
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Cysteine protease inhibitors as chemotherapy: Lessons from a parasite target
PAUL M. SELZER*†, SABINE PINGEL‡, IVY HSIEH*, BERNHARD UGELE§, VICTOR J. CHAN*, JUAN C. ENGEL*, MATTHEW BOGYO¶, DAVID G. RUSSELL||, JUDY A. SAKANARI*, AND JAMES H. MCKERROW*,**,††
Departments of *Pathology, **Pharmaceutical Chemistry, ¶Biochemistry, and ‡Medicine, University of California, San Francisco, CA 94143; |Washington University, St. Louis, MO 63110; and §I.Frauenklinik, Klinikum Innenstadt, Ludwig-Maximilians Universität München, 80337 Munich, Germany
ABSTRACT Papain family cysteine proteases are key factors in the pathogenesis of cancer invasion, arthritis, osteoporosis, and microbial infections. Targeting this enzyme family is therefore one strategy in the development of new chemotherapy for a number of diseases. Little is known, however, about the efficacy, selectivity, and safety of cysteine protease inhibitors in cell culture or in vivo. We now report that specific cysteine protease inhibitors kill Leishmania parasites in vitro, at concentrations that do not overtly affect mammalian host cells. Inhibition of Leishmania cysteine protease activity was accompanied by defects in the parasite’s lysosome/endosome compartment resembling those seen in lysosomal storage diseases. Colocalization of anti-protease antibodies with biotinylated surface proteins and accumulation of undigested debris and protease in the flagellar pocket of treated parasites were consistent with a pathway of protease trafficking from flagellar pocket to the lysosome/endosome compartment. The inhibitors were sufficiently absorbed and stable in vivo to ameliorate the pathology associated with a mouse model of Leishmania infection.
Leishmaniasis is a parasitic infection caused by various species of the protozoan Leishmania. Transmitted by the bite of sand flies, Leishmania infects 12 million people and is endemic in tropical regions of America, Africa, and the Indian subcontinent, as well as in the subtropics of Southeast Asia and the Mediterranean. Three hundred and fifty million people live in areas where the disease is common, and large epidemics affecting hundreds of thousands have occurred as recently as 1991 (1). The severe visceral form of leishmaniasis may also be an opportunistic disease in AIDS patients (1). The problem of leishmaniasis is compounded by the inadequacy of current chemotherapy. The first-line drugs are antimonial derivatives that were developed more than 40 years ago. They produce serious side effects, and refractory cases are a problem. Second-line drugs are even more toxic, and require long, repeated doses with close observation (1).
To address the need for new, cost-effective leads for the chemotherapy of leishmaniasis, we have applied strategies of structure-based drug design (2). An attractive target for new chemotherapy is a family of cathepsin L-like (cpL) and cathepsin B-like (cpB) cysteine proteases found inallspecies of Leishmania examined, and required for parasite growth or virulence (3–5). In studies with Leishmania mexicana, elimination of selected cysteine protease genes by homologous recombination showed that null mutants of the cpL gene array designated “cpb” had reduced virulence in highly susceptible BALB/c mice, and they produced no lesions atallin C57BL/6 or CBA/Ca mice (3, 4). Double null mutants of the cpL gene families “cpb” and “cpa” produced no lesions even in BALB/c mice (3). Deletion of the cpB gene “cpc” led to reduced survival of parasites in macrophages (3, 6). While structurally distinct, Leishmania cpL and cpB overlap in substrate specificity (2). Inhibitors that would effectively target both types of cysteine proteases in Leishmania, while maintaining some selectivity versus homologous host enzymes, would be ideal drug leads.
We have identified both reversible and irreversible cysteine protease inhibitors that meet these criteria. Reversible inhibitors were discovered through a structure-based drug design screen and subsequent combinatorial synthetic optimization using models of both Leishmania major cpB and cpL (2). The irreversible inhibitors are pseudopeptide substrate analogues that take advantage of the unique reactivity of the active site sulfhydryl of cysteine proteases to confer specificity for this enzyme family but maintain activity against both cpL and cpB proteases (7, 8).
METHODS
Inhibitors. The reversible inhibitors ZLIII43A and ZLIII115A are derivatives of oxalic bis[(2-hydroxy-1-naphthyl)methylene]hydrazide, a cysteine protease inhibitor lead compound found in a computer graphics screen of the Fine Chemicals Directory (9). Several of the synthetic derivatives of that lead, produced by combinatorial synthetic chemistry, proved to be potent inhibitors of homologous cpLs of malaria (10) and the Leishmania cpB (2). The irreversible inhibitor used was the pseudopeptide substrate analogue morpholine urea-phenylalanine-homophenylalanine-vinylsulfonyl-benzene (K11002, Arris Pharmaceuticals, South San Francisco, CA). Inhibitors were prepared as 20 mM stocks in dimethyl sulfoxide (DMSO) and stored at –20°C.
Protease Assays. The native L.major cpB was a gift of Jacques Bouvier (Novartis, St. Aubin, Switzerland). Papain (EC 3.4.22.2) and mammalian cathepsin B (bovine spleen; EC 3.4.22.1) were from Sigma. Recombinant cruzain was produced as previously described (11). All proteases were assayed at 25°C with an automated microtiter plate spectrofluorometer (Labsystem FluoroScan II; Northbrook, IL). Activity was detected by the liberation of 7-amino-4-methylcoumarin
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviations: cpL, cathepsin L-like cysteine protease; cpB, cathepsin B-like cysteine protease; AMC, 7-amino-4-methylcoumarin. |
|
† |
To whom reprint requests may be addressed at present address: Hoechst Roussel Vet GmbH, Research Pharmaceuticals, Building H 811, D-65926 Frankfurt/Main, Germany. E-mail: PSelzer@hrvet.com. |
|
†† |
To whom reprint requests may be addressed at: Department of Pathology, University of California, Tropical Disease Research Unit, VAMC, 4150 Clement Street 113B, San Francisco, CA 94121. E-mail: jmck@cgl.ucsf.edu. |
(AMC) (excitation wavelength=355 nm and emission wave-length=460 nm) from the synthetic peptide substrate Z-Phe-Arg-AMC (Z=benzyloxycarbonyl) (Enzyme Systems Products, Livermore, CA). The enzyme concentrations were determined by active site titration. Reversible inhibitors at various concentrations were preincubated with the respective enzyme for 5 min before the reaction was started by adding the substrate. Enzyme activities were expressed in percent of residual activity compared with an uninhibited control, and were plotted versus increasing inhibitor concentrations to calculate the IC50. Assay conditions were as follows: L.major cpB: 100 mM sodium acetate at pH 5.5, 10 mM dithiothreitol (DTT), 1 mM EDTA, 0.1% Triton X-100, 50 µM Z-Phe-Arg-AMC final concentration (from a 10 mM stock solution in DMSO); Km=7 µM. Papain and mammalian cathepsin B: 100 mM sodium acetate at pH 5.5, 10 mM DTT, 100 µM Z-Phe-Arg-AMC final concentration; Km=50 µM and 110 µM, respectively. Cruzain: The assay conditions were the same as for papain except that the substrate concentration was 20 µM; Km=1 µM. Km values were determined by nonlinear regression using the software ULTRAFIT (Biosoft, Ferguson, MO). Irreversible inhibitors were assayed in a time-based inactivation assay. The inactivation process was based on the following scheme
where E=enzyme, I=inhibitor, EI=noncovalent enzyme inhibitor complex, E-I=inactivated enzyme, k1 and k–1= noncovalent rate constants (Ki=k–1/k1), and kinact= first-order inactivation constant. The values of Ki and kinact were determined from progress curves in the presence of substrate and inhibitor. These curves were fit to a first-order equation (ULTRAFIT) to produce kobs (observed inactivation constant) values, where kobs=kinact[I]/Ki, app+[I], where Ki, app= apparent Ki). Plotting 1/kobs versus 1/[I] gives the values for Ki,app and kinact. Taking the substrate into consideration, the true Ki was calculated by Ki=Ki, app/(1+[S]/Km). At least six different inhibitor concentrations were determined in duplicate for a minimum of three independent experiments. The reaction was started by adding the enzyme, and the time-dependent inactivation was monitored. Enzyme (E) and substrate (S) concentrations: L.major cpB, E=1–2 nM, S=2.5 µM; cruzain, E=5 nM, S=5 µM; papain, E=6 nM, S= 15 µM; and cpB, E=10 nM, S=10 µM.
Cell Culture Assays. L. major promastigotes LV39(MRHO/ SU/59/P) were grown at 27°C in 5 ml (25-cm2 cell culture flask; Costar, Cambridge, MA) of RPMI medium 1640 containing 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) and 20% brain heart infusion tryptose. Parasites were maintained in the exponential growth phase by passing them twice a week. For inhibitor studies, 106 cells per ml were inoculated in new cultures, and cell growth was determined by counting the parasites with a Neubauer hemocytometer (A.O. Instruments, Buffalo, NY). The mouse macrophage cell line J774 was maintained in 75-cm2 cell culture flasks (Costar) at 37°C in RPMI medium 1640 containing 5% FBS (12 ml total volume) and passed once a week. Irradiated J774 cells (10 min, 2,700 rad, 24 h before infection) were cultured on glass coverslips in six-well cluster plates (Costar) and infected with stationary-phase promastigotes in a ratio of 1:10 for 12 h. After the infected macrophage monolayers had been washed three times with RPMI 1640, inhibitors were added to the culture and plates were incubated for 5 days at 32°C in a 5% CO2/95% air atmosphere. To determine the number of amastigotes per macrophage, cells were fixed in 100% methanol and stained with Giemsa stain. At least 200 macrophages per experiment were examined to monitor the effect of the inhibitors. Inhibitors dissolved in DMSO were from 20 mM stock solutions. DMSO concentrations up to 0.5% showed no effect on promastigotes, amastigotes, or J774 cells.
Electron Microscopy and ImmunoGold Localization. One to 5×108 promastigote parasites, treated or untreated, were washed twice with PBS (4°C, 10 min, 3,000 rpm in a Beckman Accuspin-FR centrifuge). Cells were fixed in 0.1 M sodium cacodylate buffer at pH 7.4 containing 1.5% glutaraldehyde (0.25% for ImmunoGold labeling) and 1% sucrose. Epon embedding, LR white embedding, and thin sectioning were performed according to standard protocols (12–14).
For ImmunoGold labeling, a polyclonal antiserum raised against the native L.major cpB was used in a 1:20 or 1:100 dilution, followed by a secondary antibody conjugated with 10-nm gold particles (goat antibody to rabbit IgG, 1:50, Amersham Life Sciences). Serum from the rabbit before immunization, BSA, and bovine serum were used for specificity controls. Photographs were taken with a Zeiss EM10C.
Alternatively, promastigotes of L.major were surface labeled with 500 µg/ml N-hydroxysuccinimide-biotin in PBS (pH 7.6) for 20 min on ice. The cells were washed and placed in medium at 25°C for 60 min. They were fixed in 200 mM Pipes with 4% paraformaldehyde, frozen, and processed for immunoelectron microscopy as described previously (15, 16). The thawed cryosections were probed with streptavidin (1 µg/ml), followed by mouse monoclonal anti-streptavidin and rabbit antibody to L.major cathepsin B. The antibodies were revealed by 12-nm gold-conjugated goat anti-mouse IgG and 18-nm gold-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch).
Promastigote Extracts and Western Blot Analysis. Five× 109 promastigotes were washed twice with PBS at pH 7.4. Cells were sonicated (Sonic Dismembranator 300, Fisher Scientific) on ice (three×10 sec, relative output 0.6) and adjusted with sodium acetate buffer at pH 5.5 to 1×109 cells per ml. Aliquots were stored at –20°C for 4 months without any loss of cysteine protease activity. Samples of 100 µl were solubilized by adding 20 µl of 6-fold concentrated Laemmli buffer. Samples were subjected to SDS/10% PAGE and transferred to nitrocellulose sheets. The immunoblots were incubated in 2.5% (wt/vol) blocking reagent (Boehringer Mannheim) in 100 mM maleic acid buffer (pH 7.5) for 60 min at room temperature, and then incubated overnight at 4°C with a rabbit polyclonal antiserum raised against L.major cpB or L.mexicana cpL that had been diluted 1:1000 or 1:500, respectively, in 100 mM Tris-HCl, pH 7.5, with 0.05% Tween 20 and 1% FCS. After incubation with horseradish peroxidase-conjugated secondary antibodies (1:3000; goat anti-rabbit IgG; Gibco BRL Life Technologies) for 60 min at room temperature, the blots were developed using ECL (Amersham Life Science). For active site labeling of cysteine proteases, promastigote extracts were incubated either with 50 µM 14C-labeled K11002 for 15 min at room temperature or with 125I-labeled p-nitrophenyl-derivatized E-64 and vinyl sulfone as previously described (17). Samples of 100 µl were subjected to SDS/PAGE and analyzed by fluorography.
Animal Model of Infection. All procedures were approved by the University of California, San Francisco Committee on
Table 2. Inhibition of cysteine proteases with K11002
Animal Research. Two×105 met acyclic L.major (WHOM/ IR/173) were obtained by peanut agglutinin selection as previously described (18) and were injected into each hind footpad of female BALB/c mice (18–20 g, Simonsen Laboratories, Gilroy, CA). Strain WHOM/IR/173 was used because of its higher virulence in mice compared with strain LV39(MRHO/SU/59/P). Compounds were dissolved in 100% DMSO and stored at –20°C. The final concentrations were adjusted with sterile water to give a 70:30 (vol/vol) DMSO/H2O mixture. Twenty-four hours after infection, mice were treated with 100 μl of K11002 or ZLIII115A (100 mg/kg per day, every day, 4 weeks, intraperitoneal) in either a single dose or split into two treatments per day. Each set tested consisted of five mice, including an untreated control, a DMSO-treated control, as well as uninfected mice treated with the appropriate compound. To monitor the course of the infection, the thickness of the footpads was measured once a week by a standard method using a metric caliper (dial thickness gauge no. 7305; Mitutoyo, Kawasaki, Japan) (19). To quantify parasite burden, whole footpad histology as well as limited parasite dilution assays from footpad tissues (20) were performed at the end of the experiments. To investigate side effects of the compounds, mouse liver tissue was embedded in paraffin and 5-μm sections were stained with hematoxylin. Sections of treated tissues were then compared with control liver tissue.
Cytokine Assays. IL-4 and IFN-γ were assayed in inhibitor-treated and untreated mice by monoclonal-based ELISA and normalized to standard controls as previously described (20).
RESULTS
The reversible hydrazide inhibitors, ZLIII115A and ZLIII43A, were tested against the L.major cpB, cruzain (the major cpL of Trypanosoma cruzi), papain, and mammalian cathepsin B. There was 2- to 10-fold higher inhibitory activity toward the L. major cpB versus the plant or mammalian proteases (Table 1). In the case of the irreversible pseudopeptide inhibitor K11002 (7), the first-order inactivation constant (kinact) was similar for the four enzymes. However, differences of up to 100-fold were observed for the second-order rate constants (kinact/Ki); (Table 2). The differences in activity of the mammalian cathepsin B versus the plant or parasite proteases are mainly due to differences in Ki. The L.major cpB, cruzain, and papain were inhibited to a similar extent. This similarity is consistent with the paradoxical cathepsin L-like substrate preference of the L. major cpB, due to a single amino acid modification in the S2 binding pocket (2).
The three inhibitors were next tested in cell cultures of L. major promastigotes, the extracellular stage of the parasite. Inhibitors were added to replicating Leishmania as a single dose, and cell growth was monitored for 3 days. Both the irreversible and the reversible inhibitors blocked replication of the parasite. Concentrations of 5 μM inhibited parasite growth 10-fold, whereas 20 μM and 50 μM completely inhibited cell growth (Fig. 1). Exchanging the medium every day for a total of 3 days, thereby keeping inhibitor concentrations stable (20 μM and 50 μM), led to death of the parasites. After the fourth day of this latter experiment, the medium was replaced with fresh medium without inhibitor, and the flasks were again kept under culture conditions. Even after 10 days no parasites could be detected, indicating a complete cure of the Leishmania culture by the cysteine protease inhibitors.
To confirm that the inhibitors could access the intracellular cysteine proteases of L.major, promastigote parasites were harvested after treatment with 50 μM K11002 for 24 h and extracted. The residual cysteine protease activity as measured with the fluorogenic substrate Z-Phe-Arg-AMC (which de-tects both L.major cpL and cpB activity) in K11002-treated cells was 20%±7% (n=5) relative to the control parasites. Targeting of cathepsins by K11002 was also confirmed by “tagging” the target proteases with radioactively labeled inhibitor and identification of protein species by parallel Western blot (Fig. 2). The predominant cysteine protease in L. major promastigotes is cpB (21), and this species was the predominant, but not exclusive, target of the vinyl sulfone inhibitor. The mature catalytic domains of L.major cathepsins B and L

FIG. 1. Effects of K11002 (A) and ZLIII115A (B) on the growth of L. major promastigotes. The compounds were added at time point 0, and cell growth was monitored for 3 days. ZLIII43A showed very similar inhibition profiles (2). ●, Control; △, 5 μM; ○, 20 μM; and □, 50 μM. Points are means of three independent experiments.
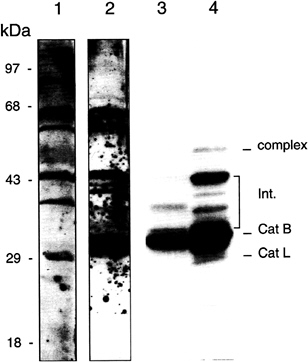
FIG. 2. Inhibitor targets and Western blot analysis of promastigote lysates. Proteins from whole promastigote extracts (Lanes 1 and 2), or whole promastigote were separated by SDS/PAGE and blotted to nitrocellulose membrane. Lane 1 was probed with a rabbit antiserum raised against a cpL from L.mexicana (a gift of Jeremy Mottram, University of Glasgow); lane 2 was probed with a rabbit antiserum raised against cpB from L.major. Extracts in lane 3 were incubated with 125I-labeled E-64, an epoxide cysteine protease inhibitor. Extracts in lane 4 were labeled with 125I-vinyl sulfone as described previously (17). Note the predominant labeling of the mature (catalytic domain) cpB in lanes 3 and 4 by both inhibitors. The vinyl sulfone also binds to the less abundant mature CpL. Both inhibitors label higher molecular weight protease precursors, which can be tentatively identified as active intermediates in protease processing (Int.) by Western blotting (lanes 1 and 2) and reexpression of specific protease genes in protease-null organisms (Sanya Sanderson and Jeremy Mottram, personal communication). The “complex” band is presumably an aggregate of protease with itself or with a carrier protein.
were labeled, as were intermediates in protease processing (Fig. 2).
To determine effects of the inhibitors on Leishmania amastigotes, the stage of the parasite that resides within mammalian host cells, irradiated macrophages (J774 cells) were infected with promastigote stationary-phase Leishmania. After 12 h, 50% of the macrophages were infected with 1 to 4 parasites per host cell. Cells were then treated with a single dose (40 µM) of inhibitor and cultured for another 5 days. At day 5, 85% of untreated J774 cells carried more than 9 parasites. This observation confirms that parasites replicate within the host cells and infect new macrophages. Replication of parasites was decreased in cultures treated with the pseudopeptide inhibitor or the hydrazide inhibitors, and few if any new macrophages were infected after treatment (Table 3). Host cell morphology was not affected by the treatment, and nonirradiated, inhibitor-treated macrophages had no difference in growth rate compared with untreated cells.
The hydrazide inhibitors and the pseudopeptide inhibitor produced very similar effects on the organelle structure within the parasites. After 24 h of treatment, myelin figures, undigested cell debris, dense bodies, and multivesicular bodies
Table 3. Treatment of amastigote parasites
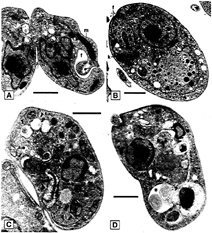
FIG. 3. Electron micrographs of Epon-embedded L.major promastigotes. Parasites were untreated (A) or treated for 24 h with 50 µM K11002 (B and C) or 50 µM ZLIII115A (D). Treatment of the parasite with either inhibitor had very similar effects, resulting in the appearance of diverse multivesicular and dense bodies (arrowheads), lipid inclusions (arrows), and myelin figures (asterisk), n, Nucleus; g, Golgi apparatus; f, flagellar pocket; m, mitochondrion; k, kinetoplast. (Bars=10 µm.)
appeared within the abnormally dilated parasite lysosomes and the flagellar pocket (Fig. 3), the site where endocytosis and exocytosis takes place (22). These abnormalities resemble alterations seen in lysosomal storage diseases caused by the deficiency or absence of specific lysosomal hydrolases (23). The nucleus and the Golgi apparatus were not affected, but some cells showed dilated mitochondria. In these latter cells the kinetoplast DNA was no longer condensed but appeared in diffuse patches. No effects on treated mammalian host cells at the light microscopic or ultrastructural level were observed.
To localize the Leishmania cysteine proteases within the parasite cell, ImmunoGold electron microscopic analysis using a L.major cpB-specific antiserum was performed. In untreated cells the gold label appeared only in lysosomes (Fig. 4 C and D). Treated cells were more heavily labeled in the dilated lysosome/endosome compartment and in the flagellar pocket (Fig. 4 E and F). Apparently empty flagellar pockets were also heavily labeled in treated parasites, but not in untreated parasites. To confirm target protease localization at the site of inhibitor-induced abnormalities, untreated promastigotes were surface-labeled with N-hydroxysuccinimide-biotin and placed back in culture to facilitate internalization of labeled proteins. This method allows visualization of the endosomal/ lysosomal network of the cells. ImmunoGold electron microscopy of these parasites revealed an abundance of cathepsin B in the flagellar pocket and in vesicles subtending that structure (Fig. 4 A and B). Some of these vesicles contained biotinylated proteins, indicating that they are endosomes or lysosomes, whereas others contained only cathepsin B, suggesting that they may be secretory vesicles.
Because of the selective arrest of parasite versus host cell growth by inhibitors added to cultures, the efficacy of the cysteine protease inhibitors in vivo was evaluated in Leishmania-infected BALB/c mice. Twenty-four hours after infection, mice received intraperitoneal injections of K11002 or ZLIII115A dissolved in DMSO/H2O (70:30). By 2 weeks, control mice had already developed footpad lesions, which progressed in size and severity. In treated animals the lesion development was significantly delayed, with no swelling of the footpads until 3–4 weeks (Figs. 5 and 6). After 4 weeks of treatment, inhibitor dosing was stopped, and lesion development paralleled that seen in control mice. At the end of the treatment period, whole footpad histology and limiting dilution assays of parasites from extracted footpad tissues showed parasite burden for treated animals was at least two logs lower than that of the untreated animals (10–7 versus 10–5). This finding is consistent with results from previous studies that documented the correlation between footpad size and numbers of parasites (18, 20). None of the compounds produced toxic effects in mice, as indicated by daily observation of weight, activity, and appearance, as well as autopsy and histologic analysis. Also, there was no evidence of a switch from the usual TH2 cytokine response to Leishmania in

FIG. 4. Immunoelectron micrographs of L.major promastigotes. (A and B) Electron micrographs of cryosections from L.major promastigotes that were surface biotinylated with N-hydroxysuccinimide-biotin and incubated in medium for 45 min prior-to fixation. The sections were probed with streptavidin/mouse anti-streptavidin mAb (12-nm gold particle conjugated to goat anti-mouse IgG) and rabbit anti-cathepsin B antibody (18-nm gold particle conjugated to goat anti-rabbit IgG). g, Golgi apparatus; e, endodome; n, nucleus; k, kinetoplast; m, mitochondrion. (Bars=0.25 µm.) (A) Cathepsin B label is observed in vesicles in the vicinity of the Golgi apparatus and in the flagellar pocket, which is strongly positive for biotinylated proteins. (B) Label is also observed in biotin-positive vesicles, or endosomes that subtend the flagellar pocket. The data indicated that cathepsin B is synthesized and proceeds through the Golgi apparatus into the secretory network, where it gains access to the flagellar pocket. (C–F) LR white-embedded and ImmunoGold-labeled (anti-L.major cpB antiserum) promastigotes. Note the specific labeling in lysosomes of untreated cells (C and D) and in multivesicular bodies (arrowheads), as well as in the flagellar pocket of treated cells (E and F). (Bars=0.5 µm.)
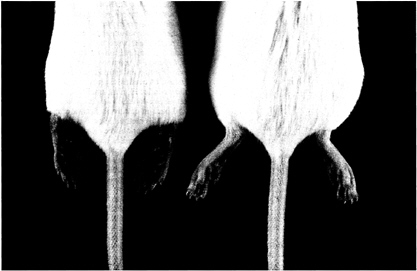
FIG. 5. Lesion development in Leishmania-infected BALB/c mice. The picture was taken after 3 weeks of treatment with K11002 (100 mg/kg per day). Note the erythema and gross edema of the footpads in the untreated mouse (left), versus no edema or erythema of the footpads in the protease inhibitor-treated mouse (right).
inhibitor-treated mice, as was reported by Maekawa et al. (24), who used the cathepsin B-specific inhibitor CA074. IFN-γ levels remained unchanged (36.7±15.8 ng/ml for 106 cells, treated mice, versus 33.4±12.3 ng/ml for 106 cells, untreated mice), and IL-4 levels remained elevated (14.4±1.0 ng/ml for 106 cells, treated mice, versus 16.7±2.1 ng/ml for 106 cells, untreated mice).

FIG. 6. Lesion sizes of treated and untreated infected BALB/c mice. Bars are means±SD of five mice (two footpads per mouse). Data shown are from one representative experiment of three independent experiments.
DISCUSSION
The results of the studies presented here suggest that cysteine protease inhibitors can have selective therapeutic effects in diseases such as leishmaniasis, where exogenous (microbial) protease activity is targeted. By inference, the lack of any significant organ or systemic toxicity of cysteine protease inhibitors also suggests that they may have utility in diseases where endogenous proteases are present in abnormal cellular or extracellular locations or at abnormally elevated levels.
Studies of cathepsin L family and cathepsin B family gene knockouts in L.mexicana (3, 4, 6) have suggested that at least two of the three cysteine protease gene families (cpa, cpb, cpc) would need to be eliminated to completely prevent parasite invasion or replication in host cells and lesion development in vivo. The inhibition of both amastigote infection of macrophages and lesion development in mice by cysteine protease inhibitors reported here is comparable to, and consistent with, the results of double cysteine protease gene knockout studies in L.mexicana (3). However, the effects seen with cysteine protease inhibitors on promastigote replication, and the flagellar pocket-endosomal pathway abnormalities seen on ultrastructural analysis were not observed in the L.mexicana double gene knockout studies. The presence of undigested debris, including myelin figures, in lysosomes or endosomes has been reported with storage diseases caused by absence of lysosomal hydrolases (23). One possibility is that, while each of the three gene families contributes to virulence of Leishmania (amastigote infection of macrophages and lesion development) in a gene dose-dependent manner, all three must be eliminated to affect promastigote replication and lysosome/endosomal function. Alternatively, the inhibitors may have prevented protease precursor processing (either autoproteolytic or by another of the three proteases) resulting in “retrograde” accumulation of unprocessed protease and organelle damage along a lysosome/endosome trafficking pathway (cf. Fig. 4). This condition would be analogous to the Golgi abnormality
observed in inhibitor-treated T.cruzi (25). In the case of L. major, the site of protease precursor processing must be later in the trafficking pathway than that of T.cruzi, probably between the flagellar pocket and the lysosome/endosome compartment. The localization of the protease in secretory vesicles destined for the flagellar pocket and in the pocket itself suggests this is one pathway by which protease may reach the lysosome. It is unclear whether the localization of cathepsin B to the flagellar pocket and subtending endosomal compartments represents the major route of delivery to the lysosomes, or whether the proteinase is also delivered directly to more mature lysosomal compartments. It is important to note that the ultrastructural abnormalities in flagellar pocket and lysosome/endosome were seen exclusively and consistently with cysteine protease inhibitors regardless of their specific chemistry (e.g., vinyl sulfone versus dihydrazide). This observation suggests that the cellular alterations seen are due specifically to inhibition of the cysteine pro teases of Leishmania.
By labeled inhibitor studies, the predominant target of the cysteine protease inhibitors is Leishmania cpB (Fig. 2). However, this conclusion probably reflects the fact that cpB is the most abundant species in L.major promastigotes. In fact, the inhibitors used effectively arrest the activity of both Leishmania cpB and cpL proteases when assayed against protease activity in either the aqueous phase or detergent phase of a Triton X-114 phase separation of promastigote extracts (P.M.S., unpublished data). Furthermore, the L.major cpB, while having sequence and structure homology to other members of the cpB family, has a substrate preference similar to that of cpL because of the absence of a glutamic acid side chain at the base of the S2 binding pocket (2). Eighty percent of the total Leishmania cysteine protease activity measured by the substrate Z-Phe-Arg-AMC could be inhibited by treatment of promastigotes with 50 μM K11002 for 24 h. This was sufficient to halt parasite replication.
A final issue concerning the Leishmania cpB and the effects of cysteine protease inhibitors in vivo arises from the results of a study by Maekawa et al. (24), who analyzed the effects of the cathepsin B-specific inhibitor CA074 on Leishmania infection in mice. Administration of this inhibitor to highly susceptible BALB/c mice resulted in a switch from the usual ineffectual TH2 cytokine response to a TH1 response that cleared the Leishmania infection. These authors concluded that inhibition of mammalian cathepsin B by CA074 resulted in altered expression of Leishmania antigens on MHC class II cells, producing the cytokine shift. This does not appear to be the mechanism contributing to the clearance of parasites in our study. As reported by Maekawa et al. (24), and confirmed by our own assays, CA074 alone does not inhibit Leishmania replication in vitro even at concentrations above 20 μM (our results) and 100 μM (24). Because CA074 does inhibit the Leishmania cpB in direct protease assays, these two results suggest that inhibition of a single type of cysteine protease is insufficient to block parasite replication. It is consistent with the results of the null mutant studies on L.mexicana cysteine protease gene families (3, 4). On the other hand, administration of the vinyl sulfone inhibitor to mice in our study did not result in a switch from TH2 to TH1 cytokines, as documented by direct measurements of IL-4 and IFN-γ levels. The vinyl sulfone inhibitor is a less effective inhibitor of mammalian cathepsin B (Table 2), whereas CA074 is a very specific and effective inhibitor of both the Leishmania and mammalian cathepsin B. We therefore conclude that the vinyl sulfone inhibitor exerts its effect by inhibiting parasite replication, as was observed in in vitro assays (Fig. 1), by virtue of its ability to inhibit both cpB and cpL Leishmania proteases.
The lack of observed toxicity to either mammalian cells in culture or mice, at the concentrations or doses of cysteine protease inhibitors used in this study, is reassuring but in some ways surprising. Tables 1 and 2 indicate that selectivity of the inhibitors versus mammalian cathepsin B, for example, is significant for the vinyl sulfone compound, but relatively less for the dihydrazides. Nevertheless, neither compound produced a significant alteration in host cells at concentrations up to 50 μM, in terms of either cell replication or ultrastructural appearance. The lack of toxicity at the doses used in mice is consistent with results of a similar study with vinyl sulfone inhibitors in the treatment of T.cruzi infection (26). We cannot rule out the possibility that inhibition of host cathepsin S by the vinyl sulfone inhibitor might affect some aspect of antigen presentation. However, a range-finding toxicology study of K11002 carried out at SRI International (Menlo Park, CA; Study M001–98, Project 1382–405, sponsored by the Developmental Therapeutics Branch of the National Institute of Allergy and Infectious Diseases) found no abnormalities in standard clinical chemistry tests and confirmed that toxicity (dsyspnea) in rats treated with this vinyl sulfone inhibitor was not seen until plasma concentrations of inhibitor exceeded 60 μM in males and 120 μM in females. The therapeutic plasma levels of inhibitor in the mouse study reported here (Figs. 5 and 6) range between 5 and 19 μM (W.Jacobsen and L.Benet, personal communication).
The selectivity of the inhibitor effects on the parasite suggests that cysteine proteases are crucial to the parasite, whereas host cells are less sensitive to cysteine protease inhibitors at the concentrations used. The lack of significant toxicity of cysteine protease inhibitors at the concentrations used in cell culture or achieved in mice may derive from several factors. First, parasites appear to take up and concentrate inhibitor much more effectively than do host cell organelles (27). Host cells also have a redundancy of protease activity not present in parasites. Even if one or more host cysteine proteases was inhibited, there may be little phenotypic effect. Finally, the concentration of proteases within host cells is substantially higher (millimolar) than that in parasites (28). Cultures of L.major parasites can be cured with inhibitors that target cysteine proteases, and, for the first time, in vivo studies suggest that disease progression can be reduced without toxicity to the host.
We thank Christopher Franklin and Elizabeth Hansell for excellent technical assistance, David Rasnick for advice on kinetic analysis, and Dan Friend for discussion of the ultrastructural studies. Jim Palmer (Arris Pharmaceuticals) kindly provided the vinyl sulfone inhibitors. This work was supported by grants from the United Nations Development Programme/World Bank/World Health Organization Special Programme for Research and Training in Tropical Diseases (T21/ 181/29) to J.A.S., by the National Institutes of Health (AI35707) to J.H.M., and by the National Institutes of Health (AI37977) to D.G.R. J.H.M. is supported by a Burroughs Wellcome Molecular Parasitology Scholar Award. P.M.S. was supported by a fellowship of the Deutsche Forschungsgemeinschaft (Se 762/1–1). M.B. is a University of California San Francisco Fellow.
1. World Health Organization (1993) UNDP/World Bank/WHO 8, Leishmaniasis, Special Programme for Research and Training in Tropical Disease. Tropical Disease Research: Progress 1991– 1992. Eleventh Programme Report, pp. 77–87.
2. Selzer, P.M., Chen, X., Chan, V.J., Cheng, M., Kenyon, G.L., Kuntz, I.D., Sakanari, J.A., Cohen, F.E. & McKerrow, J.H. (1997) Exp. Parasitol 87, 212–221.
3. Mottram, J.C, Brooks, D.R. & Coombs, G.H. (1998) Curr. Opin. Microbiol 1, 455–460.
4. Mottram, J.C., Souza, A.E., Hutchison, J.E., Carter, R., Frame, M.J. & Coombs, G.H. (1996) Proc. Natl. Acad. Sci. USA 93, 6008–6013.
5. Coombs, G.H. & Baxter, J. (1984) Ann. Trap. Med. Parasitol. 78, 21–24.
6. Bart, G., Frame, M.J., Carter, R., Coombs, G.H. & Mottram, J.C. (1997) Mol Biochem. Parasitol. 88, 53–61.
7. Palmer, J.T., Rasnick, D., Klaus, J.L. & Bromme, D. (1995) J. Med. Chem. 38, 3193–3196.
8. Bromme, D., Klaus, J.L., Okamoto, K., Rasnick, D. & Palmer, J.T. (1996) Biochem. J. 315, 85–89.
9. Ring, C.S., Sun, E., McKerrow, J.H., Lee, G.K., Rosenthal, P.J., Kuntz, I.D. & Cohen, F.E. (1993) Proc. Natl. Acad. Sci. USA 90, 3583–3587.
10. Li, R., Chen, X., Gong, B., Selzer, P.M., Li, Z., Davidson, E., Kurzban, G., Miller, R.E., Nuzum, E.O., McKerrow, J.H., et al (1996) Bioorg. Med. Chem. 4, 1421–1427.
11. Eakin, A.E., Harth, G., McKerrow, J.H. & Craik, C.S. (1992) J. Biol. Chem. 267, 7411–7420.
12. Tokuyasu, K.T. (1986) J. Microsc. 143, 139–149.
13. Selzer, P.M., Webster, P. & Duszenko, M. (1991) Eur. J. Cell Biol. 56, 104–112.
14. Bannister, L.H. & Kent, A.P. (1993) Methods Mol. Biol. 21, 415–429.
15. Russell, D.G., Xu, S. & Chakraborty, P. (1992) J. Cell Sci. 103, 1193–1210.
16. Russell, D.G. (1994) Methods Cell Biol. 45, 277–288.
17. Bogyo, M., Shin, S., McMaster, J.S. & Plough, H.L. (1998) Chem. Biol. 5, 307–320.
18. Sacks, D.L., Hieny, S. & Sher, A. (1985) J. Immunol. 135, 564–569.
19. Heinzel, F.P., Sadick, M.D. & Locksley, R.M. (1988) Exp. Parasitol. 65, 258–268.
20. Fowell, D.J., Magram, J., Turck, C.W., Killeen, N. & Locksley, R.M. (1997) Immunity 6, 559–569.
21. Sakanari, J.A., Nadler, S.A., Chan, V.J., Engel, J.C., Leptak, C. & Bouvier, J. (1997) Exp. Parasitol 85, 63–76.
22. Overath, P., Stierhof, Y.D. & Wiese, M. (1997) Trends Cell Biol 7, 27–33.
23. Ghadially, F.N. (1988) in Ultrastructural Pathology of the Cell and Matrix, ed. Ghadially, F.N. (Butterworths, London), pp. 589– 765.
24. Maekawa, Y., Himeno, K., Ishikawa, H., Hisaeda, H., Sakai, T., Dainichi, T., Asao, T., Good, R.A. & Katunuma, N. (1998) J. Immunol 161, 2120–2127.
25. Engel, J.C., Doyle, P.S., Palmer, J., Hsieh, I., Bainton, D.F. & McKerrow, J.H. (1998) J. Cell Sci. III, 597–606.
26. Engel, J.C., Doyle, P.S. Hsieh, I. & McKerrow, J.H. (1998) J. Exp. Med. 188, 725–734.
27. McGrath, M.E., Eakin, A.E., Engel, J.C., McKerrow, J.H., Craik, C.S. & Fletterick, R.J. (1995) J. Mol. Biol. 247, 251–259.
28. Xing, R., Addington, A.K. & Mason, R.W. (1998) Biochem. J. 332, 499–505.








