
4
Disorders of Copper Homeostasis
DISORDERS in copper homeostasis are discussed in this chapter. The two best-studied disorders in copper regulation, Menkes disease and Wilson disease, are described first, including the current state of knowledge of the genetics that underlie these two disorders. Occipital horn syndrome, a milder form of Menkes disease, is then discussed. Aceruloplasminemia, a newly recognized disease caused by a defect in the gene encoding ceruloplasmin, is then discussed. Other syndromes described are Tyrolean infantile cirrhosis (TIC), Indian childhood cirrhosis (ICC), and idiopathic copper toxicosis (ICT). All are associated with ingestion of high amounts of copper and might have a genetic component. Finally, disease-induced changes in copper homeostasis are discussed.
MENKES DISEASE
Menkes disease was originally diagnosed as an X-linked neurodegenerative disorder in infants characterized by poor growth and unusual "kinky" hair texture (Menkes et al. 1962). The defect is manifested primarily in males because it is X linked; eight females, however, have also been reported to have the disease (Kodama and Murata 1999). The major defect in Menkes disease is a failure to transport copper ions completely across the intestinal mucosa, ultimately leading to a severe copper deficiency in the peripheral organs (Danks et al. 1972). Transport across the blood-brain barrier is also impaired.
Clinically, Menkes patients usually have low plasma ceruloplasmin con-
centrations and decreased concentrations of copper in the liver and brain (Danks 1995). The morphological changes of the disease start in utero and are fully manifested during the perinatal period (Vulpe and Packman 1995). Premature birth and being small for gestational age are frequent characteristics of Menkes patients. Hypothermia, prolonged jaundice, feeding difficulties, and diarrhea can occur in the neonatal period (Horn et al. 1992). Developmental delays are apparent around the third month, as shown by abnormal head movement and the absence of a smiling response. Therapy-resistant convulsions also occur (Horn et al. 1992). There is no evidence that maternal copper status influences the development or expression of Menkes disease in the infant.
The primary genetic defect in Menkes disease is in the protein ATP7A, a membrane-bound Cu-ATPase that regulates the outward flow of copper ions from the interior to the exterior of the cell (Chelly et al. 1993; Mercer et al. 1993; Vulpe et al. 1993). For this reason, Menkes patients accumulated copper in the intestinal cells. Skin fibroblasts from patients with Menkes disease accumulate large amounts of copper when grown for 3 to 5 days in essential growth medium, demonstrating that the primary defect is detectable at the cellular level (Goka et al. 1976; Horn 1976). Thus, copper accumulation by fibroblasts has been used to confirm prenatal and postnatal diagnosis of Menkes disease. Unlike normal fibroblasts, fibroblasts from Menkes patients became growth-sensitive in very-low-to-medium copper concentrations during a 7-day exposure (Rayner and Suzuki 1994). No significant differences are detectable in copper accumulation by cells established from Menkes patients with different allelic variants of the disease. Thus, the severity of the phenotype cannot be determined by in vitro copper uptake assays (Masson et al. 1997).
In the case of mucosal epithelia, the ATPase transports the copper ions into the serosal capillaries as part of the absorption mechanism across the gut. Studies with animal models and isolated cell cultures suggest that the neurodegeneration is caused by a failure of the ATPase that normally transports copper, resulting in reductions in the activity of select copper-dependent enzymes (Kodama 1993; Yoshimura et al. 1995; Qian et al. 1997; Qian et al. 1998).
Efforts to treat Menkes disease by giving the patients parenteral copper in the form of copper histidine, copper acetate, or copper EDTA have had little success to date. None of these agents prevents neurological damage, although they can increase serum copper concentrations and ceruloplasmin activity. The age when copper administration begins seems to be of some consequence. In one study, a Menkes patient with a splice acceptor site mutation in a non critical region was treated with copper histidine beginning on day 8 of life. Head growth, myelination of neurons in the brain, and neurodevelopment were all normal in the patient. A half broth-
er and a cousin with the same mutation showed arrested head growth, cerebral atrophy, delayed myelination, and abnormal neurodevelopment (Kaler et al. 1996). Injections of copper histidine after 1-month of age failed to arrest the neurodegeneration in another Menkes patient (Sarkar et al. 1993). If treated early, some Menkes patients (those with only a partial gene knockout) given copper histidine survive beyond their teens (Christodoulou et al. 1998). To be effective, copper histidine must be administered at a dose of 200–1,000 µg per day, once per day or 2–3 times per week (Kaler et al. 1996). The adequacy of the dose is determined by measuring urinary copper excretion, serum copper concentrations and ceruloplasmin activity, as well as copper concentrations in the liver. An alternative and potentially effective therapy uses lipid-soluble chelators of copper, such as diethyldithiocarbamate or dimethyldithiocarbamate, administered intraperitoneally even without copper. That treatment increased the survival time of macular mutant mice, presumably by enhancing copper transport across cellular membranes (Tanaka et al. 1990). There is no record of the use of these chelators to treat Menkes patients.
The X-linked mottled locus in mice (Atp7aMo) is the homolog of the Menkes gene (Cecchi and Avner 1996). Mottled mutants exhibit a phenotype that closely resembles that seen in Menkes patients. The mottled locus spans 120 kilobase (kb) and encodes a gene for a copper-transporting ATPase that is 89.9% identical to ATP7A (Mercer et al. 1993). Spontaneous mutations at the mottled locus cause reduced fertility and viability and lead to phenotypic symptoms of classical Menkes disease and occipital horn syndrome (OHS). Of the 20 independent mottled alleles that have been identified, 10 are spontaneous and 14 arose after gamma or X irradiation (Cecchi and Avner 1996). The six allelic variants appearing most often in the literature are brindled (Atp7aMo-br), macular (A tp7aMo-ml), blotchy (Atp7aMo-bo), viable brindled (Atp7aMo-vbr), tortoiseshell (Atp7aMo-to), and dappled (Atp7aMo-dp). Males carrying the Atp7aMo-ml and Atp7aMo-br survive only a few days after birth, and males carrying Atp7aMo-vbr and Atp7aMo-blo survive for several months. Mice hemizygous for the blotchy allele have connective-tissue defects and only 30% of the normal lysyl oxidase activity (Royce et al. 1982). The mottled mutant mouse, therefore, is considered an animal model for occipital horn syndrome (OHS).
OCCIPITAL HORN SYNDROME
Mutations in the Menkes gene can also result in OHS, formerly called X-linked cutis laxia. OHS is a milder form of classical Menkes disease in which the same copper transporter, ATP7A, is affected. Because it affects the same gene locus (Xq13.3), OHS is referred to as an allelic variant of
Menkes disease (Kodama and Murata 1999). Like Menkes, OHS is recessively inherited and characterized by abnormalities in copper metabolism (Proud et al. 1996). Whereas the prevalence rate of Menkes disease appears to be 1 in 100,000–250,000 births, the prevalence rate of OHS appears to be much less. For example, only a few cases have been reported in Japan (Kodama et al. 1999).
Although Menkes and OHS have a similar abnormality in copper metabolism, the two diseases have different clinical presentations and survival potentials (Das et al. 1995). Because of late and inconsistent onset of symptoms, OHS might escape early recognition. A patient must be at least 2 years of age before a definitive diagnosis of OHS is possible (De Paepe et al. 1999). A prominent protuberance (exostoses) in the occipital bone seen on radiographs defines the condition (Proud et al. 1996). That protuberance is not seen in patients with classical Menkes disease. Additional skeletal abnormalities are short and broad clavicles, osteoporosis, laxity of the skin and joints, and bladder diverticula (Kodama et al. 1999). Serum copper and ceruloplasmin are low in some but not all patients, and cultured skin fibroblasts typically have low lysyl oxidase activity. Although disturbance in electroencephalogram patterns are seldom seen, arrested mental development and late-onset seizures often accompany the disease. Most OHS patients suffer inguinal hernias, recurrent diarrhea, urinary-tract infections, and distorted facial features, such as high foreheads and hooked noses. Radiographs show tortuous cerebral blood vessels with multiple branch occlusions (Proud et al. 1996). Although OHS patients can survive until adulthood, they have borderline intelligence. To date, no published reports have appeared describing parenteral administration of copper to correct, manage, or improve OHS patients (Kodama et al. 1999).
Studies of cells taken from OHS patients have linked MNK gene processing with the development of OHS. The human MNK gene normally has three 98-base pair (bp) tandem repeats in an upstream promoter region (Levinson et al. 1996). Genomic DNA from an OHS patient was found to have only 2 of the 98-bp repeats; no other mutations were found. The 98-bp deletion led to a dramatic decrease in the expression of a CAT reporter gene construct, suggesting that the 98-bp repeats play a role in regulating MNK mRNA expression (Levinson et al. 1996). Northern blot analysis, however, revealed no detectable reduction in MNK mRNA levels in cells derived from the patient. In another patient with OHS, an A–T transversion in intron 10 resulted in a loss of exon 10 by alternative splicing of ATP7A mRNA (Qi and Byers 1998). The mutated gene gave rise to an ATP7A variant that lacked two of the eight transmembrane domains and, for reasons not understood, remained resident in the endoplasmic reticulum and was not transported to the Golgi. To be effective in export,
the Cu-ATPase must enter into the secretory pathway by first moving from the endoplasmic reticulum to the Golgi (Francis et al. 1998). Because the error was not observed in all the mRNAs, the individual had the capacity to synthesize an intact Cu-ATPase, but at a reduced level. The observation might explain why OHS is a considered a mild as opposed to a debilitating form of Menkes disease.
Tissues from mottled mutants bearing the blotchy allele display two large sized mRNAs, demonstrating a likely defect in splicing (Mercer et al. 1993). Levinson et al. (1996) showed that the splice-site mutation in the blotchy allele is identical to that seen in mRNA from Menkes patients.
WILSON DISEASE
Wilson disease, or hepatolenticular degeneration, is an autosomal recessive disorder that results from accumulation of copper predominantly in the liver and brain (Bush et al. 1955; Strickland et al. 1969; O'Reilly et al. 1971; Frommer 1974; Gibbs and Walshe 1980). The accumulation is due to defective biliary excretion of copper. Current data indicate that adult humans need to ingest about 0.75 mg of copper daily to sustain a balance (Brewer and Yuzbasiyan-Gurkan 1992). Typically, humans ingest about 1 mg of copper per day (Holden et al. 1979; Klevay et al. 1979; Reiser et al. 1985; Hill et al. 1987; Iyengar et al. 1988; Milne 1998; Brewer and Yuzbasiyan-Gurkan 1992). The daily excess of copper averaging about 0.25 mg per day is normally excreted in the feces (Brewer and Yuzbasiyan-Gurkan 1992). However, due to a genetic defect, individuals with Wilson disease are unable to excrete the excess copper, resulting in a gradual build-up of copper in the body (Bush et al. 1955; Frommer 1974; O'Reilly et al. 1971; Strickland et al. 1969; Gibbs and Walshe 1980).
Clinically, Wilson patients begin receiving medical attention because of hepatic, neurological, or psychiatric symptoms, in roughly equal proportions (Brewer and Yuzbasiyan-Gurkan 1992; Scheinberg and Sternlieb 1984). Some individuals with Wilson disease are diagnosed before the appearance of clinical symptoms through the screening of Wilson disease siblings, who are at a 25% risk for the disease (Brewer and Yuzbasiyan-Gurkan 1992; Scheinberg and Sternlieb 1984). Wilson patients have underlying liver cirrhosis at the time of diagnosis, even if the symptoms are subclinical (Brewer and Yuzbasiyan-Gurkan 1992).
The primary genetic defect in Wilson disease is in ATP7B, which encodes a copper transport protein (Bull et al. 1993; Tanzi et al. 1993; Yamaguchi et al. 1993). The normal mechanism for biliary excretion of copper and the role of the ATP7B gene product in that excretion have yet to be elucidated. It has been hypothesized that ceruloplasmin plays a role
in copper excretion by preventing the reabsorption of copper (Iyengar et al. 1988). That hypothesis is supported by evidence of a high-molecular-weight protein in the bile that reacts with antibodies to ceruloplasmin, and contains enough copper to account for the excess copper (Iyengar et al. 1988; Chowrimootoo et al. 1996; Davis et al. 1996). There is no evidence of immunoreactive ceruloplasmin in the bile of most Wilson patients (Iyengar et al. 1988), and Wilson patients usually have low blood ceruloplasmin levels (Brewer and Yuzbasiyan-Gurkan 1992). It is not known, however, how the genetic defect in ATP7B and the effects on ceruloplasmin in Wilson patients are related. It is important to note that patients with aceruloplasminemia have normal copper concentrations (Harris et al. 1998).
Although diets low in copper have been prescribed for patients with Wilson disease in the past, more recent measurements of food copper content have indicated that only liver and shellfish are high enough in copper to warrant restriction (Brewer and Yuzbasiyan-Gurkan 1992). If the concentration of copper in drinking water at home, school, or work is greater than 0.1 mg per liter (L), most clinicians recommend that alternate sources be used in the management of Wilson disease (Brewer et al. 1998).
GENETIC CHARACTERISTICS OF WILSON AND MENKES DISEASES
The genetic mutations responsible for the loss of copper homeostasis are well characterized for Menkes and Wilson diseases. As a result, both diseases have provided insight into the genetic factors that regulate copper bioavailability and transport to organs and tissues.
The identification, sequencing, cloning, and characterization of the Wilson gene and mutations of the gene (Bull et al. 1993; Tanzi et al. 1993; Petrukhin et al. 1993; Thomas et al. 1995) and the Menkes gene (Vulpe et al. 1993; Chelly et al. 1993; Mercer et al. 1993) reveal that both genes encode P-type Cu-ATPases that are specific for copper transport (Solioz and Vulpe 1996). Cu-ATPases are complex integral membrane proteins that are part of a family of ion-transporting proteins that include the Ca2+ and Na+/K+ transport proteins. The Wilson protein (ATP7B) and the Menkes protein (ATP7A) share 57% amino acid sequence homology (Vulpe and Packman 1995) and show remarkable similarity to bacteria copper-binding proteins (Silver et al. 1993).
The Menkes gene, MNK, spans about 140–150 kb (Tumer et al. 1995; Dierick et al. 1995) and gives rise to a 8.5-kb mRNA. The Menkes mRNA encodes a protein (ATP7A) of exactly 1,500 amino acids, but additional nucleotide sequences at the 5' end are also known to occur (Tumer et al. 1995). High levels of MNK mRNA are found in muscle, kidney, lung, and
brain, and low levels are found in the placenta and pancreas, and only trace amounts are found in the liver (Chelly et al. 1993; Mercer et al. 1993). A number of mutations can result in Menkes disease. For example, out of more than 300 Menkes cases from 27 different countries, 191 unique mutations of the Menkes gene (ATP7A) were found (Tumer et al. 1995). Thirty-five of the mutations were partial gene deletions and 149 were point mutations. The largest gene deletion involved the whole gene except for the first two exons; the smallest was a deletion of exon 1. The four types of point mutations—namely, deletion/insertion, missense, nonsense, and splice site—were represented almost equally.
The Wilson transcript is 7.5 kb and encodes a protein of 1,411 amino acids. The Wilson gene is strongly expressed in the kidney, but unlike the Menkes gene, it is also strongly expressed in liver (Bull et al. 1993; Yamaguchi et al. 1993). The chromosomal locus of the Wilson gene is 13q14.3–q21. More than 40 normal allelic variants and 170 disease-associated variants have been reported for the Wilson gene (Cox and Roberts 1999).
The structure and function of the two gene products are similar. Both genes affect copper trafficking, but at different sites and in opposing directions. Immunochemical studies localized ATP7A to the perinuclear area thought to represent the Golgi (Dierick et al. 1997). From there, the Menkes protein regulates the release of copper ions into extracellular spaces by actively transporting the copper into export vesicles (Voskoboinik et al. 1998). Overexpression of the ATPase allows cells to tolerate higher than normal amounts of copper in their environment (Camakaris et al. 1995). The vesicles are not fixed but relocate to the plasma membrane in response to high extracellular copper. Some investigators postulate that ATP7A-laden vesicles continually move between the Golgi and the plasma membrane (Camakaris et al. 1995). The movement is a function of the primary structure of ATP7A. Specifically, the GMTCXXC motifs in the heavy metal binding region are important in sensing copper and triggering vesicle movement (Voskoboinik et al. 1999). In experiments using fluorescent antibodies against ATP7A, agents that disrupt Golgi structure or interfere with transport functions were observed to cause a more-dispersed pattern of fluorescence, indicating interference with the localization or aggregation of ATP7A within the secretory vesicles.
The Wilson protein (ATP7B) appears to be located on the Golgi and mitochondrial membrane (Lutsenko and Cooper 1998). In contrast to the Menkes protein, ATP7B is involved in the intracellular transport of copper (Terada and Sugiyama 1999). Given the above, it is evident that dysfunction of either ATP7A or ATP7B results in transport impairment and a disruption of copper distribution.
Regulation of Wilson and Menkes gene expression has not received much attention. Studies of a 1.3-kb segment in the 5'-flanking region of the Wilson gene found evidence of four metal response elements (MREs) and six MRE-like sequences (MLSs) that bear structural resemblance to elements found in the metallothionein gene (Oh et al. 1999). There is evidence for regulatory elements such as Spl, AP-1, AP-2, and E-box. The data suggest that the Wilson gene contains a single transcription site and has strong cis-acting elements at -811 to -653 from the transcription initiation site for high expression of the Wilson gene (Oh et al. 1999).
With a frequency of 1 per 7,000 live births, Sardinia has the highest reported incidence of Wilson disease of any population of people of Mediterranean descent (Loudianos et al. 1999). Mutation analysis in that population led to the characterization of 13 rare and 2 common mutations, together accounting for about 30% of the chromosome. The most common haplotype, however, occurs in 60.5% of the chromosome. Recently, it was learned that that mutation is not in the coded part of the gene but in regulatory elements in the 5' UTR (Loudianos et al. 1999). Sequencing the promoter GC-rich region in 92 chromosomes with the haplotype led to the discovery of a single mutation manifested as a 15-nucleotide deletion from position -441 to position -427 relative to the translation start site. A construct containing the deletion was only 25% as effective as a control in a luciferase reporter activity assay (Loudianos et al. 1999). Thus, in the Sardinian population, Wilson disease in people displaying the most common haplotype was initiated by an apparent failure to express an intact ATP7B protein at levels sufficient to maintain normal copper homeostasis.
HETEROZYGOTES FOR WILSON DISEASE
Because Wilson disease is an autosomal recessive disorder, it is exhibited only in individuals who are homozygous or compound heterozygotes1 for the gene defect. However, if the phenotype due to defects in the Wilson gene is defined as abnormal copper metabolism, then the Wilson gene can be considered a codominant gene, as abnormalities in copper homeostasis often occur in heterozygous carriers (Yuzbasiyan-Gurkan et al. 1991; Brewer and Yuzbasiyan-Gurkan 1992). Urinary copper concentrations are increased in about half of the calculated number of heterozygous siblings
of patients with Wilson disease.2 The siblings' concentrations approached the diagnostic concentration for Wilson disease (100 µg/24 hr) (see Figure 4-1) (Yuzbasiyan-Gurkan et al. 1991; Brewer and Yuzbasiyan-Gurkan 1992). Increased hepatic copper concentrations, which again approach the diagnostic concentration for Wilson disease, in siblings of Wilson patients (200 µg/g of dry weight) (see Figure 4-2) are likely due to heterozygosity for the Wilson gene (Brewer and Yuzbasiyan-Gurkan 1992). In addition, the rate of incorporation of 64Cu into ceruloplasmin in heterozygous individuals is, on average, about half that in normal individuals but can be close to the rate seen in patients with Wilson disease in some heterozygous individuals (Brewer and Yuzbasiyan-Gurkan 1992) (see Figure 4-3). The data in Figure 4-3 indicate that, with typical dietary copper intake, heterozygous carriers can accumulate copper at concentrations only slightly below those characteristic of Wilson disease. Given those data, it is possible that Wilson disease in infants could act as an autosomal dominant disease in cases where copper exposure is high (Brewer in press) (see Sensitive Populations section in Chapter 5 for a further discussion of that hypothesis).
Wilson disease has a reported incidence of about 1 in 40,000 births (Bachmann et al. 1979; Scheinberg and Sternlieb 1984; Giagheddu et al. 1985; Reilly et al. 1993). Using the Hardy-Weinberg equation (p2 + 2pq + q2 = 1), where p2 is the frequency of normal homozygotes, 2pq is the frequency of heterozygotes for the mutation, and q2 is the frequency of homozygotes for the mutation and setting q2 at 1 in 40,000, individuals who are heterozygous for Wilson disease can be calculated to make up 1% of the general population.
The above calculated frequency of Wilson-disease heterozygotes is based on the estimated incidence from Wilson-disease studies cited above. However, Wilson disease is probably underdiagnosed because of the lack of adequate screening for the disease. Currently, about 20% of chronic cirrhosis cases are idiopathic (often called ''cryptogenic"), and it is possible that Wilson disease remains undiagnosed among those individuals. In addition, liver disease in some patients who are thought to have hepatitis C might actually be due to undiagnosed Wilson disease. If a large number of patients with liver disease have undiagnosed Wilson disease, the heterozygote frequency might be as high as 2%.
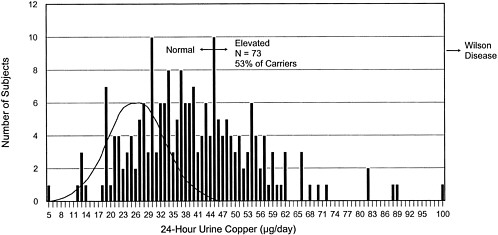
FIGURE 4-1 Frequency distribution of 24-hr urinary copper concentrations for 206 non-Wilson-disease siblings of Wilson-disease patients. This histogram shows the distribution of urinary copper values in 206 siblings of diagnosed Wilson disease patients. The siblings are not phenotypically homozygous for Wilson disease. Probabilistically, 2/3 (138) of the siblings should be carriers of the Wilson-disease gene. Of the estimated 138 carriers, 73 (53%) have increased urinary copper concentrations. The theoretical curve for urinary copper concentrations in normal individuals is superimposed on the left side of the distribution.
Source: Adapted from Brewer, in press.
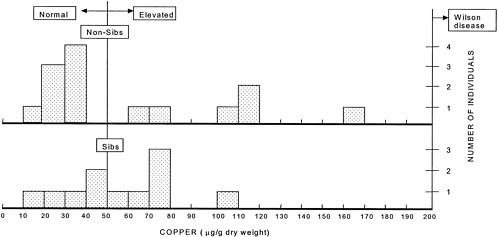
FIGURE 4-2 Hepatic copper concentrations in individuals presumed to be heterozygous for the Wilson-disease gene. The vertical line at 50 µg indicates the upper limit of the normal copper concentration range. The vertical line at 200 µg indicates the lower limit of the copper concentration at which a diagnosis of Wilson disease can be made. The lower panel shows the liver copper concentrations in the siblings (n = 11) of Wilson patients, two-thirds of whom would be expected to be heterozygotes on a probabilistic basis (with 11 individuals, 7 or 8 heterozygotes would be expected). The copper concentration is above the normal range in 6 of the individuals. The upper panel shows the liver copper concentrations in other individuals (n = 14) suspected of having Wilson disease. None of those 14 persons had Wilson disease, but hepatic copper concentrations were increased in 6 individuals. Those 6 individuals are likely heterozygous for the Wilson-gene defect.
Source: Adapted from Brewer, in press.
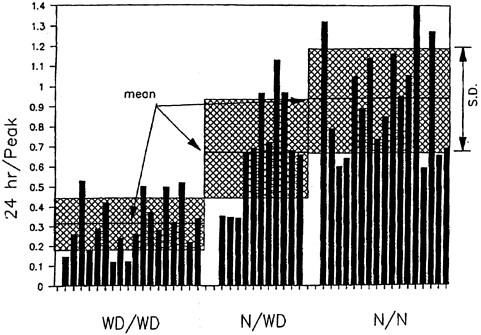
FIGURE 4-3 Incorporation of orally administered 64Cu into ceruloplasmin at 24 hr. Means and standard deviations are shown. The y axis shows the ratio of the 24-hr incorporation of radiocopper over the peak incorporation of radiocopper (at 1 or 2 hr). WD/WD, homozygous affected; N/WD, heterozygous; N/N, homozygous normal. Hatched portion = mean ± standard deviation.
Source: Brewer and Yuzbasiyan- Gurkan 1992. Reprinted with permission from Medicine; copyright 1992, Lippincott Williams & Wilkins.
ACERULOPLASMINEMIA
Aceruloplasminemia is an autosomal recessive disorder of iron metabolism characterized by a defect in the gene coding for ceruloplasmin. This disease is rare; a frequency of only 1 per 2,000,000 in cases involving non-consanguineous marriages was reported in Japan (Miyajima et al. 1999). Aceruloplasminemic individuals have no oxidase-detectable or immunoreactive ceruloplasmin in their serum (Miyajima et al. 1987). Late-onset retinal and basal ganglia degeneration, diabetes, and neurological symptoms are commonly seen in clinics. The pathogenesis of the disease has been linked to a slow accumulation of iron in tissues (Yazaki et al. 1998; Gitlin 1998). Biopsy examinations have detected unusually high amounts of iron in the pancreas, heart, kidney, spleen, and thyroid gland (Yoshida et al. 1995), and magnetic resonance imaging of the brain shows an increased iron content of the basal ganglia, thalamus, and dentate nucleus
(Yazaki et al. 1998). Tissue copper concentrations, however, tend to be unchanged and total iron-binding capacity and erythrocyte counts are normal. 59Fe administered as a trace dose tends to accumulate in the brain, heart, kidney, and liver of patients with aceruloplasminemia, confirming the biopsy reports. 64Cu administered intravenously, however, shows no such tendency, and tissue copper remains basically normal (Logan et al. 1994). Intravenous injections of human ceruloplasmin raise the serum iron in these patients (Logan et al. 1994). The lack of cardinal copper-related symptoms in aceruloplasminemic individuals challenges what has been considered the essential role of ceruloplasmin in copper transport and homeostasis and has increased the focus on its role.
Despite similarities with hemosiderosis and other iron-overload disorders, aceruloplasminemia is unusual in showing major alterations in neurological functions. Patients with aceruloplasminemia eventually succumb to the effects of increased iron in the tissues, particularly the basal ganglia (Miyajima et al. 1998). An increased susceptibility to lipid peroxidation is believed to contribute substantially to the neuropathology, which suggests that free-radical-mediated tissue injury is responsible for the basal-ganglia degeneration (Miyajima et al. 1996, 1998). Ceruloplasmin has recently been shown to function as an antioxidant in neutralizing nitric oxide via nitrosothiol formation (Inoue et al. 1999). A membrane-bound (GPI anchor) form of ceruloplasmin has been found in glia cells, which might have cytoprotective function in brain (Patel and David 1997). Lack of that form of ceruloplasmin in the aceruloplasminemic patient could also result in neurodegeneration.
Molecular genetic studies of DNA from cells from aceruloplasminemic individuals have detected specific mutations in the ceruloplasmin mRNA. Mutations that occur often lead to splicing errors and abridged forms of ceruloplasmin that cannot support normal ferroxidase function (Yazaki et al. 1998). In one patient with the disease, a 5-bp insertion in exon 7 caused an out-of-frame shift that generated a stop codon that aborted the protein from the ribosome before synthesis was completed (Harris et al. 1995). In another, a guanine-to-adenine transition at a splice acceptor site caused a similar premature termination of the protein during biosynthesis (Yoshida et al. 1995).
Despite careful clinical investigations of afflicted individuals, it is still unclear how ceruloplasmin controls iron homeostasis and tissue distribution. Copper-deficiency studies in which ceruloplasmin is lowered never achieve a total absence of the protein, as is observed in aceruloplasminemia. Such studies are further confounded by a disrupted copper status. A mouse knockout model, however, has recently been developed, and preliminary studies show no abnormalities in cellular iron uptake but do show a pronounced impairment in the movement of iron out of the reticu-
loendothelial cells and hepatocytes (Harris et al. 1998). An issue that merits investigation is the extent to which aceruloplasminemia might modulate an individual's susceptibility to copper toxicity.
TYROLEAN INFANTILE CIRRHOSIS
Between about 1900 and 1980, 138 infants and young children died in the Tyrolean area of western Austria from liver cirrhosis; that syndrome has been termed Tyrolean infantile cirrhosis (TIC) (Müller et al. 1996). The pathology of TIC is indistinguishable from Indian childhood cirrhosis and other forms of hepatic copper toxicosis (see descriptions below) (Müller et al. 1996). A common feature among the infants and young children with TIC was having been fed a one-to-one mixture of unpasteurized cow's milk with water heated for about 20 min in old copper pots (Müller et al. 1996). Such a preparation results in a high concentration of copper in the milk (Müller et al. 1996). Copper cooking utensils were common in the Tyrolean area because of an extensive copper mining industry that was present until about 1926. The disease appears to be an autosomal recessive disorder (Müller et al. 1996), whose manifestations might be due to a combination of a genetic predisposition and a high intake of copper. Reports of this syndrome have essentially disappeared since about 1980, probably as copper cooking utensils were gradually replaced in the region by modern cooking utensils in the mid-to-late 1960s.
INDIAN CHILDHOOD CIRRHOSIS
The etiology of Indian childhood cirrhosis (ICC) appears to be similar to that of TIC (Bavdekar et al. 1996; Bhave et al. 1982; Pandit and Bhave 1996; Popper et al. 1979; Tanner et al. 1983; Tanner et al. 1979). ICC occurs in India in infants and very young children fed milk stored in brass or copper containers. Copper might have a role in the disease for the following reasons: milk stored in that manner is very high in copper (Popper et al. 1979; Tanner et al. 1979; Bhave et al. 1982; Tanner et al. 1983; Bavdekar et al. 1996; Pandit and Bhave 1996); high copper concentrations have been found in the livers of ICC patients (Bavdekar et al. 1996; Bhave et al. 1982; Pandit and Bhave 1996; Popper et al. 1979; Tanner et al. 1983; Tanner et al. 1979); penicillamine treatment rapidly improves the symptoms (Bavdekar et al. 1996; Bhave et al. 1982; Pandit and Bhave 1996; Popper et al. 1979; Tanner et al. 1983; Tanner et al. 1979); and other possible causes of liver disease have been ruled out in most of the patients (Bavdekar et al. 1996; Bhave et al. 1982; Pandit and Bhave 1996; Popper
et al. 1979; Tanner et al. 1983; Tanner et al. 1979). In addition, once storage of milk in copper and brass containers was reduced, the disease began to disappear. An autosomal recessive component also seems to play a role in the disease, because the siblings, but not the parents, of ICC patients were affected (Bavdekar et al. 1996; Bhave et al. 1982; Pandit and Bhave 1996; Popper et al. 1979; Tanner et al. 1983; Tanner et al. 1979). Therefore, as with TIC, the liver disease in ICC might be due to genetic abnormalities in copper metabolism and a high copper intake.
IDIOPATHIC COPPER TOXICOSIS
Cases of idiopathic copper toxicosis (ICT) have been reported in numerous countries. Those cases may arise from a number of different etiologies. ICT has also been called ICC-like cirrhosis, copper-associated childhood cirrhosis, and copper-associated liver disease in childhood (Lefkowitch et al. 1982; Adamson et al. 1992; Aljajeh et al. 1994; Horslen et al. 1994; Müller et al. 1998). ICT has been reviewed recently by Müller et al. (1998). Most ICT cases are infants or very young children, but some are children up to 10 years of age. In many cases, increased ingestion of copper has been demonstrated, and there is either consanguinity of parents or involvement of more than one sibling. Müller et al. (1998) hypothesized that ICT is caused by a combination of an autosomal recessive inherited defect in copper metabolism and excess copper intake. In some cases, especially in infants and toddlers, the source of excess copper appears to have been drinking water (Müller et al. 1998) (see Chapter 5).
OTHER GENETIC DISORDERS
Two additional genetic conditions of humans that might enhance the susceptibility to copper toxicosis deserve brief mention. Individuals with a rare type of alloalbuminemia, "albumin Christchurch," have a mutation in the gene that encodes the primary copper-binding site of plasma albumin, and the albumin Christchurch has reduced ability to bind Cu(II) compared with normal albumin. (Brennan and Carrell 1980). As a consequence, individuals with albumin Christchurch have functional impairment in plasma capacity to transport ionic copper. Individuals with type I-A oculocutaneous albinism have mutations in the gene that encodes tyrosinase, a key copper-containing enzyme in the pathway of melanin biosynthesis (Oetting and King 1992; Passmore et al. 1999). The melanin of pigmented tissues (e.g., retina and skin) avidly binds copper, zinc, and other metal ions in vitro (Froncisz et al 1980; Andrzejczyk and Buszman 1992) and in
vivo (Bowness and Morton 1953; Horcicko et al. 1973). Melanin is postulated as a possible storage repository for essential metals (Pfeiffer and Mailloux 1988), and indications of abnormal metabolism of copper and zinc have been reported in black albino patients, compared with Caucasian albino patients and control groups (Silverstone et al 1986). Subjects with oculocutaneous albinism lack tyrosinase activity. That deficiency is associated with an absence of melanin, which can sequester and detoxify copper and other metals. Therefore, patients with oculocutaneous albinism might be prone to developing copper toxicosis.
DISEASE-INDUCED CHANGES IN COPPER HOMEOSTASIS
In addition to the above genetic disorders, there are a number of environmental and physiological conditions that can perturb copper metabolism and alter the delivery of copper to select tissues. The conditions shown to alter copper metabolism are diverse and include exercise, infection, inflammation, cirrhosis, diabetes, and hypertension (Sandstead 1995; Disilvestro et al. 1992; Turnlund 1988; Walter et al. 1991; Miesel and Zuber 1993; Keen 1993). In all of those conditions, a common finding is hypercupremia, which is due to high concentrations of plasma ceruloplasmin. The increase in plasma ceruloplasmin is the result of an increase in the synthesis of ceruloplasmin in the liver, with its subsequent release into the plasma pool. An increase in ceruloplasmin synthesis represents one component of the so-called acute-phase response. This response is triggered by cytokines and select hormones that are released in response to tissue injury (Cousins 1985; Dinarello 1989). Although the acute-phase response is transitory in most cases, it can persist for long periods if tissue injury is continuous. The long-term health consequences of hypercupremia have not been well defined; however, some investigators have reported that the occurrence of hypercupremia is a risk factor for cardiovascular disease and for some cancers (Salonen et al. 1991). Mechanistically, it can be argued that persistent hypercupremia represents an oxidative challenge, if the hypercupremia is associated with a rise in low-molecular-bound copper pools, which have the potential to be involved in redox cycling and, thus, the generation of reactive oxygen species. An increase in DNA damage, secondary to copper-induced oxidative stress, could present a carcinogenic risk. Similarly, an increase in oxidative stress could result in an increased risk for some forms of cardiovascular disease (Dubick et al. 1999; Esterbauer et al. 1992; Gey et al. 1991). Given the above, it can be hypothesized that chronic hypercupremia can be a causative factor in the
initiation of some diseases. An alternative argument to the above is that the hypercupremia is simply a marker for either early tissue damage or other metabolic abnormalities that are the primary initiators of the vascular disease or cancer. Most investigators think that hypercupremia is an effect rather than a cause of disease. Additional research on this issue is needed.
It is important to point out that the occurrence of high concentrations of copper in plasma does not necessarily translate into high concentrations in soft tissue; indeed, high plasma concentrations in some diseases, including diabetes and hypertension, have been correlated with low concentrations in muscle, liver, and aortic tissue (Dubick et al. 1999; Sjogren et al. 1986; Tilson 1982). With respect to hypertension, the low copper concentrations in aortic tissue have been correlated with low activity in copper zinc superoxide dismutase and high concentrations of tissue lipid peroxide (Dubick et al. 1987; Dubick et al. 1999). The above observations are consistent with the concept that copper deficiency is a risk factor for cardiovascular disease (Klevay 1989), although the copper deficiency occurs due to disease-induced changes in copper metabolism rather than to dietary copper deficiency.
It is not known to what extent disease-induced changes in copper metabolism alter the requirement for dietary copper or the sensitivity to copper toxicity. However, if persistent hypercupremia results in an increased excretion of copper, this condition might result in an increased dietary requirement for copper.
CONCLUSIONS
-
Menkes disease is X linked, and Wilson disease is an autosomal recessive disorder.
-
With the recent identification of the genes responsible for Menkes and Wilson diseases, considerable progress has been made in the understanding of human copper metabolism.
-
With the identification of the Menkes and Wilson genes, rapid progress has been made in the understanding of the pathophysiology, pathogenesis, and diagnosis of these disorders.
-
Heterozygous carriers of the Wilson-disease gene can have abnormal copper metabolism and, consequently, excess accumulation of copper.
-
TIC, ICC, and ICT occur in infants or young children. TIC and ICC appear to have familial components with an autosomal recessive inheritance pattern. Some cases of ICT appear to have an autosomal recessive
-
inheritance pattern, and others do not. In all three disorders, excess copper ingestion is typically found.
-
A number of diseases can result in chronic hypercupremia.
RECOMMENDATIONS
-
Research should be conducted to establish the frequency and characteristics of the Wilson-disease gene defects.
-
The influence of heterozygosity for gene defects that alter copper transport or susceptibility to copper toxicity needs to be defined.
-
Research should be conducted to establish the genetic causes of susceptibility to copper in TIC, ICC and ICT and the frequency of these genetic defects.
-
The potential risk of copper toxicity in patients with aceruloplasminemia needs to be determined.
REFERENCES
Adamson, M., B. Reiner, J.L. Olson, Z. Goodnam, L. Plotnick, I. Bernardini, and W.A. Gahl. 1992. Indian childhood cirrhosis in an American child. Gastroenterology 102(5):1771–1777.
Aljajeh, A., S. Mughal, B. al-Tahou, T. Ajrawi, E.A. Ismail, and N.C. Nayak. 1994. Indian childhood cirrhosis-like liver disease in an Arab child . A brief report. Virchows Arch. 424(2):225–227.
Andrzejczyk, J., and E. Buszman. 1992. Interaction of Fe3+, Cu2+ and Zn2+ with melanin and melanoproteins from bovine eyes. Acta Biochim. Pol. 39(1):85–88.
Bachmann, H., J. Lössner, B. Gruss, and U. Ruchholtz. 1979. The epidemiology of Wilson's disease in the German Democratic Republic and current problems from the viewpoint of population genetics [in German]. Psychiatr. Neurol. Med. Psychol. 31(7):393–400.
Bavdekar, A.R., S.A. Bhave, A.M. Pradhan, A.N. Pandit, and M.S. Tanner. 1996. Long term survival in Indian childhood cirrhosis treated with D-penicillamine. Arch. Dis. Child. 74(1):32–35.
Bhave, S.A., A.N. Pandit, A.M. Pradhan, D.G. Sidhaye, A. Kantarjian, A. Williams, I.C. Talbot, and M.S. Tanner. 1982. Liver disease in India. Arch. Dis. Child. 57(12):922–928.
Bowness, J.M., and R.A. Morton. 1953. The association of zinc and other metals with melanin and melanin-protein complex. Biochem. J. 53(4): 620–626.
Brennan, S.O. and R.W. Carrell. 1980. Functional abnormality of proalbumin Christchurch. Biochim. Biophys. Acta 621(1):83–88.
Brewer, G.J. In press. Editorial: Is heterozygosity for a Wilson's disease gene defect an important underlying cause of infantile and childhood copper toxicosis syndromes? J. Trace Elem. Exp. Med.
Brewer, G.J. and V. Yuzbasiyan-Gurkan. 1992. Wilson disease. Medicine 71(3):139–164.
Brewer, G.J., R.D. Dick, V. Johnson, J.A. Brunberg, K.J. Kluin, and J.K. Fink. 1998. Treatment of Wilson's disease with zinc: XV. Long-term follow-up studies. J. Lab. Clin. Med. 132(4):264–278.
Bull, P.C., G.R. Thomas, J.M. Rommens, J.R. Forbes, and D.W. Cox. 1993. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes' gene. Nat. Genet. 5(4):327–337.
Bush, J.A., J.P. Mahoney, H. Markowitz, C.J. Gubler, G.E. Cartwright, and M.M. Wintrobe. 1955. Studies on copper metabolism. XVI. Radioactive copper studies in normal subjects and in patients with hepatolenticular degeneration. J. Clin. Invest. 34:1766–1778.
Camakaris, J., M.J. Petris, L. Bailey, P. Shen, P. Lockhart, T.W. Glover, C. Barcroft, J. Patton, and J.F. Mercer. 1995. Gene amplification of the
Menkes (MNK; ATP7A) P-type ATPase gene of CHO cells is associated with copper resistance and enhanced copper efflux. Hum. Mol. Genet. 4(11):2117–2123.
Cecchi, C., and P. Avner. 1996. Genomic organization of the mottled gene, the mouse homologue of the human Menkes disease gene. Genomics 37(1):96–104.
Chelly, J., Z. Tumer, T. Tonnesen, A. Petterson, Y. Ishikawa-Brush, N. Tommerup, N. Horn, and A.P. Monaco. 1993. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 3(1):14–19.
Chowrimootoo, G.F., H.A. Ahmed, and C.A. Seymour. 1996. New insights into the pathogenesis of copper toxicity in Wilson's disease: Evidence for copper incorporation and defective canalicular transport of caeruloplasmin. Blochem. J. 315(Pt 3):851–855.
Christodoulou, J., D.M. Danks, B. Sarkar, K.E. Baerlocher, R. Casey, N. Horn, Z. Tumer, and J.T. Clarke. 1998. Early treatment of Menkes disease with parenteral copper-histidine: Long-term follow-up of four treated patients. Am. J. Med. Genet. 76(2):154–164.
Cousins, R.J. 1985. Absorption, transport and hepatic metabolism of copper and zinc. Physiol. Rev. 65(2):238–309.
Cox, D.W., and E.A. Roberts. 1999. Wilson Disease. GeneChnics, University of Washington, Seattle. Online. Available: http://www.genechnics.org/profiles/wilson/details.html
Danks, D.M. 1995. Disorders of copper transport. Pp. 2211–2235 in The Metabolic and Molecular Basis of Inherited Disease, 7th Ed., C.R. Scriver, A.L. Beaudet, W.M. Sly, and D. Valle, eds. New York: McGrawHill.
Danks, D.M., P.E. Campbell, B.J. Stevens, V. Mayne, and E. Cartwright. 1972. Menkes's kinky hair syndrome. An inherited defect in copper absorption with widespread effects. Pediatrics 50(2):188–201.
Das, S., B. Levinson, C. Vulpe, S. Whitney, J. Gitschier, and S. Packman S. 1995. Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am. J. Hum. Genet. 56(3):570–576.
Davis, W., G.F. Chowrimootoo, and C.A. Seymour. 1996. Defective biliary excretion in Wilson's disease: The role of caeruloplasmin. Eur. J. Clin. Invest. 26(10):893–901.
De Paepe, A., B. Loeys, K. Devriendt, and J.P. Fryns. 1999. Occipital Horn syndrome in a 2-year-old boy. Clin. Dysmorphol. 8(3):179–183.
Dierick, H.A., A.N. Adam, J.F. Escara-Wilke, and T.W. Glover. 1997. Immunocytochemical localization of the Menkes copper transport protein (ATP7A) to the trans-Golgi network. Hum. Mol. Genet. 6(3): 409–416.
Dierick, H.A., L. Ambrosini, J. Spencer, T.W. Glover, and J.F. Mercer. 1995. Molecular structure of the Menkes disease gene (ATP7A). Genomics 28(3):462–469.
Dinarello, C.A. 1989. The endogenous pyrogens in host-defense interactions. Hosp. Pract. (Off Ed) 24(11):111—5, 118, 121 passim.
DiSilvestro, R.A., J. Marten, and M. Skehan. 1992. Effects of copper supplementation on ceruloplasmin and copper-zinc superoxide dismutase in free-living rheumatoid arthritis patients. J. Am. Coll. Nutr. 11(2):177–80.
Dubick, M.A., G.C. Hunger, S.M. Casey, and C.L. Keen. 1987. Aortic ascorbic acid, trace elements, and superoxide dismutase activity in human aneurysmal and occlusive disease. Proc. Soc. Exp. Biol. Med. 184(2):138–143.
Dubick, M.A., C.L. Keen, R.A. DiSilvestro, C.D. Eskelson, J. Ireton, and G.C. Hunter. 1999. Antioxidant enzyme activity in human abdominal aortic aneurysmal and occlusive disease. Proc. Soc. Exp. Biol. Med. 220(1):39–45.
Esterbauer, H., J. Gebicki, H. Puhl, and G. Jurgens. 1992. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic. Biol. Med. 13(4):341–390.
Francis, M.J., E.E. Jones, E.R. Levy, S. Ponnambalam, J. Chelly, and A.P. Monaco. 1998. A Golgi localization signal identified in the Menkes recombinant protein. Hum. Mol. Genet. 7(8):1245–1252.
Frommer, D.J. 1974. Defective binary excretion of copper in Wilson's disease. Gut 15(2):125–129.
Froncisz, W., T. Sarna, and J.S. Hyde. 1980. Cu2+ probe of metal-ion binding sites in melanin using electron paramagnetic resonance spectroscopy. I. Synthetic melanins. Arch. Biochem. Biophys. 202(1):289–303.
Gey, K.F., P. Puska, P. Jordan, and U.K. Moser. 1991. Inverse correlation between plasma vitamin E and mortality from ischemic heart disease in cross-cultural epidemiology. Am. J. Clin. Nutr. 53(1 Suppl):326S–334S.
Giagheddu, A., L. Demelia, G. Puggioni, A.M. Nurchi, L. Contu, G. Pirari, A. Deplano, and M.G. Rachele. 1985. Epidemiologic study of hepatolenticular degeneration (Wilson's disease) in Sardinia (1902–1983). Acta Neurol. Scand. 72(1):43–45.
Gibbs, K., and J.M. Walshe. 1980. Biliary excretion of copper in Wilson's disease. Lancet 2(8193):538–539.
Gitlin, J.D. 1998. Aceruloplasminemia. Pediatr. Res. 44(3):271–276.
Goka, T.J., R.E. Stevenson, P.M. Hefferan, and R.R. Howell. 1976. Menkes disease: A biochemical abnormality in cultured human fibroblasts. Proc. Natl. Acad. Sci. (USA) 73(2):604–606.
Harris, Z.L., L.W. Klomp and J.D. Gitlin. 1998. Aceruloplasminemia: an
inherited neurodegenerative disease with impairment of iron homeostasis. Am. J. Clin. Nutr. 67(5 Suppl):972S–977S.
Harris, Z.L., Y. Takahashi, H. Miyajima, M. Serizawa, R.T. MacGillivray, and J.D. Gitlin. 1995. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. (USA) 92(7):2539–2543.
Hill, G.M., G.J. Brewer, A.S. Prasad, C.R. Hydrick, and D.E. Hartmann. 1987. Treatment of Wilson's disease with zinc: I. Oral zinc therapy regimens. Hepatology 7(3):522–528.
Holden, J.M., W.R. Wolf, and W. Mertz. 1979. Zinc and copper in self selected diets. J. Am. Diet Assoc. 75(1):23–28.
Horcicko, J., J. Borovansky, J. Duchon and B. Prochazkova. 1973. Distribution of zinc and copper in pigmented tissues. Hoppe Seylers Z. Physiol. Chem. 354(2):203–204.
Horn, N. 1976. Copper incorporation studies on cultured cells for prenatal diagnosis of Menkes' disease. Lancet 1(7970):1156–8.
Horn, N., T. Tonnesen, and Z. Tumer. 1992. Menkes disease: An X-linked neurological disorder of the copper metabolism. Brain Pathol. 2(4):351–362.
Horslen, S.P., M.S. Tanner, T.D. Lyon, G.S. Fell, and M.F. Lowry. 1994. Copper associated childhood cirrhosis. Gut 35(10):1497–1500.
Inoue, K., T. Akaike, Y. Miyamoto, T. Okamoto, T. Sawa, M. Otagiri, S. Suzuki, T. Yoshimura, and H. Maeda. 1999. Nitrosothiol formation catalyzed by ceruloplasmin. Implication for cytoprotective mechanism in vivo. J. Biol. Chem. 274(38):27069–27075.
Iyengar, V. G.J. Brewer, R.D. Dick, and O.Y. Chung. 1988. Studies of cholecystokinin-stimulated biliary secretions reveal a high molecular weight copper-binding substance in normal subjects that is absent in patients with Wilson's disease. J. Lab. Clin. Med. 111(3):267–274.
Kaler, S.G., S. Das, B. Levinson, D.S. Goldstein, C.S. Holmes, N.J. Patronas, S. Packman, and W.A. Gahl. 1996. Successful early copper therapy in menkes disease associated with a mutant transcript containing a small In-frame deletion. Biochem. Mol. Med. 57(1):37–46.
Keen, C.L. 1993. Effects of exercise and heat on mineral metabolism and requirements. Pp. 117–135 in Nutritional Needs in Hot Environments. B.M. Marriott, ed. Washington, DC: National Academy Press.
Klevay, L.M. 1989. Ischemic heart disease as copper deficiency. Adv. Exp. Med. Biol. 258:197–208.
Klevay, L.M., S.J. Reck, and D.F. Barcome. 1979. Evidence of dietary copper and zinc deficiencies. JAMA 241(18):1916–1918.
Kodama, H. 1993. Recent developments in Menkes disease. J. Inherit. Metab. Dis. 16(4):791–799.
Kodama, H., and Y. Murata. 1999. Molecular genetics and pathophysiology of Menkes disease. Pediatr. Int. 41(4):430–435.
Kodama, H., Y. Murata, and M. Kobayashi. 1999. Clinical manifestations and treatment of Menkes disease and its variants. Pediatr. Int. 41(4): 423–429.
Lefkowitch, J.H., C.L. Honig, M.E. King, and J.W. Hagstom. 1982. Hepatic copper overload and features of Indian childhood cirrhosis in an American sibship. N. Engl. J. Med. 307(5):271–277.
Levinson, B., R. Conant, R. Schnur, S. Das, S. Packman, J. Gitschier. 1996. A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome. Hum. Mol. Genet. 5(11):1737–42.
Logan, J.I., K.B. Harveyson, G.B. Wisdom, A.E. Hughes and G.P. Archbold. 1994. Hereditary caeruloplasmin deficiency, dementia and diabetes mellitus. QJM 87(11):663–70.
Loudianos, G., V. Dessi, M. Lovicu, A. Angius, A. Figus, F. Lilliu, S. De Virgiliis, A.M. Nurchi, A. Deplano, P. Moi, M. Pirastu, and A. Cao. 1999. Molecular characterization of wilson disease in the Sardinian population—Evidence of a founder effect. Hum. Mutat. 14(4):294–303.
Lutsenko, S., and M.J. Cooper. 1998. Localization of the Wilson's disease protein product to mitochondria. Proc. Natl. Acad. Sci. (USA) 95(11): 6004–6009.
Masson, W., H. Hughes, D. Papworth, Y. Boyd and N. Horn. 1997. Abnormalities of copper accumulation in cell lines established from nine different alleles of mottled are the same as those found in Menkes disease. J. Med. Genet. 34(9):729–732.
Menkes, J.H., M. Alter, G.K. Steigleder, D.R. Weakley, and J.H. Sung. 1962. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics 29:764–779.
Mercer, J.F., J. Livingston, B. Hall, J.A. Paynter, C. Begy, S. Chandrasekharappa, P. Lockhart, A. Grimes, M. Bhave, and D. Siemieniak. 1993. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 3(1):20–25.
Miesel, R. and M. Zuber. 1993. Copper-dependent antioxidase defenses in inflammatory and autoimmune rheumatic diseases. Inflammation 17(3):283–294.
Milne, D.B. 1998. Copper intake and assessment of copper status. Am. J. Clin. Nutr. 67(5 Suppl.):1041S–1045S.
Miyajima, H., M. Fujimoto, S. Kohno, E. Kaneko, and J.D. Gitlin. 1998. CSF abnormalities in patients with aceruloplasminemia. Neurology 51(4):1188–1190.
Miyajima, H., S. Kohno, Y. Takahashi, O. Yonekawa and T. Kanno. 1999. Estimation of the gene frequency of aceruloplasminemia in Japan. Neurology 53(3):617–619.
Miyajima, H., Y. Nishimura, K. Mizoguchi, M. Sakamoto, T. Shimizu, and
N. Honda. 1987. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology 37(5):761–767.
Miyajima, H., Y. Takahashi, M. Serizawa, E. Kaneko, and J.D. Gitlin. 1996. Increased plasma lipid peroxidation in patients with aceruloplasminemia. Free Radic. Biol. Med. 20(5):757–760.
Müller, T., H. Feichtinger, H. Berger, and W. Müller. 1996. Endemic Tyrolean infantile cirrhosis: An exogenetic disorder. Lancet 347(9005):877–880.
Müller, T., W. Müller, and H. Feichtinger. 1998. Idiopathic copper toxicosis. Am. J. Clin. Nutr. 67(5 Suppl.):1082S–1086S.
Oetting, W.S., and R.A. King. 1992. Molecular analysis of type I-A (tyrosinase negative) oculocutaneous albinism. Hum. Genet. 90(3):258–262.
Oh, W.J., E.K. Kim, K.D. Park, S.H. Hahn, and O.J. Yoo. 1999. Cloning and characterization of the promoter region of the Wilson disease gene. Biochem. Biophys. Res. Commun. 259(1):206–211.
O'Reilly, S., P.M. Weber, M. Oswald and L. Shipley. 1971. Abnormalities of the physiology of copper in Wilson's disease. 3. The excretion of copper. Arch. Neurol. 25(1):28–32.
Pandit, A., and S. Bhave. 1996. Present interpretation of the role of copper in Indian childhood cirrhosis. Am. J. Clin. Nutr. 63:930S-835S.
Passmore, L.A., B. Kaesmann-Kellner, B.H.F. Weber. 1999. Novel and recurrent mutations in the tyrosinase gene and the P gene in the German albino population. Hum. Genet. 105(3):200–210.
Patel, B.N., and S. David. 1997. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 272(32):20185–190.
Petrukhin, K., S.G. Fischer, M. Pirastu, R.E. Tanzi, I. Chernov, M. Devoto, L.M. Brzustowicz, E. Cayanis, E. Vitale, and J.J. Russo. 1993. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat. Genet. 5(4):338–343.
Pfeiffer, C.C., R.J. Mailloux. 1988. Hypertension: heavy metals, useful cations and melanin as a possible repository. Med. Hypotheses 26(2): 125–130.
Popper, H., S. Goldfischer, I. Sternlieb, N.C. Nayak and T.V. Madhavan. 1979. Cytoplasmic copper and its toxic effects. Studies in Indian childhood cirrhosis. Lancet 1(8128):1205–1208.
Proud, V.K., H.G. Mussell, S.G. Kaler, D.W. Young, and A.K. Percy. 1996. Distinctive Menkes disease variant with occipital horns: delineation of natural history and clinical phenotype. Am. J. Med. Genet. 65(1):44–51.
Qi, M., and P.H. Byers. 1998. Constitutive skipping of alternatively spliced exon 10 in the ATP7A gene abolishes Golgi localization of the Menkes protein and produces the occipital horn syndrome. Hum. Mol. Genet. 7(3):465–469.
Qian, Y., E. Tiffany-Castighoni, and E.D. Harris. 1997. A Menkes P-type ATPase involved in copper homeostasis in the central nervous system of the rat. Brain Res. Mol. Brain. Res. 48(1):60–66.
Qian, Y., E. Tiffany-Castighoni, J. Welsh, and E.D. Harris. 1998. Copper efflux from murine microvascular cells requires expression of the Menkes disease Cu-ATPase. J. Nutr. 128(8):1276–1282.
Rayner, M.H. and K.T. Suzuki. 1994. Effect of medium copper concentration on the growth, uptake and intracellular balance of copper and zinc in Menkes' and normal control cells. Biometals 7(3):253–260.
Reilly, M., L. Daly and M. Hutchinson. 1993. An epidemiological study of Wilson's disease in the Republic of Ireland . J. Neurol. Neurosurg. Psychiatry 56(3):298–300.
Reiser, S., J.C. Smith Jr., W. Mertz, J.T. Holbrook, D.J. Scholfield, A.S. Powell, W.K. Canfield and J.J. Canary. 1985. Indices of copper status in humans consuming a typical American diet containing either fructose or starch. Am. J. Clin. Nutr. 42(2):242–251.
Royce, P.M., J. Camakaris, J.R. Mann, and D.M. Danks. 1982. Copper metabolism in mottled mouse mutants. The effect of copper therapy on lysyl oxidase activity in brindled (Mobr) mice. Blochem. J. 202(2):369–371.
Salonen, J.T., R. Salonen, H. Korpela, S. Suntioinen, and J. Tuomilehto. 1991. Serum copper and the risk of acute myocardial infarction: A prospective population study in men in eastern Finland. Am. J. Epidemiol. 134(3):268–276.
Sandstead, H.H. 1995. Requirements and toxicity of essential trace elements, illustrated by zinc and copper. Am. J. Clin. Nutr. 61(3 Suppl): 621S–624S.
Sarkar, B., K. Lingertat-Walsh and J.T. Clarke. 1993. Copper-histidine therapy for Menkes disease. J. Pediatr. 123(5):828–830.
Scheinberg, I. H. and I. Sternlieb. 1984. Wilson's disease. Major Problems in Internal Medicine, Vol. 23, L.H. Smith, ed. Philadelphia: W.B. Saunders.
Silver, S., G. Nucifora, and L.T. Phung. 1993. Human Menkes X-chromosome disease and the staphylococcal cadmium-resistance ATPase: A remarkable similarity in protein sequences. Mol. Microbiol. 10(1):7–12.
Silverstone, B.Z., I. Nawratzki, D. Berson and L. Yanko. 1986. Zinc and copper metabolism in oculocutaneous albinism in the Caucasian. Metab. Pediatr. Syst. Ophthalmol. 9(1):589–591.
Sjögren, A., L. Edvinsson, C.H. Florén, and M. Abdulla. 1986. Zinc and copper in striated muscle and body fluids from subjects with diabetes mellitus type I. Nutr. Res. 6(2):147–154.
Solioz, M., and C. Vulpe. 1996. Cpx-type ATPases: A class of P-type ATPases that pump heavy metals. Trends. Biochem. Sci. 21(7):237–241.
Strickland, G.T., W.M. Beckner, M.L. Leu, and S. O'Reilly. 1969. Copper-
67 studies in Wilson's disease patients and their families. Clin. Res. 17(2):396.
Tanaka, K., K. Kobayashi, Y. Fujita, C. Fukuhara, S. Onosaka, and K. Min. 1990. Effects of chelators on copper therapy of macular mouse, a model animal of Menkes' kinky disease. Res. Commun. Chem. Pathol. Pharmacol. 69(2):217–227.
Tanner, M.S., A.H. Kantarjian, S.A. Bhave, and A.N. Pandit. 1983. Early introduction of copper-contaminated animal milk feeds as a possible cause of Indian childhood cirrhosis. Lancet 2(8357):992–995.
Tanner, M.S., B. Portmann, A.P. Mowat, R. Williams, A.N. Pandit, C.F. Mills, and I. Bremner. 1979. Increased hepatic copper concentration in Indian childhood cirrhosis. Lancet 1(8128):1203–1205.
Tanzi, R.E., K. Petrukhin, J.L. Pellequer, W. Wasco, B. Ross, D.M. Romano, E. Parano, L. Pavone, and L.M. Brzustowicz. 1993. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 5(4):44–50.
Terada, K., and T. Sugiyama. 1999. The Long-Evans Cinnamon rat: an animal model for Wilson's disease. Pediatr. Int. 41(4):414–418.
Thomas, G.R., J.R. Forbes, E.A. Roberts, J.M. Walshe, and D.W. Cox. 1995. The Wilson disease gene: Spectrum of mutations and their consequences. Nat. Genet. 9(2):210-7.
Tilson, M.D. 1982. Decreased hepatic copper levels. A possible chemical marker for the pathogenesis of aortic aneurysms in man. Arch. Surg. 117(9):1212–1213.
Tumer, Z., B. Vural, T. Tonnesen, J. Chelly, A.P. Monaco, and N. Horn. 1995. Characterization of the exon structure of the Menkes disease gene using vectorette PCR. Genomics 26(3):437–442.
Turnlund, J.R. 1988. Copper nutriture, bioavailability, and the influence of dietary factors . J. Am. Diet Assoc. 88(3):303–308.
Voskoboinik, I., H. Brooks, S. Smith, P. Shen, and J. Camakaris. 1998. ATP-dependent copper transport by the Menkes protein in membrane vesicles isolated from cultured Chinese hamster ovary cells. FEBS Lett 435(2–3):178–182.
Voskoboinik I., D. Strausak, M. Greenough, H. Brooks, M. Petris, S. Smith, J.F. Mercer, and J. Camakaris. 1999. Functional analysis of the N-terminial CXXC metal-binding motifs in the human Menkes copper-transporting P-type ATPase expressed in cultured mammalian cells. J. Biol. Chem. 274(31):22008–22012.
Vulpe, C.D., and S. Packman. 1995. Cellular copper transport. Annu. Rev. Nutr. 15:293–322.
Vulpe, C., B. Levinson, S. Whitney, S. Packman and J. Gitschier. 1993. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 3(1):7–13.
Walter, R.M. Jr., J.Y. Uriu-Hare, K.L. Olin, M.H. Oster, B.D. Anawalt,
J.W. Critchfield, and C.L. Keen. 1991. Copper, zinc, manganese, and magnesium status and complications of diabetes mellitus. Diabetes Care 14(11):1050–1056.
Yamaguchi, Y., M.E. Heiny and J.D. Gitlin. 1993. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson's disease. Blochem. Biophys. Res. Commun. 197(1):271–277.
Yazaki, M., K. Yoshida, A. Nakamura, K. Furihata, M. Yonekawa, T. Okabe, N. Yamashita, M. Ohta and S. Ikeda. 1998. A novel splicing mutation in the ceruloplasmin gene responsible for hereditary ceruloplasmin deficiency with hemosiderosis. J. Neurol. Sci. 156(1):30–34.
Yoshida, K., K. Furihata, S. Takeda, A. Nakamura, K. Yamamoto, H. Morita, S. Hiyamuta, S. Ikeda, N. Shimizu, and N. Yanagisawa. 1995. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat. Genet. 9(3):267–72.
Yoshimura, N., K. Kida, S. Usutani, and M. Nishimura. 1995. Histochemical localization of copper in various organs of brindled mice after copper therapy. Pathol. Int. 45(1):10–18.
Yuzbasiyan-Gurkan, V., V. Johnson, and G.J. Brewer. 1991. Diagnosis and characterization of presymptomatic patients with Wilson's disease and the use of molecular genetics to aid in the diagnosis. J. Lab. Clin. Med. 118(5):458–465.



























