
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Chaperone rings in protein folding and degradation
ARTHUR L. HORWICH*†‡, EILIKA U. WEBER-BAN*, AND DANIEL FINLEY§
*Department of Genetics and †Howard Hughes Medical Institute, Yale School of Medicine, New Haven, CT 06510; and §Department of Cell Biology, Harvard Medical School, Boston, MA 02115
ABSTRACT Chaperone rings play a vital role in the opposing ATP-mediated processes of folding and degradation of many cellular proteins, but the mechanisms by which they assist these life and death actions are only beginning to be understood. Ring structures present an advantage to both processes, providing for compartmentalization of the substrate protein inside a central cavity in which multivalent, potentially cooperative interactions can take place between the substrate and a high local concentration of binding sites, while access of other proteins to the cavity is restricted sterically. Such restriction prevents outside interference that could lead to nonproductive fates of the substrate protein while it is present in non-native form, such as aggregation. At the step of recognition, chaperone rings recognize different motifs in their substrates, exposed hydrophobicity in the case of protein-folding chaperonins, and specific “tag” sequences in at least some cases of the proteolytic chaperones. For both folding and proteolytic complexes, ATP directs conformational changes in the chaperone rings that govern release of the bound polypeptide. In the case of chaperonins, ATP enables a released protein to pursue the native state in a sequestered hydrophilic folding chamber, and, in the case of the proteases, the released polypeptide is translocated into a degradation chamber. These divergent fates are at least partly governed by very different cooperating components that associate with the chaperone rings: that is, cochaperonin rings on one hand and proteolytic ring assemblies on the other. Here we review the structures and mechanisms of the two types of chaperone ring system.
Almostallproteins proceed through a life cycle circumscribed by their folding and degradation. Because both processes are exergonic, it was long assumed that they occur through straightforward molecular mechanisms or simply spontaneously, in the case of folding. Independent studies of these two processes, however, have recently revealed their dependence in vivo on large and remarkably intricate molecular machines (refs. 1 and 2; Fig. 1). These complexes, like many other protein machines, are driven by ATP, but their common physical feature is a ring structure. The ATPase subunits within these machines form symmetric or pseudosymmetric rings of 6–9 members, enclosing a central cavity (Fig. 2). The cavity defines the substrate binding site, and the substrate can enter or exit this cavity by moving perpendicular to the plane of the ring. Folding substrates leave such rings by retracing their original path of entry whereas proteolytic substrates appear to pass through the ring into a second, ATP-independent ring compartment containing proteolytic active sites.
ATP-dependent chaperone rings have proven to be evolutionarily ubiquitous and include well studied protein-folding chaperonins, such as bacterial GroEL (3), the archaebacterial thermosome (4), and the eukaryotic CCT complex (ref. 5; Figs. 1 and 2). Chaperone rings serving as proteolytic assistants include the bacterial ClpA (6), ClpX (7), and HslU (8) and the eukaryotic 19S proteasome cap structure (regulatory particle), also known as PA700 (refs. 9 and 10; Figs. 1 and 2). In the case of chaperonins, their overall function is well established: namely, assisting proteins to fold to their native form. In the case of the ring chaperones involved in proteolytic degradation, their action appears to involve recognition of specific proteins, destabilization of their structure, and translocation of unfolded polypeptide chains into associated proteolytic cylinders (see ref. 11).
The functional similarities between the ATPase rings of the chaperonins and the ATP-dependent proteases may be an example of evolutionary convergence. In any case, there is no significant sequence similarity between these two types of ATPase rings. All known ATP-dependent proteases belong to the Walker family of ATPases, a vast and functionally diverse collection of enzymes (12). By contrast, the design of the ATPase domain of the chaperonins appears to be specific to chaperonins themselves (see, e.g., ref. 13). In both families of ATPases, large-scale conformational changes are dictated by the presence or absence of the γ phosphate of the bound adenine nucleotide. Thus, both systems are to a first approximation two-state systems, although, in the case of GroEL, anticooperative interplay between the two rings and asymmetric binding of GroES provide for at least one additional substate that is critical to the forward movement of the reaction cycle (see below). A detailed understanding of how the ATPase cycle drives proteolysis of protein substrates has not yet been achieved for the ATP-dependent proteases.
The chaperone rings of the ATP-dependent proteases appear to play a preparative role, recognizing proteins slated for turnover and promoting their unfolding, actions that the proteolytic cylinders cannot by themselves carry out. Indeed, in the absence of the associating chaperone ring, proteolytic cylinders, such as bacterial ClpP or the eukaryotic 20S proteasome, degrade small peptides inefficiently and are inactive on physiological protein substrates (see, e.g., refs. 14–16).
In contrast, some of the associating chaperone assemblies, when assayed in the absence of their proteolytic cylinders, retain the ability to recognize physiological substrates and, moreover, appear to be able to dissociate oligomeric proteins or low order protein aggregates (refs. 17–19; see also ref. 20). For example, in the case of ClpX-mediated dissociation of MuA transposase tetramer from recombined DNA (refs. 19 and 21; Fig. 1), the cognate ClpP protease is apparently prevented from acting on MuA at the transposase complex, perhaps by inability to associate with ClpX in this setting. Thus, the relationship between the two actions of protease-associating ring assemblies, assistance to degradation and
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviation: EM, electron microscopy. |
|
‡ |
To whom reprint requests should be addressed. E-mail: horwich@csbmet.csb.yale.edu. |
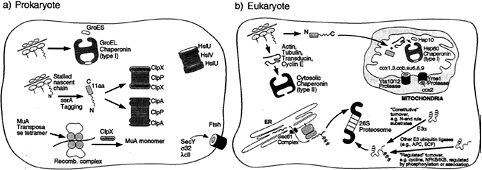
FIG. 1. Schematic illustration of the role of chaperone rings in ATP-dependent protein folding and unfolding/degradation in prokaryotic and eukaryotic cells. Protein folding chaperonins are illustrated in the upper portion of each “cell,” and proteolytic chaperones and the associated proteolytic cylinders are shown in the lower portion. In the case of the prokaryotic Clp components, the homohexameric ATPases, ClpA or ClpX, form coaxial associations with the termini of the double ring cylindrical serine protease, ClpP, delivering recognized substrates to it for degradation (see text). In the absence of association with ClpP, however, ClpA or ClpX can mediate disassembly of oligomeric substrate proteins, exemplified by ClpX-mediated disassembly of the MuA transposase tetramer. Note the two chaperonin classes in the eukaryotic cell (cytosolic and mitochondrial). In the case of the eukaryotic proteasome, the general pathways of ubiquitination to direct proteins for degradation by the proteasome are shown. Not shown is the presence of the proteasome in the nuclear compartment, where similar pathways of turnover appear to be operative.
oligomeric dissociation, may be governed by whether the ring is associated with a cognate proteolytic cylinder.
Notably, other ring-shaped proteolytic assemblies in the cell have covalently linked the ATPase and protease functions in one polypeptide, as in the FtsH bacterial membrane metalloprotease or the related AAA-ATPase containing proteases of the mitochondrial inner membrane, Yta10–12 and Yme1 (refs. 22–26; Fig. 1). Joining of the two functions within one polypeptide is not restricted to the membrane proteases; the soluble bacterial protease Lon and its mitochondrial homolog, PIM1, are similarly designed (25). The principles of action of these proteases may be the same as those assemblies composed of distinct chaperone and proteolytic rings, but we confine our discussion here to the latter situation, in which the chaperone moiety is amenable to analysis both on its own and in a binary complex with the proteolytic component.
Architecture-Function Considerations
Both chaperonins and the protease-associating chaperone rings, the latter often referred to as regulatory complexes, are radiajly symmetric (or pseudosymmetric) assemblies of ˜110– 140 Å diameter, housing axial cavities (refs. 6 and 27; Fig. 2). Chaperonins are composed of two back-to-back rings whose axial cavities are blocked at the equatorial “base” of each ring by the collective of COOH termini of the surrounding subunits, which protrude into the central space (28). (The COOH termini are not resolvable crystallographically because of disorder from a GGM repeat sequence, but the collective of termini is visible as a mass in cryoEM.) Thus, chaperonins contain two noncontiguous cavities, 45–65 Å in diameter, one at each end of the cylindrical structure. The cavities are formed by surrounding apical domains, attached on hinges to small intermediate domains, hinged in turn to the equatorial base (Fig. 2). The central cavities have been identified by electron microscopy (EM) and functional studies as the sites of binding of non-native polypeptide, which, at least in the case of the bacterial chaperonin, GroEL, occurs through hydrophobic side chains exposed on the cavity wall (see ref. 29). These side chains apparently bind exposed hydrophobic surfaces specifically present in non-native proteins.
The folding-active state of GroEL is produced when both ATP and the cochaperonin GroES bind to the polypeptide-containing ring; the apical domains of the bound ring undergo large conformational movements, 60° upward rotation and 90° clockwise twisting motion, that move the hydrophobic binding sites away from the cavity, releasing the bound protein into what is now a sequestered space that is “capped” by GroES and enlarged 2-fold in volume (refs. 3 and 30; Fig. 2). The walls of the cavity assume a hydrophilic character that favors burial of hydrophobic residues in the folding substrate protein and exposure of hydrophilic residues, promoting folding to the native state.
Protease-associated chaperone rings also exhibit axial cavities but, in contrast with those of chaperonins, these seem likely to be, in the active state, continuous channels through which recognized substrate proteins can be translocated into the central space of the associated proteolytic cylinder (11). The diameter of such channels is somewhat uncertain, lacking crystallographic resolution so far, but recent cryoEM studies approximate the cavity in bacterial ClpA to 70–80 Å at the widest point, narrowing down to a 10- to 20-Å passageway at the end that interfaces with ClpP (6). For its own part, ClpP, in a stand-alone crystal structure, exhibits a central opening at its terminal ends of ˜10 Å (ref. 31; Fig. 2). This opens into a cavity of >50 Å height and diameter. In the case of the crystal structure of the yeast 20S proteasome (32), there is no detectable axial opening into the chamber, with the NH2 termini of the a-subunits obstructing passage (Fig. 2). This implies a gating action by the ATP-dependent association of the 19S “cap” complex with the proteolytic cylinder. Indeed, in the case of the proteasome, a substitution in the ATP binding site of one of six ATPases in the 19S complex (Rpt2) results in a strong inhibition of the peptidase activity of the proteasome, suggesting that even peptides cannot traverse the channel without involving an ATP-directed gating mechanism (33). The small size and apparent gating of the passageways into the proteolytic cylinders appear likely to exclude the bulk of cellular proteins from the lumen of the proteolytic cyliner. At the same time, a requirement is imposed that proteins must be unfolded before their translocation into the proteolytic cylinder. In fact, ClpA alone has been shown to act as an unfoldase in vitro, globally unfolding a monomeric substrate
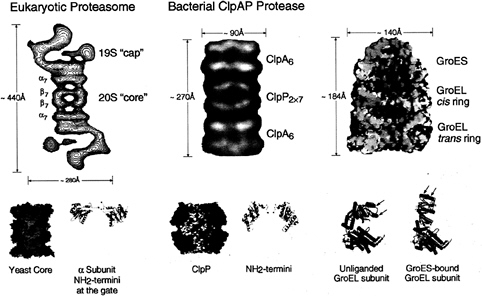
FIG. 2. Architecture of the eukaryotic proteasome and bacterial ClpAP chaperone-protease complexes and of the bacterial GroEL-GroES chaperonin pair. Side views from electron microscopy of the eukaryotic 26S proteasome (Left) and bacterial ClpAP (Center) showing the respective chaperone assemblies associated with the respective proteolytic cylinders (taken from ref. 11). The stoichiometries of the constitutent oligomeric rings are designated by subscripts; note that the eukaryotic proteasome is composed of seven distinct a. subunits and seven distinct ß subunits arranged 2-fold symmetrically to compose the four rings. Shown below are space-filling cutaway images of the proteolytic cylinders, derived from the crystal structures of Wang et al. (31) and Groll et al. (32), with active sites shown as red dots, as well as ribbon diagrams of their entryways, also taken from ref. 11. A space-filling view of the GroEL-GroES-ADP7 asymmetric chaperonin complex is shown (Upper Right), taken from Xu et al. (3), illustrating the differences between GroEL rings in the polypeptide-accepting and folding-active states. The open trans ring of the asymmetric complex exposes hydrophobic residues (shown in yellow) that can capture a non-native polypeptide. Subsequent GroES/ATP binding to the ring with polypeptide replaces this surface with a hydrophilic one (shown in blue), enlarges the cavity 2-fold in volume, and encapsulates the space in which a polypeptide, released from the hydrophobic binding sites, pursues folding in solitary confinement. Below, the rigid body movements of apical (red) and intermediate (green) domains of GroEL that occur on GroES binding are shown, taken from Xu et al. (3). The apical peptide binding surfaces of helices H and I (arrows), as well as an underlying segment, are removed from facing the central cavity to a position rotated upward 60° and twisted 90° clockwise (see text and ref. 3 for details).
protein (79). Translocation through the channel may constitute the first committed step in proteolysis by these ATP-dependent proteases.
In the case of both the bacterial and eukaryotic chaperone components, the rings apposed coaxially to the proteolytic cylinder are composed of six ATPase-containing subunits (6, 33, 34). Considering that the cognate proteolytic cylinders are 7-membered double or quadruple rings (see, e.g., refs. 31, 32, 36), with the exception of six-fold symmetric HslV (35), there is an obvious symmetry mismatch. With such a 6-on-7 interface, the chaperone subunits cannot form a 1-to-1 match with proteolytic subunits in the same way that, for example, GroEL subunits match up exactly with subunits of the GroES cochaperonin partner (3). It is unclear how this unusual and evolutionarily preserved behavior may translate into a functional role. Is it designed to inherently weaken the association between the two components? This seems unlikely, because most chaperone/protease complexes appear to be stable as long as ATP is present. The symmetry mismatch may dispose to rotational sliding or ratcheting of the faces of the respective rings across each other (6). Perhaps it is a manifestation of a mechanism of translocation of substrate protein down the axial channel, such that a polypeptide chain is “spooled” through a narrow opening into the proteolytic chamber by a rotational or ratcheting motion (see, e.g., ref. 36). This model cannot apply toallATP-dependent proteases, however. As mentioned above, the ATPase and proteolytic domains are contained within a single polypeptide in the Lon and membrane-bound metalloproteases, where linking of these domains would prevent relative rotation. Interestingly, in EM images of the eukaryotic proteasome, the two asymmetric 19S complexes are observed in a 2-fold rotational orientation with respect to each other, potentially requiring coupled rotation to satisfy a ratchet model (e.g., ref. 10; see Fig. 2)
In the case of the proteasomal cap structure, not only is there an eight-subunit “base” containing six ATPase subunits, but also an ˜400-kDa “lid” structure, comprising eight subunits in yeast, connected to the base by what looks like a “hinge” in EM images (ref. 37; Fig. 2). When the lid is removed from the yeast proteasome by a mutation eliminating a protein supporting the connection to the base (Rpn10), ubiquitinated proteins can no longer be degraded. Thus, the lid appears to be specifically required for recognition of ubiquitin conjugates. By contrast, with only the base structure remaining attached to the 20S proteasome rings, a nonubiquitinated protein, casein, can still be efficiently degraded (37). These observations would seem to support a model of recognition wherein the lid structure binds the ubiquitin moiety of a ubiquitinated protein while the
base either simultaneously or subsequently binds the adjoined substrate moiety.
It should be pointed out that ubiquitin conjugation of a protein per se is not associated with its unfolding, although in many cases ubiquitin conjugation may be activated by unfolding of a protein, which exposes sequences or non-native structures recognizable by the ubiquitin conjugation system (see, e.g., ref. 38). In other cases, a ubiquitin-conjugated native substrate structure is presented to the proteasomal regulatory particle and must somehow be unfolded by it, or perhaps trapped in a spontaneously unfolded state. The mechanism of such unfolding remains unclear. Does ubiquitin itself participate in the unfolding process? If ubiquitin and substrate bind at multiple points to the lid and base, then ATP-mediated conformational change could exert a shearing force on the attached substrate protein that would act to unfold it. It seems that the lid structure would, at a minimum, allow retention of ubiquitinated proteins in proximity to the base apparatus, kinetically favoring interaction with it and the consequent ATP-dependent unfolding and translocation into the proteolytic cylinder.
The use by the proteasome of a tag by which to hold the substrate in place while it is exposed to an unfolding machinery is a mechanistic feature quite distinct from anything used by the chaperonins or other classical molecular chaperones. If a proteasome substrate undergoes a failed trial of unfolding, it is unlikely to dissociate because it will presumably remain tethered to the proteasome via the ubiquitin tag. This arrangement may account for the remarkable observation that the proteasome will degrade almost any soluble protein if it is ubiquitinated. However, stabilization of the folded state of the ubiquitinated protein can apparently prevent unfolding and degradation, as was indicated by an experiment with a dihydrofolate reductase variant recruited to the proteasome via the N-end rule pathway. When the folded state of the dihydrofolate reductase was stabilized by binding its ligand, methotrexate, it was no longer subject to degradation (39).
In the same way that the lid presumed to recognize ubiquitin lies atop the ATPase base in the proteasome cap, domains that may have analogous function seem to be present in the bacterial system in some cases. For example, subunits of the ClpA ring contain a second major domain attached through a hinge to the base (refs. 6 and 7; Fig. 2). A second ATPase motif is present in this domain, which probably corresponds to the NH2-terminal portion of ClpA because the COOH-terminal portion is homologous to ClpX, which also associates with ClpP (40). How these domains participate in binding, unfolding, and translocation will require structural study—both EM and crystallographic—as well as functional analyses.
Substrate Protein Recognition
Because ubiquitin is clearly the major recognition determinant for the eukaryotic proteasome, the specificity of its substrate protein recognition is accounted for mainly at the level of the ubiquitin conjugation system. The remarkable set of E3 ubiquitin protein ligases involved in this process appears to be large in number, and the nature of molecular recognition by these gatekeepers is under intensive study (for reviews, see refs. 38 and 41; see Fig. 1). The subunits involved in recognition of ubiquitinated proteins remain to be identified. Subunit S5a/ Rpn10 of the proteasome regulatory particle specifically binds multiubiquitin chains in vitro (42). This has been observed with the subunit derived from a number of species, including human, Drosophila melanogaster, Saccharomyces cerevisiae, and Arabidopsis thaliana. Yet studies in vivo in S.cerevisiae, and more recently in plants, show that the ubiquitin chain binding site in S5a/Rpn10 does not have a significant involvement in the degradation of ubiquitinated proteins (see, e.g., refs. 43 and 44; R.Vierstra, personal communication).
In the case of the bacterial chaperone/protease pairs, at least one means of designation for proteolysis involves a tagging mechanism reminiscent of ubiquitination, although it is cotranslational (45, 46). A peptide encoded by the ssrA gene is used to mark for degradation incomplete nascent polypeptide chains that become stalled at the ribosome because the encoding messages have been prematurely transcriptionally terminated or nuclease-cleaved (ref. 46; Fig. 1). This RNA is a remarkable 362-base hybrid RNA whose 5′ end resembles tRNA-ala and whose 3′ end encodes a 10-residue peptide (ANDENYALAA) followed by an ochre terminator. A working model of R.T.Sauer and coworkers (46) suggests that alanine-charged ssrA RNA enters the unoccupied P site of a stalled nascent chain ribosome complex that has reached the 3′ end of a truncated message, adding an alanine (unencoded) to the nascent chain. The ribosome then switches to translation of the ssrA RNA encoding the 10-residue adduct, which is added as an extension of the incomplete nascent chain. This COOH-terminal amino acid sequence comprises an element for recognition and proteolytic degradation by ClpAP or ClpXP (47). In particular, when the ssrA peptide was added at the coding sequence level to λ represser (amino acids 1–93), it led to rapid turnover of the fusion protein in vivo. Conversely, such fusion proteins were no longer rapidly degraded in ClpP deletion mutants or in ClpA-ClpX double deletion mutants.
Interestingly, another signal for recognition by the ClpAP complex resides at the other, NH2-terminal, end of a potential set of test proteins that followed the N-end rule for degradation in bacteria (48). The presence of arginine, lysine, leucine, phenylalanine, tyrosine, or tryptophan conferred short half-life (<2 min) on the test proteins exposing one of these residues at the NH2 terminus. This short half-life compares to proteins exposing other residues, measuring >10 hr. For proteins bearing the destabilizing residues, deletion of ClpA resulted in alteration of half-life to that of proteins bearing the stabilizing residues. So far, such observations have not been extended to physiological substrates. The recognition event has not been reconstituted in vitro with purified ClpA and could possibly involve additional factors. It remains a fascinating question as to how ClpA specifically recognizes the NH2-terminal residue in the test protein studied.
Recognition of the COOH-terminal sequences in substrates by ClpX (and likely ClpA as well) appears to be mediated through a COOH-terminal domain in the chaperone, distal to the ATPase domain, that contains a tandem motif (49). The two motifs have been suggested to resemble PDZ domains, which are modular 100-residue structures shown to recognize COOH-terminal tetrapeptides with a characteristic primary sequence, X-Thr/Ser-X-Val-COO− (50–52). The similarity remains, however, to be established by structural studies. Nevertheless, when one or both of these motifs of ClpX were expressed independently, they were able to efficiently bind an Arc-MuA fusion bearing the COOH-terminal 10-residue sequence of MuA (LEQNRRKKAI), which is required for recognition and disassembly of tetrameric MuA by ClpX (21). Likewise, the isolated motifs recognized an Arc-ssrA fusion. The COOH-terminal tetrapeptide sequences recognized by PDZ domains are unstructured until they become bound, whereupon they are incorporated as an additional β-strand at the edge of a sheet in the PDZ domain. Consistently, here, the COOH-terminal region of MuA in an Arc-MuA fusion was shown to be unstructured in one-dimensional NMR studies whereas the NH2-terminal Arc region behaved as a native structure. In sum, the ssrA and MuA COOH-terminal recognition systems in bacteria bear some degree of resemblance to the ubiquitin system, with the signals themselves not leading to dissociation/unfolding of the substrate protein until the signal recruits the protein to the cap structure. Baker and coworkers have proposed that the tandem substrate recognition domains of ClpX may disassemble oligomeric substrates by forming two
points of contact with separate subunits of the tetramer; subsequent ATP-directed conformational change of the ClpX might then pry apart the subunits (49).
The mechanism of recognition of other targets of ClpA and ClpX is less clear. Surprisingly, though, dimers of the plasmid P1 initiator protein, RepA, can associate with unassembled ClpA subunit monomers or dimers (under conditions of absence of nucleotide; assembly of ClpA into hexamer requires adenine nucleotide). These complexes, however, are unstable (53). By contrast, complexes formed with ClpA hexamer at 23°C in ATPγS were stable and “committed,” such that they could only release RepA after exposure to ATP, discharging it as the DNA binding-competent monomer.
In the case of chaperonins, it is clear that specific primary sequences in non-native substrate proteins are not involved with recognition but, rather, that structures with exposed hydrophobic surfaces, such as collapsed states that can bind in the central cavity of the chaperonin, are recognized (see ref. 29). Binding seems likely to be multivalent; i.e., it involves multiple contacts between the polypeptide and the surrounding apical domains. There is some uncertainty concerning whether the action of polypeptide binding is associated with partial unfolding of kinetically trapped substrate proteins. Whereas the small protein, barnase, can be transiently globally unfolded by GroEL (54), other natural substrate proteins that require the complete GroEL/GroES/ATP system for folding are not subject to global unfolding in association with binding, as determined by deuterium exchange experiments (refs. 55 and 56, and S.Walter, personal communication). More generally, GroEL may favor binding of less-folded states and, as such, may shift an equilibrium between non-native species toward less-folded ones (57).
Action of ATP
The role of ATP, for both chaperonins and the ring chaperones involved in proteolysis, is to galvanize the components into an active association with their respective cochaperonins and proteolytic cylinders and to commence the particular actions of folding or degradation. The specific roles of ATP binding and hydrolysis are better understood for chaperonins but are beginning to be dissected for the proteolytic complexes as well. In the case of both machineries, it appears that ATP binding is sufficient to drive formation of the active complexes. In the case of GroEL, for example, binding of ATP to a ring occurs cooperatively atallseven sites (58, 59) and enables rapid and high-affinity binding of GroES to the same ring (60). This association is accompanied by the large conformational changes mentioned above, which are associated with release, possible transient unfolding (80), and subsequent folding of a bound polypeptide in the encapsulated cavity of the ring (3, 30, 61). At the same time, binding of ATP to one GroEL ring is specifically anticooperative for binding of ATP in the opposite ring (62), and, because ATP occupancy is required for efficient GroES binding, this sets up the inherent asymmetry of the chaperonin system, such that only one ring is folding-active at a time (refs. 30 and 63; see Fig. 3).
In the case of the proteasome, ATP binding drives the stable association of cap structures at both ends of the catalytic cylinder, a step that appears to be cooperative (6, 64). In the case of the ClpA chaperone, the presence of ATP or a nonhydrolyzable analogue, such as ATPγS, is required for stable assembly of the hexamer ring and for its association with ClpP (65). Yet here, in contrast with the chaperonin system, ATP binding alone is probably not sufficient to initiate proteolysis. For example, in the presence of ATPγS, the ClpA substrate, RepA, remained stably bound by ClpA hexamer (53). Only on subsequent addition of ATP was it released as monomer. Concordantly, RepA was not degraded in the presence of ATPγS, ClpA, and ClpP. These observations suggest that ATP hydrolysis is likely to be required for both actions associated with RepA proteolysis: dissociation of the RepA subunits from each other and unfolding/translocation. Nevertheless, it remains possible that ATPγS fails to produce the same stereochemistry of binding and resultant allosteric effects as ATP. This has proven to be the case for the GroEL system, for example, where AMP-PNP was found to be able neither to promote folding in association with formation of the cis complex nor to productively discharge the cis ligands from a cis ADP ternary complex on binding to the trans ring (30).
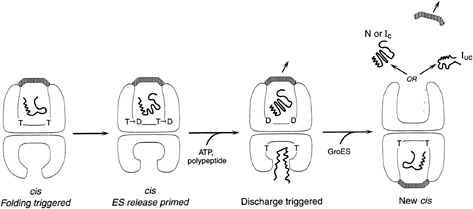
FIG. 3. GroEL-GroES reaction cycle-rings alternate in formation of folding-active cis ternary complexes. Folding is triggered when ATP and GroES bind to the same (cis) ring as polypeptide, releasing it into the GroES-encapsulated, enlarged, and now hydrophilic cavity. This very stable complex is the longest-lived state of the chaperonin system in the presence of non-native polypeptide (63), and it is weakened and prepared for dissociation by hydrolysis in the cis ring, which allows entry of ATP and non-native polypeptide into the trans ring (30). These in turn accelerate the dissociation of the cis ligands, including polypeptide. GroES binds to the ATP/polypeptide-liganded trans ring, completing formation of a new cis complex on this ring. Thus, GroEL rings alternate back and forth as folding-active (see text for additional detail).
In contrast, both of these actions were readily promoted by binding of ATP, in the absence of hydrolysis (see Fig. 3). The stereochemical differences between ATP and its analogues were revealed by study of GroEL with a catalytic mutation that did not affect its affinity for ATP but reduced turnover to a level ≈2% of wild-type. Additionally, real-time substrate fluorescence measurements with wild-type GroEL in the presence of ATP/GroES showed immediate onset of fluorescence changes reflective of folding well before any cis ATP hydrolysis occurred (30). Yet with the proteolytic assistants, it seems more likely that hydrolysis is required, because multiple turnovers of the chaperone ring seem likely to be necessary to unfold and translocate the protein substrate into the proteolytic cylinder. Further studies, such as single ATP turnover experiments, will be necessary to address such action.
The nature of the conformational change exerted on the ClpA ring by ATP binding and hydrolysis is as yet unknown. Correspondingly, it is unclear whether the ATP-mediated conformational change of ClpA affects ClpP, either directly, by opening its orifice, for example, or indirectly, by allosterically influencing its active sites (see ref. 66 for discussion). In the case of the proteasome cap, a “wagging” or rocking motion of the 19S cap relative to the 20S core has been reported, although whether this is linked to ATP binding and turnover has yet to be resolved (10). Thus, for these components, the means by which ATP turnover translates into unfolding and translocation of substrate remains to be seen. Notably, ATP turnover proceeds in these rings regardless of whether the proteolytic cylinder or substrate polypeptide is associated.
There seem to be two general models for how ATP-driven translocation between chaperone ring and protease might occur. One involves ATP-dependent unfolding of substrate in the chaperone ring, associated with threading of an extended polypeptide chain through a narrow passage into the proteolytic cylinder which itself engages and may “pull” the substrate via contacts with it, degrading it in a more or less processive manner. Such translocation resembles that of ER and mitochondrial precursor proteins traversing Sec and Tom/Tim membrane complexes in extended states. In those settings, an Hsp70 chaperone functions with expenditure of ATP to “ratchet” or “motor” the chain across the membrane from inside. Here, ATP is expended “behind” the passageway into the proteolytic cylinder, suggesting that a “pushing” action could be involved. This more closely resembles the system of export through the bacterial membrane, where the SecA chaperone utilizes ATP (binding) to drive a segment of both itself and polypeptide substrate through the translocon (67). In the case of the proteolytic cylinders, alternatively, perhaps the energy of peptide bond cleavage at the inside aspect could be coupled to forward movement. A second model invokes an ATP-directed conformational change that associates unfolding of substrate in the chaperone ring with a conformational switch amounting to opening of a “trap door” into the proteolytic component that allows the entire substrate, or perhaps domain-sized portions, to drop into the proteolytic cylinder for multipoint proteolytic processing. This would presumably involve opening of both the axial exitway in the base of the chaperone ring and the entryway of the proteolytic cylinder. Although it seems clear, for example, that the 20S proteasome must be gated, as yet the gate has not been observed in an open state, so the size of the opening is unknown.
It is also unclear whether nucleotide binding and turnover in the proteolytic chaperone rings is cooperative or synchronous or whether it occurs in some sequential manner that could be linked to rotational motion. In the case of chaperonins, cooperative ATP binding is used within a ring to enable it to function as a uniform 7-fold symmetric unit in binding the 7-fold symmetric GroES cochaperone. ATP hydrolysis is used in a folding-active ring to weaken the stable association of GroES with the ring (ref. 30; see Fig. 3). Such weakening “primes” the ring for dissociation that is allosterically triggered by binding of ATP and non-native polypeptide in the open opposite (trans) ring (refs. 30 and 63; see Fig. 3). At the same time that it primes the cis ring, cis hydrolysis sends an allosteric signal to the trans ring that adjusts its apical domains from a conformational orientation that cannot accept ligands to one that is now fully open and available for binding (ref. 63; see Fig. 3). Thus, ATP hydrolysis allosterically primes the GroEL machine to switch rings, signaling the end of a folding reaction in one ring and the preparation for a new one on the opposite ring.
In the case of GroEL, but also the proteolytic chaperones, there seems to be no requirement at any point in the cycle to produce symmetric complexes bearing a ring at both ends of the assembly simultaneously (63, 65, 68). For example, a second GroES does not have to bind to discharge the one present in a folding-active complex (30, 69, 70). Recent kinetic studies indicate, in fact, that GroES cannot bind to an available ATP-bound trans ring until a slow transition occurs within the cis ADP chaperonin complex that leads to the departure of the cis GroES (63). Thus, at most, one GroES is arriving while the other is departing. By contrast, stable 2:1 assemblies of chaperone-protease complexes can be isolated from cells and are readily formed in vitro; yet, these assemblies appear to be no more catalytically active than 1:1 complexes (65, 68), raising such issues as whether only one side of a 2:1 complex can be occupied with substrate and proteolytically active at any time, whether there is alternation between sides, and how occupancy of one side could inhibit access of substrate to, proteolytic activity of, or departure of product peptides from the other side.
Commitment of Substrate
In the case of RepA dimer, a single round of association with ClpA followed by ATP-mediated release is sufficient to produce the DNA binding-competent RepA monomer (53). This was demonstrated either by supplying an excess of casein substrate competitor that would block rebinding of RepA or by diluting the RepA-ClpA binary complex formed in ATPγS before ATP addition, such that rebinding would be disfavored. These studies indicate commitment of dimeric RepA substrate to dissociation into monomers in one round of interaction with the chaperone. Such committed behavior with respect to substrate protein differs considerably from that of chaperonins, which appear to eject substrate proteins after a timed period of folding in the cis chamber, regardless of whether substrate has reached the native state or not (see ref. 29; Fig. 3). For many substrate proteins, this results in a requirement for multiple rounds of release and rebinding by chaperonin in order for a population of molecules to reach the native state. Such release of non-native forms allows a kinetic partitioning to occur, whence non-native forms not only can be rebound by chaperonin but may be recognized by other chaperones, or even proteases (70–73). This prevents the chaperonin system from becoming engorged with misfolded or defective proteins that are not able to reach the native state. This behavior, attractive for chaperonins, would not be as appealing for the proteolytic system—in general, the release of partly degraded proteins would have little or no functional value.
On the other hand, it has been suggested that some substrates may be only partially processed by the proteasome: for example, the transcription factor NF-κB (74). It has been shown that removal of a COOH-terminal cytoplasmic anchoring domain from the NFκB precursor protein, p105, enables the NH2-terminal domain to enter the nucleus to activate transcription. One model for how this occurs.involves preferential unfolding and translocation of the COOH-terminal domain into the proteolytic cylinder, with action of the pro-
teasome somehow aborted before the NH2-terminal transcriptional activation domain enters the cylinder. A resistance of the NH2-terminal domain to unfolding may underlie its resistance to degradation, but there seems also the possibility that removal of a ubiquitin tag from the polypeptide could provide a means of escape, as discussed below. Alternative to partial processing, however, is a model in which initial cleavage by an endopeptidase separates the two domains and is followed by proteasome-mediated degradation of the COOH-terminal fragment (75).
Although the proteolytic system appears generally to be a committed one, it seems to have evolved a fail-safe mechanism or “editor” that prevents inappropriate commitment to turning over potentially active substrate proteins. The PA700 isopeptidase, an integral component of the mammalian 19S cap, enables removal of ubiquitin monomers from polyubiquitinated proteins, affording the chance to rescue those proteins that bear only short lengths of ubiquitin chains (ref. 76; see also ref. 77). The bias against degradation of short chains would predispose the proteasome to preferentially degrade proteins that have efficiently interacted with E3 enzymes to produce processive formation of long chains. Given that polyubiquitin chains are disassembled by the proteasome from their distal ends (76), translocation into the proteolytic cylinder of substrates carrying long polyubiquitin chains is likely to be favored kinetically over chain removal. On the other hand, those proteins with only short ubiquitin chains can be relieved of them and thus protected from proteasomal turnover.
Isopeptidases are also crucial for the recycling of ubiquitin, insofar as ubiquitin itself is not degraded by the proteasome. So, for both aborted and “productive” substrates, there may be an obligatory, but presumably late, step of ubiquitin removal. Indeed, ubiquitin removal is probably also necessary to allow the entire substrate polypeptide to pass through the channel into the proteolytic cylinder because the folded state of ubiquitin is remarkably stable (e.g., ref. 78), and it is thus likely to present a barrier to translocation. In addition, the ubiquitin chain is likely to be inaccessible to translocation because it is anchored to the ubiquitin receptor of the regulatory particle. These considerations suggest that the last key step in the proteasome’s reaction cycle occurs within the regulatory particle and consists in the release of the ubiquitin chain from the substrate. This step is presumably operative forallsubstrates, both “typical” ones as well as “nonsubstrates” that carry too few ubiquitin groups and are released after being edited by the PA700 isopeptidase.
Prospects for Further Mechanistic Understanding
In the short term, we can look forward to crystallographic views of states of the proteolytic chaperone rings, which will allow deductions about ATP-mediated unfolding and translocation, and further mechanistic studies. For both the chaperonin and proteolytic ring systems, however, attention must focus ultimately on the fate of the substrate polypeptide. This represents a challenge in both cases because the substrate occupies, or comes to occupy in the case of the proteolytic machines, a non-native conformation that does not exhibit the structural order and symmetry of the machines themselves. Indeed, substrates seem likely to occupy an ensemble of conformations, as compared with the uniformity from molecule to molecule of the states of the machines. Understanding this system will require facing some of the same problems that the chaperonin system currently confronts related to the location and conformation of substrate during binding and folding, both of which are difficult to examine at high resolution. Spectroscopic approaches seem likely to yield the most definitive answers but will be stretched to their limits to get at these questions.
We thank W.Fenton for critical reading of the manuscript. E.U.W. is supported by a Jane Coffin Childs Fellowship and A.L.H. by the Howard Hughes Medical Institute.
1. Baumeister, W., Walz, J., Zühl & Seemüller E. (1998) Cell 92, 367–380.
2. Bukau, B. & Horwich, A.L. (1988) Cell 92, 351–366.
3. Xu, Z., Horwich, A.L. & Sigler, P.B. (1997) Nature (London) 388, 741–750.
4. Ditzel, L., Löwe, J., Stock, D., Stetter, K.-O., Huber, H., Huber, R. & Steinbacher, S. (1998) Cell 93, 125–138.
5. Lewis, V.A., Hynes, G. M, Zheng, D., Saibil, H. & Willison, K. (1992) Nature (London) 358, 249–252.
6. Beuron, F., Maurizi, M.R., Belnap, D.M., Kocsis, E., Booy, F.P., Kessel, M. & Steven, A.C. (1998) J. Struct. Biol 123, 248–259.
7. Grimaud, R., Kessel, M., Beuron, F., Steven, A.C. & Maurizi, M.R. (1998) J. Biol Chem. 273, 12476–12481.
8. Rohrwild, M., Pfeifer, G., Santarius, U, Müller, S. A, Huang, H.-C, Engel, A, Baumeister, W. & Goldberg, A.L. (1997) Nat. Struct. Biol. 4, 133–139.
9. DeMartino, G.N., Moomaw, C.R., Zagnitko, O.P., Proske, R.J., Chu-Ping, M., Afendis, S.J., Swaffield, J.C. & Slaughter, C.A. (1994) J. Biol Chem. 269, 20878–20884.
10. Walz, J., Erdmann, A., Kania, M., Typke, D., Koster, A.J. & Baumeister, W. (1998) J. Struct. Biol. 121, 19–29.
11. Larsen, C.N. & Finley D. (1997) Cell 91, 431–434.
12. Confalonieri, F. & Duguet, M. (1995) BioEssays 17, 639–650.
13. Boisvert, D.C., Wang, J., Otwinowski, Z., Horwich, A.L. & Sigler, P.B. (1996) Nat. Struct. Biol. 3, 170–177.
14. Hwang, B.J., Woo, K.M., Goldberg, A.L. & Chung, C.H. (1998) J. Biol. Chem. 263, 8727–8734.
15. Arrigo, A.-P., Tanaka, K., Goldberg, A.L. & Welch, W.J. (1988) Nature (London) 331, 192–194.
16. Chu-Ping, M., Vu, J.H., Proske, R.J., Slaughter, C.A. & DeMartino, G.N. (1994) J. Biol. Chem. 269, 3539–3547.
17. Wickner, S., Gottesman, S., Skowyra, D., Hoskins, J., McKenney, K. & Maurizi, M.R. (1994) Proc. Natl. Acad. Sci. USA 91, 12218–12222.
18. Wawrzynow, A, Wojtkowiak, D., Marszalek, J., Banecki, B., Jonsen, M., Graves, B., Georgopoulos, C. & Zylicz, M. (1995) EMBO J. 9, 1867–1877.
19. Levchenko, I., Luo, L. & Baker, T.A. (1995) Genes Dev. 9, 2399–2408.
20. Glover, J.R. & Lindquist, S. (1998) Cell 94, 73–82.
21. Levchenko, I., Yamauchi, M. & Baker, T.A. (1997) Genes Dev. 11, 1561–1572.
22. Leonhard, K., Herrmann, J.M., Stuart, R.A, Mannhaupt, G., Neupert, W. & Langer, T. (1996) EMBO J. 15, 4218–4229.
23. Arlt, H., Tauer, R., Feldmann, H., Neupert, W. & Langer, T. (1996) Cell 85, 875–885.
24. Gottesman, S., Maurizi, M.R. & Wickner, S. (1997) Cell 91, 435–438.
25. Suzuki, C.K., Rep, M., Maarten van Dijl, J., Suda, K., Grivell, L.A. & Schatz, G. (1997) Trends Biochem. Sci. 22, 118–122.
26. Arlt, H., Steglich, G., Perryman, R., Guiard, B., Neupert, W. & Langer, T. (1998) EMBO J. 17, 4837–4847.
27. Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D.C., Joachimiak, A., Horwich, A.L. & Sigler, P.B. (1994) Nature (London) 371, 578–586.
28. Chen, S., Roseman, A.M., Hunter, A.S., Wood, S.P., Burston, S.G., Ranson, N.A., Clarke, A.R. & Saibil, H.R. (1994) Nature (London) 371, 261–264.
29. Fenton, W.A. & Horwich, A.L. (1997) Protein Sci. 6, 743–760.
30. Rye, H.S., Burston, S.G., Fenton, W. A, Beechem, J.M., Xu, Z., Sigler, P.B. & Horwich, A.L. (1997) Nature (London) 388, 792–798.
31. Wang, J., Hartling, J.A. & Flanagan, J.M. (1997) Cell 91, 447–456.
32. Groll, M., Ditzel, L., Löwe, J., Stock, D., Bochtler, M., Bartunik, H.D. & Huber, R. (1997) Nature (London) 386, 463–471.
33. Rubin, D.M., Glickman, M.H., Larsen, C.N., Dhruvakumar, S. & Finley, D. (1998) EMBO J. 17, 4909–4919.
34. Glickman, M.H., Rubin, D.M., Fried, V.A. & Finley, D. (1998) Mol Cell. Biol. 18, 3149- 3162.
35. Bochtler, M., Ditzel, L., Groll, M. & Huber, R. (1997) Proc. Natl. Acad. Sci. USA 94, 6070–6074.
36. Löwe, J., Stock, D., Jap, B., Zwickl, P., Baumeister, W. & Huber, R. (1995) Science 268, 533–539.
37. Glickman, M.H., Rubin, D.M., Coux, O., Wefes, I., Pfeifer, G., Cjeka, Z., Baumeister, W., Fried, V.A. & Finley, D. (1998) Cell 94, 615–623.
38. Varshavsky, A. (1997) Trends Biochem. Sci. 22, 383–387.
39. Johnston, J.A., Johnson, E.S., Waller, P.R.H. & Varshavsky, A. (1995) J. Biol Chem. 270, 8172–8178.
40. Gottesman, S., Clark, W.P., Crecy-Lagard, V. & Maurizi, M.R. (1993) J. Biol. Chem. 268, 22618–22626.
41. Ciechanover, A. (1998) EMBO J. 17, 7151–7160.
42. Deveraux, Z., Ustrell, V., Pickart, C. & Rechsteiner, M. (1994) J. Biol. Chem. 269, 7059–7061.
43. van Nocker, S., Sadis, S., Rubin, D.M., Glickman, M.H., Fu, H., Coux, O., Wefes, I., Finley, D. & Vierstra, R.D. (1996) Mol. Cell. Biol. 16, 6020–6028.
44. Fu, H., Sadis, S., Rubin, D.M., Glickman, M., van Nocker, S., Finley, D. & Vierstra, R.D. (1998) J. Biol. Chem. 273, 1970–1981.
45. Tu, G.-F., Reid, G.E., Zhang, J.-G., Moritz, R.L. & Simpson, R.J. (1995) J. Biol. Chem. 270, 9322–9326.
46. Keiler, K.C., Waller, P.R.H. & Sauer, R.T. (1996) Science 271, 990–993.
47. Gottesman, S., Roche, E., Zhou, Y.N. & Sauer, R.T. (1998) Genes Dev. 12, 1338–1347.
48. Tobias, J.W., Shrader, T.E., Rocap, G. & Varshavsky, A. (1991) Science 254, 1374–1377.
49. Levchenko, L, Smith, C.K., Walsh, N.P., Sauer, R.T. & Baker, T.A. (1997) Cell 91, 939–947.
50. Kim, E., Niethammer, M., Rothschild, A., Jan, Y.N. & Sheng, M. (1995) Nature (London) 378, 85–88.
51. Kornau, H.-C., Schenker, L.T., Kennedy, M.B. & Seeburg, P.H. (1995) Science 269, 1737–1740.
52. Doyle, D.A., Lee, A., Lewis, J., Kim, E., Sheng, M. & MacKinnon, R. (1996) Cell 85, 1067–1076.
53. Pak, M. & Wickner, S. (1997) Proc. Natl. Acad. Sci. USA 94, 4901–4906.
54. Zahn, R., Perrett, S., Stenberg, G. & Fersht, A.R. (1996) Science 271, 642–645.
55. Gross, M., Robinson, C.V., Mayhew, M., Hartl, F.U. & Radford, SE (1996) Protein Sci. 5, 2506–2513.
56. Goldberg, M.S., Zhang, J., Matthews, C.R., Fox, R.O. & Horwich, A.L. (1997) Proc. Natl. Acad. Sci. USA 94, 1080–1085.
57. Walter, S., Lorimer, G.H. & Schmid, F.X. (1996) Proc. Natl. Acad. Sci. USA 93, 9425- 9430.
58. Gray, T.E. & Fersht, A.R. (1991) FEBS Lett. 292, 254–258.
59. Bochkareva, E.S., Lissin, N.M., Flynn, G.C., Rothman, J.E. & Girshovich, A.S. (1992) J. Biol Chem. 267, 6796–6800.
60. Jackson, G.S., Staniforth, R.A., Halsall, D.J., Atkinson, T., Holbrook, J.J., Clarke, A.R. & Burston, S.G. (1993) Biochemistry 32, 2554–2563.
61. Kad, N.M., Ranson, N.A., Cliff, M.J. & Clarke, A.R. (1998) J. Mol Biol 278, 267–278.
62. Yifrach, O. & Horovitz, A. (1995) Biochemistry 34, 5303–5308.
63. Rye, H.S., Roseman, A.M., Chen, S., Furtak, K., Fenton, W.A., Saibil, H.R. & Horwich, A.L. (1999) Cell 97, 325–338.
64. Adams, G.M., Falke, S., Goldberg, A.L., Slaughter, C.A., DeMartino, G.N. & Gogol, E.P. (1997) J. Mol Biol 273, 646–657.
65. Maurizi, M.R., Singh, S.K., Thompson, M.W., Kessel, M. & Ginsburg, A. (1998) Biochemistry 37, 7778–7786.
66. Gottesman, S., Wickner, S. & Maurizi, M.R. (1997) Genes Dev. 11, 815–823.
67. Economou, A. & Wickner, W. (1994) Cell 78, 835–843.
68. Adams, G.M., Crotchett, B., Slaughter, C.A., DeMartino, G.N. & Gogol, E.P. (1998) Biochemistry 37, 12927–12932.
69. Hayer-Hartl, M., Martin, J. & Hartl, F.-U. (1995) Science 269, 836–841.
70. Burston, S.G., Weissman, J.S., Farr, G.W., Fenton, W.A. & Horwich, A.L. (1996) Nature (London) 383, 96–99.
71. Farr, G.W., Scharl, E.C., Schumacher, R.J., Sondek, S. & Horwich, A.L. (1997) Cell 89, 927–937.
72. Ranson, N.A., Burston, S.G. & Clarke, A.R. (1997) J. Mol. Biol. 266, 656–664.
73. Kandror, O., Busconi, L., Sherman, M. & Goldberg, A.L. (1994) J. Biol Chem. 269, 23575–23582.
74. Palombella V.J., Rando, O.J., Goldberg, A.L. & Maniatis, T. (1994) Cell 78, 773–785.
75. Lin, L. & Ghosh, S. (1996) Mol. Cell Biol. 16, 2248–2254.
76. Lam, Y.A., Xu, W., DeMartino, G.N. & Cohen, R.E. (1997) Nature (London) 385, 737–740.
77. Hochstrasser, M. (1996) Annu. Rev. Genet. 30, 405–439.
78. Cary, P.D., King, D.S., Crane-Robinson, C., Bradbury, E.M., Rabbani, A., Goodwin, G.H. & Johns, E.W. (1980) Eur. J. Biochem. 112, 577–580.
79.Weber-Ban, E.U., Reid, B.G., Miranker, A.D. & Horwich,A. L. (1999) Nature (London), in press.
80. Shtilerman, M., Lorimer, G.H. & Englander, S.W. (1999) Science 284, 822–825.








