
B— Homeostatic Processes in Brain Aging: The Role of Apoptosis, Inflammation, and Oxidative Stress in Regulating Healthy Neural Circuitry in the Aging Brain
Carl W. Cotman
INTRODUCTION
Research over the past several years has shown that the brain regions most vulnerable to mechanisms of inflammation, apoptosis, and free radical generation are those that serve primary functions in cognition. This must mean that mechanisms serving cognition and plasticity leave these circuits more vulnerable to injury and dysfunction. Curiously, many of the same molecules and molecular pathways can serve either beneficial or detrimental functions, depending on the exact cell, level of the factor, and acute versus chronic state. The mechanism can be local or can spread from the very local cellular microenvironment to cellular units and to entire systems. The progressive nature of the challenge faced can increasingly compromise cognitive functions.
During brain aging, the brain accumulates a series of insults and injuries and therefore must compensate through the activation of homeostatic mechanisms. Some of these mechanisms, however, once activated, may become part of degenerative cascades. Thus, the aging brain must accomplish homeostatic maintenance without the engagement of unregulated degenerative mechanisms. Over time, the compromising of circuitry integrity and function will take an inevitable toll on cognitive function. This paper presents the hypothesis that brain aging at a mechanistic level is resolvable into a series of distinct phases. These phases are associated with discrete molecular events that evolve into cascades, and, importantly, each phase may require different intervention strategies and certainly must be resolved in order to understand
the overall aging process. The literature discussed in support of this hypothesis has been limited due to space constraints and thus some citations may have been unintentionally omitted.
BRAIN AGING, A MULTIPHASE PROCESS: INITIATION AND PROPAGATION PHASES
Several investigators have suggested that Alzheimer's disease may represent an accelerated decline of the normal processes of brain aging. Thus, for example, the normal aged brain appears to accumulate plaques and tangles. This hypothesis suggests that Alzheimer's disease then is simply a further progression in the accumulation of these hallmarks and that relative risk factors would determine the nature of when and how fast accumulation and cognitive decline occur. Hence, all individuals would be subject to the same basic mechanism, and only the rate constant would differ with aging. While this hypothesis is one possibility at a mechanistic level, it is imprecise and does not address the current body of data suggesting that Alzheimer's disease results from a series of mechanisms and cascades that, over time, drive progressive pathology.
I suggest that the aging process can be resolved into a series of distinct states (Figure B-1). Let us assume that under the arbitrary age of 120 it is
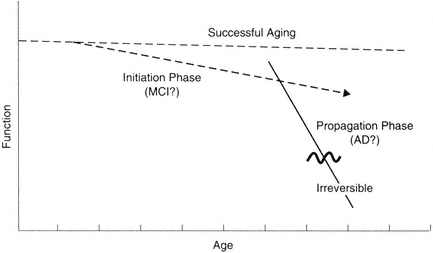
FIGURE B-1 Model for distinct phases in brain aging. It is becoming increasingly clear that at a mechanistic level, the primary driving mechanisms leading to progressive loss are multiphasic. The implication is that different therapeutic strategies will be needed at different phases of the processes.
possible to maintain normal brain function. This must be true, since some individuals maintain function for this period of time. Indeed, it appears that cognitive function can be preserved in some individuals even though some sensory functions may be compromised. Thus, as illustrated in Figure B-1, the successful aging line can be relatively flat.
Let us further postulate that the mechanism of decline can be divided into two general phases: initiation and propagation (Johnson et al., 1998). The initiation phase is distinct in terms of relative risk factors (e.g., APOE genotype) and protective factors. According to this hypothesis, the phase is relatively reversible and very amenable to interventions; it is relatively stable even though decline does occur. This idea is consistent with epidemiology data (e.g., Breitner, 1996) and histochemical data on factors affecting the accumulation of β-amyloid (see Johnson et al., 1998). For example, APOE at low levels of amyloid accumulation can determine the average age of decline onset. Once amyloid accumulation (or a similar process) reaches a certain level, however, the propagation phase is set in motion.
The propagation phase is distinct in that self-reinforcing molecular cascades are the net driving force; these cascades supersede contributing risk factors and accelerate pathogenesis. In terms of β-amyloid accumulation, this phase is largely independent of APOE-ε4, although it may have gender factors. Examples of possible autocatalytic cascades that could contribute to a propagation phase include the ability of amyloid to induce the amyloid precursor protein (APP) and chronic inflammation (see Cotman and Su, 1996). Such a propagation phase could be reversible up to a threshold point; however, once past this point, it may be irreversible.
This hypothetical model must also be considered in the context of the microenvironment. Thus, in the initiation phase, one would postulate that pathology is focused on vulnerable regions, but within those regions is largely confined to local domains, perhaps even individual cells. Subsequent transition/entry into the propagation phase results in the spread of pathology throughout the network. This propagation mechanism is unknown but may represent a breakdown of the local microenvironment. This is an important issue and is not addressed in the context of most current molecular mechanisms.
The progression of pathology is embodied in the Braak and Braak staging, in which the induction of tangles spreads through the limbic system network and is in essence the morphological equivalent of this set of events. Recently, my colleagues and I have established morphological evidence for transsynaptic propagation of neurofibrillary tangle pathology. We examined a series of neuropathologically staged cases and traced the temporal induction of AT8 and PHF staining in a well-established trisynaptic pathway: entorhinal stellate neurons, dentate gyrus granule cells, and CA4 pyramidal neurons. Cellular changes along this circuit appeared to initiate in the entorhinal cor-
tex in early Alzheimer's disease and progress through the circuit as the disease progressed, suggestive of a transsynaptic mechanism of propagation. The stimulus/agent is, of course, unknown.
In summary, I suggest that a separation of mechanisms may occur in the evolution of Alzheimer's disease. These stages may parallel various clinical stages. The relatively flat progression has been referred to as successful aging, optimal aging, etc. The initiation phase may be similar to the phase called mild cognitive impairment. The propagation phase may be the entry into Alzheimer's disease and related dementias. Of course, the correlation between brain changes and functional state is challenging due in no small part to the reserves and functional plasticity of circuits, particularly in the successful aging and initiation phases. Thus, for example, it would be anticipated that changes that occur in the initiation phase are subclinical for many measures, even though from a mechanistic viewpoint these changes are signatures of progression.
The implication of this concept is that it suggests that different therapeutics will be necessary to abort or slow the mechanism, depending on the stage of progression. Thus the initiation phase may be amenable to such interventions as nonsteroidal anti-inflammatory drugs, estrogen, education, antioxidants, etc. The later propagation phase may be much less sensitive or even insensitive to such interventions, although there is evidence to suggest that antioxidants such as Vitamin E can modulate progression at this point.
THE INITIATION PHASE AND EARLY EVENTS IN A PATHOLOGICAL CASCADE
To study the initiation phase in vitro or in vivo, it is necessary to develop a model in which subthreshold insults occur that do not cause overt cell death but rather impair cellular function. An appropriate stimulus to promote cell dysfunction is Aβ, since this neurotoxic protein accumulates in the form of senile plaques in the aged human and canine brain. Exposing cell cultures to sufficient levels of Aβ causes cell death in neurons and glia (see Cotman et al., 1999, for a review). Neurons die by initiating programmed cell death pathways, the up-regulation of pro-apoptotic proteins or the down-regulation of anti-apoptotic proteins (Paradis et al., 1996). To identify events associated with the initiation phase, we are now using subthreshold levels of Aβ that do not cause overt neuronal death to determine the sequence of events that occur early in response to an injury. These events may be subtle indicators of neuron dysfunction that develops prior to the classic forms of pathology found in the aged brain, such as senile plaques, neurofibrillary tangles, and cell death. Once neurons are exposed to a potentially toxic stimulus, signal transduction pathways are activated that initiate a cascade of events leading to neuronal dysfunction. This hypothesis led us to examine the role of signal
transduction pathways in neurons as early mediators of neuron dysfunction and subsequent death.
Signal Transduction Pathways May Be Compromised Much Sooner Than Degeneration Develops
One of the components that is critical in signal transduction is CREB (cyclic AMP response element binding protein), a molecule that mediates a plethora of responses involving gene transcription. Briefly, the pathway leading to the activation of CREB starts with an increase in Ca2+ or cAMP, which leads to the activation of calcium calmodulin-dependent (CAM) Kinase IV or cAMP-dependent protein kinase (protein kinase A). CAM Kinase IV or protein kinase A translocates into the nucleus and phosphorylates CREB, thereby activating this protein. Once CREB is phosphorylated, it can bind to cyclic AMP response element (CRE) in the promoter region of specific genes and increase transcription, leading to increased RNA and protein levels. One of these proteins is brain-derived neurotrophic factor (BDNF), which promotes neuron survival and plasticity (Cellerino et al., 1996; Galuske et al., 1996; Ma et al., 1998). As the actions of CREB become more elucidated, it is becoming apparent that CREB is functionally important for neuroplasticity (Ahn et al., 1999; Bailey et al., 1996; Glazewski et al., 1999; Schultz et al., 1999; Segal et al., 1998).
Recent data suggest that transgenic mice that do not express CREB or mice treated with antisense mRNA to CREB show impaired long-term potentiation, a physiological mechanism thought to underlie short-term memory (Glazewski et al., 1999; Schultz et al., 1999). A recent study also indicates that brains of humans diagnosed with Alzheimer's disease have decreased levels of phosphorylated CREB (pCREB) (Yamamoto-Sasaki et al., 1999), and it is hypothesized that this decline may have a role in memory decrements. While a direct mechanism or causal effect of declines in pCREB has not been established in normal aging in humans or animals, it is hypothesized that short-term memory deficits that occur as mild cognitive impairment in humans or as memory impairments in individual old canines may be a consequence of neuronal dysfunction associated with decreased phosphorylation of CREB signal transduction mechanisms or other transcription factors.
This leads to the question of whether a similar series of events occurs with sublethal exposures to Aβ, as would be expected in the early initiation phase. Depolarization of cells, as would occur in vivo with long-term potentiation, induces the phosphorylation of CREB. However, in the presence of sub-threshold levels of Aβ, there is a significant reduction in pCREB (Tong et al., in press). One interpretation of these results is that transcriptional activation by CREB could be compromised in dysfunctional neurons prior to overt cell death. If the neuron can reverse dysfunction, such as a diminished CREB
signaling pathway or an Aβ insult, then it can be returned to a functional state. Otherwise, apoptotic degenerative pathways may be initiated and the neuron removed from the system entirely.
Does the loss of pCREB regulation affect the encoding of proteins that are involved in neuroplasticity? The answer appears to be yes. CREB is involved with the transcription of BDNF, which is involved with learning and memory mechanisms, encoding long-term potentiation, cell health and survival, and protection from injury. In our culture model system, Aβ decreases depolarization-mediated induction of BDNF transcription (Tong et al., in press). Thus, subthreshold Aβ exposure can lead to some remarkable changes in neuron function, including decreased transcription of BDNF, a protein important for promoting neuron survival in the absence of any overt pathological change. In fact, BDNF is decreased in brains with Alzheimer's disease.
The proinflammatory cytokines are another illustration of the ability of the same molecule to support neuronal functions or contribute to dysfunction/degeneration. Experimentally, TNFα and IL-1β have been implicated in multiple examples of neurodegeneration (Feuerstein et al., 1998; Griffin et al., 1989; Martin et al., 1997). The engagement of these receptors can govern such diverse cellular responses as cellular proliferation, differentiation, and effector functions, or drive cells into apoptosis (Baker and Reddy, 1996, 1998). Therefore, the same signals that induce proliferation and differentiation can also induce cell death under different conditions, such as different activation states, developmental states, or cellular associations (Kang et al., 1992; Lenardo, 1991; Radvanyi et al., 1993). TNFα represents an excellent example of the pleiotrophic nature of these cytokines. TNFα can directly trigger apoptosis but has also been found to be neuroprotective in certain instances (Bruce et al., 1996). This difference in the response to TNFα may depend on the metabolic state of the cells and tissue being exposed to TNFα, because when TNFα is applied to a healthy brain, it typically does not induce neurodegeneration. However when TNFα is combined with an insult such as ischemia, TNF induces a robust increase in neuronal death (Rothwell and Hopkins, 1995).
In addition to having a direct effect on cellular physiology, TNFα has been shown to disrupt the signal transduction pathways induced by other physiological ligands, such as insulin (Peraldi et al., 1996; Paz et al., 1997). More recently, a new mechanism resulting in neurodegeneration involving TNFα has been proposed by Venters et al. (1999). They noted that in addition to an increase in TNFα during an inflammation in the central nervous system, there is also an increased expression of the hormone insulin-like growth factor (IGF-I). Activation of the IGF-I receptor on neurons is neuroprotective, inhibiting apoptosis (Dudek et al., 1997; Russell et al., 1998). Previous reports have shown that simply changing the ratio of IGF-1 and TNFα can shift the balance between survival and neuronal death (Barone et
al., 1997; Loddick et al., 1998). Venters et al. found that TNRα significantly reduces the ability of IGF-I to promote survival in cerebellar granule neurons. These investigators proceeded to show that TNFα inhibits the ability of IGF-I to initiate tyrosine phosphorylation of the insulin receptor substrate 2 (IRS-2), thereby blocking the activation of downstream P13-kinase. Thus diseases of the central nervous system that have an inflammatory component involving TNFα, such as multiple sclerosis, AIDS-dementia complex, and Alzheimer's disease, may use intracellular cross-talk between TNFα and IGF-I receptors to inhibit survival signaling by IGF-I and perhaps other neurotrophic factors. Thus some molecules promote homeostasis by multiple mechanisms, and in diseases such as Alzheimer's disease, these mechanisms may be recruited into cascades that trigger disease progression.
In summary, neurons that experience pro-apoptotic insults appear to shut down key signal transduction pathways. At the single cell level, this is probably a wise strategy, because it potentially removes dysfunctional cells from the network. This would then, in turn, allow the fully functional cells to maintain brain function and possibly activate auxiliary use-dependent plasticity mechanisms (Figure B-2).
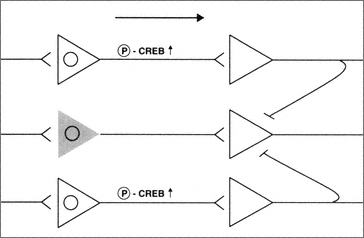
FIGURE B-2 Neurons subjected to sublethal pro-apoptotic insults may be deficient in their ability to regulate key signal transduction pathways serving functional plasticity. The presence of sublethal concentrations of B-amyloid results in an inability to phosphorylate CREB and thus control of essential transcriptional mechanisms. Thus these neurons are partially removed from the circuit (shaded cell).
Interventions during the Initiation Stage
What types of interventions could be implemented in the initiation stage of brain aging to promote successful aging? Clearly, the issue is to discover interventions that will in turn establish the validity of mechanistic predictions. One possibility is to regulate the expression of BDNF, a downstream product of the CREB signal transduction pathway. In general, behavioral modifications and exercise may have merit. Several years ago, we began to examine the possibility that simple behavioral interventions, such as voluntary running, could promote neuron function via increased expression of BDNF. The experimental design of these studies included providing rats with access to a running wheel and recording the distance traveled while allowing each animal to run voluntarily. Voluntary exercise increased BDNF mRNA in hippocampal areas after several hours (Oliff et al., 1998) or days (Neeper et al., 1995, 1996). Recently, we also have examined the possible involvement of CREB-mediated signal transduction and found that pCREB increased in response to the voluntary running paradigm during a period of seven days, while total levels of CREB were unchanged (Shen et al., in press). Recent data also suggest that exercise improves memory in aged humans (Binder et al., 1999; Grealy et al., 1999; Williams et al., 1997), but the cause of this improvement in cognition has not yet been determined. However, together these data suggest that exercise can be a driving force on plasticity mechanisms by enhancing the activation of factors that promote transcription of genes involved with neuron function and ultimate survival.
Other interventions may also increase the expression of BDNF and have functional consequences. Several studies have been examining this question; the work on environmental enrichment is particularly important (Kempermann et al., 1997, 1998). Three weeks of environmental enrichment significantly stimulated cell proliferation, BDNF expression and resistance to insults, and inhibited apoptotic cell death (Young et al., 1999). Proliferating cell nuclear antigen (PCNA) levels were increased and double-stranded DNA breaks (TUNEL) were decreased in the enrichment group relative to controls, suggesting that neurogenesis occurred in response to environmental enrichment. Furthermore, rats in the enriched environment were resistant to kainate-induced seizures, and neuron death in response to seizures was ameliorated (Young et al., 1999). Finally, the expression levels of BDNF were higher in the enrichment group relative to controls. Upstream of BDNF expression, the authors also showed that environmental enrichment increased the expression of CREB and pCREB, particularly in the proliferating zone of new neurons.
To summarize, many studies indicate that environmental enrichment and exercise, relatively modest interventions, can promote successful aging by modifying brain health at the single neuron level. Studies on behavioral
interventions and brain aging are currently understudied in humans. Behavioral interventions in brain aging are popular at the community level, but clinical studies in patient populations are greatly lacking, as are mechanistic studies in animal models.
PROPAGATION PHASE: MANY INDUCERS OF APOPTOSIS ACCUMULATE IN THE AGING BRAIN
In the propagation phase, mechanisms including apoptosis, inflammation, and oxidation are activated either in combination or chronically beyond a certain level. Homeostatic balances are exceeded and dyshomeostasis prevails. This phase illustrates the importance of homeostasis in brain aging and the identification of mechanisms that can lead into dyshomeostasis. The discussion below illustrates the key principles. Apoptosis normally serves during development to remove excess cells, and in disease or injury it serves to destroy damaged cells. This is a well-accepted concept in all basic biological systems. With age, a variety of stimuli accumulate in the brain that may induce apoptotic pathways in neurons. In the aging and Alzheimer's-affected brain, β-amyloid, a 40–42 amino acid peptide, accumulates in the extracellular space as small deposits and senile plaques. Based on the observation that neurites surrounding β-amyloid deposits exhibit both sprouting and degenerative responses, we proposed that this peptide is not metabolically inert, but rather possesses biological activity. Our findings established two key principles: β-amyloid induces neurotoxicity in a conformation-specific manner, and apoptotic mechanisms underlie this toxicity (Cotman and Anderson, 1995; Anderson et al., 1995; Loo et al., 1995, 1993; Watt et al., 1994); these observations have since been confirmed by many others. Interestingly, prior to causing cell death, β-amyloid also induces the formation of dystrophic-like neurite morphology in cultured neurons (Pike et al., 1992; Fraser et al., 1994).
Oxidative insults also readily initiate apoptosis (Whittemore et al., 1994), and oxidative damage is known to occur in the aging and Alzheimer's-affected brain (Benzi and Moretti, 1995). Similarly, reductions in glucose metabolism have been suggested to contribute to neurodegeneration in Alzheimer's disease (Beal et al., 1993; Goto et al., 1993; Haxby and Rapoport, 1986; Hoyer et al., 1988; McGeer et al., 1986), and β-amyloid has been shown to exacerbate neurodegeneration in cultured neurons when glucose levels are reduced (Copani et al., 1991). Furthermore, excitotoxic damage can, under some conditions, initiate apoptosis, and many investigators have suggested that excitotoxic damage contributes to neurodegenerative diseases, including Alzheimer's disease (Dodd et al., 1994). Recent studies have also shown that glutamate transport proteins may be greatly reduced in the Alzheimer's-affected brain (Masliah et al., 1996; Simpson et al., 1994), which could exacerbate excitotoxic mechanisms. The profile of initiating factors strongly sug-
gests that in the course of aging and age-related neurodegenerative disease, neurons are increasingly subjected to apoptosis inducers. In some cases, these factors may act synergistically. For example, neuronal apoptosis may be significantly potentiated by the addition of subthreshold doses of β-amyloid and either excitotoxic or oxidative insults (Dornan et al., 1993; Koh et al., 1990; Mattson et al., 1992; Pike et al., 1997). Finally, mitochondrial damage may contribute to apoptosis as an intracellular effector. Mitochondria are a major source of free radicals and the release of cytochrome c is a potent inducer of caspase activation. Indeed, this organelle may be a prime target of aging and thus a contributor to the apoptosis cascade.
Some genetic risk factors also increase the probability that cells will engage apoptotic mechanisms. Overexpression of presenilin (PS) 1 or 2 results in an increased susceptibility of cells to apoptotic insults. PS mutations sensitize neurons to apoptosis by trophic factor withdrawal, metabolic insults and β-amyloid (Deng et al., 1996; Wolozin et al., 1996; Kim et al., 1997). It has been suggested that PS mutations cause perturbed calcium release from the endoplasmic reticulum and increased levels of oxidative stress (Mattson et al., 1998). Indeed, introduction of PS-1 into oocytes results in enhanced release of intracellular calcium and this is further increased by the presence of a PS-1 mutation. The effect appears to be downstream from the inositol trisphosphate receptor, because inositol trisphosphate injected directly into the cell elicits the increased release. Thus, because calcium homeostasis contributes to apoptosis, these gene products increase the probably that neurons may degenerate via apoptosis. Thus, in patients carrying PS mutations, apoptosis is likely to be one of the mechanisms of neuronal degeneration. The amyloid precursor protein itself appears capable of initiating apoptosis. There is growing evidence that the amyloid precursor protein is a receptor resembling a polypeptide hormone receptor (Nishimoto et al., 1997). The cytoplasmic portion of the protein contains a G-protein activator sequence (H657–K676) and will bind and activate G0. It has been suggested that the mutations result in a constitutively active G0 and that this causes apoptosis (Nishimoto et al., 1997; Yamatsuji et al., 1996).
Clearly then, there is ample potential for the induction of apoptosis mechanisms in the aging and Alzheimer's-affected brain. In this context, it is essential to determine if such pathways are activated in the Alzheimer's-affected brain. Indeed, a growing body of evidence supports this hypothesis (see Cotman et al., 1999).
In general, it appears as if brain aging acute phase responses often become chronic and escape the local microenvironment. The same mechanisms that are normally adaptive can become dysfunctional. This is ''dysfunctional plasticity," in which the same adaptive mechanisms turn against the system as overcompensation evolves, safety margins decline, and redundancy is lost.
Multiple-level cascades can shift the balance between beneficial and
nonbeneficial functions: The significance of cellular change to cognitive function evolves in a hierarchy from the cell, to cellular units, to systems.
-
Each participating brain region in an overall system is selectively vulnerable to select genetic and/or environmental/disease-related conditions.
-
Dysfunction in one part of the system can compromise the entire connectionist network.
-
Dyshomeostasis is encoded into the network and alters input/output profiles, which may be optimal for the residual system, but the system now operates at another state function. Systems homeostasis/plasticity is understudied in aging, and that which is represented is largely limited to rodents.
-
Progressively more network plasticity and more good cells are required to maintain even normal baseline functions. This further weakens the linkages. One process affects the others.
Inflammatory Mechanisms May Convert a Precarious State into Net Degeneration
Acute injury initiates inflammatory mechanisms. Inflammatory mechanisms include the activation of complement pathways that lead to cell lysis and the up-regulation of death receptors and their respective ligands. These death receptors in the immune system serve to maintain homeostasis through selective cell death by way of apoptosis. In the brain, acute inflammatory responses are part of the natural repair process, but chronic inflammation probably drives degeneration, much like a chronic infection. Thus, inflammatory mechanisms are another example of the delicate balance. Clearly, inflammatory mechanisms suppress the initiation phase as anti-inflammatory medications delay the age of onset for Alzheimer's disease. It is, of course, unknown at the present time whether the same interventions will be effective during the propagation phase, but there are strong arguments to indicate they will probably be ineffective. Inflammation and beneficial actions have also been dramatically brought to the forefront by the discovery that antibodies developed against amyloid can activate the immune system to cause the remove of senile plaques (Schenk et al., 1999). Thus the balance of immune activity in the nervous system is highly critical.
Some age-related risk factors such as Aβ, oxidative damage, and imbalances in glutamate may contribute to the emergence of inflammation and place cells at further risk for degeneration. Recent evidence suggests that reactive oxygen intermediates (ROIs) are potent inducers of FasL and that antioxidants suppress this transcriptional dependent process. Inhibition of FasL expression appears associated with decreased binding of nuclear factor NF-kB, an important redox-controlled transcription factor (Bauer et al., 1998). In response to oxidative stress, there is an increase in FasL expression
on microglia cells. Importantly, compared with classical mediators of microglia activation (e.g., TNFα, LPS), oxidative stress was the most potent. TNRα can render microglia sensitive to FasL apoptosis by inducing Fas expression and down-regulation of Bcl-2 and Bcl-xL. (Spanaus et al., 1998). Further, hypoxia followed by re-oxygenation resulted in increased FasL expression (Vogt et al., 1998). Because the temporal and spatial patterns of microglia activation in injuries such as hypoxia coincides with the onset of DNA degradation and apoptosis in regions of selective neuronal loss, it has been suggested that microglia play a possible role in apoptosis.
Other risk factors can also induce FasL. In the Alzheimer's-affected brain, there is a reduction in the glutamate transporter (Masliah et al., 1996) that may contribute to neurodegeneration due to additional activation of glutamate receptors. Activation of NMDA receptors may also cause an increase in FasL. After a single injection of NMDA, there is an increase in FasL that begins after about 10 days and persists for up to 5 months. This increase may participate in long-term degeneration as part of a mechanism in the balance between repair and synaptic turnover/remodeling (Shin et al., 1998).
In summary, the gradual age-dependent increase of cytokines and their receptors capable of inducing apoptosis in the central nervous system could place neurons at increased risk for degeneration. It is possible that microglia and other cells in the brain also participate in the production of death ligands and thereby enhance the risk. Thus either autocrine or paracrine mechanisms may become active. Figure B-3 summarizes a model of the possible mechanism. At present a detailed study on the aging and Alzheimer's-affected brain has not been conducted, nor in fact has a detailed study at the anatomical level been reported in animal models. The development of many new reagents and the continued articulation of the mechanisms of Fas/caspase regulation provide a window of opportunity for the pursuit of this research direction.
Oxidative Stress Is a Major Risk Factor for Brain Aging
Oxidative stress is a candidate for causing neuron dysfunction through a molecular cascade (Figure B-4). Oxidative stress is problematic for a number of reasons. Oxidation of proteins and enzymes within cells can interfere with their normal function (Stadtmann, 1992). Free radicals can also damage DNA, and a 50 percent increase in DNA oxidative damage has been reported in the human brain (Lyras et al., 1997; Gabbita et al., 1998; Mecocci et al., 1994; Lovell et al., 1999). Indeed, extensive DNA damage appears to accumulate with age and is particularly prominent in the aged dog brain and in humans (Su et al., 1997; Anderson et al., in press). Lipids are also vulnerable to oxidative stress and the levels of lipid peroxidation are elevated in the human brain, which in turn may induce membrane disturbances and loss of homeostasis within cells (Balazs and Leon, 1994; Palmer and Burns, 1994). In fact,
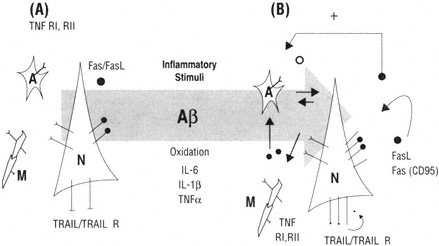
FIGURE B-3 The immune responses in the brain are a critical part of homeostasis but when chronically activated may contribute to the evolution of brain pathology. Neurons and glia express receptors and ligands in the TNF superfamily (A). In the presence of inflammatory stimuli such as Aβ, oxidation and select cytokines would be predicted to be up-regulated (B) and thus would make cells at risk for activating pro-apoptotic mechanisms similar to that reported in the aging immune system.
one lipid peroxidation product, 4-hydroxynonenal (HNE), is elevated in the brains and cerebrospinal fluids of cases of Alzheimer's disease (Lovell et al., 1997; Markesbery and Lovell, 1998), is toxic in vitro and in vivo, and impairs visuospatial memory in rats at physiological levels (Bruce-Keller et al., 1998). Isoprostane, a chemically stable peroxidation product of arachidonic acid, increases as well and is used as a marker for the extent of lipid peroxidation in vivo. In particular, Praticao et al. (1998) demonstrated that isoprostanes were elevated in the brains and cerebrospinal fluids of patients with Alzheimer's disease.
Oxidative stress can also lead to the misprocessing of APP to form amyloidogenic products. Several in vitro experiments suggest that energy-related metabolic stress leads to reduced levels of secreted APP mediated by β-secretase and in fact, may lead to increased production of amyloidogenic fragments (Gabuzda et al., 1994; Gasparini et al., 1997; Multhaup et al., 1997). Oxidative stress increases the production of both APP and β-amyloid (Frederikse et al., 1996). β-amyloid itself can lead to the generation of reactive oxygen species (ROS), superoxide radicals, hydroxynonenal (HNE), and membrane lipid peroxidation (Mark et al., 1997; Behl et al., 1992; Pereira et
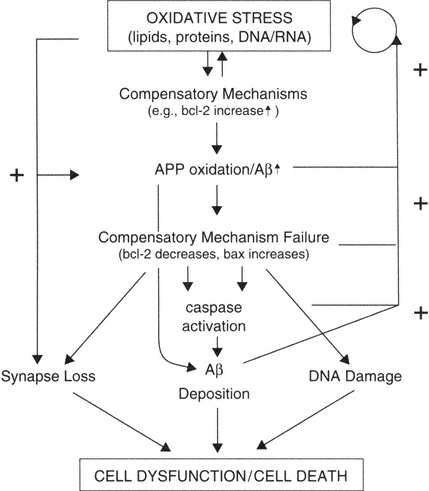
FIGURE B-4
Oxidative stress is an early and continued event in brain aging, leading to increases in expression of proteins involved with promoting cell survival (bcl-2). Oxidation causes damage to lipids, proteins, and DNA/RNA. Oxidative stress also induces the expression of APP and can contribute to the misprocessing of APP, leading to generation of amyloidogenic fragments. The production of Aβ fragments may lead to a loss of compensatory ability (decreased bcl-2, increased bax). All of these factors in turn contribute to more Aβ deposition, possibly synapse loss, and DNA damage. Ultimately the system converges and results in neuron dysfunction and/or in some neurons' death. At several points in this proposed framework, positive feedback loops exist such that the deposition of Aβ exacerbates oxidative stress and oxidative stress itself results in decreased levels of bcl-2 and increased levels of bax.
al., 1999; McDonald et al., 1997). There is also evidence that β-amyloid can itself be oxidized resulting in enhanced aggregation (Dyrks et al., 1992).
The accumulation of amyloidogenic fragments, in turn, accelerates existing molecular cascades associated with oxidative stress. β-amyloid also promotes cell dysfunction by increasing the expression levels of bax, decreasing levels of bcl-2 (Paradis et al., 1996), and, in many systems, activating caspases. Thus, decreases in bcl-2 from oxidative stress and Aβ insults can leave cells particularly vulnerable to oxidative stress (Hochman et al., 1998). Finally, activation of caspases cleaves APP, producing additional β-amyloid fragments (Gervais et al., 1999; Barnes et al., 1998; LeBlanc et al., 1999). These are exciting leads, as they suggest alternative mechanisms for the production of β-amyloid.
Oxidative stress may lead to an initial increase in cellular compensatory mechanisms mediated by the expression of pro-and anti-apoptotic proteins. One family of proteins, the bcl-2 family, serves as an intracellular checkpoint and determines whether or not a cell engages in an apoptotic program (Oltvai et al., 1993). Bcl-2 can be inactivated by the formation of heterodimers with a second highly homologous protein, bax. Bax promotes apoptotic cell death (Oltvai et al., 1993; Reed, 1994). Thus, the balance between levels of bcl-2 and bax can serve as an indicator of cellular state. Ultimately, we hypothesize, this compensatory mechanism is inadequate and eventually leads to decreased bcl-2 levels with a corresponding increase in bax, shifting the system to a neurodegenerative status. Exposing neuronal or endothelial cells to oxidative stress decreases levels of bcl-2 and increases the expression levels of bax (Longoni et al., 1999; Maroto and Perez-Polo, 1997).
Thus, we can hypothesize that oxidative stress causes a cascade of events in which there are multiple positive feedback loops that amplify the cascade. For example: (1) oxidative damage within mitochondria leads to further free radical production, (2) increased oxidation leads to additional Aβ, which in turn generates additional APP, (3) Aβ and accumulating oxidative damage activates caspases, which in turn cleaves APP and generates additional Aβ and oxidative damage, and (4) progressive oxidative damage and Aβ decreases bcl-2, leaving neurons more vulnerable to oxidative damage and other insults. Common to each of these events is oxidative damage. Thus, an antioxidant intervention should, in principle, suppress the progression of brain pathology at one or more steps in the cascade. This is consistent with a vast and somewhat unappreciated literature on the efficacy of antioxidants in the aging process.
Antioxidants Are Effective Interventions
There is a growing body of literature indicating that administration of antioxidants to aged animals and individuals can have dramatic effects on
TABLE B-1 Antioxidant Interventions and Improvements in Cognitive Function—A Summary of the Existing Literature
|
Antioxidant |
Subjects |
Duration of Study |
Cognition |
|
PBN |
Gerbils |
14 days |
Improved spatial memory in radial arm maze |
|
Blueberry spinach strawberry |
19-month-old rats |
8 weeks |
Improved one-trial learning in the water maze Improved vitamin E levels in the hippocampus |
|
PBN vitamin E |
24-month-old rats |
4–5 months |
Improved memory retention on spatial memory task Improved learning of a spatial memory task |
|
Strawberry spinach vitamin E |
6-month-old rats |
8 months |
Enhanced retention in a spatial water maze task |
|
PBN |
24-month-old rats |
9.5 months |
Improved memory retention on spatial memory task Faster acquisition of a one-way activate avoidance task Improved learning of a spatial memory task |
|
PBN |
Senescence-accelerated mice |
12 months |
Significantly lengthened life span |
|
Vitamin E |
Rats receiving intraventricular Aβ |
7 days |
Reduced spatial memory impairments induced by Aβ |
|
Vitamin E |
Patients with Alzheimer's disease |
2 years |
Significantly delayed institutionalization |
behavior and age-related brain oxidative status. Furthermore, in normal aging, reducing oxidative stress through the use of nutritional supplements including vitamin E is beneficial for cognition and immune system function (Fryer, 1998; Reidel and Jorissen, 1998; Blumberg and Halpner, 1999; Meydani et al., 1995; Perrig et al., 1997). Table B-1 summarizes the striking efficacy of antioxidant interventions.
Antioxidants can also significantly aid the human brain. Over 4 years ago, the inclusion of Vitamin E into a clinical trial of patients with mild to moderate Alzheimer's disease resulted in the finding that Vitamin E supple-
ments given to patients significantly delayed the time to institutionalization (Sano et al., 1997). Antioxidants also enhance cognition in rat models, and antioxidant application or diet supplementation can improve spatial memory (Joseph et al., 1999; Socci et al., 1995). Currently, an Alzheimer's Disease Cooperative Study using a randomized, double-blind, placebo-controlled trial is evaluating the safety and efficacy of 2,000 IU of Vitamin E to delay the clinical progression of elderly populations from mild cognitive impairment to Alzheimer's disease. Thus, there is strong rationale for the further evaluation of this intervention in an aging model in which the time interval between cognitive assessment and biochemical/neuroanatomical study can be tightly controlled. We need to evaluate dietary interventions using both short-term and long-term clinical trials.
APOPTOSIS CHECKPOINT CASCADE IN THE ALZHEIMER'S-AFFECTED BRAIN: THE SEARCH FOR HOMEOSTASIS
The presence of a mechanism to hold degeneration in check may provide an explanation for one of the seeming controversies in the Alzheimer's disease literature. As we have described, TUNEL labeling provides evidence for active apoptosis in a large subset of neurons in the Alzheimer's-affected brain. However, many more Alzheimer's-affected neurons exhibit evidence for DNA damage in the absence of morphological changes, indicative of terminal apoptosis, for example, the formation of nuclear apoptotic bodies. In classical apoptosis, cells die within hours or days of the initial insult. If TUNEL labeling in the Alzheimer's-affected brain reflected the true initiation of classical apoptosis, then it follows that most TUNEL-positive neurons would die in a few days. However, in many mild cases of Alzheimer's disease (MMSE above 16), over 50 percent of the neurons exhibit TUNEL labeling. Thus, most neurons should have degenerated within a few days if apoptosis is actively in progress in these cells, a prediction that is inconsistent with the progression of neuronal loss in Alzheimer's disease. In addition, most TUNEL-positive neurons do not exhibit morphological markers of apoptosis, such as nuclear apoptotic bodies or other key molecular factors (Su et al., 1994; Lucassen et al., 1997) This apparent inconsistency has led some to the conclusion that neurons in the Alzheimer's-affected brain die primarily by necrosis (Stadelmann et al., 1998).
On the other hand, it is possible that neurons have developed a series of counteractive measures to repair damage and delay death, in other words, a kind of molecular counterattack in order to minimize unnecessary cell loss. This concept of an apoptosis checkpoint cascade may help to understand an apparent puzzle in the neuronal apoptosis literature: the prolonged presence of indices of DNA damage and apoptotic regulatory protein expression may be a result of a counteractive strategy that neurons mobilize to hold apoptosis
in check, delay death, and attempt repair. In this context, it is possible that cell cycle proteins could contribute to cellular repair in neurons. Repair of DNA damage may be a particularly key example of such a mechanism. For example, many neurons in vulnerable regions of the Alzheimer's-affected brain show an up-regulation of GADD45, a protein that is associated with DNA checkpoint repair at the Gl transition. Alzheimer's-affected neurons that express GADD45 often also show DNA damage and increased levels of Bcl-2. In addition, in support of a role in promoting cell survival, GADD45 transfected cells show improved survival after DNA damage (Torp et al., 1998). Thus, Bcl-2, GADD45, and other protective molecules such as PCNA could serve to help repair DNA damage and assist in neuronal survival. Similarly, p16, p21, and other negative regulators of the cell cycle could participate in delaying degeneration. That is, many checkpoints in the cell death pathway may exist and perhaps prevent the unnecessary loss of irreplaceable cells. This possibility may make the study of signal transduction pathways particularly critical, because it may provide an opportunity for early interventions.
Taken together, this hypothesis may suggest that neurons could activate an "apoptosis checkpoint cascade," in which injured neurons may regulate the activation of pro-apoptotic proteins such as bax with anti-apoptotic proteins such as Bcl-2. In addition, it can be hypothesized that damaged neurons could activate the cell cycle and perhaps employ checkpoint molecules in a similar pro-and anti-apoptotic regulation point (Figure B-5).
CHANGES IN CYTOKINES AND THEIR RECEPTORS IN LYMPHOCYTES OCCUR IN AGING AND, SURPRISINGLY, MANY OF THE MOLECULAR PROFILES IN THE AGING BRAIN ARE SIMILAR TO THESE AND TO THE IMMUNE SYSTEM
Normal aging is associated with a decrease in the proportion of lymphocytes in the blood (lymphopenia) and a progressive decline in T-cell function, although the underlying mechanisms that cause these changes are largely unknown (Miller, 1996; Nagel et al., 1988; Proust et al., 1987; Thoman and Weigle, 1989). The reductions in T-cell functions include a decrease in response to mitogens and soluble antigens, as well as defects in signal transduction. The initial investigations in this area indicated that lymphocytes from elderly individuals were significantly more sensitive to activation-induced apoptosis (Phelouzat et al., 1996; Herndon et al., 1997). The relationships between increased cytokine expression, sensitivity to apoptosis, and lymphopenia led Gupta and colleagues to investigate whether there is an increased expression of death receptors and associated adapter and initiator proteins. An increased expression of Fas and FasL was accompanied by a decrease in Bcl-2 expression in memory cells of both CD4+ and CD8+T-cells from aged versus younger individuals. In addition, FasL caused an increased
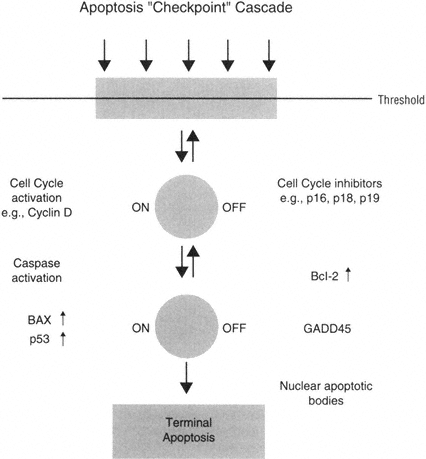
FIGURE B-5 Neurons as nondividing cells seem to delay or check the activation of apoptosis processes in order to attempt repair.
proportion of both T-cell subsets from older individuals to undergo apoptosis, indicating that the death receptors are functional in the aging immune system (Aggarwal and Gupta, 1998). In a follow-up study, two additional members of the Fas family of receptors, TNFRI and TNFRII, were examined in lymphocytes from aged individuals (Aggarwal et al., 1999). Once again, the investigators found increased sensitivity to undergo apoptosis, this time induced by TNFα. Moreover, they found that the TNFRI was expressed at elevated levels in aged lymphocytes and that the TNFRII was expressed at lower levels than
in young individuals. Coinciding with the changes in the TNFRs was an increase in TNFR-associated death domain protein (TRADD) and a decrease in TNFR-associated factor 2 (TRAF-2). These changes mirror those previously reported by Ware et al. (1991) following T-cell activation. However, in the case of lymphocytes from aged individuals, there was no increase in the expression of activation antigens, thus indicating that the T-cells were not in an activated state but rather reflect changes associated with an aging immune system. The potential importance of the change in the ratio of the two TNF receptors becomes obvious when one considers that the TNFRI contains a death domain and, when oligomerized by TNFα binding, recruits TRADD and FADD, resulting in the activation one of the initiator caspases, caspase-8. The TNFRII and TRAF-2 are involved in activation of NFkB and JNK, which are believed to mediate the anti-apoptotic effects of TNFα (Baker and Reddy, 1996, 1998). Based on these observations, Gupta and colleagues have proposed that the cellular and subcellular basis of this age-related immuno-senescence appears to at least partially involve increases in receptors linked with apoptosis and decreases in related compensatory receptors (Aggarwal and Gupta, 1998, 1999; Aggarwal et al., 1999).
Age-related changes may extend to other organs and may be displayed in the ratio of gene expression patterns. The recent introduction of gene chip technology into the field may have particular application to the field of aging. Thus, for example, the gene expression profile of aging in muscle tissue and the influence of caloric restriction have been described (Lee et al., 1999). In essence, using high-density oligonucleotide arrays representing 6,347 genes, it was shown that aging resulted in a differential gene expression pattern that reflected increased cellular stress and lower expression of metabolic and bio-synthetic genes. Some of these, such as DNA repair enzymes, are the same as those induced in the nervous system with age and degeneration. An example of an up-regulated gene found in the muscle is GADD45, which as discussed above, we have found is also induced in Alzheimer's disease.
Importantly, caloric restriction, which is the only really true intervention known to retard aging in mammals, almost completely prevented the gene expression pattern changes that occur with aging. This is a technology that should find particular use in the study of brain aging and cognition but will probably be difficult to get funded through peer review panels because it will be considered just a ''fishing expedition." These gene patterns summarize in one experiment the literature for the past 10 years of individual gene expression patterns. These expression patterns can be envisioned in essence as a fingerprint of homeostasis versus dyshomeostasis. In fact, this pattern of gene expression can be looked at as a view of the cells to engage homeostasis and plasticity mechanisms to compensate for age-related change.
CELLULAR PLASTICITY MECHANISMS AND THE MAINTENANCE OF HOMEOSTASIS
As cells degenerate and their numbers are reduced in circuitry, other mechanisms become engaged at a cell and systems level beyond those of the molecular level. Examples include the sprouting of new synapses in response to nearby cell loss and the sprouting of dendrites in neighboring cells. In addition, whole networks respond in terms of altered processing using somewhat redundant, but maybe initially suboptimal strategies. These, oftentimes, can increase the time for cognitive processing, but will still accomplish the task. There is also a growing body of literature indicating that, in the course of brain aging, more of the brain has to be involved in a task that would normally require only minimal activation of circuits; thus, the circuits are working much harder to accomplish the same task. This would indicate that use-dependent change and practice effects, together with appropriate pharmaceuticals, might have a rational basis for cognitive rehabilitation.
STRATEGIES AND SOLUTIONS FOR THE FUTURE
In conclusion, there are several principles that appear to be evolving in the field that are in need of additional testing:
-
Brain aging is not a linear process; the aging process passes through phases.
-
The initiation phase can compromise neuronal function and is probably reversible as it represents functional homeostasis.
-
Interventions include antioxidants, use-dependent plasticity (behavioral/physical/cognitive stimulation), regulation of inflammation, estrogen replacement therapy, etc., and are most effective in this phase.
-
The propagation phase is initiated through a series of molecular cascades driven by accumulating failures and compensation mechanisms and is less readily reversible.
-
Interventions may be phase-dependent, and effective interventions at one phase may be inappropriate/inadequate at others.
Strategies must and can be developed to identify weak molecular linkages and to assist cells in correcting them prior to irreversible losses and the development of cascades.
-
There is a clear significant and major gap in supported research in the essential hierarchical areas, and circuit-based analyses at a systems level are needed.
-
Transgenic animals offer great promise, but there is a great need for
-
aged animals and standard behavioral protocols. Other animal models should be supported.
-
The single-variable approach inherent in most molecular studies at present is too limited; there is a great need to explore complex interactions at a molecular level.
-
Many behavioral theories, particularly in the practice of neuropsychology, lack solid mechanistic foundations and quantitative support, handicapping the growth of the field.
-
There is, in general, a gap between cognitive research and molecular mechanistic studies in brain aging. The National Institute on Aging should be encouraged continually to stimulate innovative approaches.
In summary, the key may very well be to create a shift in the intellectual environment in brain aging and cognition as well as pursue the leads already defined.
REFERENCES
Aggarwal, S., S. Gollapudi, and S. Gupta 1999 Increased TNF-alpha-induced apoptosis in lymphocytes from aged humans: Changes in TNF-alpha receptor expression and activation of caspases. Journal of Immunology 162(4):2154–2161.
Aggarwal, S., and S. Gupta 1998 Increased apoptosis of T cell subsets in aging humans: Altered expression of fas (CD95), fas ligand, Bcl-2, and bax. Journal of Immunology 160(4):1627–1637.
1999 Increased activity of caspase 3 and caspase 8 in anti-fas-induced apoptosis in lymphocytes from ageing humans. Clinical and Experimental Immunology 117(2):285–290.
Ahn, S., et al. 1999 A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron 23(3):559–568.
Anderson, A.J., C.J. Pike, and C.W. Cotman 1995 Differential induction of immediate early gene proteins in cultured neurons by beta-amyloid (A beta): Association of c-jun with a beta-induced apoptosis. Journal of Neurochemistry 65(4):1487–1498.
Anderson, A.J., C.J. Pike, and C.W. Cotman 1995 Differential induction of immediate early gene proteins in cultured neurons by beta-amyloid (A beta): Association of c-jun with a beta-induced apoptosis. Journal of Neurochemistry 65(4):1487–1498.
Anderson, A.J., W.W. Ruehl, L.K. Fleischman, K. Stenstrom, T.L. Entriken, and B.J. Cummings In press DNA damage is correlated with a beta deposition and unrelated to cytoskeletal neuropathology in the canine model of Alzheimer's disease. Progress in Neuro-Pharmacology and Biological Psychiatry 24.
Azizeh, B., F.L. Van Muiswinkel, D.H. Cribbs, A.J. Tenner, and C.W. Cotman 1999 Non-filbrillar β-amyloid: Priming the respiratory burst of rat microglial cells and human monocytes. Neurobiology of Aging (submitted).
Bailey, C.H., et al. 1996 Toward a molecular definition of long-term memory storage. Proceedings of the National Academy of Sciences of the United States of America 93(24):13445–13452.
Baker, S.J., and E.P. Reddy 1996 Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene 12(1):1–9.
1998 Modulation of life and death by the TNF receptor superfamily. Oncogene 17(25): 261–270.
Balazs, L., and M. Leon 1994 Evidence of an oxidative challenge in the Alzheimer's disease brain. Neurochemical Research 19(9):1131–1137.
Barnes, N.Y., L. Li, K. Yoshikawa, L.M. Schwartz, R.W. Oppenheim, and C.E. Milligram 1998 Increased production of amyloid precursor protein provides a substrate for caspase-3 cleavage in dying motoneurons. Journal of Neuroscience 18(15):5869–5880.
Barone, F.C., et al. 1997 Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 28(6):1233–1244.
Bauer, M.K.A., et al. 1998 Role of reactive oxygen intermediates in activation-induced CD95 (APO-1/fas) ligand expression. Journal of Biological Chemistry 273(14):8048–8055.
Beal, M.F., B.T. Hyman, and W. Koroshetz 1993 Do deficits in mitochendrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends in Neurosciences 16(4):178–184.
Behl, C., J. Davis, G.M. Cole, and D. Schubert 1992 Vitamin E protects nerve cells from amyloid-beta protein toxicity. Biochemical and Biophysical Research Communications 186:944–950.
Benzi, G., and A. Moretti 1995 Are reactive oxygen species involved in Alzheimer's disease? Neurobiology of Aging 16(4):661–674.
Binder, E.F., et al. 1999 The relation between psychometric test performance and physical performance in older adults. Journal of Gerontology. Series A, Biological Sciences and Medical Sciences 54(8):M428–M432.
Blumberg, J., and A. Halpner 1999 Antioxidant status and function: Relationships to aging and exercise. Pp. 251–275 in Antioxidant Status, Diet, Nutrition and Health, A. Papas, ed. New York: CRC Press LLC.
Breitner, J.C.S. 1996 Inflammatory processes and anti-inflammatory drugs in Alzheimer's Disease: A current appraisal. Neurobiology of Aging 17:789–794.
Bruce, A.J., et al. 1996 Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nature Medicine 2(7):788–794.
Bruce-Keller, A.J., Y.-J. Li, M.A. Lovell, P.J. Kraemer, D.S. Gary, R.R. Brown, W.R. Markesbery, and M.P. Mattson 1998 4-hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. Journal of Neuropathology and Experimental Neurology 57(3):257–267.
Cellerino, A., et al. 1996 The action of neurotrophins in the development and plasticity of the visual cortex. Progress in Neurobiology 49(1):53–71. [Published erratum appears in Progress in Neurobiology 50(2–3):333].
Copani, A., J.-Y. Koh, and C.W. Cotman 1991 ß-amyloid increases neuronal susceptibility to injury by glucose deprivation. NeuroReport 2(12):763–765. $
Cotman, C.W., and A.J. Anderson 1995 A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Molecular Neurobiology 10(1):19–45.
Cotman, C.W., K.J. Ivins, and A.J. Anderson 1999 Apoptosis in Alzheimer disease. Chapter 23 in Alzheimer Disease , 2nd ed., R.D. Terry, R. Katzman, K.L. Bick, and S.S. Sisodia, eds. Philadelphia: Lippincott, Williams & Wilkins.
Cotman, C.W., and J.H. Su 1996 Mechanisms of neuronal death in Alzheimer's disease. Brain Pathology 6:493–506.
Deng, G., J. Su, and C.W. Cotman 1996 Gene expression of Alzheimer's associated presenilin-2 in the frontal cortex of Alzheimer and aged control brain. FEBS Letters 3994:17–20.
Dodd, P.R., H.L. Scott, and R.I. Westphalen 1994 Excitotoxic mechanisms in the pathogenesis of dementia. Neurochemistry International 25(3):203–219.
Doman, W.A., et al. 1993 Bilateral injections of ßA(25–35)+IBO into the hippocampus disrupts acquisition of spatial learning in the rat. NeuroReport 5:165–168.
Dudek, H., et al. 1997 Regulation of neuronal survival by the serine-threonine protein kinase akt. Science 275(5300):661–665.
Dyrks, T., E. Dyrks, T. Hartmann, C. Masters, and K. Beyreuther 1992 Amyloidogenicity of beta A4 and beta A4-bearing amyloid protein precursor fragments by metal-catalyzed oxidation. Journal of Biological Chemistry 267(25):18210–18217.
Feuerstein, G.Z., X. Wang, and F.C. Barone 1998 The role of cytokines in the neuropathology of stroke and neurotraurna. Neuroirnmunomodulation 5(3–4):143–159.
Fraser, P.E., L. Levesque, and D.R. McLachlan 1994 Alzheimer AB amyloid forms an inhibitory neuronal substrate. Journal of Neurochemistry 62(3):1227–1230.
Frederikse, P.H., D. Garland, J.S. Zigler, and J. Piatigorsky 1996 Oxidative stress increases production of β-amyloid precursor protein and b-amyloid (Aβ) in mammalian lenses, and Aβ has toxic effects on lens epithelial cells. Journal of Biological Chemistry 271(17):10169–10174.
Fryer, M.J. 1998 Vitamin E status and neurodegenerative disease. Nutritional Neuroscience 1:327–351.
Gabbita, S.P., M.A. Lovell, and W.R. Markesbery 1998 Increased nuclear DNA oxidation in the brain in Alzheimer's disease. Journal of Neurochemistry 71:2034–2040.
Gabuzda, D., J. Busciglio, L.B. Chen, P. Matsudaira, and B.A. Yankner 1994 Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. Journal of Biological Chemistry 269(18):13623–13628.
Galuske, R.A., et al. 1996 Brain-derived neurotrophic factor reversed experience-dependent synaptic modifications in kitten visual cortex. European Journal of Neuroscience 8(7): 1554–1559.
Gasparini, L., M. Racchi, L. Benussi, D. Curti, G. Binetti, A. Bianchetti, M. Trabucchi, and S. Govoni 1997 Effect energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neuroscience Letters 231:113–117.
Gervais, F.G., et al. 1999 Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic a beta peptide formation. Cell 97(3):395–406.
Glazewski, S., et al. 1999 Impaired experience-dependent plasticity in barrel cortex of mice lacking the alpha and delta isoforms of CREB. Cerebral Cortex 9(3):249–256.
Goto, I., et al. 1993 Positron emission tomographic (PET) studies in dementia. Journal of the Neurological Sciences 114(1):1–6.
Grealy, M.A., et al. 1999 Improving cognitive function after brain injury: The use of exercise and virtual reality. Archives of Physical Medicine and Rehabilitation 80(6):661–667.
Griffin, W.S., et al. 1989 Brain interieukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America 86:7611–7615.
Haxby, J.V., and S.I. Rapoport 1986 Abnormalities of regional brain metabolism in Alzheimer's disease and their relation to functional impairment. Progress in Neuro-Psychopharmacology and Biological Psychiatry 10(3–5):427–438.
Herndon, F.J., H.C. Hsu, and J.D. Mountz 1997 Increased apoptosis of CD45RO-T cells with aging. Mechanisms of Ageing and Development 94(1–3):123–134.
Hochman, A., et al. 1998 Enhanced oxidative stress and altered antioxidants in brains of bcl-2-deficient mice. Journal of Neurochemistry 71(2):741–748.
Hoyer, S., K. Oesterreich, and O. Wagner 1988 Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type. Journal of Neurology 235:143–148.
Johnson, J.K., R. McCleary, M.H. Oshita, and C.W. Cotman 1998 Initiation and propagation stages of beta-amyloid are associated with distinctive apolipoprotein e, age, and gender profiles. Brain Research 798(1–2):18–24.
Joseph, J.A., B. Shukitt-Hale, N.A. Denisova, D. Bielinski, A. Martin, J.J. McEwen, and P.C. Bickford 1999 Reversals of age-related declines in neuronal signal transduction, cognitive and motor behavioral deficits with blueberry, spinach, or strawberry dietary supplementation. Journal of Neuroscience 19(18):8114–8121.
Kang, S.M., et al. 1992 Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science 257(5073):1134–1138.
Kempermann, G., H.G. Kuhn, and F.H. Gage 1997 More hippocampal neurons in adult mice living in an enriched environment. Nature 386:493–495.
1998 Experience-induced neurogenesis in the senescent dentate gyrus. Journal of Neuroscience 18(9):3206–3212.
Kim, T.W., et al. 1997 Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science 277(5324):373–376.
Koh, J.Y., L.L. Yang, and C.W. Cotman 1990 ß-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Research 533(2):315–320.
Korotzer, A.R., et al. 1993 Beta-amyloid peptides induce degeneration of cultured rat microglia. Brain Research 624(1–2):121–125.
LeBlanc, A., H. Liu, C. Goodyer, C. Bergeron, and J. Hammond 1999 Caspase-6 role in apoptosis of human neurons, amyloidgenesis, and Alzheimer's disease. Journal of Biological Chemistry 274 (33):23426–23436.
Lee, C.K., R.G. Klopp, R. Weindruch, and T.A. Prolla 1999 Gene expression profile of aging and its retardation by caloric restriction. Science 285:1390–1393.
Lenardo, M.J. 1991 Interteukin-2 programs mouse alpha beta t lymphocytes for apoptosis. Nature 353(6347):858–861.
Loddick, S.A., et al. 1998 Displacement of insulin-like growth factors from their binding proteins as a potential treatment for stroke . Proceedings of the National Academy of Sciences of the United States of America 95(4):1894–1898.
Longoni, B., E. Boschi, G.C. Demontis, P.L. Marchiafava, and F. Mosca 1999 Regulation of bcl-2 protein expression during oxidative stress in neuronal and endothelial cells. Biochemical and Biophysical Research Communications 260:522–526.
Loo, D.T., M.C. Althoen, and C.W. Cotman 1995 Differentiation of serum-free mouse embryo cells into astrocytes is accompanied by induction of glutamine synthetase activity. Journal of Neuroscience Research 42:184–191.
Loo, D., A. Copani, C.I. Pike, E. Whittemore, A.J. Walencewicz, and C.W. Cotman 1993 Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proceedings of the National Academy of Sciences of the United States of America 90(17):7951–7955.
Lovell, M.A., W.D. Ehmann, M.P. Mattson, and W.R. Markesbery 1997 Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiology of Aging 18(5):457–461.
Lovell, M.A., S.P. Gabbita, and W.R. Markesbery 1999 Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF . Journal of Neurochemistry 72:771–776.
Lucassen, P.J., W.C. Chung, W. Kamphorst, and D.F. Swaab 1997 DNA damage distribution in the human brain as shown by in situ end labeling; Area-specific differences in aging and Alzheimer disease in the absence of apoptotic morphology. Journal of Neuropathology and Experimental Neurology 56:887–900.
Lyras, L., N.J. Cairns, A. Jenner, P. Jenner, and B. Halliwell 1997 An assessment of oxidative damage to proteins, lipids and DNA in brain from patients with Alzheimer's disease. Journal of Neurochemistry 68:2061–2069.
Ma, Y.L., et al. 1998 Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience 82(4):957–967.
Mark, R.J., Z. Pang, J.W. Geddes, K. Uchida, and M.P. Mattson 1997 Amyloid β peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. Journal of Neuroscience 17(3):1046–1054.
Markesbery, W.R., and M.A. Lovell 1998 Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiology of Aging 19(1):33–36.
Maroto, R., and J.R. Perez-Polo 1997 BCL-2-related protein expression in apoptosis: Oxidative stress versus serum deprivation in PC12 cells. Journal of Neurochemistry 69(2):514–523.
Martin, D., et al. 1997 Role of IL-1 in neurodegeneration: Preclinical findings with IL-Ira and ICE inhibitors. P. 392 in Neuroinflammation: Mechanisms and Management, P.L. Wood, ed. Totowa, NJ: Humana Press Inc.
Masliah, E., et al. 1996 Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Annals of Neurology 40(5):759–766.
Mattson, M.P., et al. 1992 B-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. Journal of Neuroscience 12(2):376–389.
1998 Presenilins, the endoplasmic reticulum, and neuronal apoptosis in Alzheimer's disease. Journal of Neurochemistry 70(1):1–14.
McDonald, D.R., K.R. Brunden, and G.E. Landreth 1997 Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. Journal of Neuroscience 17(7):2284–2294.
McGeer, P.L., et al. 1986 Positron emission tomography in patients with clinically diagnosed Alzheimer's disease. Canadian Medical Association Journal 134(6):597–607.
Mecocci, P., U. MacGarvey, et al. 1994 Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Annals of Neurology 36(5):747–751.
Meydani, S., D. Wu, M. Santos, and M. Hayek 1995 Antioxidants and immune response in aged persons: Overview of present evidence. American Journal of Clinical Nutrition 62(Supplement): 1462S–1476S.
Miller, R.A. 1996 The aging immune system: Primer and prospectus. Science 273(5271):70–74.
Multhaup, G., T. Ruppert, A. Schlicksupp, L. Hesse, D. Beher, C.L. Masters, and K. Beyreuther 1997 Reactive oxygen species and Alzheimer's disease. Biochemical Pharmacology 54:533–539.
Nagel, J.E., et al. 1988 Decreased proliferation, interleukin 2 synthesis, and interleukin 2 receptor expression are accompanied by decreased mRNA expression in phytohemagglutinin-stimulated cells from elderly donors. Journal of Clinical Investigation 81(4):1096–1102.
Neeper, S.A., F. Gomez-Pinilla, J. Choi, and C.W. Cotman 1995 Exercise and brain neurotrophins. Nature 373:109.
1996 Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Research 726:49–56.
Nishimoto, I., et al. 1997 Apoptosis in neurodegenerative diseases. Advances in Pharmacology 41(5266):337–368.
Oliff, H.S., N.C. Berchtold, P. Isackson, and C.W. Cotman 1998 Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Molecular Brain Research 61:147–153.
Oltvai, Z.N., C.L. Milliman, and S.J. Korsmeyer 1993 Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell 74(4):609–619.
Palmer, A.M., and M.A. Burns 1994 Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer's disease. Brain Research 645(1–2):338–342.
Paradis, E., H. Douillard, M. Koutroumanis, C. Goodyer, and A. LeBlanc 1996 Amyloid beta peptide of Alzheimer's disease downregulates bcl-2 and upregulates bax expression in human neurons . Journal of Neuroscience 16(23):7533–7539.
Paz, K., et al. 1997 A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. Journal of Biological Chemistry 272(47):29911–29918.
Peraldi, P., et al. 1996 Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. Journal of Biological Chemistry 271(22):13018–13022.
Pereira, C., M.S. Santos, and C. Oliveira 1999 Involvement of oxidative stress on the impairment of energy metabolism induced by Aβ peptides on PC12 cells: Protection by antioxidants. Neurobiology of Disease 6:209–219.
Perrig, W.J., P. Perrig, and H.B. Stahelin 1997 The relation between antioxidants and memory performance in the old and very old. Journal of the American Geriatrics Society 45:718–724.
Phelouzat, M.A., et al. 1996 Excessive apoptosis of mature T lymphocytes is a characteristic feature of human immune senescence. Mechanisms of Ageing and Development 88(1–2):25–38.
Pike, C.J., et al. 1994 Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer's disease. Neuroscience 63(2):517–531.
Pike, C.J., B.J. Cummings, and C.W. Cotman 1992 ß-amyloid induces neuritic dystrophy in vitro: similarities with Alzheimer pathology. Neuroreport 3:769–772.
Pike, C.J., N. RamezanArab, and C.W. Cotman 1997 Beta-amyloid neurotoxicity in vitro: Evidence of oxidative stress but not protection by antioxidants. Journal of Neurochemistry 69(4):1601–1611.
Praticáo, D., M.Y. Lee, J.Q. Trojanowski, J. Rokach, and G.A. Fitzgerald 1998 Increased F2-isoprostanes in Alzheimer's disease: evidence for enhanced lipidperoxidation in vivo. FASEB Journal 12(15):1777–1783.
Proust, J.J., et al. 1987 Age-related defect in signal transduction during lectin activation of murine T lymphocytes. Journal of Immunology 139(5):1472–1478.
Radvanyi, L.G., G.B. Mills, and R.G. Miller 1993 Relegation of the T cell receptor after primary activation of mature T cells inhibits proliferation and induces apoptotic cell death. Journal of Immunology 150(12):5704–5715.
Reed, J.C. 1994 Bcl-2 and the regulation of programmed cell death. Journal of Cell Biology 124(1):1–6.
Reidel, W.J., and B.L. Jorissen 1998 Nutrients, age and cognitive function. Current Opinion in Clinical Nutrition and Metabolic Care 1:579–585.
Rothwell, N.J., and S.J. Hopkins 1995 Cytokines and the nervous system II: Actions and mechanisms of action. Trends in Neurosciences 18(3):130–136.
Russell, J.W., et al. 1998 Insulin-like growth factor-I prevents apoptosis in neurons after nerve growth factor withdrawal. Journal of Neurobiology 36(4):455–467.
Sano, M., C. Ernesto, R.G. Thomas, M.R. Klauber, K. Schafer, M. Grundman, P. Woodbury, J. Growdon, C.W. Cotman, E. Pfeiffer, L.S. Schneider, and L.J. Thal 1997 A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The New England Journal of Medicine 336:1216–1222.
Schenk, D., R. Barbour, W. Dunn, G. Gordon, H. Grajeda, T. Guido, K. Hu, J. Huang, K. Johnson-Wood, K. Khan, D. Kholodenko, M. Lee, Z. Liao, 1. Lieberburg, R. Motter, L. Mutter, F. Soriano, G. Shopp, N. Vasquez, C. Vandevert, S. Walker, M. Wogulis, T. Yednock, D. Games, and P. Seubert 1999 Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177.
Schultz, S., et al. 1999 Direct evidence for biphasic cAMP responsive element-binding protein phosphorylation during long-term potentiation in the rat dentate gyrus in vivo. Journal of Neuroscience 19(13):5683–5692.
Segal, M., et al. 1998 CREB activation mediates plasticity in cultured hippocampal neurons. Neural Plasticity 6(3):1–7.
Shen, H., L. Tong, and C.W. Cotman In press Effects of exercise on CREB phosphorylation and CRE DNA-binding activity in the rat hippocampus.
Shin, S.W., et al. 1998 Persistent expression of fas/fasL mRNA in the mouse hippocampus after a single NMDA injection. Journal of Neurochemistry 71(4):1773–1776.
Simpson, I.A., et al. 1994 Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer's disease. Annals of Neurology 35(5):546–551.
Socci, D.J.C., B.M. Crandall, and G.W. Arendash 1995 Chronic antioxidant treatment improves the cognitive performance of aged rats. Brain Research 693(1–2):88–94.
Spanaus, K.S., R. Schlapbach, and A. Fontana 1998 TNF-alpha and IFN-gamma render microglia sensitive to fas ligand-induced apoptosis by induction of fas expression and down-regulation of bcl-2 and bcl-xL. European Journal of Immunology 28(12):4398–4408.
Stadelmann, C., W. Bruck, C. Bancher, K. Jellinger, and H. Lassmann 1998 Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. Journal of Neuropathology and Experimental Neurology 57:456–464.
Stadtmann, E.R. 1992 Protein oxidation and aging. Science 257:1220–1224.
Su, J.H., A.J. Anderson, B.J. Cummings, and C.W. Cotman 1994 Immunohistochemical evidence for apoptosis in Alzheimer's disease. NeuroReport 5:2529–2533.
Su, J.H., G. Deng, and C.W. Cotman 1997 Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer's disease brain . Brain Research 774:193–199.
Thoman, M.L., and W.O. Weigle 1989 The cellular and subcellular bases of immunosenescence. Advances in Immunology 46(3):221–261.
Tong, L., P.L. Thornton, and C.W. Cotman In press β-amyloid (1–42) impairs neuronal activity-dependent CREB signaling.
Torp, R., J.H. Su, G. Deng, and C.W. Cotman 1998 GADD45 is induced in Alzheimer's disease, and protects against apoptosis in vitro. Neurobiology of Disease 5:245–252.
Venters, H.D., et al. 1999 A new mechanism of neurodegeneration: A proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proceedings of the National Academy of Sciences of the United States of America 96(17):9879–9884.
Vogt, M., et al. 1998 Oxidative stress and hypoxia/reoxygenation trigger CD95 (APO-1/fas) ligand expression in microglial cells. FEBS Letters 429(1):67–72.
Ware, C.F., et al. 1991 Tumor necrosis factor (TNF) receptor expression in T lymphocytes. Differential regulation of the type I TNF receptor during activation of resting and effector T cells. Journal of Immunology 147(12):4229–4238.
Watt, J.A., C.j. Pike, A.j. Walencewicz, and C.W. Cotman 1994 Ultrastructural analysis of beta-amyloid-induced apoptosis in cultured hippocampal neurons. Brain Research 661:147–156.
Whittemore, E.R., D.T. Loo, and C.W. Cotman 1994 Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. NeuroReport 5(13):1585–1588.
Williams, P., et al. 1997 Effects of group exercise on cognitive functioning and mood in older women. Australia and New Zealand Journal of Public Health 21(1):45–52.
Wolozin, B., et al. 1996 Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science 274(5293):1710–1713.
Yamamoto-Sasaki, M., et al. 1999 Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Research 824(2):300–303.
Yamatsuji, T., et al. 1996 G protein-mediated neuronal DNA fragmentation induced by familial Alzheimer's disease-associated mutants of APP. Science 272(5266):1349–1352.
Young, D., et al. 1999 Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nature Medicine 5(4):448–453.






























