
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, at the Arnold and Mabel Beckman Center in Irvine, CA.
A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood
(sterol regulatory element-binding proteins/transcription/Site-1 protease/Site-2 protease/sterol-sensing domain)
MICHAEL S. BROWN* AND JOSEPH L. GOLDSTEIN*
Department of Molecular Genetics, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75235
ABSTRACT The integrity of cell membranes is maintained by a balance between the amount of cholesterol and the amounts of unsaturated and saturated fatty acids in phospholipids. This balance is maintained by membrane-bound transcription factors called sterol regulatory element-binding proteins (SREBPs) that activate genes encoding enzymes of cholesterol and fatty acid biosynthesis. To enhance transcription, the active NH2-terminal domains of SREBPs are released from endoplasmic reticulum membranes by two sequential cleavages. The first is catalyzed by Site-1 protease (S1P), a membrane-bound subtilisin-related serine protease that cleaves the hydrophilic loop of SREBP that projects into the endoplasmic reticulum lumen. The second cleavage, at Site-2, requires the action of S2P, a hydrophobic protein that appears to be a zinc metalloprotease. This cleavage is unusual because it occurs within a membrane-spanning domain of SREBP. Sterols block SREBP processing by inhibiting S1P. This response is mediated by SREBP cleavage-activating protein (SCAP), a regulatory protein that activates S1P and also serves as a sterol sensor, losing its activity when sterols overaccumulate in cells. These regulated proteolytic cleavage reactions are ultimately responsible for controlling the level of cholesterol in membranes, cells, and blood.
Cholesterol has long been known to play an important role in modulating fluidity and phase transitions in the plasma membranes of animal cells (1). Recently, a new role for cholesterol has been appreciated. Cholesterol, together with sphingomyelin, forms plasma membrane rafts or caveolae that are sites where signaling molecules are concentrated (2, 3). To perform these functions, membrane cholesterol must be maintained at a constant level. This homeostasis is achieved by a feedback regulatory system that senses the level of cholesterol in cell membranes and modulates the transcription of genes encoding enzymes of cholesterol biosynthesis and uptake from plasma lipoproteins. The modulators are a family of membrane-bound transcription factors called sterol regulatory element-binding proteins (SREBPs), which must be released proteolytically from membranes to act (4). This article summarizes recent progress in understanding the SREBPs and the sterolregulated proteases that release them.
Three SREBPs are currently recognized. Two are produced from a single gene through the use of alternate promoters that produce transcripts with different first exons (5). The cDNAs for these proteins, designated as SREBP-1a and SREBP-1c, were cloned from human and mouse cells (6–8). SREBP-1c was cloned independently from rat adipocytes and was designated ADD-1 (9). The third isoform, SREBP-2 is produced from a separate gene (5, 10).
The SREBPs are three-domain proteins of ˜1,150 amino acids that are bound to membranes of the endoplasmic reticulum (ER) and nuclear envelope in a hairpin orientation (4) (see Fig. 1). The NH2-terminal domain of ˜480 amino acids and the COOH-terminal domain of ˜590 amino acids project into the cytosol. They are anchored to membranes by a central domain of ˜80 amino acids that comprises two membrane-spanning sequences separated by a short 31-aa loop that projects into the lumen of the ER and nuclear envelope.
The NH2-terminal domains of SREBPs are transcription factors of the basic-loop-helix-leucine zipper (bHLH-Zip) family (4, 11). The extreme NH2 terminus contains a stretch of acidic amino acids that recruits transcriptional coactivators, including CBP (12). In SREBP-1a and SREBP-2, these acidic sequences are relatively long. In SREBP-1c, the acidic sequence is shorter, and this protein is a much weaker activator than the other two SREBPs (7, 8, 13). The NH2-terminal domains ofallthree SREBPs also contain a bHLH-Zip motif that mediates dimerization, nuclear entry, and DNA binding. Within the basic region of this motif, the SREBPs contain a tyrosine in place of an arginine that is conserved in nearly aII of the other bHLH family members (11, 14). This substitution allows SREBPs to recognize decanucleotide segments of DNA called sterol regulatory elements (SREs) (14). In contrast to the usual binding sites for bHLH proteins, which are palindromic, SREs are nonpalindromic, and they usually contain one or two copies of the sequence CAC (6, 11). When tested for binding activity against random sequences of DNA (14), the SREBPs show a strong preference for the SRE sequence that was originally defined in the enhancers of the genes encoding the low density lipoprotein (LDL) receptor and 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) synthase, namely, TCACCCCACT (15, 16). In other promoters, the SREBPs recognize different sequences, and a clear consensus has not been defined (17).
In sterol-depleted cells, the NH2-terminal domains of the SREBPs are released from membranes by two sequential proteolytic cleavages that must occur in the proper order (18). The NH2-terminal domain then travels to the nucleus, where it binds to SREs in the enhancers of multiple genes encoding enzymes of cholesterol biosynthesis, unsaturated fatty acid biosynthesis, triglyceride biosynthesis, and lipid uptake (reviewed in ref. 19). In the cholesterol biosynthetic pathway, well defined target genes include HMG-CoA synthase, HMG-CoA
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviations: bHLH-Zip, basic-helix-loop-helix-leucine zipper; CHO, Chinese hamster ovary; ER, endoplasmic reticulum; HMGCoA, 3-hydroxy-3-methylglutaryl CoA; LDL, low density lipoprotein; PLAP, placental alkaline phosphatase; SRE, sterol regulatory element; SREBP, sterol regulatory element-binding protein; SCAP, SREBP cleavage-activating protein; S1P; Site-1 protease; S2P; Site-2 protease. |
|
* |
E-mail: mbrow1@mednet.swmed.edu or jgolds@mednet.swmed.edu. |
reductase, farnesyl diphosphate synthase, and squalene synthase (20). The targets in the fatty acid and triglyceride biosynthetic pathways include acetyl CoA carboxylase, fatty acid synthase, stearoyl CoA desaturase, and glycerol-3-phosphate acyltransferase (4, 17, 20). The SREBPs also enhance transcription of the LDL receptor, which mediates cholesterol uptake from plasma lipoproteins. Overexpression of the NH2-terminal nuclear domains of SREBPs also elevates mRNAs encoding many other enzymes required for lipid synthesis, including enzymes that generate acetyl CoA and reduced pyridine nucleotides (21).
When sterols build up within cells, the proteolytic release of SREBPs from membranes is blocked. The NH2-terminal domains that have already entered the nucleus are rapidly degraded in a process that is blocked by inhibitors of proteasomes (22). As a result of these events, transcription ofallof the target genes declines. This decline is complete for the cholesterol biosynthetic enzymes, whose transcription is entirely dependent on SREBPs. The decline is less complete for the fatty acid biosynthetic enzymes whose basal transcription can be maintained by other factors (13, 23).
Two-Step Proteolytic Release of SREBPs
The two-step proteolytic release of the NH2-terminal domains is illustrated schematically in Fig. 1. The process begins when a protease, termed Site-1 protease (S1P), cleaves the SREBPs
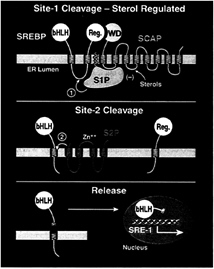
FIG. 1. Model for the sterol-mediated proteolytic release of SREBPs from membranes. (Top) Release is initiated by Site-1 protease (S1P), a sterol-regulated protease that recognizes the SCAP/ SREBP complex and cleaves SREBP in the luminal loop between two membrane-spanning sequences. SCAP allows Site-1 cleavage to be activated when cells are deprived of sterols, and it inhibits this process when sterols are abundant. (Middle) Once the two halves of SREBP are separated, a second protease, Site-2 protease (S2P), cleaves the NH2-terminal bHLH-Zip domain of SREBP at a site located within the membrane-spanning region. (Bottom) After the second cleavage, the NH2-terminal bHLH-Zip domain leaves the membrane, carrying three hydrophobic residues at its COOH-terminus. The protein enters the nucleus, where it activates target genes controlling lipid synthesis and uptake.
at a site within the hydrophilic loop that projects into the lumen of the ER (Fig. 1 Top). In SREBP-2, this cleavage occurs between the leucine and serine of the sequence RSVLS (24). S1P absolutely requires a basic residue at the P4 position, and it strongly prefers a leucine at the P1 position. The residues at the P2, P3, and P1′ positions can be substituted freely without affecting cleavage (24).
Cleavage by S1P separates the SREBPs into two halves, both of which remain membrane-bound (Fig. 1 Middle). The separation can be detected by immunoprecipitation experiments; after cleavage, an antibody against the COOH-terminal domain no longer precipitates the membranebound NH2-terminal domain. The membrane-bound NH2-terminal domain is termed the intermediate fragment of SREBP (18).
After the two halves of the SREBP have separated, a second protease, designated Site-2 protease (S2P), cleaves the NH2-terminal intermediate fragment at a site that is just within its membrane-spanning domain (Fig. 1 Middle). In SREBP-2, this cleavage occurs between the leucine and cysteine of the sequence DRSRILLC (25). The second arginine of this sequence is believed to represent the boundary between the hydrophilic NH2-terminal domain and the hydrophobic membrane-spanning segment. Thus, the cleavage occurs three residues within the membrane-spanning segment. When the NH2-terminal fragment leaves the membrane to enter the nucleus, it carries the three hydrophobic ILL residues at its COOH-terminus (Fig. 1 Bottom). Studies of intact cells showed that recognition by S2P requiresallor part of the DRSR sequence. The exact recognition sequence has not been defined. Each of the ILLC residues can be replaced singly with alanines without affecting cleavage (25).
Sterols block the proteolytic release process by selectively inhibiting cleavage by S1P (Fig. 1 Top). Current evidence indicates that S2P is not regulated directly by sterols, but it is regulated indirectly because the enzyme cannot act until the two halves of SREBP have been separated through the action of S1P (18).
SREBP Cleavage-Activating Protein (SCAP)
The first advance in understanding SREBP regulation came with the isolation of a cDNA encoding SREBP cleavage-activating protein (SCAP), a regulatory protein that is required for cleavage at Site-1 (26). SCAP is an integral membrane protein of 1,276 amino acids with two distinct domains. The NH2-terminal domain of ≈730 amino acids consists of alternating hydrophilic and hydrophobic sequences that appear to form eight membrane-spanning helices (27). This domain anchors SREBP to membranes of the ER. The COOH-terminal domain of ≈550 amino acids projects into the cytosol. It contains five WD-repeats. Similar repeats, each ≈40 residues in length, are found in many intracellular proteins, where they often mediate protein-protein interactions (28). The crystal structure of one such protein, the β-subunit of heterotrimeric G proteins, revealed that the WD-repeats form the blades of a propeller-like structure that bridges the α- and γ-subunits (29, 30).
Within cells, SCAP is found in a tight complex with SREBPs (31, 32). The association is mediated by an interaction between the COOH-terminal regulatory domain of the SREBP and the WD-repeat domain of SCAP. Formation of this complex is required for Site-1 cleavage, as revealed by the following experiments (31, 32): (i) truncation of the COOH-terminal domain of SREBP-2 prevents interaction with SCAP and abolishes susceptibility to cleavage by S1P; (ii) Overexpression of a cDNA encoding the membrane-anchored COOH-terminal domain of either SCAP or SREBP-2 competitively disrupts the formation of the complex between endogenous
SCAP and endogenous SREBP-2, and this abolishes Site-1 cleavage. This block can be overcome by overexpressing full-length SCAP or SREBP-2. Based on these findings, we hypothesized that the SCAP/SREBP complex is the true substrate for S1P (Fig. 1 Top).
SCAP as a Sterol Sensor
In addition to its requirement for Site-1 cleavage, SCAP is also the target for sterol suppression of this cleavage. This conclusion emerged from studies of mutant Chinese hamster ovary (CHO) cells that were selected for resistance to oxysterol-mediated feedback suppression of SREBP activity (26). When added to the medium surrounding cultured cells, certain oxysterols, including 25-hydroxycholesterol, block the Site-1 cleavage of SREBPs and thereby abolish cholesterol synthesis (4). These oxysterols cannot replace the functions of cholesterol in cell membranes, and the cells therefore die unless they are given a usable exogenous source of cholesterol. Oxysterol-resistant mutants survive under these conditions because they fail to respond to oxysterols by turning off cholesterol synthesis, and this forms the basis of a genetic selection (33).
Oxysterol-resistant mutant CHO cells fall into two complementation classes, both of which are genetically dominant. Class 1 mutants are sterol-resistant because they produce a truncated form of SREBP-2 that encodes the complete NH2-terminal segment but terminates before the membrane attachment domain (34, 35). The truncated protein goes directly to the nucleus without a requirement for proteolysis, and thus it cannot be suppressed by oxysterols.
Class 2 mutants produce normal full-length SREBP-1 and SREBP-2 and proteolyze them normally, but they cannot turn off proteolysis in response to sterol overload. We identified the defective gene in the Class 2 mutants by preparing a cDNA library from one of the mutant cell lines, transfecting pools of cDNAs into cultured human embryonic kidney 293 cells, and assaying for a relief of the oxysterol-dependent inhibition of expression of a reporter gene driven by an SRE-containing promoter. One cDNA was found to confer the oxysterol resistance phenotype, and this turned out to encode a mutant version of SCAP (26). The gene had undergone a C-to-G substitution, which changed amino acid 443 from aspartic acid to asparagine (Fig. 2). The identical point mutation was found in two other independently isolated mutant cell lines (36). In a fourth cell line, a point mutation in the SCAP gene changed a tyrosine at amino acid 298 to cysteine (37) (Fig. 2). When any of these mutant SCAP cDNAs is transfected into wild-type
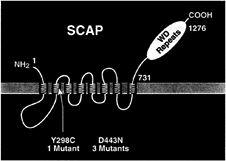
FIG. 2. Membrane topology of SCAP, showing the location of two point mutations that produce a sterol-resistant phenotype in mutant cells. The yellow region denotes the putative sterol-sensing domain of SCAP.
cells, it abolishes the susceptibility of S1P to inhibition by oxysterols, including 25-hydroxycholesterol (26). We interpret these findings to indicate that sterols normally suppress S1P activity by interacting with SCAP, either directly or indirectly. The mutant forms of SCAP are resistant to sterol inhibition, and therefore they continue to facilitate S1P activity even when sterols are present. The ability of the mutant SCAP to act in the presence of oxysterols represents a gain of function, and this explains the dominant defect in the oxysterol-resistant cells.
The remarkable aspect of the oxysterol-resistant forms of SCAP is that both of the sterol resistance mutations fall within a 160-aa segment of the membrane domain of SCAP (Fig. 2). This segment, which comprises five of the eight membrane-spanning sequences of SCAP, has been termed the sterolsensing domain. A similar stretch of five membrane-spanning sequences has been identified in three other proteins, each of which is influenced by cholesterol (Fig. 3). A sterol-sensing domain is found in the membrane attachment region of the ER enzyme, HMG-CoA reductase (26). This domain is responsible for the enhanced degradation of HMG-CoA reductase that occurs when oxysterols are added to cells (38, 39). A similar sterol-sensing domain is found in the Niemann-Pick type C1 protein, which is required for the movement of LDL-derived cholesterol from the lysosome to the ER (40). A sterol-sensing domain also has been identified in Patched, a polytopic membrane protein that serves as the receptor for the mor-
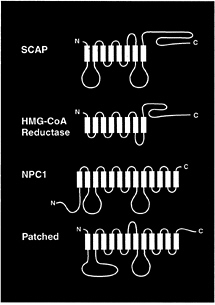
FIG. 3. Membrane proteins that contain sterol-sensing domains. The identified proteins are Chinese hamster SCAP (1,276 amino acids), Chinese hamster HMG-CoA reductase (887 amino acids), mouse Niemann-Pick type C1 (NPC1) (1,278 amino acids), and mouse Patched (1,434 amino acids). The sterol-sensing domains of these proteins, denoted in yellow, correspond to the following residues: SCAP, amino acids 280–446; HMG-CoA reductase, amino acids 57–224; NPC1, amino acids 617–691; and Patched, amino acids 420–589. The sequence alignments of the four sterol-sensing domains are published in Fig. 2 of ref. 37.
phogenic protein Hedgehog (41), the only known protein to which cholesterol is covalently attached (42). Whether the sterol-sensing domains interact directly with sterols, or whether they recognize other proteins that are in turn influenced by sterols, is not known.
Candidate Gene for Site-2 Protease
In addition to yielding SCAP, somatic cell genetics has also yielded candidate genes for the Site-2 and Site-1 proteases. The first of these, termed S2P, was isolated from a mutant line of CHO cells that is unable to produce LDL receptors, cholesterol biosynthetic enzymes, or fatty acid desaturases (43). The molecular defect was traced to a specific inability to carry out Site-2 cleavage of SREBPs (18, 44). The cells cleave the SREBPs at Site-1, but the NH2-terminal domain remains membrane-bound, owing to the failure of cleavage at Site-2. These cells are therefore auxotrophs that require cholesterol and unsaturated fatty acids for growth.
Hasan et al. at Dartmouth (43) found that the defect in one cholesterol auxotrophic cell line (M19 cells) was recessive, and they corrected the defect by transfecting genomic DNA from normal human cells and selecting for the ability to grow in the absence of cholesterol. Genomic DNA from the transfected cells was used to transfect fresh M19 cells, and this procedure was repeated several times, both at Dartmouth and at the University of Texas Southwestern Medical Center. Each repetition led to the elimination of extraneous human DNA, and eventually the cells retained only a small amount of human DNA that included the complementing gene. The human DNA from these cells was detected by PCR using repetitive human Alu elements as primers. Eventually, we were able to identify the human gene that complemented the defect in the M19 cell. Transfection of a cDNA encoded by this gene restores Site-2 cleavage in M19 cells and abolishes cholesterol auxotrophy (44).
The gene that complements the defect in M19 cells was called S2P (44). This gene encodes a protein that is necessary for Site-2 cleavage of SREBPs. Although circumstantial evidence suggests that S2P may be the Site-2 protease (see below), we have no direct biochemical evidence to support this contention. S2P might also be an auxiliary factor that is necessary in order for the true Site-2 protease to act.
The human S2P gene encodes an extremely hydrophobic protein of 519 amino acids (Fig. 4B). Most of the protein is hydrophobic, but there are two hydrophilic stretches, one of which is cysteine-rich and the other of which contains a stretch of 23 consecutive serines. Current evidence indicates that these two hydrophilic sequences project into the lumen of the ER and the remainder of the protein is embedded in the membrane itself (N.Zelenski, R.B.Rawson, J.L.G., and M.S.B., unpublished work).
One of the hydrophobic segments of S2P contains the sequence HEIGH, which conforms to the HEXXH consensus for the active site of zinc metalloproteases. This large and well studied family has members in every living organism from Archaea to humans (45, 46). One particularly well studied example is the bacterial enzyme thermolysin (47). In these proteases, the two histidines form covalent bonds with a zinc molecule, and the glutamic acid polarizes a water molecule so that it can make a nucleophilic attack on the peptide bond. The two X amino acids are variable among family members, but in several cases they are isoleucine-glycine, thus conforming to the exact sequence in S2P. Mutagenesis experiments confirmed that the HEXXH sequence is required in order for S2P to restore Site-2 cleavage in M19 cells (44). When either of the histidines or the glutamic acid was changed to alanine, the protein lost the ability to restore Site-2 cleavage. Computer-based searches of DNA databases revealed fragments of DNA encoding parts of proteins with significant resemblances to S2P
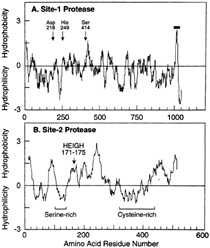
FIG. 4. Hydropathy plots of hamster Site-1 protease (A) and human Site-2 protease (B). The residue-specific hydropathy index was calculated over a window of 20 residues by the method of Kyte and Doolittle (60) as described (44, 51). For Site-1 protease, arrows denote the three amino acids of S1P that correspond to the catalytic triad for subtilisin-like serine proteases. For Site-2 protease, the arrow denotes the sequence in S2P corresponding to the consensus HEXXH pentapeptide metal binding site for zinc metalloproteases. The one transmembrane sequence in S1P is denoted by the horizontal bar. The serine- and cysteine-rich regions in S2P are indicated.
in Drosophila melanogaster (33% identity over 197 residues); Caenorhabditis elegans (43% identity over 199 residues); Schistosoma mansoni (27% identity over 117 residues); and Sulfolobus solfataricus (25% identity over 366 residues). All of these proteins share the HEXXH consensus except S.mansoni, whose available sequence does not extend into this region. All of these proteins also share the overall hydrophobic character of human S2P (44).
The mutagenesis data are consistent with the idea that S2P is indeed the Site-2 protease, but so far our multiple attempts to demonstrate in vitro protease activity for isolated S2P have failed. It is likely that these failures relate to the formidable technical difficulty in producing an active form of a membrane-embedded enzyme, especially one whose putative substrate is a leucine-cysteine bond that is sequestered within the membrane-spanning region of another protein (25). Getting the enzyme and substrate together in a test tube has proven extremely difficult.
If S2P is indeed a zinc metalloprotease, its hydrophobicity distinguishes it from other members of this family. Although the family includes membrane-bound enzymes such as matrix metalloproteases and the converting enzymes for angiotensin and endothelin, their structures differ fundamentally from that of S2P. In these other enzymes, the active sites are contained within hydrophilic domains that resemble those of soluble zinc metalloproteases (46). The catalytic domain is simply attached to the membrane by a hydrophobic extension. In S2P the putative active site is contained within an otherwise hydrophobic sequence that appears to be embedded in the membrane (Fig. 4B). If S2P is a protease, it will be the first identified protease whose substrate is a membrane-spanning region of another protein. Proteolysis within a lipid bilayer may require
a hydrophobic enzyme. How such an enzyme would function in such an environment is unknown.
Inasmuch as the S2P gene was cloned by complementation of the defect in M19 cells, it was important to demonstrate that this gene was indeed mutated in this cell line. Northern gel analysis showed that the S2P mRNA was detectable in wild-type CHO cells and inallorgans studied, but it was not detectable in M19 cells (44). The S2P gene was mapped to the X chromosome (44). Although wild-type CHO-K1 cells should have two copies of this gene, Southern blotting data suggested that the cells had only one copy. In the M19 cells, which were derived from CHO-K1 cells, this single copy had undergone a complex rearrangement, precluding transcription (44).
Candidate Gene for Site-1 Protease
The somatic cell genetic approach that permitted the cloning of S2P initially presented obstacles when we tried to use it for cloning S1P. The difficulty arose because of the presence of only a single copy of the S2P gene in the parental CHO-K1 cells. Whenever we mutated CHO cells and selected for cholesterol auxotrophy, we always isolated cells with mutations in S2P. We reasoned that this was because of the high likelihood of obtaining a mutation in a single-copy gene as compared with the low likelihood of obtaining simultaneous mutations in two copies of a gene, as was presumably the case for the S1P gene.
To circumvent this problem, we transfected CHO-K1 cells with an expressible cDNA encoding S2P and isolated a permanent cell line that contains multiple copies of this cDNA, thereby reducing the likelihood of obtaining S2P-deficient mutants (48). After mutagenesis, several approaches were used to isolate cells that were deficient in S1P (48). In the most successful approach, we first attempted to enrich for mutants that were haploinsufficient for S1P by incubating the cells with LDL that had incorporated a fluorescent cholesteryl ester, pyrene-methyl cholesteryl oleate (PMCA-oleate). We reasoned that cells with only a single copy of S1P would produce fewer LDL receptors because they would have lower amounts of nuclear SREBPs. Cells that were incubated with fluorescent LDL were separated by a fluorescence-activated cell sorter, and the cells with the lowest uptake were selected.
The sorted cells were subjected to a second round of mutagenesis in an attempt to inactivate the single remaining copy of the S1P gene (48). The cells then were selected for complete cholesterol auxotrophy by using a modification of the amphotericin resistance approach originally developed by Limanek et al. (49). In this procedure, cells are incubated briefly in a low concentration of LDL as the sole source of cholesterol. Cells that have normal SREBP activity will maintain their cholesterol levels as a result of enhanced cholesterol synthesis and uptake of LDL through LDL receptors. Cells with blocks in SREBP processing cannot obtain cholesterol from either of these sources, and they therefore become depleted in cholesterol. The cells then are treated with amphotericin, a polyene antibiotic that disrupts plasma membranes by forming complexes with cholesterol (50). Whereas wild-type cells are killed by amphotericin, cholesterol-deficient cells are resistant. After this selection, the cholesterol auxotrophs are rescued by addition of a mixture of cholesterol (dissolved in ethanol), small amounts of mevalonate to supply nonsterol products, and oleate to counteract the anticipated block in synthesis of unsaturated fatty acids (48).
The two-step mutagenesis approach described above and a modified one-step version of this approach yielded several cell lines that were auxotrophic for cholesterol because they failed to cleave SREBPs at Site-1. Cell fusion studies showed that these defects were recessive (48). We then used these cells as recipients in a transient transfection protocol designed to clone the defective gene. As a reporter in these assays, we designed a vector that encodes a fusion protein whose secretion from cells depends on cleavage by S1P. The fusion protein consists of human placental alkaline phosphatase (PLAP) joined to the COOH-terminal half of SREBP-2 (51) (Fig. 5). PLAP is a membrane-bound enzyme that is normally translocated to the plasma membrane with its catalytic domain facing the extracellular space. It is anchored to the membrane by a COOH-terminal glycophospholipid anchor. The PLAP/BP2 fusion protein begins with the signal sequence of alkaline phosphatase followed by the catalytic domain. The PLAP is truncated to eliminate its COOH-terminal membrane anchor, and the truncated PLAP is fused to the luminal loop of SREBP-2 just to the NH2-terminal side of the RSVL recognition sequence for S1P.
When the PLAP/BP2 fusion protein is expressed in wild-type cells, the catalytic domain is translocated into the ER lumen by virtue of the PLAP signal sequence. The NH2-terminal end of PLAP is freed from its membrane attachment by signal peptidase. The COOH-terminal end remains attached to the membrane by virtue of its connection to the COOH-terminal half of SREBP-2. Cleavage by S1P releases the catalytic domain into the lumen and allows it to be secreted into the medium where its activity can be measured by a sensitive chemiluminescence assay (51).
Validation experiments showed that wild-type CHO cells secreted PLAP into the medium when transfected with the cDNA encoding the PLAP/BP2 fusion protein (51). Secretion required cotransfection with a vector encoding SCAP, apparently because the endogenous SCAP was not sufficient to yield high-level cleavage of the protein. Secretion was suppressed by sterols, and it also was abolished when the arginine of the RSVL sequence was changed to alanine. All of these findings strongly suggested that secretion of PLAP required S1P. This was confirmed when we produced the PLAP/BP2 fusion protein in the mutant SRD-12B cells that lack S1P activity.
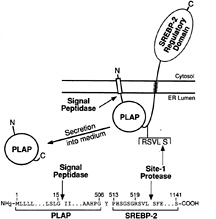
FIG. 5. Proteolytic processing and secretion of the PLAP/BP2 fusion protein used for the complementation cloning of S1P. The details of the construction of the plasmid encoding this fusion protein are described in ref. 51. In brief, the plasmid was generated by fusing the sequence encoding the signal peptide and soluble catalytic domain of human placental alkaline phosphatase (amino acids 1–506) with the sequence encoding amino acids 513–1,141 of human SREBP-2. Secretion of the catalytic domain of PLAP requires cleavage by signal peptidase and Site-1 protease. [Figure reproduced with permission from ref. 51 (Copyright 1998, Cell Press).])
These cells were unable to secrete PLAP even when they were cotransfected with the SCAP-producing vector.
To clone the S1P gene, we transiently transfected the mutant SRD-12B cells with the PLAP/BP2 expression vector, a plasmid encoding SCAP, and pools of cDNAs from an expression library derived from CHO cells that produce S1P (51). To control for transfection efficiency, we included a vector encoding ß-galactosidase driven by the cytomegalovirus promoter. After transfection, the medium was assayed for PLAP activity, and the cells were assayed for ß-galactosidase. We tested 300 pools of 1,000 cDNAs per pool, and identified two pools that were able to restore the secretion of PLAP in the SRD-12B cells. Subdivision of these positive pools eventually led to the purification of a single positive cDNA.
The positive cDNA encoded a protein of 1,052 amino acids whose sequence hadallof the properties expected for an enzyme that cleaves the luminal RSVL sequence at Site-1 of SREBPs (51). We therefore named this protein S1P. The protein begins with a hydrophobic stretch with the typical properties of a signal sequence, indicating that it is translocated into the ER lumen (Fig. 4A). The signal sequence is followed by domain that identifies it as a member of the large family of subtilisin-related serine proteases. This is followed by a COOH-terminal extension that also is predicted to lie within the lumen, followed by a hydrophobic putative transmembrane domain and a short sequence that is predicted to lie on the cytoplasmic side of the membrane. This COOH-terminal tail has a strikingly basic character.
Subtilisin-related enzymes, or subtilases, are serine proteases that contain a catalytic site with the classic triad of serine, aspartic acid, and histidine residues as well as a remote asparagine that contributes to a so-called oxyanion hole (52). Although they share the catalytic triad with the other large family of serine proteases, the trypsin-like enzymes, the subtilases are believed to have evolved independently. Members of the subtilisin family are found inallliving cells from bacteria to humans. In mammals, the previously characterized members of this family consist of the prohormone convertases, of which furin is the prototype. These enzymes function within the lumen of organelles in the secretory pathway, where they cleave membrane-bound or secretory proteins (such as the insulin pro-receptor, pro-von Willebrand factor, and proopiomelanocortin) before their transport to the cell surface or secretion from the cell (53, 54). All of the mammalian prohormone convertases cleave after basic residues, usually after dibasic sequences, and most of them also require a basic residue at the P4 site. The classic recognition sequence is RX(R/K)R (54). Prokaryotic members of this family, typified by Savinase from Bacillus lentus, cleave after hydrophobic residues without a requirement for any basic residue (55). The sequence of the catalytic domain of mammalian S1P more closely resembles that of bacterial Savinase than that of mammalian subtilisins. This observation is consistent with the predicted ability of S1P to cleave after a hydrophobic residue: i.e., the leucine of the RSVL sequence of SREBPs (24).
The sequence of human S1P was first reported by a Japanese group who sequenced random cDNAs from a human myeloid cell library (56). By virtue of its DNA sequence, the encoded protein was recognized as a member of the subtilisin family, and the catalytic triad residues were predicted. However, the putative enzyme was not assayed, and nothing was known of its physiologic function. The hamster S1P that we cloned by complementation is 97% identical to the human sequence (51). Using reverse transcriptase-PCR and degenerate oligonucleotides corresponding to the catalytic-site residues of bacterial subtilisin, Seidah et al. (57) recently cloned a cDNA, designated SKI-1, from mouse and rat cells whose amino acid sequences are 97% identical to those of hamster and human S1P. SKI-1 thus appears to be the murid ortholog of hamster and human S1P.
Northern blotting showed that the S1P mRNA is produced in wild-type CHO cells and in all 15 human tissues that were examined. The mRNA was not detectable in the mutant SRD-12B cells. Genomic blots showed that these cells contain one copy of a rearranged S1P gene and a second copy that has a normal restriction pattern but is presumably mutated not to produce detectable mRNA (51).
When we introduced an expression vector encoding S1P into SRD-12 B cells, we restored the ability of these cells to cleave SREBP-1 and SREBP-2 at Site-1 in a sterol-regulated manner (51). The cells were now able to synthesize their own cholesterol, and all of their auxotrophies were abolished. Transfected S1P could not restore any of these functions when we replaced any one of the three residues that were predicted to form the catalytic triad, further supporting the notion that this protein is indeed a serine protease (51). This conclusion was supported by the finding of Seidah et al. (57), who showed that the culture medium from cells overexpressing S1P (or SKI-1) could cleave pro-brain-derived neurotrophic factor after the threonine of an RGLTS sequence.
Cell fractionation experiments confirmed that S1P is an intrinsic membrane protein (51). The protein was shown to contain N-linked carbohydrates that remained in the endoglycosidase H-sensitive form, suggesting that the protein did not reach the medial-Golgi apparatus (51). Seidah et al. (57) used immunofluorescence techniques to study the distribution of S1P (or SKI-1) in cells stably overexpressing the protein. They found the protein in structures that resembled the ER, the Golgi complex, and small vesicles. Whether this reflects the distribution of the endogenous native protein remains unknown. Like other members of the subtilisin family, S1P is predicted to have an NH2-terminal propeptide that must be cleaved in order for it to form an active enzyme. The site of this cleavage and its mechanism remain to be determined.
Unresolved Questions
From the standpoint of physiologic regulation, the crucial unresolved questions relate to the requirement for SCAP in the S1P cleavage reaction and the mechanism by which SCAP activity is abolished by sterols. All of the known members of the subtilisin family function independently, and they do not require a membrane-bound cofactor like SCAP. Does SCAP play a role in the direct recognition of SREBP by S1P? Or does SCAP play a more indirect role, perhaps by transporting SREBPs to the places in the cell where the active form of S1P resides?
Some evidence in favor of the latter mechanism has come from a study of the carbohydrate composition of SCAP. When CHO cells were grown in the presence of sterols and SCAP activity was suppressed, the N-linked carbohydrates of SCAP remained in the endoglycosidase H-sensitive form, suggesting that SCAP remained in the ER (37). However, after cells were switched to sterol-depleted medium and cleavage of SREBPs was inaugurated, the carbohydrates of SCAP were converted to the endoglycosidase H-resistant form. The latter observation indicates that SCAP had reached the medial-Golgi complex (37), yet our preliminary cell fractionation experiments show that the bulk of the endoglysidase H-resistant protein was still in the ER. We interpret these data to indicate that, in sterol-depleted cells, SCAP cycles from the ER to the Golgi and back again. Inasmuch as SCAP is in a complex with full-length SREBP, these data suggest that SCAP may escort SREBP to some post-ER compartment where cleavage takes place. When sterols are added to cells, SCAP remains in the ER, presumably in a complex with SREBP. This may prevent SREBP from reaching the organelle that contains active S1P, thereby precluding cleavage. This hypothesis should be testable now that SCAP and S1P have been identified.
A second unresolved question relates to potential roles of S2P and S1P in proteolytic processing of other proteins in addition to SREBPs. As noted above, hydrophobic proteins that resemble S2P, including the putative zinc-binding site, are found as far back as Archaea. This suggests that S2P may play more general housekeeping roles in addition to processing SREBPs.
S1P also may play a more general role in proteolytic cleavage. S1P is the first vertebrate subtilisin whose sequence more closely resembles the bacterial members of this family as compared with the mammalian members. This finding is consistent with the observation that S1P cleaves SREBP after a hydrophobic leucine residue rather than after a basic residue. S1P also appears to act in a pre-Golgi compartment, which differs from the prohormone convertases, which generally act in the Golgi or in post-Golgi compartments (53, 54). The requirement for SCAP suggests that the activity of S1P may be restricted to SREBPs because no other proteins are known to require SCAP for cleavage. Moreover, cells that lack S1P grow normally as long as they are supplied with the end-products of the SREBP pathway (48). On the other hand, the finding that S1P (or SKI-1) can cleave pro-brain-derived neurotrophic factor when overexpressed in intact cells or in vitro raises the possibility that the protease may have broader actions. This argument is rendered less persuasive by the observation that the site in pro-brain-derived neurotrophic factor that is cleaved by S1P does not correspond to the major site of physiologic pro-brain-derived neurotrophic factor processing in vivo (58, 59).
Clearly, the intense study of S1P and S2P is only beginning. Given the rich scientific experience with other proteases, all of the unresolved questions about these two reactions will likely be answered in the near future. These answers should markedly advance our knowledge of cholesterol homeostasis.
This work was supported by research grants from the National Institutes of Health (HL20948) and the Perot Family Foundation.
1. Devaux, P.F. (1993) Curr. Opin. Struct. Biol. 3, 489–494.
2. Simons, K. & Ikonen, E. (1997) Nature (London) 387, 569–572.
3. Anderson, R.G.W. (1998) Annu. Rev. Biochem. 67, 199–225.
4. Brown, M.S. & Goldstein, J.L. (1997) Cell 89, 331–340.
5. Hua, X., Wu, J., Goldstein, J.L., Brown, M.S. & Hobbs, H.H. (1995) Genomics 25, 667–673.
6. Yokoyama, C, Wang, X., Briggs, M.R., Admon, A., Wu, J., Hua, X., Goldstein, J.L. & Brown, M.S. (1993) Cell 75, 187–197.
7. Shimano, H., Horton, J.D., Shimomura, I., Hammer, R.E., Brown, M.S. & Goldstein, J.L. (1997) J. Clin. Invest. 99, 846–854.
8. Shimomura, I., Shimano, H., Horton, J.D., Goldstein, J.L. & Brown, M.S. (1997) J. Clin. Invest. 99, 838–845.
9. Tontonoz, P., Kim, J.B., Graves, R.A. & Spiegelman, B.M. (1993) Mol. Cell Biol. 13, 4753–4759.
10. Hua, X., Yokoyama, C., Wu, J., Briggs, M.R., Brown, M.S., Goldstein, J.L. & Wang, X. (1993) Proc. Natl. Acad. Sci. USA 90, 11603–11607.
11. Parraga, A, Bellsolell, L., Ferre-D’Amare, A.R. & Burley, S.K. (1998) Structure (London) 6, 661–672.
12. Näär, A.M., Beaurang, P.A., Robinson, K.M., Oliner, J.D., Avizonis, D., Scheek, S., Zwicker, J., Kadonaga, J.T. & Tjian, R. (1998) Genes Dev. 12, 3020–3031.
13. Pai, J., Guryev, O., Brown, M.S. & Goldstein, J.L. (1998) J. Biol. Chem. 273, 26138–26148.
14. Kim, J.B., Spotts, G.D., Halvorsen, Y.-D., Shih, H.-M, Ellenberger, T., Towle, H.C. & Spiegelman, B.M. (1995) Mol. Cell. Biol. 15, 2582–2588.
15. Smith, J.R., Osborne, T.F., Brown, M.S., Goldstein, J.L. & Gil, G. (1988) J. Biol. Chem. 263, 18480–18487.
16. Smith, J.R., Osborne, T.F., Goldstein, J.L. & Brown, M.S. (1990) J. Biol. Chem. 265, 2306–2310.
17. Magana, M.M. & Osborne, T.F. (1996) J. Biol. Chem. 271, 32689–32694.
18. Sakai, J., Duncan, E.A.. Rawson, R.B., Hua, X., Brown, M.S. & Goldstein, J.L. (1996) Cell 85, 1037–1046.
19. Horton, J.D. & Shimomura, I. (1999) Curr. Opin. Lipidol. 10, 143–150.
20. Edwards, P.A. & Ericsson, J. (1998) Curr. Opin. Lipidol. 9, 433–440.
21. Shimomura, I., Shimano, H., Korn, B.S., Bashmakov, Y. & Horton, J.D. (1998) J. Biol. Chem. 273, 35299–35306.
22. Wang, X., Sato, R., Brown, M.S., Hua, X. & Goldstein, J.L. (1994) Cell 77, 53–62.
23. Horton, J.D., Shimomura, I., Brown, M.S., Hammer, R.E., Goldstein, J.L. & Shimano, H. (1998) J. Clin. Invest. 101, 2331–2339.
24. Duncan, E.A., Brown, M.S., Goldstein, J.L. & Sakai, J. (1997) J. Biol. Chem. 272, 12778–12785.
25. Duncan, E.A., Davé, U.P., Sakai, J., Goldstein, J.L. & Brown, M.S. (1998) J. Biol. Chem. 273, 17801–17809.
26. Hua, X., Nohturfft, A, Goldstein, J.L. & Brown, M.S. (1996) Cell 87, 415–426.
27. Nohturfft, A., Brown, M.S. & Goldstein, J.L. (1998) J. Biol. Chem. 273, 17243–17250.
28. Neer, E.J., Schmidt, C.J., Nambudripad, R. & Smith, T.F. (1994) Nature (London) 371, 297–300.
29. Wall, M. A, Coleman, D.E., Lee, E., Iniguez-Lluhi, J.A., Posner, B.A., Gilman, A.G. & Sprang, S.R. (1995) Cell 83, 1047–1058.
30. Lambright, D.G., Sondek, J., Bohm, A., Skiba, N.P., Hamm, H.E. & Sigler, P.B. (1996) Nature (London) 379, 311–319.
31. Sakai, J., Nohturfft, A., Cheng, D., Ho, Y.K., Brown, M.S. & Goldstein, J.L. (1997) J. Biol. Chem. 272, 20213–20221.
32. Sakai, J., Nohturfft, A., Goldstein, J.L. & Brown, M.S. (1998) J. Biol. Chem. 273, 5785–5793.
33. Metherall, J.E., Ridgway, N.D., Dawson, P.A., Goldstein, J.L. & Brown, M.S. (1991) J. Biol. Chem. 266, 12734–12740.
34. Yang, J., Sato, R., Goldstein, J.L. & Brown, M.S. (1994) Gene Dev. 8, 1910–1919.
35. Yang, J., Brown, M.S., Ho, Y.K. & Goldstein, J.L. (1995) J. Biol. Chem. 270, 12152–12161.
36. Nohturfft, A., Hua, X., Brown, M.S. & Goldstein, J.L. (1996) Proc. Natl. Acad. Sci. USA 93, 13709–13714.
37. Nohturfft, A., Brown, M.S. & Goldstein, J.L. (1998) Proc. Natl. Acad. Sci. USA 95, 12848–12853.
38. Gil, G., Faust, J.R., Chin, D.J., Goldstein, J.L. & Brown, M.S. (1985) Cell 41, 249–258.
39. Kumagai, H., Chun, K.T. & Simoni, R.D. (1995) J. Biol. Chem. 270, 19107–19113.
40. Loftus, S. K, Morris, J. A, Carstea, E.D., Gu, J.Z., Cummings, C., Brown, A., Ellison, J., Ohno, K., Rosenfeld, M.A., Tagle, D.A, et al. (1997) Science 277, 232–235.
41. Tabin, C.J. & McMahon, A.P. (1997) Trends Cell Biol. 7, 442–446.
42. Porter, J.A., Young, K.E. & Beachy, P.A. (1996) Science 274, 255–259.
43. Hasan, M.T., Chang, C.C.Y. & Chang, T.Y. (1994) Somatic Cell Mol. Genet. 20, 183–194.
44. Rawson, R.B., Zelenski, N.G., Nijhawan, D., Ye, J., Sakai, J., Hasan, M.T., Chang, T.-Y., Brown, M.S. & Goldstein, J.L. (1997) Mol. Cell 1, 47–57.
45. Hooper, N.M. (1994) FEBS Lett. 354, 1–6.
46. Rawlings, N.D. & Barrett, A.J. (1995) Methods Enzymol. 248, 183–228.
47. Holmes, M.A. & Matthews, B.W. (1982) J. Mol. Biol. 160, 623–639.
48. Rawson, R.B., Cheng, D., Brown, M.S. & Goldstein, J.L. (1998) J. Biol. Chem. 273, 28261–28269.
49. Limanek, J.S., Chin, J. & Chang, T.Y. (1978) Proc. Natl. Acad. Sci. USA 75, 5452–5456.
50. DeKruijff, B., Gerritsen, W.J., Oerlemans, A., Demel, R.A. & Van Deenen, L.L.M. (1974) Biochim. Biophys. Acta 339, 30–43.
51. Sakai, J., Rawson, R.B., Espenshade, P.J., Cheng, D., Seegmiller, A.C., Goldstein, J.L. & Brown, M.S. (1998) Mol. Cell 2, 505–514.
52. Siezen, R.J. & Leunissen, J.A.M. (1997) Protein Sci. 6, 501–523.
53. Seidah, N.G. & Chretien, M. (1994) Methods Enzymol. 244, 175–188.
54. Nakayama, K. (1997) Biochem. J. 327, 625–635.
55. Sørensen, S.B., Bech, L.M., Meldal, M. & Breddam, K. (1993) Biochemistry 32, 8994–8999.
56. Nagase, T., Miyajima, N., Tanaka, A., Sazuka, T., Seki, N., Sato, S., Tabata, S., Ishikawa. K.-i., Kawarabayasi, Y., Kotani, H. & Nomura, N. (1995) DNA Res. 2, 37–43.
57. Seidah, N.G., Mowla, S.J., Hamelin, J., Mamarbachi, A.M., Benjannet, S., Touré, B.B., Basak, A., Munzer, J.S., Marcinkiewicz, J., Zhong, M., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 1321–1326.
58. Leibrock, J., Lottspeich, F., Hohn, A., Hofer, M., Hengerer, B., Masiakowski, P., Thoenen, H. & Barde, Y.-A. (1989) Nature (London) 341, 149–152.
59. Rosenfeld, R.D., Zeni, L., Haniu, M., Talvenheimo, J., Radka, S.F., Bennett, L., Miller, J.A. & Welcher, A.A. (1995) Protein Expression Purif. 6, 465–471.
60. Kyte, J. & Doolittle, R.F. (1982) J. Mol. Biol. 157, 105–132.








