
This paper was presented at the National Academy of Sciences colloquium “Proteolytic Processing and Physiological Regulation,” held February 20–21, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Cellular mechanisms of β-amyloid production and secretion
SUKANTO SINHA* AND IVAN LIEBERBURG
Elan Pharmaceuticals, South San Francisco, CA 94080
ABSTRACT The major constituent of senile plaques in Alzheimer’s disease is a 42-aa peptide, referred to as β-amyloid (Aβ). Aβ is generated from a family of differentially spliced, type-1 transmembrane domain (TM)-containing proteins, called APP, by endoproteolytic processing. The major, relatively ubiquitous pathway of APP metabolism in cell culture involves cleavage by α-secretase, which cleaves within the Aβ sequence, thus precluding Aβ formation and deposition. An alternate secretory pathway, enriched in neurons and brain, leads to cleavage of APP at the N terminus of the Aβ peptide by β-secretase, thus generating a cell-associated β-C-terminal fragment (β-CTF). A pathogenic mutation at codons 670/671 in APP (APP “Swedish”) leads to enhanced cleavage at the β-secretase scissile bond and increased Aβ formation. An inhibitor of vacuolar ATPases, bafilomycin, selectively inhibits the action of β-secretase in cell culture, suggesting a requirement for an acidic intracellular compartment for effective β-secretase cleavage of APP. β-CTF is cleaved in the TM domain by γ-secretase(s), generating both Aβ 1–40 (90%) and Aβ 1–42 (10%). Pathogenic mutations in APP at codon 717 (APP “London”) lead to an increased proportion of Aβ 1–42 being produced and secreted. Missense mutations in PS-1, localized to chromosome 14, are pathogenic in the majority of familial Alzheimer’s pedigrees. These mutations also lead to increased production of Aβ 1–42 over Aβ 1–40. Knockout of PS-1 in transgenic animals leads to significant inhibition of production of both Aβ 1–40 and Aβ 1–42 in primary cultures, indicating that PS-1 expression is important for γ-secretase cleavages. Peptide aldehyde inhibitors that block Aβ production by inhibiting γ-secretase cleavage of β-CTF have been discovered.
Aβ Is Derived from APP. Alzheimer’s disease is a wide-spread, neurodegenerative, dementia-inducing disorder of the elderly that has been estimated to affect more than 4 million people in the United States alone. The disease is characterized by synaptic loss and neuronal death in the cerebral cortex and the hippocampus, with the presence of extensive extracellular amyloid plaques and intracellular neurofibrillary tangles (1). The pathology of Alzheimer’s disease has been studied extensively for the last 20 years, but it was not until about 15 years ago that the first molecular handle in understanding this complex degenerative disease was obtained, when the protein sequence of the extracellular amyloid was determined (2). The cloning of APP, achieved in 1987 (3), established that the fibrillar, ≈40-aa-long amyloid peptide deposited as the major constituent of both senile and cerebrovascular plaques is derived from a type-1 TM protein. The parsimonious hypothesis, immediately arising as a consequence of the schematic shown in Fig. 1, was that two separate endoproteolytic events released the smaller Aβ peptide from its precursor.
APP was also found to be expressed in a variety of tissues as a family of differentially spliced forms, the transcripts ranging in predicted size from 695 to 770 aa. The two longer forms, known as APP751 and APP770, contained a 56-aa domain with homology to the Kunitz family of serine protease inhibitors (4). APP695, the splicing variant lacking the Kunitz domain, was preferentially expressed in neuronal tissue, leading to the speculation that the production of Aβ from APP could be regulated by a protease that is inhibited by this domain.
The demonstration that a secreted, soluble form of APP was functionally identical to a previously isolated serine protease inhibitor called protease nexin II (5), together with the finding that the Kunitz domain showed restricted inhibitory activity toward a number of serine proteases (6), strengthened the hypothesis that the soluble ectodomain of APP functions as a circulating protease inhibitor.
Secreted APP (sAPP) Production: α-Secretase. Transfection of the various forms of APP into mammalian cells showed that newly synthesized APP, N-glycosylated in the endoplasmic reticulum, matures in the secretory pathway by the addition of O-glycosyl residues and tyrosine sulfation in the trans-Golgi network (7); cellular turnover of full-length, membrane-bound, mature APP is accompanied by the release in the conditioned medium (CM) of the soluble ectodomain of the protein and the appearance of a truncated cell-associated CTF (8). The soluble sAPP is detected, not only in the CM of transfected cells, but is also found in plasma and cerebrospinal fluid, suggesting a conserved metabolic pathway. Direct sequencing of the CTF obtained from APP-transfected cells showed that the endoproteolytic cleavage generating the sAPP and the corresponding CTF occurs primarily by cleavage between amino acids 16 and 17 of the Aβ sequence (9), i.e., inside the Aβ sequence. Analysis of the metabolism of various site-specific mutants of APP led to the conclusion that the cleavage site of this unidentified cellular enzyme, named α-secretase, was relatively nonspecific, with distance from the TM being a more important parameter than the actual identity of amino acids at the cleavage site(s) (10). The ubiquity of this pathway, which by definition could not produce Aβ, led to the proposition that the “normal” cellular metabolism of APP precludes the formation of Aβ. The corollary, that the production of Aβ is caused by abnormal or “aberrant” cleavages in the FL-APP molecule, came to be accepted as well.
Further, it was recognized that α-secretase activity could be stimulated in cells by using phorbol esters, leading to the activation of protein kinase C (11). The demonstration that muscarinic agents mimic this effect (12) indicated that stimulated α-cleavage could be linked in neuronal cells to the activity of cholinergic agents. This demonstration lent more credence to the hypothesis that α-secretory processing of APP is a “good” pathway that is diminished in brain with Alzhei-
|
|
PNAS is available online at www.pnas.org. |
|
|
Abbreviations: Aβ, β-amyloid; TM, transmembrane; CTF, C-terminal fragment; sAPP, secreted APP; Wt, wild type; CHO, Chinese hamster ovary; CM, conditioned medium. |
|
* |
To whom reprint requests should be addressed. E-mail: ssinha@elanpharma.com. |
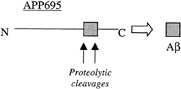
FIG. 1. Aβ is generated from precursor protein, APP. N, N terminus; C, C terminus.
mer’s disease, perhaps as a consequence of loss of cholinergic stimulation. The “uncleaved” APP could then be cleaved by aberrant proteolytic events, perhaps mediated by lysosomal enzymes, generating Aβ.
sAPP Production: β-Secretase. The first piece of evidence that Aβ production may not be aberrant after all was provided by the observation that both APP-transfected HEK293 cells (13) as well as fetal neuronal cultures (14) constitutively release Aβ 1–40 into the culture medium, i.e., Aβ generation and extracellular release are by-products of normal cellular metabolism of APP. This conclusion, dramatic at the time, has since been confirmed by many investigators and has come to be widely accepted.
Shortly thereafter, it was shown that a truncated form of sAPP was released from HEK293 cells transfected with APP, as well as from primary fetal human neuronal cultures (15). Using a neoepitope-specific antibody, these investigators showed that the truncated sAPP ended precisely at Met-596, a marker of specific endoproteolytic cleavage immediately N-terminal to the Aβ sequence. This β-sAPP made up a much larger proportion of total sAPP in the neuronal culture CM than in the HEK293 cell CM, suggesting that this alternative secretory cleavage, by the so-called β-secretase, was more prominent in cells derived from the central nervous system.
The consequences of these two pivotal observations were that it became possible to measure three key metabolites of APP (α-sAPP, β-sAPP, and Aβ) in a cellular context and especially to look for both inhibitors and potential stimulators of Aβ release under defined conditions.
Stimulated Release of sAPP: Effect on Aβ. Phorbol esters, such as phorbol 12-myristate 13-acetate or phorbol dibutyrate, have been used widely to stimulate sAPP release in a variety of cellular systems. Early results suggested that stimulation of sAPP release was accompanied, reciprocally, by a decrease in Aβ release (16). However, subsequent analysis in a neuroblastoma cell line in culture showed that stimulated release of sAPP was not always accompanied by decreased Aβ (17). Although phorbol 12-myristate 13-acetate virtually universally stimulates α-sAPP production, there is little, if any, effect on β-sAPP levels, and the reduction of Aβ is often only transient (J.Knops and S.S., unpublished observations). No effect on synthesis of APP was seen in these experiments. Thus, there is not necessarily a mutually exclusive relationship between α-and β-secretory cleavages, a conclusion that has become more apparent as other pharmacological agents for affecting APP metabolism have become available.
Bafilomycin and β-sAPP Inhibition. A double mutation of codons 670/671 of APP, replacing the Lys-Met sequence with Asn-Leu (18) and segregating with very early-onset Alzheimer’s disease with classic pathologic hallmarks, was described in 1992. Transfection of HEK293 cells with cDNA constructs coding for the mutated protein led to a 6-fold increase in extracellularly released Aβ (19) compared with wild-type (Wt) APP. Concurrent analysis of the sAPP species released showed that there was also a substantial increase in the β-sAPP being released from such cells. The so-called Swedish mutation in APP thus seems to exert its pathogenic effect via an increased production of Aβ, mediated by increased β-secretase cleavage in the mutated protein. This observation provided, not only a mechanistic explanation for a pathogenic mutation, but also a cellular system, relevant to the underlying disease model, in which to study pharmacological agents that can selectively inhibit the formation of Aβ.
A specific and potent inhibitor of vacuolar ATPases, bafilomycin, was shown to inhibit β-sAPP selectively, but not α-sAPP, both from HEK293 cells transfected with APP Swedish mutants and from fetal neuronal cultures (20). This effect was ascribed to the known pharmacological activity of bafilomycin, treatment with which leads to the elevation of intravesicular pH in a variety of acidic organelles, including, but not restricted to, endosomes and lysosomes (21). The concordance of the data obtained from studies with both the mutant APP-transfected cells and fetal neuronal cultures metabolizing endogenous Wt APP showed (i) that selective inhibition of β-secretase cleavage results in inhibition of Aβ release and (ii) that α-sAPP release is not affected under these conditions. Further, these data provided indirect but convincing evidence that acidic intracellular conditions are most conducive to efficient β-secretase processing of APP.
Like APP, a number of other membrane-bound proteins are “shed” from the cell surface, often in response to stimulation by phorbol esters (22). A pathologically important protein in this regard is pro-tumor necrosis factor-α (proTNF-α), which undergoes cell-surface proteolysis by an “α-secretase-like” enzyme to release circulating TNF. The purification and identification of the TNF-α-converting enzyme (TACE) as a membrane-bound metalloprotease (23) led to speculation that, like TACE, APP α-secretase is also a member of the adamalysin protease family. Cells deficient in TACE do not show any defect in constitutive α-cleavage of APP (24); however, no stimulated release of sAPP is evident on treatment with phorbol esters, suggesting that TACE plays a key role in regulated, but not constitutive, α-cleavage of APP. Metalloprotease inhibitors directed toward such proteases inhibit α-sAPP release from Chinese hamster ovary (CHO) cells in a dose-dependent manner (25), but such treatments have no significant effect on either β-sAPP or Aβ (E.Goldbach, S. Suomensaari, J.Knops, and S.S., unpublished observations).
The results of the phorbol ester, bafilomycin, and metalloprotease inhibitor studies strongly suggest that a simple reciprocal relationship does not exist between α- and β-cleavage or between sAPP production and Aβ release. It seems most likely that α-secretase and β-secretase are cellularly segregated, mechanistically distinct enzymes, and it is the direct action of the latter that correlates most with Aβ release.
Pathogenic Mutations in APP. Three separate missense mutations in APP, occurring at codon 717 (London mutations), also cause early-onset Alzheimer’s (26) but do so by a mechanism very different from that of the Swedish mutation.
After β-secretase cleavage, the C terminus of the β-peptide has to be generated by a further proteolytic event, which takes place in the TM domain of APP. In keeping with the imaginative and sequential nomenclature for the enzymes postulated to be involved in cellular APP proteolysis and Aβ generation, the enzyme cleaving in the TM domain to generate the C terminus of the Aβ peptide has been named γ-secretase.
It has been shown that most of the Aβ released from both cell lines derived from tissues other than those from the central nervous system and from neuronal cells terminates at residue 40. However, a small proportion (5–10%) extends to residue 42 (27). It has been postulated that the major pathologic culprit in Alzheimer’s disease is this subpopulation of Aβ, because this longer, more aggregation-prone species deposits preferentially in both sporadic and familial Alzheimer’s disease brains. Careful measurement of the Aβ released from cells transfected with the various London mutations revealed that although
total Aβ released was unaffected, the proportion of Aβ 1–42 increased by 50–90%, i.e., from about 10% of the total to about 20% (28). The London mutations thus shift the balance of γ-secretase cleavage slightly toward the 42 over the 40 cleavage site, which is sufficient, apparently, to cause disease. These observations have led to the proposition that there are at least two separate γ-secretases for the Aβ40 and Aβ42 sites. In the absence of definitive information, this subject lies at the heart of a current debate.
Both the London and the Swedish mutations have been used to develop transgenic models of the pathology seen in Alzheimer’s disease. The so-called PDGF promoter APP mouse was developed with the Val717Phe mutation (29). As the animals age, Aβ 1–42 deposits preferentially in the hippocampus and the cortex, mirroring the pattern seen in Alzheimer’s disease. Like Alzheimer’s disease, no senile plaques are seen in the cerebellum, in spite of expression of the transgene in this region. In addition to plaques, one may observe neuritic dystrophy, microglial activation, and astrocytic activation (30), following closely on the heels of the amyloid deposition. The major hallmarks of the disease are thus preserved in these models, which will be invaluable in evaluating the efficacy of compounds targeting the production or aggregation of the Aβ peptide.
Presenilins and Alzheimer’s Disease. APP mutations, as illuminating as they have been in both the causative role of Aβ in Alzheimer’s disease and in underscoring the importance of both β- and γ-secretase-mediated cleavages for Aβ generation and release, are relatively rare and confined to only a few familial pedigrees. A much larger number of familial Alzheimer’s disease pedigrees cluster to chromosome 14, and the product of this gene, S182, was revealed to be a multiple-membrane-spanning protein (31) imaginatively called presenilin-1. At least 37 separate missense mutations have been documented in this protein. A related gene, STML2, on chromosome 1, the protein product of which is called presenilin-2 (32), has also been shown to have missense mutations that cause Alzheimer’s disease, and two of these mutations have been documented thus far. The pathology seen in the brains of the pedigrees examined invariably show dramatic deposition of amyloid, virtually all of which are in the 1–42 form (33). Disease caused by the PS-1 mutations is aggressive, early-onset, and fully penetrant.
Cotransfection of presenilin mutants along with APP revealed the same phenomenon seen with the London mutations, i.e., the presenilin mutants invariably increase the proportion of x–42 forms between 50–100% over that seen with Wt presenilins (34). No significant effects on sAPP release or on the levels of total Aβ released are seen in these experiments. Cotransfection of APP carrying one of the London mutations along with a mutant PS-1 leads to an additive effect on the increased Aβ40/42 ratio.
Thus, the majority of familial Alzheimer’s mutants cluster to a gene, the protein product of which somehow modulates the γ-secretase cleavage with the same consequences resulting from London mutations. The homology of the PS-1 to sel-12, a Caenorhabditis elegans gene that facilitates signaling by Notch (35), has led to speculation about cellular mechanisms that might underlie the increased γ-secretase cleavage at residue 42.
The most telling data have emerged from an attempt to create PS-1 –/– animals. The homozygous animals die in utero with severe developmental abnormalities reminiscent of Notch –/– animals. However, the introduction, via viral vectors, of Wt and mutant APPs into cortical cultures produced from these embryos (36) showed that, although normal APP maturation and sAPP release were unaffected, the cells were deficient in γ-secretase cleavage of the α- and β-CTFs generated by the action of α- and β-secretases; both Aβ and p3 (the α-CTF-derived γ-secretase cleavage product) ending at residue 40 or 42 decreased by 80%, with a corresponding increase in the ambient levels of the corresponding CTFs. These results strongly suggest that the expression of PS-1 is needed for the majority of functional γ-secretase activity in vivo. Perhaps the residual production of Aβ and p3 is mediated by PS-2.
Peptide Aldehyde Inhibitors of Aβ Release. It has been known for some time that, in cell lines derived from peripheral tissues, such as HEK293, much of the full-length mature APP is degraded via a lysosomal pathway. The application of lysosomotropic agents, such as chloroquine and NH4Cl, or cysteine protease inhibitors, such as E-64 and leupeptin, led to enhanced recovery of full-length membrane-bound APP and the visualization of degradation intermediates (37). However, neither E-64 nor leupeptin have any effect on the release of Aβ under such conditions, indicating that the so-called “endosomal-lysosomal” degradation pathway was probably not involved in the generation of Aβ. However, Z-Val-Phe-CHO, a dipeptide aldehyde originally identified as a potent inhibitor of a number of intracellular cysteine proteases, such as cathepsin B, cathepsin L, and calpain (38), was shown to inhibit Aβ release at low micromolar levels in a dose-dependent manner (39). A number of other dipeptide aldehydes, with ED50 values varying between 1 and 25 μM, were also shown to be active as inhibitors of cellular Aβ release in HEK293 cells transfected with either Wt or Swedish APP.
Analysis of the cellular pattern of metabolites indicated that the release of both p3 and Aβ was being inhibited by such compounds, with concomitant increases in the levels of the corresponding CTFs. The mechanism of the action of such compounds is therefore via inhibition of γ-secretase cleavage, either as direct inhibitors of the enzyme or through indirect effects on events critical to γ-secretase cleavage. As shown in Table 1, some closely related compounds in this series have differential effects on their relative potency toward Aβx-40 vs. Aβx-42 inhibition in HEK293 cells stably transfected with the APP Swedish mutants. These effects have led some investigators to propose that different γ-secretases are involved in the two cleavages.
However, it has been suggested that Aβ 1–40 is produced at greater proximity to the cell surface than is Aβ 1–42 (40); if this suggestion is accurate, variations in intracellular compound levels in different intracellular compartments may explain the differential inhibitory susceptibilities with some of these compounds.
Although the peptide aldehydes seem to point to the role of an intracellular cysteine or serine protease as pivotal to γ-secretase processing of CTFs, direct evidence for such an enzyme target for these compounds is still lacking. In this regard, a recent publication (41) has put forward a quite remarkable proposition as to the possible nature of γ-secretase. In this report, the mutation of either of two separate TM aspartic acid residues in PS-1, Asp-257 in TM6 and Asp-385 in TM7, leads to a lowering of Aβ and increases the amounts of the α- and β-CTFs, as seen in the PS-1 –/– mice-derived neuronal cultures. The authors suggest that PS-1 may be γ-secretase, with the two aspartic acid residues forming a catalytic system analogous to that conserved in the aspartic proteinase family. It should be noted that no discernible amino acid sequence homology exists between PS-1 and any aspartic
Table 1. Effect of dipeptide aldehydes on cellular Aβ release
|
|
ED50, μM |
|
|
Compound |
Aβ x–40 |
Aβ x–42 |
|
Z-Val-Phe-CHO |
15.5 |
67.4 |
|
2-Napthyl-Val-Phe-CHO |
2.6 |
2.7 |
|
Z-Phe-Val-CHO |
Not inhibitory |
|
|
Z-Leu-Phe-CHO |
5.0 |
– |
proteinase, even around the putative “active-site” Asp-257 and Asp-385 residues, and more direct evidence is needed in support of this concept.
β-Secretase: Rate-Limiting Enzyme for Aβ Production. The London mutations in APP and the missense mutations in PS-1 that lead to Alzheimer’s disease have in common their alteration of the relative cleavage at the –40 and –42 sites in the TM domain of APP. The specificity of these γ-secretase cleavages were analyzed further by sequentially replacing amino acids 35–48 in the TM domain with Phe (42), akin to the “Ala scan” used for other scanning mutagenesis approaches. The production of Aβ and the relative ratios of x-40 vs. x-42 forms were then analyzed in the CM of cells transfected with these mutant forms. Although position 45 was identified as being critical for –42 cleavage, there was little specificity at the γ-cleavage sites; although there were alterations in the relative ratios, total Aβ formation was relatively unaffected by the scanning mutagenesis, suggesting that the precise identity of the amino acid residues at or near the γ-cleavage sites was not critical to total cleavage.
In sharp contrast, site-directed mutagenesis at the Met-Asp cleavage site on the β-end leads to dramatic effects on Aβ production (43). Although the substitution of Leu for Met at the P1 position (akin to the Swedish mutation) leads to enhancement of Aβ formation, substitution at this site by most other amino acids leads to a suppression of Aβ release in the extracellular medium, presumably by inhibition of β-secretase cleavage. Effective β-secretase cleavage is thus a prerequisite for formation and secretion of Aβ. In the case of some of the mutants, the fact that shorter Aβ peptides are secreted at a lower rate may represent the effect of an alternate cleavage site exposed as a result of conformational change in the mutated protein.
In conjunction with the results obtained with bafilomycin, it seems that β-secretase cleavage is a rate-limiting event for the formation of the “substrate” for γ-secretase. The latter enzymatic process is quite capable of turning over even the 5- to 6-fold excess β-CTFs generated in APP Swedish-transfected HEK293 cells, over that produced with Wt alone. Further, the Swedish mutation, unfortunately for the pedigree, causes disease by presenting a preferred β-cleavage site to the cellular enzyme.
β-Secretase: Isolation and Characterization. The search for enzymes that specifically cleave at the β-cleavage site in APP was initiated long before there was any cellular evidence for the presence of such a metabolic pathway. Although enzymes such as the metalloendopeptidase (EC 3.4.24.15) and cathepsin D were proposed to be candidate β-secretases, primarily as a result of cleavage specificity shown by using short peptide substrates (44), neither enzyme has passed the tests of being able to cleave full-length APP specifically, generating both the N- and C-terminal fragments. Cotransfection of these enzymes along with APP into cells such as HEK293 did not lead to the overproduction of either Aβ or β-sAPP (45).
The existence of the β-secretase pathway of APP cleavage, enriched in neuronal cells, leads to specific cleavage of APP at the N terminus of the Aβ peptide sequence. This cleavage leads to the formation of the soluble β-sAPP, as well as the membrane-associated β-CTF, the immediate precursor to Aβ. The compilation of the cellular results obtained by studying APP processing thus suggests that a true candidate β-secretase should have, at a minimum, the following characteristics. (i) It should specifically cleave APP at the Met-Asp site to generate the corresponding β-sAPP and β-CTF fragments, (ii) A true candidate β-secretase should show preferential cleavage toward Swedish over Wt sequence at the cleavage site. (iii) A true candidate β-secretase should function optimally at an acidic pH. (iv) A true candidate β-secretase also would be enriched in brain and neuronal tissue but present in cell lines such as HEK293 as well. The isolation and enzymatic characterization of a membrane-bound protease from human brain that meets these criteria (46) has been made possible by using APP-based fusion proteins incorporating both Wt and Swedish sequences, as well as the development of very specific ELISA-based quantitative assays for measuring cleavage at the β-cleavage site(s) in these fusion proteins. Although the identity of this enzymatic activity is not yet published, recombinant expression and cotransfection with APP would establish whether such an enzyme fulfills the additional cellular criteria of showing enhanced, specific cleavage in APP proteins at the β-cleavage sites.
1. Selkoe, D.J. (1991) Neuron 6, 487–498.
2. Glenner, G.G. & Wong, C.W. (1984) Biochem. Biophys. Res. Commun. 3, 885–890.
3. Kang, J., Lemaire, H.G., Unterbeck, A., Salbaum, J.M., Masters, C.L., Grzeschik, K.H., Multhaup, G., Beyreuther, K. & Muller-Hill, B. (1987) Nature (London) 325, 733–736.
4. Ponte, P., Gonzalez-DeWhitt, P., Schilling, J., Miller, J., Hsu, D., Greenberg, B., Davis, K., Wallace, W., Lieberburg, I. & Fuller, F. (1988) Nature (London) 331, 525–527.
5. Oltersdorf, T., Fritz, L.C., Schenk, D.B., Lieberburg, I., Johnson-Wood, K.L., Beattie, E.C., Ward, P.J., Blacher, R.W., Dovey, H.F. & Sinha, S. (1989) Nature (London) 341, 144–147.
6. Sinha, S., Dovey, H.F., Seubert, P., Ward, P.J., Blacher, R.W., Blaber, M., Bradshaw, R.A., Arici, M., Mobley, W.C. & Lieberburg, I. (1990) J. Biol. Chem. 265, 8983–8985.
7. Weidemann, A., Konig, G., Bunke, D., Fischer, P., Salbaum, J.M., Masters, C.L. & Beyreuther, K. (1989) Cell 57, 115–126.
8. Oltersdorf, T., Ward, P.J., Henriksson, T., Beattie, E.C., Neve, R., Lieberburg, I. & Fritz, L.C. (1990) J. Biol. Chem. 265, 4492–4497.
9. Esch, F.S., Keim, P.S., Beattie, E.C., Blacher, R.W., Culwell, A.R., Oltersdorf, T., McClure, D. & Ward, P.J. (1990) Science 248, 1122–1124.
10. Sisodia, S.S. (1992) Proc. Natl. Acad. Sci. USA 89, 6075–6079.
11. Buxbaum, J.D., Gandy, S.E., Cicchetti, P., Ehrlich, M.E., Czernik, A.J., Fracasso, R.P., Ramabhadran, T.V., Unterbeck, A.J. & Greengard, P. (1990) Proc. Natl. Acad. Sci. USA 87, 6003–6006.
12. Nitsch, R.M., Slack, B.E., Wurtman, R.J. & Growdon, J.H. (1992) Science 258, 304–307.
13. Seubert, P., Oltersdorf, T., Lee, M.G., Barbour, R., Blomquist, C., Davis, D.L., Bryant, K., Fritz, L.C., Galasko, D., Thal, L.J., et al. (1993) Nature (London) 361, 260–263.
14. Haass, C., Schlossmacher, M.G., Hung, A.Y., Vigo-Pelfrey, C., Mellon, A., Ostaszewski, B.L., Lieberburg, I., Koo, E.H., Schenk, D., Teplow, D.B., et al. (1992) Nature (London) 359, 322–325.
15. Seubert, P., Vigo-Pelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D., Sinha, S., Schlossmacher, M., Whaley, J., Swindlehurst, C., et al. (1992) Nature (London) 359, 325–327.
16. Buxbaum, J.D., Koo, E.H. & Greengard, P. (1993) Proc. Natl. Acad. Sci. USA 90, 9195–9180.
17. Dyrks, T., Monning, U., Beyreuther, K. & Turner, J. (1994) FEBS Lett. 349, 210–214.
18. Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B. & Lannfelt, L. (1992) Nat. Genet. 1, 345–347.
19. Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A.Y., Seubert, P., Vigo-Pelfrey, C., Lieberburg, I. & Selkoe, D.J. (1992) Nature (London) 360, 672–674.
20. Knops, J., Suomensaari, S., Lee, M., McConlogue, L., Seubert, P. & Sinha, S. (1995) J. Biol Chem. 270, 2419–2422.
21. Yoshimori, T., Yamamoto, A., Moriyama, Y., Futai, M. & Tashiro, Y. (1991) J. Biol Chem. 266, 17707–17712.
22. Hooper, N.M., Karran, E.H. & Turner, A.J. (1997) Biochem. J. 321, 265–279.
23. Black, R.A., Rauch, C.T., Kozlosky, C.J., Peschon, J.J., Slack, J.L., Wolfson, M.F., Castner, B.J., Stocking, K.L., Reddy, P., Srinivasan, S., et al. (1997) Nature (London) 385, 729–733.
24. Buxbaum, J.D., Liu, K.N., Luo, Y., Slack, J.L., Stocking, K.L., Peschon, J.J., Johnson, R.S., Castner, B.J., Cerretti, D.P. & Black, R.A. (1998) J. Biol Chem. 273, 27765–27767.
25. Arribas, J., Coodly, L., Vollmer, P., Kishimoto, T.K., Rose-John, S. & Massague, J. (1996) J. Biol Chem. 271, 11376–11382.
26. Goate, A.M. (1998) Cell Mol. Life Sci. 54, 897–901.
27. Dovey, H.F., Suomensaari-Chrysler, S., Lieberburg, I., Sinha, S. & Keim, P.S. (1993) NeuroReport 4, 1039–1042.
28. Suzuki, N., Cheung, T.T., Cai, X.D., Odaka, A., Otvos, L., Jr., Eckman, C., Golde, T.E. & Younkin, S.G. (1994) Science 264, 1336–1340.
29. Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., Carr, T., Clemens, J., Donaldson, T., Gillespie, F., et al. (1995) Nature (London) 373, 523–527.
30. Chen, K.S., Masliah, E., Grajeda, H., Guido, T., Huang, J., Khan, K., Motter, R., Soriano, F. & Games, D. (1998) Prog. Brain Res. 117, 327–334.
31. Sherrington, R., Rogaev, E.I., Liang, Y., Rogaeva, E.A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., et al. (1995) Nature (London) 375, 754–760.
32. Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D.M., Oshima, J., Pettingell, W.H., Yu, C.E., Jondro, P.D., Schmidt, S.D., Wang, K., et al. (1995) Science 269, 973–977.
33. Lemere, C.A., Lopera, F., Kosik, K.S., Lendon, C.L., Ossa, J., Saido, T.C., Yamaguchi, H., Ruiz, A., Martinez, A., Madrigal, L., et al. (1996) Nat. Med. 2, 1146–1150.
34. Citron, M., Westaway, D., Xia, W., Carlson, G., Diehl, T., Levesque, G., Johnson-Wood, K., Lee, M., Seubert, P., Davis, A., et al. (1997) Nat. Med. 3, 67–72.
35. Levitan, D. & Greenwald, I. (1995) Nature (London) 377, 351– 354.
36. De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., Von Figura, K. & Van Leuven, F. (1998) Nature (London) 391, 387–390.
37. Knops, J., Lieberburg, I. & Sinha, S. (1992) J. Biol. Chem. 267, 16022–16024.
38. Mehdi, S., Angelastro, M.R., Wiseman, J.S. & Bey, P. (1988) Biochem. Biophys. Res. Commun. 157, 1117–1123.
39. Higaki, J., Quon, D., Zhong, Z. & Cordell, B. (1995) Neuron 14, 651–659.
40. Hartmann, T., Bieger, S.C., Bruhl, B., Tienari, P.J., Ida, N., Allsop, D., Roberts, G.W., Masters, C.L., Dotti, C.G., Unsicker, K., et al. (1997) Nat. Med. 3, 1016–1020.
41. Wolfe, M.S., Xia, W., Ostaszewski, B.L., Diehl, T.S., Kimberley, W.T. & Selkoe, D.J. (1999) Nature (London) 398, 513–517.
42. Lichtenthaler, S.F., Wang, R., Grimm, H., Uljon, S., Masters, C.L. & Beyreuther, K. (1999) Proc. Natl. Acad. Sci. USA 96, 3053–3058.
43. Citron, M., Teplow, D.B. & Selkoe, D.J. (1995) Neuron 14, 661–670.
44. Brown, A.M., Tummolo, D.M., Spruyt, M.A., Jacobsen, J.S. & Sonnenberg-Reines, J. (1996) J. Neurochem. 66, 2436–2445.
45. Thompson, A., Grueninger-Leitch, F., Huber, G. & Malherbe, P. (1997) Brain Res. Mol. Brain Res. 48, 206–214.
46. Sinha, S., Suomensaari, S., Keim, P., Jacobson-Croak, K., Zhao, J., Hu, K., Tan, H., Tatsuno, G., McConlogue, L., Lieberburg, I., et al. (1997) Soc. Neurosci. Abstr. 23, 4.





