). We reasoned that, although this reaction is probably facilitated by the especially nucleophilic thiolateimidazolium ion pair in papain-like cysteine proteases, suitably activated Michael acceptors might also undergo addition by the presumably less nucleophilic catalytic cysteine of 3C. A trans-a, ß-unsaturated ethyl ester incorporated into a CBZ protected tripeptide corresponding to the N-terminal portion of a canonical 3C protease cleavage sequence (Fig. 1, compound III) afforded a compound with relatively potent irreversible inhibition of 3C (22). The compound had moderate antiviral activity in HeLa cells infected with rhinovirus serotype 14, was nontoxic to the limit of its solubility, and was not inactivated by short exposure to DTT. These results encouraged us to initiate additional studies of Michael acceptors to enhance their activity against 3C protease further.
Fig. 3 shows the 2.3-Å x-ray structure of compound III bound to serotype 2 3C protease. The peptidic portion of the molecule closely resembles that of the aldehyde I and binds similarly to the enzyme active site (24). Unlike compound I, the P1 side chain of compound III is identical to that for Gln, the P1 residue in the vast majority of 3C cleavage sequences. The carboxamide oxygen accepts hydrogen bonds from the side chains of His-161 and Thr-142, and the amide nitrogen donates hydrogen bonds to the backbone carbonyl oxygen of Thr-142 and to an ordered water molecule. Thus, all possible hydrogen bonding interactions for a Gln side chain are fully satisfied within the complimentary S1 binding site. The geometrical specificity conferred by these highly directional hydrogen bonds is important in orienting the inhibitor’s vinyl group (or in the case of a substrate, the susceptible carbonyl carbon) for nucleophilic attack by Cys-147. Cys-147 is covalently linked to the inhibitor’s electrophilic ß-carbon with the carbonyl oxygen of the ethyl ester positioned above the oxyanion hole, where it makes a hydrogen bond to the backbone amide of Cys-147. As observed for aldehyde inhibitors bound to 3C protease, the 144–145 peptide linkage has the backbone amide pointing away from the oxyanion hole, although low occupancy (˜20%) of the other conformer having NH (residue 145) directed toward the oxyanion hole is seen in this and several other P1 Gln-containing Michael acceptors for which we have obtained high-resolution x-ray cocrystal structures. The ethyl ester portion of the Michael acceptor extends into the leaving group side of the protease active site formed by residues 22–25 and by the tight loop connecting ß-strands ßA2 and ßB2. The leaving group pocket is of sufficient size to accommodate the ethyl ester group easily in an extended low-energy Z conformation.
As noted previously, the stretch of amino acids 142–146 immediately N-terminal to the catalytic cysteine is important in 3C proteases for both substrate recognition and stabilization of the tetrahedral intermediate-transition state. In the absence of bound ligands, the corresponding residues in rhinovirus, poliovirus, and hepatitis A 3C proteases exist in multiple conformations and/or are highly mobile, as evidenced by average temperature factors of 50–60 Å2. In rhinovirus 3C protease cocrystal structures with inhibitors having Gln-like side chains, the segment 142–146 adopts a well defined conformation (except for the 144–145 peptide linkage, which has either of two conformations) with temperature factors below the average for the remainder of the protein. Thus, Gln side-chain recognition in the S1 pocket is tightly coupled with a disorder-to-order transition in a crucial region of the protein involved in transition-state stabilization. The available crystallographic evidence suggests that peptides lacking Gln-like functionality at P1 are unable to select the catalytically relevant conformation for the protein segment 142–146 from an ensemble of accessible states, providing a structural explanation for the observation that proteolysis of short 7- to 11-aa peptides by 3C protease has an absolute requirement for Gln at the P1 position (12). This observation also underscores the probable importance of P1 Gln functionality in mechanism-based activation of Michael acceptors as inhibitors of 3C protease.
Covalent irreversible inactivation of 3C by Michael acceptors proceeds according to a kinetic mechanism that can be broken down into two parts (Scheme 1).
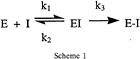

FIG. 3. Compound III bound to serotype 2 human rhinovirus 3C protease. Color coding is the same as in Fig. 2.
The inhibitor initially forms a reversible encounter complex with 3C, which can then undergo a chemical step (nucleophilic attack by Cys-147) leading to stable covalent-bond formation. The observed second-order rate constant for inactivation (kobs/I) depends on both the equilibrium binding constant k2/k1 and the chemical rate for covalent bond formation k3 (23). We anticipated that Michael-acceptor inhibitors with specificity for 3C protease would likely achieve high rates of enzyme inactivation by combining good equilibrium binding with a modest rate of covalent-bond formation. The rate of chemical inactivation presumably depends on not only the intrinsic electrophilic character of the inhibitor, but on how the reactive vinyl group is oriented in the active site relative to Cys-147 before nucleophilic attack and on the extent to which the transition state for the reaction can be stabilized by the enzyme. Mechanism-based activation of an inherently weak Michael acceptor as a means of increasing the rate of the chemical step, and thus kobs/I, is conceptually more attractive than attempting to achieve a similar effect by simply increasing intrinsic electrophilic reactivity, which would likely impart undesirable properties to such compounds.
Within this conceptual framework, we experimented first with the effect of varying the Michael-acceptor electron-withdrawing group and then, for a subset of electrophiles with suitable antiviral and toxicity profiles, proceeded to a second level of optimization involving the 3C protease recognition portion of compound III.
Michael-Acceptor Inhibitors of 3C Protease: Structure-Activity Studies
Variation of the Michael Acceptor. Recently, an extensive structure-activity study exploring modification of the Michael-acceptor portion of compound III has been published (24). The results can be summarized as follows. (i) A series of ester-derived Michael acceptors with substituted alcohol groupsallshowed good inhibitory activity with kobs/I values of 3,000 to 40,000 M–1.s–1. The benzyl ester had higher anti-3C protease activity than the parent compound (kobs/I=39,400 compared with 25,000 M–1.s–1 for compound III) but performed worse in the antiviral assay (EC50=3.2 vs. 0.54 µM for compound III). cis-a,ß-Unsaturated esters or trans-a,ß-unsaturated esters substituted at the a-position had reduced activity compared with the benchmark compound III. (ii) Amide-containing Michael acceptors in general had reduced activity against 3C protease, poorer antiviral activity, and/or increased toxicity compared with the corresponding esters. (iii) Aliphatic and aryl a,ß-unsaturated ketones were extremely potent anti-3C protease agents with kobs/I values between 120,000 and 500,000 M–1.s–1. However, these molecules had reduced antiviral activity (EC50 > 2 µM) and were toxic to cells. The ketones were also inactivated by short exposure to DTT, consistent with their expected high electrophilicity. (iv) Vinyl sulfones, nitriles, phosphonates, oximes, and several vinyl heterocycles had weak (kobs/I < 600 M–1.s–1) or no detectable inhibitory activity, (v) Michael acceptors with acyl lactam, acyl oxazolidinone, and acyl urea functionalities were potent 3C protease inhibitors but, like the corresponding ketones, were inactivated by exposure to nonenzymatic thiols.
As a consequence of their good inhibitory activity against 3C protease, their encouraging antiviral activity, stability in the presence of nonenzymatic thiols, low cellular toxicity, and ease of synthesis, trans-a,ß-unsaturated esters emerged as the Michael acceptors of choice with which to initiate the process of optimizing the peptidic portion of compound III.
Variation of 3C Protease Recognition Elements. Analogs of compound III truncated after the P1 Gin or after the P2 Phe were poor 3C protease inhibitors with kobs/I values of 4.5 and 400 M–1.s–1, respectively. Therefore, structure-activity studies were conducted with tripeptide-derived molecules (24).
Substitutions at P1. Michael acceptors incorporating any variation in the γ-carboxamide portion of the P1 side chain had weak or no 3C protease inhibitory activity. Inclusion of various heteroatoms in the aliphatic portion of the glutamine side chain also reduced activity compared with the benchmark molecule III (24). As described above, the serotype 2 3C protease cocrystal structure with compound III indicates that the P1 side-chain cis-NH is exposed to solvent. Selective alkylation of the amide was viewed as a means of reducing inhibitor peptide character without compromising binding. We enforced cis-amide geometry by incorporating a P1 lactam moiety into the inhibitor design. Based on modeling, we predicted that (S) stereochemistry would be required at the lactam α-carbon to position correctly lactam side-chain hydrogen bonding functionality, which is essential for recognition and binding in the S1 pocket. The resulting molecule was 10-fold more potent than compound III against type 14 3C protease and more than 5-fold better as an antiviral agent in cell culture (25).
Substitutions at P2. Replacement of the P2 benzyl side chain generally leads to reduced inhibitory properties. Smaller aliphatic side chains having fewer van der Waals contacts with the large S2 specificity pocket are particularly poor inhibitors. In the case of type 14 3C protease, additional functionality at the 4-position can lead to modestly higher kobs/I values; however, the same compounds when tested against 3C from other rhinovirus serotypes were often less inhibitory than compound III. The 4-fluoroPhe analog was moderately more potent than the parent compound in assays against 3C protease from serotypes 2, 14, and 16 (24). The P2 backbone amide of compound III donates a hydrogen bond to the side-chain oxygen of invariant Ser-128. Ser-128 is located in a turn on an exposed, somewhat flexible loop forming one side of the S2 specificity pocket (Fig. 3). Various 3C protease cocrystal structures indicate that this loop can undergo small (≈1.5-Å) inhibitor-specific conformational changes. We reasoned that replacement of the P2–P3 peptide bond with ketomethylene functionality would reduce the peptidic character of the resulting molecule, whereas loss of the exposed surface hydrogen bond might not impact inhibitory activity severely. The ketomethylene inhibitor showed slightly reduced 3C protease inhibition (17,400 M–1.s–1), compared with that of compound III, but had improved antiviral properties (26).
Substitutions at P3. The leucine side chain of compound III is solvent exposed. As expected, a wide variety of functionality is tolerated at this position with minor effects on enzyme inhibitory activity (24).
Substitutions at P4. Attempts to optimize the N-terminal (P4) functionality focused initially on modifications to the benzyl portion of the CBZ group to enhance binding in the hydrophobic S4 specificity pocket. We were also interested in exploring replacements for the carbamate oxygen atom adjacent to the benzyl group. The cocrystal structure of compound III with serotype 2 3C protease (Fig. 3) reveals that this inhibitor oxygen atom is positioned partially inside the S4 pocket with a gap between it and the side chain of Phe-170 (24). The thiocarbamate analog of CBZ had significantly increased inhibitory activity (kobs/I=280,000 M–1.s–1) and improved antiviral properties (EC50=0.27 μM). A 1.9-Å crystal structure of the thiocarbamate analog of compound III bound to serotype 2 3C protease indicated that the thiocarbamate sulfur atom lies 1.5 Å deeper in the S4 pocket than the corresponding oxygen of compound III and is in van der Waals contact with Phe-170 (24). Replacement of oxygen with the larger, more easily polarized, and more easily dehydrated S atom probably accounts for much of the increase in kobs/I by enhancing equilibrium binding of the inhibitor to 3C protease before covalent-bond formation.
Concerns about possible metabolic instability of P4 thiocarbamate containing 3C protease inhibitors prompted a more systematic search for other N-terminal amides with improved activity compared with compound III. Tripeptidyl ethyl propenoate Michael acceptors of sequence Leu-Phe-Gln were assembled on solid supports. The N-terminal amine was coupled to a variety of carboxylic acids and acid chlorides to yield approximately 500 N-terminal protected tripeptide Michael acceptors. These compounds were screened subsequently against type 14 3C protease by using high-throughput assay techniques (27). Accordingly, the N-terminal 5-methylisoxazole-3-carboxamide analog was identified as a potent 3C protease inhibitor (kobs/I=260,000 M–1.s–1) with improved antiviral activity (EC50=0.25 μM) compared with that of compound III.
AG7088, a 3C Protease Inhibitor with Potent Antiviral Activity Against Multiple Human Rhinovirus Serotypes
For each position in the N-terminal protected tripeptide portion of compound III, modifications were identified that imparted increased activity against 3C protease and better antiviral properties compared with those of the parent molecule. We anticipated that by combining several of these individually beneficial modifications into a single molecule, further improvements in enzyme inhibition and antiviral activity could be achieved. Below, the inhibitory, antiviral, and enzyme-specificity properties of one such compound, AG7088, are described further.
Activity Against Rhinovirus 3C Protease. The covalent structure of AG7088 is shown in Fig. 1. The compound has excellent activity against serotype 14 3C protease (kobs/I=1, 470,000 M–1.s–1) and is a potent antiviral agent with low toxicity in the HeLa cell assay (EC50=0.013 μM; toxic concentration, 50% > 100 μM; ref. 28). AG7088 is highly specific for picornavirus 3C proteases, having negligible inhibitory activity against a panel of mammalian cysteine and serine proteases, including cathepsin B, elastase, chymotrypsin, trypsin, thrombin, and calpain (25). Direct inhibition of rhinovirus 3C proteolytic activity in virally infected H1-HeLa cells treated with AG7088 can be inferred from dose-dependent accumulations of viral precursor proteins shown by SDS/PAGE analysis of radiolabeled polyproteins (28). A crystal structure of AG7088 bound to serotype 2 3C protease was determined at 1.85-Å resolution (Fig. 4).
The overall binding mode of AG7088 to 3C protease is generally similar to that described for compound III; however, the structurally distinct N-terminal protecting groups are oriented differently in the protein’s S4 binding subsite. As anticipated, the five-member lactam ring at P1 makes three hydrogen bonds with the protease similar to those for com-pound III. However, as a result of constraints imposed on the internal geometry of the lactam ring, the hydrogen bond between the lactam amide NH and the backbone carbonyl of Thr-142 is longer (3.2 Å) and the geometry less favorable than in the case of compound III, in which optimal positioning of the P1 carboxamide by rotation about the Cδ–Cγ bond is less hindered. Why then does replacement of the P1 Gin in compound III with a five-member lactam ring increase kobs/I against type 14 3C protease by almost a factor of 10?
The relatively rigid lactam side chain at P1 stands to lose less conformational entropy on binding in the S1 pocket than the more flexible Gin and therefore probably binds tighter to 3C protease than its acyclic counterpart. Another favorable effect of binding on entropy may result from the manner in which the lactam affects the conformation of unbound AG7088 in solution. We have determined the small-molecule crystal structure
FIG. 4. AG7088 bound to serotype 2 rhinovirus 3C protease. The protein is rendered as a semitransparent solvent-accessible surface color coded at each residue according to amino acid conservation among the 10 serotypically distinct rhinovirus ß C proteases of known primary structure. Residues indicated in dark blue are identically conserved among the 10 known sequences. Increasing amino acid variation at a particular residue is indicated by progressively warmer coloring, with purple signifying two differences and red signifying seven differences among the 10 known 3C protease sequences. Other color coding is the same as in Fig. 2, except that the fluorine atom of AG7088 is purple.
of AG7088 (T.L.Hendrixson, unpublished results) and find that its conformation is very similar to that observed for AG7088 in complex with 3C protease. In both cases, the two lactam (CH2) groups pack against the side chain of the P3 valine, which may help stabilize the active conformer in solution thus reducing entropy loss on inhibitor binding. The two lactam (CH2) groups also create additional van der Waals contacts with backbone atoms of residues 143 and 144, which, compared with a P1 Gln, may further reduce the flexibility and conformational heterogeneity that is observed for this region in the absence of bound inhibitors. Particularly noteworthy is that, for AG7088 bound to 3C protease, the peptide bond 144–145 has its NH pointing in toward the oxyanion hole where it may play a role in hydrogen bonding to the carbonyl oxygen of the Michael acceptor in the transition state for Michael addition. We have determined cocrystal structures for five P1 cyclic lactam-containing 3C protease inhibitors, and in each case, the 144–145 peptide is in what we believe to be the active conformation. In contrast, more that 20 cocrystal structures of P1 Gln-containing irreversible 3C protease inhibitors aII show this peptide bond turned around with the backbone NH group pointing out into solvent (see Fig. 3). These results suggest that the greater ability of a P1 lactam to stabilize the catalytically active conformation of residues N-terminal to the nucleophilic Cys-147 may accelerate the chemical step and thus contribute to the increase in kobs/I compared with P1 Gln-containing analogs.
In compound III, the P2 backbone amide donates a hydrogen bond to the side-chain hydroxyl of Ser-128. As a consequence of replacing this group with a methylene moiety in AG7088, surface-exposed Ser-128 moves 0.7 Å where it can interact preferentially with bulk solvent. The isoxazole group of AG7088 is more buried in the S4 pocket than the CBZ of compound III and is oriented orthogonal to the CBZ benzene ring. The isoxazole oxygen is positioned close to the side chain of Phe-170, which moves on average about 0.6 Å compared with its position in the complex with compound III. Deeper penetration of this group into S4 also causes positional changes (˜0.8 Å) centered around the backbone and side-chain atoms of Asn-165 with somewhat smaller displacements for Gly-166 as well. One consequence of these induced protein movements is that the shape of the S1 pocket changes slightly, particularly in the region proximate to the P1 side-chain amide and its attached methylene, suggesting that alterations in the N-terminal blocking group can affect binding of the P1 substituent.
Antiviral Activity of AG7088 Against Rhinovirus Serotypes. In H1-HeLa or MRC-5 cell protection assays, AG7088 inhibited replication of all 48 rhinovirus serotypes tested to date (28), including representative virus strains derived from minor and major receptor groups (29). The mean EC50 and EC90 values are 0.023 µM (range: 0.003–0.081 µM) and 0.082 µM (range: 0.018–0.261 µM), respectively (28). Pirodavir and pleconaril are antipicornaviral agents that bind to viral capsids, preventing receptor attachment and/or viral uncoating. Pirodavir inhibited the replication of 42 of 47 rhinovirus serotypes tested with a mean EC50 value of 0.32/µM (range: 0.003–4.770 µM), whereas pleconaril inhibited replication of 42 of 45
serotypes tested with a mean EC50 value of 0.822 µM (range: 0.003–8.112 µM) (28). The 50% cytotoxic concentration of AG7088 is > 1,000 µM compared with 150 µM and 77 µM for pirodavir and pleconaril, respectively (28). These studies establish AG7088 as a highly potent, nontoxic antirhinoviral agent with broad efficacy against multiple virus serotypes. The compound has been formulated for intranasal delivery and has recently entered clinical trials.
Experimental Crystal Structure of AG7088 Bound to Serotype 2 Rhinovirus 3C Protease. Serotype 2 human rhinovirus 3C protease was incubated with a 3-fold molar excess of AG7088 in the presence of 2% (vol/vol) DMSO for 24 h at 4°C. The complex was concentrated to 6.8 mg/ml and then passed through a 0.22-µm cellulose-acetate filter. Crystals were grown at 13°C by using a hanging-drop vapor-diffusion method in which equal volumes (3 µl) of the protein-ligand complex and reservoir solution were mixed on plastic coverslips and sealed over individual wells filled with 1 ml of reservoir solution containing 20% (vol/vol) polyethylene glycol (molecular weight 10,000) and 0.1 M Hepes (pH 7.5).
A single crystal measuring 0.3×0.1×0.1 mm (space group P212121; a=34.32, b=65.68, c=77.89 Å) was prepared for low-temperature data collection by transfer to an artificial mother liquor solution consisting of 400 µl of the reservoir solution mixed with 125 µl of glycerol and then flash frozen in a stream of N2 gas at –170°C. X-ray diffraction data were collected with a MAR Research 345-mm imaging plate and processed with DENZO. Diffraction data were 89.2% complete to a resolution of 1.85 Å with R(sym)=1.9%. Protein atomic coordinates from the cocrystal structure of type 2 3C protease with compound I (15) were used to initiate rigid-body refinement in X-PLOR followed by simulated annealing and conjugate gradient minimization protocols. Placement of the inhibitor, addition of ordered solvent, and further refinement proceeded as described in ref. 15. The final R factor was 21.8% [12,184 reflections with F > 2s(F)]. The root-mean-square deviations from ideal bond lengths and angles were 0.016 Å and 2.9°, respectively. The final model consisted of all atoms for residues 1–180 (excluding the side chain of residues 12, 21, 45, and 65) plus 221 water molecules.
1. Kräusslich, H.G. & Wimmer, E. (1988) Annu. Rev. Biochem. 57, 701–754.
2. Kay, J. & Dunn, B.M. (1990) Biochem. Biophys. Acta 1048, 1–8.
3. Lawson, M.A. & Semler, B.L. (1990) Curr. Top. Microbiol. Immunol. 161, 49–87.
4. Roehl, H.H., Parsley, T.B., Ho, T.V. & Semler, B.L. (1997) J.Virol. 71, 578–585.
5. Leong, L.E.C., Walker, P.A. & Porter, A.G. (1993) J. Biol Chem. 268, 25735–25739.
6. Andino, R., Rieckhof, G.E., Achacoso, P.L. & Baltimore, D. (1993) EMBO J. 12, 3587–3598.
7. Xiang, W., Harris, K.S., Alexander, L. & Wimmer, E. (1995) J. Virol. 69, 3658–3667.
8. Sperber, S.J. & Hayden, F.G. (1988) Antimicrob. Agents Chemother. 32, 409–419.
9. Matthews, D.A., Smith, W.W., Ferre, R.A., Condon, B., Budahazi, G., Sisson, W., Villafranca, J.E., Janson, C.A., McElroy, H.E., Gribskov, C.L., et al. (1994) Cell 77, 761–771.
10. Allaire, M., Chernaia, M.M., Malcolm, B.A. & James, M.N.G. (1994) Nature (London) 369, 72–76.
11. Mosimann, S.C., Cherney, M.M., Sia, S., Plotch, S. & James, M.N.G. (1997) J. Mol. Biol. 273, 1032–1047.
12. Long, L.A, Orr, D.C., Cameron, J.M., Dunn, B.M. & Kay, J. (1989) FEBS Lett. 258, 75–78.
13. Malcolm, B.A, Lowe, C., Shechosky, S., Mckay, R.T., Yang, C.C., Shah, V.J., Simon, R.J., Vederas, J.C. & Santi, D.V. (1995) Biochemistry 34, 8172–8179.
14. Shepherd, T.A., Cox, G.A., McKinney, E., Tang, J., Wakulchik, M., Zimmerman, R.E. & Villarreal, E.C. (1996) Bioorg. Med. Chem. Lett. 6, 2893–2896.
15. Webber, S.E., Okano, K., Little, T.L., Reich, S.H., Xin, Y., Fuhrman, S.A., Matthews, D.A., Love, R.A., Hendrickson, T.F., Patick, A.K., III, et al. (1998) J. Med. Chem. 41, 2786–2805.
16. Kaldor, S.W., Hammond, M., Dressman, B.A, Labus, J.M., Chadwell, F.W., Kline, A.D. & Heinz, B.A. (1995) Bioorg. Med. Chem. Lett. 5, 2021–2026.
17. Burley, S.K. & Petsko, G.A. (1988) Adv. Protein Chem. 39, 125–153.
18. Webber, S.E., Tikhe, J., Worland, S.T., Fuhrman, S.A, Hendrickson, T.F., Matthews, D.A., Love, R.A., Patick, A.K., Meador, J.W., Ferre, R.A., et al. (1996) J. Med. Chem. 39, 5072–5082.
19. Sanderson, P.E.J. & Naylor-Olsen, A.M. (1998) Curr. Med. Chem. 5, 289–304.
20. Hanzlik, R.P. & Thompson, S.A. (1984) J. Med. Chem. 27, 711–712.
21. Liu, S. & Hanzlik, R.P. (1992) J. Med. Chem. 35, 1067–1075.
22. Dragovich, P.S., Webber, S.E., Babine, R.E., Fuhrman, S. A, Patick, A.K., Matthews, D.A., Lee, C.A., Reich, S.H., Prins, T.J. & Marakovits, J.T. (1998) J. Med. Chem. 41, 2806–2818.
23. Meara, J.P. & Rich, D.H. (1995) Bioorg. Med. Chem. Lett. 5, 2277–2282.
24. Dragovich, P.S., Webber, S.E., Babine, R.E., Fuhrman, S.A., Patick, A.K., Matthews, D.A., Reich, S.H., Marakovits, J.T., Prins, T.J. & Zhou, R. (1998) J. Med. Chem. 41, 2819–2834.
25. Dragovich, P.S., Webber, S.E., Babine, R.E., Fuhrman, S.A., Patick, A.K., Matthews, D.A, Reich, S.H., Marakovits, J.T., Prins, T.J. & Zhou, R. (1999) J. Med. Chem. 42, 1213–1224.
26. Dragovich, P.S., Prins, T.J., Zhou, R., Fuhrman, S.A., Patick, A.K., Matthews, D.A., Ford, C.E., Meador, J.W., Ferre, R.A. & Worland, S.T. (1999) J. Med. Chem. 42, 1203–1212.
27. Dragovich, P.S., Zhou, R., Skalitzky, D.J., Fuhrman, S.A., Patick, A.K., Ford, C.E., Meador, J.W. & Worland, S.T. (1999) Bioorg. Med. Chem. Lett. 7, 589–598.
28. Patick, A.K., Binford, S.L., Brothers, M.A., Jackson, R.L., Ford, C.E., Diem, M.D., Maldonado, F., Dragovich, P.S., Zhou, R., Prins, T.J., et al. (1999) Antimicrob. Agents Chemother, in press.
29. Uncapher, C.R., DeWitt, C.M. & Colonno, R.J. (1991) Virology 180, 814–817.








