
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
How the protease thrombin talks to cells
SHAUN R. COUGHLIN*
Cardiovascular Research Institute and Departments of Medicine and Cellular and Molecular Pharmacology, University of California, San Francisco, CA 94143–0130
ABSTRACT How does a protease act like a hormone to regulate cellular functions? The coagulation protease thrombin (EC 3.4.21.5) activates platelets and regulates the behavior of other cells by means of G protein-coupled protease-activated receptors (PARs). PAR1 is activated when thrombin binds to and cleaves its amino-terminal exodomain to unmask a new receptor amino terminus. This new amino terminus then serves as a tethered peptide ligand, binding intramolecularly to the body of the receptor to effect transmembrane signaling. The irreversibility of PAR1’s proteolytic activation mechanism stands in contrast to the reversible ligand binding that activates classical G protein-coupled receptors and compels special mechanisms for desensitization and resensitization. In endothelial cells and fibroblasts, activated PAR1 rapidly internalizes and then sorts to lysosomes rather than recycling to the plasma membrane as do classical G protein-coupled receptors. This trafficking behavior is critical for termination of thrombin signaling. An intracellular pool of thrombin receptors refreshes the cell surface with naïve receptors, thereby maintaining thrombin responsiveness. Thus cells have evolved a trafficking solution to the signaling problem presented by PARs. Four PARs have now been identified. PAR1, PAR3, and PAR4 canallbe activated by thrombin. PAR2 is activated by trypsin and by trypsin-like proteases but not by thrombin. Recent studies with knockout mice, receptor-activating peptides, and blocking antibodies are beginning to define the role of these receptors in vivo.
Among their myriad roles, extracellular proteases can function like hormones to regulate cellular behaviors. Perhaps the best-studied example of such a process is activation of platelets by the coagulation protease thrombin (EC 3.4.21.5). This article briefly reviews our current understanding of the receptors that mediate protease signaling in platelets and other cells and points out some of the interesting questions they raise.
How Does a Protease Talk to a Cell?
Because platelets and thrombin are important in myocardial infarction and other thrombotic processes, understanding how thrombin activates platelets has long been an important goal (1). How does thrombin talk to platelets? Thrombin signaling is mediated at least in part by a family of G protein-coupled protease-activated receptors (PARs), for which PAR1 is the prototype (2, 3). Thrombin activates PAR1 by binding to and cleaving its amino-terminal exodomain to unmask a new receptor amino terminus (2). This new amino terminus then serves as a tethered peptide ligand, binding intramolecularly to the body of the receptor to effect transmembrane signaling (Fig. 1) (2, 4, 5). The synthetic peptide SFLLRN, which mimics the first six amino acids of the new amino terminus unmasked by receptor cleavage, functions as an agonist for PAR1 and
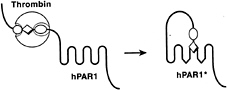
FIG. 1. Mechanism of PAR1 activation. Thrombin (large sphere) recognizes the amino-terminal exodomain of the G protein-coupled thrombin receptor PAR1. This interaction utilizes sites both amino-terminal (P1–P4, small sphere) and carboxyl-terminal (P9'–P14', small oval) to the thrombin cleavage site. Thrombin cleaves the peptide bond between receptor residues Arg-41 and Ser-42. This serves to unmask a new amino terminus beginning with the sequence SFLLRN (diamond) that functions as a tethered ligand, docking intramolecularly with the body of the receptor to effect transmembrane signaling. hPAR1, human PAR1; the asterisk indicates the activated form. Synthetic SFLLRN peptide will function as an agonist, bypassing the requirement for receptor cleavage.
activates the receptor independently of thrombin and proteolysis (2, 6, 7). Beyond supporting the tethered ligand model of receptor activation, such peptides have been useful as agonists for probing PAR function in various cell types and as a starting point for antagonist development.
Thus PAR1 is a peptide receptor that carries its own ligand. The ligand remains hidden until it is revealed by selective cleavage of PAR1’s amino-terminal exodomain. This proteolytic switch removes amino-terminal sequence that sterically hinders ligand function and generates a new protonated amino group at the amino terminus created by receptor cleavage. In the SFLLRN peptide, the cognate protonated amino group is critical for agonist activity (7, 8). Parallels with zymogen activation in serine proteases are apparent (2, 9). In conversion of trypsinogen to trypsin, precise proteolytic cleavage generates a new amino terminus that bears a new protonated amino group, which then docks intramolecularly to trap the protease in its active conformation (9).
Irreversible Activation, Disposable Receptors, and Intracellular Reserves
The mechanism of PAR1 activation is strikingly irreversible. Cleavage of PAR1 by thrombin is irrevocable, and the tethered ligand generated cannot diffuse away from the receptor. In the absence of the reversible ligation that characterizes most receptor systems, how is PAR1 shut off? The ß2-adrenergic receptor has served as a prototype for dissecting the molecular events responsible for G protein-coupled receptor desensiti-
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviation: PAR, protease-activated receptor. |
|
* |
To whom reprint requests should be addressed. E-mail: shaun_coughlin@quickmail.ucsf.edu. |
zation and resensitization (10–13). Upon activation, ß2-adrenergic receptor is rapidly phosphorylated. It then binds arrestin, preventing further interaction with G proteins. Arrestin also mediates internalization of ß2-adrenergic receptors via clathrin-coated pits (14, 15). Within an endosomal compartment, receptors dissociate from ligand, are dephosphorylated, and recycle back to the cell surface competent to signal again. Thus trafficking serves to remove activated ß2-adrenergic receptors from the cell surface and to return the receptors to the surface in an off state, ready to respond again to ligand.
Like the ß2-adrenergic receptor, PAR1 is rapidly phosphorylated and uncoupled from signaling after activation (16, 17). PAR1 is also internalized after activation (18–20). However, instead of efficiently recycling after internalization, activated PAR1 sorts predominantly to lysosomes (18, 19, 21). Indeed, in transfected fibroblast cell lines, activation decreased the half-life of PAR1 from 8 hr to 30 min (22). Recent studies that employed chimeras between PAR1 and the substance P receptor were informative regarding the role of PAR1’s distinct sorting pattern in signal termination (22, 23). Wild-type substance P receptor internalized and recycled after activation like ß2-adrenergic receptor; PAR1 bearing the substance P receptor’s cytoplasmic tail (P/S) behaved similarly. By contrast, wild-type PAR1 and a substance P receptor bearing PAR1’s cytoplasmic carboxyl tail (S/P) sorted to lysosomes after activation. Consistent with these observations, PAR1 and the S/P chimera were effectively down-regulated by their respective agonists as assessed by both receptor protein levels and signaling. By contrast, substance P receptor and the P/S chimera showed little down-regulation. Strikingly, cells expressing the P/S chimera signaled indefinitely after exposure to thrombin, apparently due to “resignaling” by cleaved and activated thrombin receptors returning to the cell surface (23). These data suggest that the cytoplasmic tails of PAR1 and substance P receptor specify distinct intracellular sorting patterns in a single cell type. More importantly, the “irreversible” thrombin signaling seen in cells expressing the P/S chimera suggests that lysosomal sorting is indeed necessary to prevent persistent signaling by activated PAR1.
When some cell types were exposed to thrombin for a prolonged period, a steady-state level of cleaved receptors was detected on the cell surface (16, 18). In such a state, cells were refractory to thrombin but responded to the PAR1-activating peptide SFLLRN (16, 18). Such responses were mediated by a subset of PAR1 molecules in which the tethered ligand was modified or otherwise prevented from functioning (24, 25). The significance of this phenomenon is unclear; it may represent a mechanism for dealing with the minority of activated PAR1 molecules that escape sorting to lysosomes.
Termination of PAR1 signaling thus occurs at several levels. The initial uncoupling of PAR1 depends on phosphorylation and may involve arrestin binding, as for other G proteincoupled receptors. Activated PAR1 is prevented from recycling and “resignaling” mainly by its sorting to lysosomes—a trafficking solution to a signaling problem. Such mechanisms for maintaining the temporal fidelity of thrombin signaling are presumably important in fibroblasts and vascular endothelial cells; both cell types express PAR1 and may need to respond to thrombin accurately over time.
While assuming special significance in the case of proteolytically activated PAR1, internalization and degradation of activated receptors is important for long-term down-regulation in many receptor systems. PAR1 may be useful as a model system for characterizing this sorting process in mammalian cells.
The finding that each PAR1 molecule is used once and discarded raises the question of how cells maintain responsiveness to thrombin over time. In fibroblasts and endothelial cells, unactivated PAR1 appears to cycle slowly between the cell surface and an intracellular compartment, such that at steady state approximately one-half of PAR1 molecules are inside the cell and protected from thrombin cleavage (19, 21). This intracellular “reserve” can repopulate the cell surface with naïve receptors without new receptor synthesis, thereby restoring or maintaining responsiveness to thrombin. Slow agonist-independent internalization of PAR1 is required for maintaining this intracellular reserve (20, 26). Hence, the irreversibility of PAR1’s proteolytic activation mechanism is accommodated by special desensitization and resensitization machinery. Like recycling and lysosomal sorting, tonic and agonist-triggered internalization of PAR1 were separable by mutation (20, 26). This observation suggests that distinct machinery may recognize naïve vs. activated PAR1 and that elucidating the molecular basis for PAR1’s trafficking behavior might reveal new mechanisms.
A Protease-Activated Receptor Family
Recognition and cleavage of PAR1 by thrombin is specified by two short stretches of amino acids in PAR1’s amino-terminal exodomain. LDPR/S binds thrombin’s active center, and the “hirudin-like” sequence DKYEPF binds thrombin’s fibrinogen-binding exosite (4, 27–30). Thrombin’s role in activating PAR1 appears limited to cleaving the receptor (4, 30). Indeed, replacing the PAR1 thrombin cleavage site LDPR/S with the enteropeptidase cleavage site DDDDK/S produced a receptor that signaled to enteropeptidase but not thrombin (4). A trypsin cleavage site was similarly effective (25). It is noteworthy that such a discrete sequence dictates receptor specificity. One might expect that it would be relatively easy to generate a family of receptors with distinct protease specificities once one protease-activated receptor had evolved.
Four PARs are now known (Fig. 2). PAR1, PAR3, and PAR4 are thrombin receptors (2, 3, 31–33). PAR1 and human PAR3 respond to thrombin at subnanomolar concentrations (2, 3, 31, 33). PAR4 requires higher but probably still physiological levels of thrombin for activation (see below) (32, 33), perhaps because it lacks the hirudin-like thrombin-binding
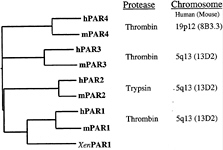
FIG. 2. Protease-activated receptor family. Four PARs are known. Amino acid sequence identity between human (h-) and mouse (m-) homologues of each is approximately 60%, but identity between different PARs within a single species falls to approximately 30%. Xen indicates Xenopus. Human PAR1, PAR3, and PAR4 can be activated by thrombin, and sensing thrombin is likely, at least in part, their role in vivo (see text). One receptor, PAR2, is activated by trypsin and tryptase but not by thrombin. Its roles in vivo remain to be explored. The four PAR genes share a common two-exon structure. In essence, the first exon encodes a signal peptide and the second the mature receptor protein. The genes encoding PARs 1, 2, and 3 are adjacent in the mouse and human genomes, whereas the PAR4 gene resides at a separate location (32, 65, 66).
sequence that is present in PAR1 and PAR3. PAR2 is activated by trypsin and tryptase, not by thrombin (34, 35).
It is interesting to note that a Xenopus thrombin receptor (36) is clearly identifiable as a PAR1 homologue (Fig. 2), suggesting that several PAR genes may have existed before amphibians and mammals diverged. How and in what context did PARs evolve? It was relatively easy to “evolve” a tethered ligand in vitro for the formyl peptide receptor (37). However, the identity of the common ancestor of PARs and other G protein-coupled receptors, the temporal relationship of the appearance of PAR genes vs. that of various protease cascades, and the function of the first PAR are unknown.
Given the importance of thrombin and platelets in myocardial infarction and other thrombotic processes, identification of the receptors responsible for thrombin signaling in platelets has been a high priority. Recent studies outlined below provide a model for the roles of the known PARs in this process. The roles of PARs in other cell types and processes are just beginning to be explored.
PARs and Platelet Activation
Our understanding of the role of PARs in platelet activation is evolving rapidly. PAR1 mRNA and protein were detected in human platelets (2, 38–40). PAR1-activating peptides activated human platelets (2, 6, 7). PAR1-blocking antibodies inhibited human platelet activation by low but not high concentrations of thrombin (38, 39). These data suggested a role for PAR1 in activation of human platelets by thrombin but held open the possibility that other receptors contribute. Curiously, in mouse platelets, PAR1 appeared to play no role. PAR1 expression was difficult to detect and PAR1-activating peptides did not activate rodent platelets (41–43). Moreover, platelets from PAR1-deficient mice responded like wild-type platelets to thrombin (43). The latter observation prompted a search for additional thrombin receptors and led to the identification of PAR3 (31). PAR3 was indeed expressed in mouse platelets (31) but could not be detected in human platelets (44). Inhibition of PAR3 function with antibodies that bound to PAR3’s hirudin-like domain or by gene knockout prevented mouse platelet activation by low but not high concentrations of thrombin (33, 45). These results established that PAR3 is necessary for normal thrombin signaling in mouse platelets but also pointed to the existence of another platelet thrombin receptor. Such a receptor, PAR4, was recently identified (32, 33). PAR4 appears to function in both mouse and human platelets (32, 33, 44). Thus in both mouse and human, platelets utilize two thrombin receptors. A “high-affinity” thrombin receptor (PAR1 in human, PAR3 in mouse) is necessary for responses to low concentrations of thrombin, whereas a “low-affinity” receptor (PAR4 in both species) mediates responses at higher concentrations of thrombin. Do these receptors account for thrombin activation of platelets? Addressing this question at the genetic level awaits generation of a mouse deficient in both PAR3 and PAR4. In the meantime, pharmacological studies of human platelets suggest that the answer might be yes (44). Inhibition of PAR1 function alone—whether by blocking antibody, antagonist, or desensitization—inhibited platelet responses at 1 nM thrombin but only slowed responses at 30 nM thrombin. Inhibition of PAR4 function alone with a blocking antibody had no effect at either concentration. Strikingly, combined inhibition of PAR1 and PAR4 signaling profoundly inhibited platelet responses even at high concentrations of thrombin (44).
Available data suggest that PAR4 activation is not necessary for robust responses in human platelets when PAR1 function is intact. Why do platelets have two receptors? Aside from providing a backup signaling device, PAR4 might allow platelets to respond to proteases other than thrombin, mediate thrombin signaling to distinct effectors or with a tempo different from that of PAR1, or function in platelet responses beyond simple secretion and aggregation. The existence of two genes and gene products also raises the possibility of differential regulation at many levels in platelets or other cell types. Most interestingly, it is possible that PARs interact. These issues remain to be explored.
The identification of the receptors that mediate platelet activation by thrombin raises important questions regarding strategies for the development of antithrombotic therapies. Clearly PAR antagonists can be developed (44, 46). The observation that PAR1 inhibition blocked platelet responses to low concentrations of thrombin and slowed responses to high concentrations raises the question of whether PAR1 inhibition alone might be sufficient for an antithrombotic effect (44, 47). Alternatively, it may be necessary to block both PAR1 and PAR4 to prevent or arrest thrombosis in vivo. Whether such strategies should be pursued can now be determined by using receptor blocking reagents in appropriate animal models.
A Role for Thrombin Signaling in Embryonic Development and Other Processes?
The role of PARs in cell types other than platelets is under active investigation in a number of laboratories. Several attractive hypotheses focus on possible roles for PARs in protease signaling to the blood vessel wall. In the adult, PAR1 is expressed by vascular endothelial cells and smooth muscle cells and is thus opportunely positioned to mediate communication between blood and the cells comprising the vessel wall. In cell culture, thrombin causes endothelial cells to deliver the leukocyte adhesion molecule P-selectin to their surfaces (48), to secrete von Willebrand factor (48), to elaborate growth factors and cytokines (49, 50), and to change shape and increase permeability (51). Thrombin is also a mitogen for fibroblasts (52) and vascular smooth muscle cells (53) and has a variety of metabolic effects on these cells. Vascular injury in any form, whether metabolic, mechanical, immune-mediated, or infectious, is likely to promote local thrombin generation at some level. These considerations prompt the hypothesis that thrombin might participate in acute and/or chronic inflammatory and proliferative responses to vascular injury. One might also imagine a role for thrombin signaling in the setting of angiogenesis, where leaky nascent vessels might trigger local thrombin activity. PAR-deficient mice will be invaluable for testing such hypotheses.
We are particularly interested in the role of PAR1 in embryonic development because it may reveal unanticipated roles for the coagulation cascade that are independent of platelet activation and fibrin formation. Approximately half of PAR1-deficient embryos die between embryonic days 9.5 and 10.5 (43, 54). Histological examination of these embryos revealed embryonic blood cells in the pericardial, amniotic, and exocoelomic cavities, suggesting a defect in hemostatic mechanisms or vascular integrity (C.Griffin and S.R.C., unpublished results). Deficiency of pro thrombin or factor V, which is necessary for thrombin generation, caused grossly similar developmental defects (55–57). Although one might ascribe bleeding in these knockouts to failed fibrin generation and/or platelet activation, fibrinogen (58) and platelets (59) are not necessary for normal embryonic development. Moreover, PAR1 is not expressed in mouse platelets, at least in the adult, and platelets from the PAR1-deficient mice that survived to adulthood had no defect in their response to thrombin (43). The relationships of the developmental phenotypes of PAR1, factor V, and prothrombin deficiency have not been formally tested, and it is certainly possible, even likely, that thrombin acts on targets other than PAR1 and/or that PAR1 has activators other than thrombin during development. Nonetheless, it is tempting to postulate that the “vascular integrity
defect” common to PAR1-, prothrombin-, and factor V-deficient embryos is due at least in part to defective thrombin signaling in cells other than platelets. Although PAR1 is expressed in a variety of cell types at embryonic day 9.5 (E9.5), in situ hybridization of E9.5 embryos revealed PAR1 mRNA to be most abundant in endothelial cells (ref. 60 and data not shown). This prompts the working hypothesis that PAR1 signaling in endothelial cells is important for normal vascular development. Thrombin generation is triggered when factor VIIa in plasma meets extravascular tissue factor, hence the coagulation protease cascade can be viewed in part as a “leak detector” for blood vessels. Perhaps developing blood vessels use this system to monitor their functional status as they grow and remodel. Studies designed to test the role of endothelial PAR1 in vascular development are ongoing.
Summary
PARs provide one mechanism by which proteases can act as hormones and talk directly to cells. In PARs, nature has utilized a mechanism analogous to zymogen activation to trigger ligation of a G protein-coupled receptor. The irreversibility of this activation mechanism poses an unusual problem for receptor desensitization and resensitization, a problem solved by specialized receptor trafficking. Such trafficking bells and whistles raise the question of how long PARs have had to evolve and how broad their spectrum of activities might be. Four PARs are now known. Given the myriad of membrane-anchored and soluble extracellular proteases, it would not be surprising if more existed. Indeed, because only a few amino acids in their amino-terminal exodomains dictate the specificity of PARs for their activating proteases, one might predict that new PARs with new protease specificities might “easily” evolve. Thrombin’s cellular actions motivated the search for PAR1 (2, 3) and descriptions of cellular responses to trypsin that were independent of PAR1 presaged the identification of PAR2 (61). Cathepsin G and tissue factor/ VIIa each elicit interesting signaling phenomena (62–64), as do a variety of other proteases; whether known or new PARs will account for such signaling remains to be determined. Similarly, defining the roles of the known PARs in vivo in normal and disease states remains an important challenge. These receptors have already provided useful insights into regulation of platelet function and are likely to provide surprises regarding the regulatory roles of proteases in other cell types and processes.
1. Davey, M. & Luscher, E. (1967) Nature (London) 216, 857–858.
2. Vu, T.-K. H., Hung, D.T., Wheaton, V.I. & Coughlin, S.R. (1991) Cell 64, 1057–1068.
3. Rasmussen, U.B., Vouret-Craviari, V., Jallat, S., Schlesinger, Y., Pages, G., Pavirani, A., Lecocq, J.P., Pouyssegur, J. & Van Obberghen-Schilling, E. (1991) FEBS Lett. 288, 123–128.
4. Vu, T.-K. H, Wheaton, V.I., Hung, D.T. & Coughlin, S.R. (1991) Nature (London) 353, 674–677.
5. Chen, J., Ishii, M., Wang, L., Ishii, K. & Coughlin, S.R. (1994) J. Biol Chem. 269, 16041–16045.
6. Vassallo, R.J., Kieber, E.T., Cichowski, K. & Brass, L.F. (1992) J. Biol Chem. 267, 6081–6085.
7. Scarborough, R.M., Naughton, M.A., Teng, W., Hung, D.T., Rose, J., Vu, T.K., Wheaton, V.I., Turck, C.W. & Coughlin, S.R. (1992) J. Biol. Chem. 267, 13146–13149.
8. Coller, B.S., Ward, P., Ceruso, M., Scudder, L.E., Springer, K., Kutok, J. & Prestwich, G.D. (1992) Biochemistry 31, 11713– 11720.
9. Bode, W., Schwager, P. & Huber, R. (1978) J. Mol Biol. 118, 99–112.
10. Yu, S.S., Lefkowitz, R.J. & Hausdorff, W.P. (1993) J. Biol. Chem. 268, 337–341.
11. Krueger, K.M., Daaka, Y., Pitcher, J.A. & Lefkowitz, R.J. (1997) J. Biol Chem. 272, 5–8.
12. Lohse, M., Benovic, J., Codina, J., Caron, M. & Lefkowitz, R. (1990) Science 248, 1547–1550.
13. Freedman, N.J. & Lefkowitz, R.J. (1996) Recent Prog. Horm. Res. 51, 319–351; Discussion 352–353.
14. Ferguson, S.S., Downey, W.R., Colapietro, A.M., Barak, L.S., Menard, L. & Caron, M.G. (1996) Science 271, 363–366.
15. Goodman, O.J., Krupnick, J.G., Santini, F., Gurevich, V.V., Penn, R.B., Gagnon, A.W., Keen, J.H. & Benovic, J.L. (1996) Nature (London) 383, 447–450.
16. Ishii, K., Hein, L., Kobilka, B. & Coughlin, S.R. (1993) J. Biol. Chem. 268, 9780–9786.
17. Ishii, K., Chen, J., Ishii, M., Koch, W.J., Freedman, N.J., Lefkowitz, R.J. & Coughlin, S.R. (1994) J. Biol Chem. 269, 1125–1130.
18. Hoxie, J.A., Ahuja, M., Belmonte, E., Pizarro, S., Parton, R. & Brass, L.F. (1993) J. Biol Chem. 268, 13756–13763.
19. Hein, L., Ishii, K., Coughlin, S.R. & Kobilka, B.K. (1994) J. Biol Chem. 269, 27719–27726.
20. Shapiro, M.J., Trejo, J., Zeng, D.W. & Coughlin, S.R. (1996) J. Biol. Chem. 271, 32874–32880.
21. Woolkalis, M.J., DeMelfi, T.J., Blanchard, N., Hoxie, J.A. & Brass, L.F. (1995) J. Biol Chem. 270, 9868–9875.
22. Trejo, J., Hammes, S.R. & Coughlin, S.R. (1998) Proc. Natl. Acad. Sci. USA 95, 13698–13702.
23. Trejo, J. & Coughlin, S.R. (1999) J. Biol. Chem. 274, 2216–2224.
24. Trejo, J., Connolly, A.J. & Coughlin, S.R. (1996) J. Biol. Chem. 271, 21536–21541.
25. Hammes, S.R. & Coughlin, S.R. (1999) Biochemistry 38, 2486–2493.
26. Shapiro, M.J. & Coughlin, S.R. (1998) J. Biol. Chem. 273, 29009–29014.
27. Liu, L., Vu, T.-K. H., Esmon, C.T. & Coughlin, S.R. (1991) J. Biol Chem. 266, 16977–16980.
28. Mathews, I. L, Padmanabhan, K.P., Ganesh, V., Tulinsky, A., Ishii, M., Chen, J., Turck, C.W., Coughlin, S.R. & Fenton, J.N. (1994) Biochemistry 33, 3266–3279.
29. Hung, D.T., Vu, T.-K. H., Wheaton, V.I., Charo, I.F., Nelken, N.A., Esmon, C.T. & Coughlin, S.R. (1992) J. Clin. Invest. 89, 444–450.
30. Ishii, K., Gerszten, R., Zheng, Y.-W., Turck, C.W. & Coughlin, S.R. (1995) J. Biol Chem. 270, 16435–16440.
31. Ishihara, H., Connolly, A.J., Zeng, D., Kahn, M.L., Zheng, Y.W., Timmons, C., Tram, T. & Coughlin, S.R. (1997) Nature (London) 386, 502–506.
32. Xu, W.F., Andersen, H., Whitmore, T.E., Presnell, S.R., Yee, D.P., Ching, A., Gilbert, T., Davie, E.W. & Foster, D.C. (1998) Proc. Natl. Acad. Sci. USA 95, 6642–6646.
33. Kahn, M.L., Zheng, Y.W., Huang, W., Bigornia, V., Zeng, D., Moff, S., Farese, R.V., Jr., Tam, C. & Coughlin, S.R. (1998) Nature (London) 394, 690–694.
34. Nystedt, S., Emilsson, K., Wahlestedt, C. & Sundelin, J. (1994) Proc. Natl. Acad. Sci. USA 91, 9208–9212.
35. Nystedt, S., Emilsson, K., Larsson, A.K., Strombeck, B. & Sundelin, J. (1995) Eur. J. Biochem. 232, 84–89.
36. Gerszten, R.E., Chen, J., Ishii, M., Ishii, K., Wang, L., Nanevicz, T., Turck, C.W., Vu, T.-H. K. & Coughlin, S.R. (1994) Nature (London) 368, 648–651.
37. Chen, J., Bernstein, H.S., Chen, M., Wang, L., Ishii, M., Turck, C.W. & Coughlin, S.R. (1995) J. Biol. Chem. 270, 23398–23401.
38. Hung, D.T., Vu, T.K., Wheaton, V. L, Ishii, K. & Coughlin, S.R. (1992) J. Clin. Invest. 89, 1350–1353.
39. Brass, L.F., Vassallo, R.R., Belmonte, E., Ahuja, M., Cichowski, K. & Hoxie, J.A. (1992) J. Biol. Chem. 267, 13795–13798.
40. Molino, M., Bainton, D.F., Hoxie, J.A., Coughlin, S.R. & Brass, L.F. (1997) J. Biol. Chem. 272, 6011–6017.
41. Derian, C.K., Santulli, R.J., Tomko, K.A., Haertlein, B.J. & Andrade-Gordon, P. (1995) Thromb. Res. 6, 505–519.
42. Connolly, T.M., Condra, C., Feng, D.M., Cook, J.J., Stranieri, M.T., Reilly, C.F., Nutt, R.F. & Gould, R.J. (1994) Thromb. Haemostasis 72, 627–633.
43. Connolly, A.J., Ishihara, H., Kahn, M.L., Farese, R.V. & Coughlin, S.R. (1996) Nature (London) 381, 516–519.
44. Kahn, M.L., Nakanishi-Matsui, M., Shapiro, M.J., Ishihara, H. & Coughlin, S.R. (1999) J. Clin. Invest. 103, 879–887.
45. Ishihara, H., Zeng, D., Connolly, A.J., Tam, C. & Coughlin, S.R. (1998) Blood 91, 4152–4157.
46. Bernatowicz, M.S., Klimas, C.E., Hartl, K.S., Peluso, M., Allegretto, N.J. & Seiler, S.M. (1996) J. Med. Chem. 39, 4879–4887.
47. Cook, J.J., Sitko, G.R., Bednar, B., Condra, C., Mellott, M.J., Feng, D.M., Nutt, R.F., Shafer, J.A., Gould, R.J. & Connolly, T.M. (1995) Circulation 91, 2961–2971.
48. Hattori, R., Hamilton, K.K., Fugate, R.D., McEver, R.P. & Sims, P.J. (1989) J. Biol Chem. 264, 7768–7771.
49. Daniel, T.O., Gibbs, V.C., Milfay, D.F., Garavoy, M. & Williams, L.T. (1986) J. Biol. Chem. 261, 9579–9582.
50. Colotta, F., Sciacca, F.L., Sironi, M., Luini, W., Rabiet, M.J. & Mantovani, A. (1994) Am. J. Pathol. 144, 975–985.
51. Lum, H. & Malik, A.B. (1994) Am. J. Physiol. 267, L223-L241.
52. Chen, L.B. & Buchanan, J.M. (1975) Proc. Natl. Acad. Sci. USA 72, 131–135.
53. McNamara, C.A., Sarembok, I.J., Gimple, L.W., Fenton, J.W., II, Coughlin, S.R. & Owens, G.K. (1992) J. Clin. Invest. 91, 94–98.
54. Darrow, A.L., Fung, L.W., Ye, R.D., Santulli, R.J., Cheung, W.M., Derian, C.K., Burns, C.L., Damiano, B.P., Zhou, L., Keenan, C.M., et al. (1996) Thromb. Haemostasis 76, 860–866.
55. Sun, W.Y., Witte, D.P., Degen, J.L., Colbert, M.C., Burkart, M.C., Holmback, K., Xiao, Q., Bugge, T.H. & Degen, S.J. (1998) Proc. Natl. Acad. Sci. USA 95, 7597–7602.
56. Xue, J., Wu, Q., Westfield, L.A., Tuley, E.A., Lu, D., Zhang, Q., Shim, K., Zheng, X. & Sadler, J.E. (1998) Proc. Natl. Acad. Sci. USA 95, 7603–7607.
57. Cui, J., O’Shea, K.S., Purkayastha, A., Saunders, T.L. & Ginsburg, D. (1996) Nature (London) 384, 66–68.
58. Suh, T.T., Holmback, K., Jensen, N.J., Daugherty, C.C., Small, K., Simon, D.I., Potter, S. & Degen, J.L. (1995) Genes Dev. 9, 2020–2033.
59. Shivdasani, R.A., Rosenblatt, M.F., Zucker, F.D., Jackson, C.W., Hunt, P., Saris, C.J. & Orkin, S.H. (1995) Cell 81, 695–704.
60. Soifer, S.J., Peters, K.G., O’Keefe, J. & Coughlin, S.R. (1993) Am. J. Pathol 144, 60–69.
61. Levine, L. (1994) Prostaglandins 47, 437–449.
62. Selak, M. (1994) Biochem. J. 297, 269–275.
63. Røttingen, J.A., Enden, T., Camerer, E., Iversen, J.G. & Prydz, H. (1995) J. Biol Chem. 270, 4650–4660.
64. Camerer, E., Røttingen, J.A., Iversen, J.G. & Prydz, H. (1996) J. Biol. Chem. 271, 29034–29042.
65. Schmidt, V.A., Nierman, W.C., Maglott, D.R., Cupit, L.D., Moskowitz, K.A., Wainer, J.A. & Bahou, W:F. (1998) J. Biol Chem. 273, 15061–15068.
66. Kahn, M.L., Hammes, S.R., Botka, C. & Coughlin, S.R. (1998) J. Biol. Chem. 273, 23290–23296.





