
5
Human Genetics and the Human Genome Project
Genes are the fundamental units of heredity, and the genome is the organism’s ensemble of genes. The genotype is the individual organism’s unique set of all the genes. In a complex manner, the genotype governs the phenotype, which is the ensemble of all traits of the organism’s appearance, function, and behavior. Genes are now known to be deoxyribonucleic acid (DNA) sequences from which ribonucleic acid (RNA) is transcribed. The transcripts of most, but not all, genes are then translated into proteins, which are composed of amino acid sequences and which perform most of the cell’s functions by virtue of their catalytic activity and the interactions occurring at their specific binding sites. Hence, the gene is required for a phenotypic trait, because it encodes a protein involved in the generation of the trait.
It is not known precisely how many genes the human genome contains, but estimates range from 61,000 to 140,000 (Dickson 1999; Dunham et al. 1999). By comparison, the nematode Caenorhabditis elegans has 19,000 genes. In humans, only 5% of the DNA of the genome actually encodes proteins. The rest serves either as regulatory sequences that specify the conditions under which a gene will be transcribed, as introns (sequences that are transcribed but not translated), or as spacer DNA of yet unknown function. Each gene is located at a particular site on a chromosome, and in the diploid phase of the life cycle of humans and other metazoa, there are two chromosomes with that gene site in each nucleus. These two gene copies are called alleles. Particularly relevant to this report, many variant alleles of each gene have arisen during human evolution, and different alleles often confer slight or great differences in some particular trait of the organism, when members of a population are compared.
In this chapter, the committee describes the fields of human genetics and genomics. The role of molecular epidemiology in detecting developmental toxicants is discussed, as well as the difficulties in the detection of complex genotype-environment interactions.
GENOTYPE, PHENOTYPE, AND MULTIFACTORIAL INHERITANCE
In his classic experiments of the mid-nineteenth century, Gregor Mendel (1865) chose the pea plant (Pisum sativum) in which to study the segregation and assortment of particulate determinants of phenotypic traits. He was fortunate to choose several traits, each of which was controlled by a single genetic locus. The alleles at each locus, when inherited, acted in either a dominant or recessive manner, and their action was not significantly influenced by other genes or by environmental factors under his conditions of testing. Consequently, he observed precise and interpretable mathematical ratios for the phenotypes of the progeny in each breeding experiment. Traits of phenotype that show such easily interpretable patterns of inheritance are called simple, or Mendelian, traits, and these generally are governed by a single genetic locus.
However, the relationship between genotype and phenotype is almost always very complex. Even when scientists consider one particular gene and know its particular allelic form, its effect on phenotype is often subject to either or both of two variables: (1) the different alleles of various other major and modifier genes in the organism’s genome, and (2) various environmental conditions. Such traits display a multifactoral pattern of inheritance (also called complex or non-Mendelian inheritance) and are termed complex traits or multiplex phenotypes (for a recent review, see Lander and Schork 1994). Multifactorial inheritance is much more common than simple inheritance. Such traits entail the interaction of two or more genes (a polygenic trait). The genes can contribute to the phenotypic trait in a quantitative and additive manner (e.g., genes A, B, and C might contribute 20%, 30%, and 50%, respectively, to a trait such as birth weight). These genes are called “quantitative trait loci,” and genetic methods for analyzing their contributions are powerful. Alleles of the BRCA1 gene, for example, appear to contribute about 5% to the overall risk of breast cancer (for a review, see Brody and Biesecker 1998), but several other contributors, which are analytically believed to exist, have not yet been identified. Still more complex patterns of inheritance can be traced to multiple genes acting in nonadditive manners. Segregated alleles might be neither dominant nor recessive. Finally, a gene might show incomplete penetrance (only some members of the population show the trait) or variable expressivity (members of the population vary in the extent of the trait) or both.
Other traits are modifiable by the environment. Such traits are not at all unusual and might overlap with polygenic traits. Studies in model organisms, such as the fruit fly Drosophila melanogaster, have long shown that a gene’s effect on a trait can be modified by such extrinsic factors as temperature, chemi-
cals, nutrition, and crowding. Geneticists who are particularly interested in evolution have argued that gene-environment interactions are so pervasive and important that one should not speak of a “phenotypic trait” of the organism but of its “norm of reaction,” which is a set of phenotypes produced by an individual genotype when it is exposed to different environmental conditions (Stearnes 1989). The relationship between genotype, environment, and phenotype, which is sometimes called the gene-environment interaction, can be expressed as
Genotype + Environment → Phenotype.
Although multifactorial inheritance is a nuisance to geneticists, it describes most human heritable diseases and virtually all susceptibilities. As mentioned in Chapter 2, approximately 25% of human developmental defects possibly follow multifactorial inheritance. Humans and experimental animals are notoriously heterogeneous in their responses to drugs or environmental pollutants. The favored explanation at present is that the heterogeneity reflects a combination of the heterogeneous exposure circumstances (extrinisic conditions) and heterogeneous genotypes for susceptibility (intrinsic conditions). Examples of exposure plus susceptibility would be the age of onset of lung cancer in cigarette smokers or the likelihood of asthma induced by urban pollution. The gene-environment relationship is further confounded in developmental toxicology by the need to consider the genotype of both the mother and the embryo or fetus, how and where a toxicant is metabolized, and the developmental stage at which a toxicant crosses the placenta. Gene-environment interactions are obviously relevant to the fields of molecular epidemiology and developmental toxicology.
POLYMORPHISMS
A polymorphism denotes the presence of two or more alleles of a particular gene within a population of organisms; the minority allele is present at a gene frequency of at least 1% (Hartl and Taubes 1998). That frequency is a somewhat arbitrary cutoff set by population geneticists and minority alleles of still lower frequency are called “rare alleles.” In keeping with the Hardy-Weinberg distribution (p2 + 2pq + q2) for two alleles at a single locus, if the minority allele is present at a 1% gene frequency, it is then present in heterozygous form in about 2% of the members of that population and in homozygous form in 0.01% of the population.
Whatever the frequency, alleles are now defined in the most general way, namely, as different nucleotide sequences of the same gene—that is, as changes of one or more bases (adenine, thymine, cytosine, and guanine) relative to the reference DNA base sequence. However, finding such a difference does not in itself reveal much about an effect on phenotype. If the sequence difference oc-
curs in a coding region of the gene, protein activity or stability might be affected. If the change is synonymous (i.e., the amino acid is not altered), conservative, or located in a region of the protein where any of several amino acids is acceptable, protein activity or stability might not be affected. If a DNA sequence change occurs in the transcribed region of the gene but not in the coding region, it might affect the reading frame, splicing, mRNA stability, translation efficiency, or transcriptional regulation. If outside the transcription region of the gene, the change still might affect the time, place, and level of expression of the gene, although not the protein’s sequence. Additional work has to be done to identify the effect of the particular DNA change on protein function or level.
Polymorphisms reaching the 1% allelic frequency level are generally expected to have a selectively advantageous phenotypic consequence of altered protein activity or amount. However, a variant might be represented below the 1% level or close to 1%, because it arose in a small founder group of organisms that, because of local fortuitous circumstances, proliferated to a large population relative to other members of that species. Such a polymorphism might have no effect on protein activity or amount. It would just be a marker of that lineage of organisms. It might even have a negative selective effect.
Modern sequencing methods have greatly increased the capacity of researchers to detect alleles. For a particular gene sequence, any two unrelated people within a population are likely to have sequence differences. A gene sequence is taken to include all regulatory and transcribed regions of the DNA. When a base change first arises, due to oxidative hits, replication errors, ultraviolet-induced thymine dimers or other forms of DNA damage, one round of new DNA synthesis is usually required to become “fixed” in a double stranded form that is immune to repair and to count as a mutation. Before this synthesis, the base change often results in a mismatch in the DNA double helix, and a number of mismatch repair enzymes remove such errors (Snow 1997). However, out of every million or more DNA sites that become damaged, an error occasionally escapes uncorrected. Unrepaired mutations are thought to occur naturally at frequencies of once per 106-108 bases per generation. Because humans have such a large genome, roughly 75 new mutations accumulate per human individual per lifetime. Most of these are probably not deleterious. Many do not occur in protein coding regions (5% of the human DNA sequence) or, if they do, do not change the particular amino acid (synonymous substitutions). Some are deleterious, however, and the deleterious mutation rate in humans (nonsynonymous amino acid changes affecting activity) has recently been estimated to be at least 1.6 new deleterious mutations per diploid genome per generation. The authors conclude that this rate “is close to the upper rate tolerable for a species such as humans that has a low reproductive rate” (Eyre-Walker and Keightley 1999). It is likely that the human population is full of genetic variation, and this variation must be considered and appraised in any evaluation of an individual’s susceptibility to developmental toxicants.
THE HUMAN GENOME PROJECT
The genome of an organism is the total genetic content of the organism, or more broadly, it is the organism’s entire DNA content—including nontranscribed, non-cis-regulatory regions of DNA such as centromeres and telomeres. The study of the genomes of organisms, which is called genomics, includes areas of research determining the genetic and physical maps of genomes, the DNA sequences of genomes, the functions of genes and proteins, the cis-regulatory elements of genes, and the time, place, and conditions of expression of genes. A prominent part of genomics has become the managing of the massive amount of gathered information (a field referred to as bioinformatics) and the analysis of data with regard to, for example, aspects of the organization of the genome, the comparison of genomes of different organisms, and the global patterns of expression of genes.
The Human Genome Project (HGP) was launched in October 1990 by the National Institutes of Health (NIH) as a federally funded initiative. The immediate goal was then, as it is now, to complete the accurate sequencing of the approximately 3.5 billion human DNA base pairs (the haploid amount) by the end of 2003 (F.S. Collins et al. 1998). A “rough draft sequence”, comprising approximately 90% of the human genome, was completed in mid-2000 (www.ornl.gov/hgmis/project/progress.html). In the longer term, a goal is to identify all human genes. Identification is difficult. In an organism such as yeast, which is favorable for the identification of genes by mutational genetic analysis, more than half the genes had gone undetected until the genome sequence became available (Brown and Botstein 1999). The lack of detection was in part due to large redundant regions of the yeast genome. In vertebrates, mutational genetic analysis is much more difficult, and redundancy might be more widespread. Therefore, initial gene identification by sequencing is the approach of choice. A gene is initially identified as an open reading frame (ORF), which can be discerned directly by looking at the sequence, or it is initially identified as an expressed sequence tag (EST) site, a sequence complementary to a known piece of transcribed RNA (see below). Thereafter, the goal is to identify each gene as a sequence encoding a full-length RNA and a protein of known function. The functions of nontranscribed regions, such as the numerous large cis-regulatory regions setting conditions for gene expression, will have to be elucidated as well. This task is still more difficult, currently involving a number of approaches, including the construction of transgenic animal lines carrying portions of the regulatory region in conjunction with a reporter gene (e.g., green fluorescent protein, GFP).
The functional analysis of the genome, in terms of the time and place of expression of genes and the functions of the gene products, is sometimes called “functional genomics” or even “post-genomics.” The analysis of a protein’s function might be simple if the protein sequence resembles that of another well-understood protein and might be difficult if sequence motifs are absent. The analysis of
the organism’s protein composition (called the “proteome”) and function is sometimes called “proteomics.” Targeted areas of the HGP currently include genetic and physical mapping of the human genome, DNA sequencing, analysis of the genomes of numerous important nonhuman organisms, informatics to handle the tremendous increase in the rate of information generated, resource and technology development, and the ethical, legal, and social implications (ELSI) of genetic research for individuals and for society (F.S. Collins et al. 1998).
The HGP is supported by NIH and U.S. Department of Energy (DOE) at 22 specialized genome research centers in the United States and in many university, national, and private-sector laboratories. At NIH, the name was changed to the National Center for Human Genome Research in 1993 and, since late 1996, has become the National Human Genome Research Institute (NHGRI). At least 14 other countries also have programs for analyzing the genomes of various organisms—ranging from microbes and economically important plants and animals to humans.
The explosion of genomics information has occurred sooner than the most daring scientists would have predicted. Following the first complete genome sequence, that of Haemophilus influenzae in 1995, seven more genomes were completed in the next 18 months, namely, four more eubacterial genomes, two archaebacterial genomes, and one unicellular eukaryote genome—that of the yeast Saccharomyces cerevisiae. In December 1998, the genome of the first multicellular eukaryote, Caenorhabditis elegans, was completed (100 kilobases of DNA sequence and 19,000 genes identified at least as ORFs). As of the end of 1999, more than 30,000 human genes had been partially identified, located, and sequenced. Human chromosome 22 has been sequenced and is projected to contain at least 679 genes (Dunham et al. 1999). In the mouse, at least 14,000 genes have been described. The fruit fly (Drosophila melanogaster) sequence was completed in 1999 (Adams et al. 2000). In mid-2000, an approximate (“working draft”) human sequence was completed. By 2003, numerous nonhuman genomes will be sequenced as well, including the mouse Mus musculus, the zebrafish Danio rerio, the silkworm Bombyx mori, the rat, dog, cat, chicken, rice, corn, wheat, barley, cotton, the plant Arabidopsis thaliana, and probably also the cow, sheep, pig, and horse. The sequencing of the mouse genome is running well ahead of schedule.
New technologies, resources, and applications have become increasingly available to researchers of many diverse scientific fields, including cancer research, drug discovery, medical genetics, and environmental genetics, and their availability should also accelerate numerous major advances in developmental toxicology in the next decade, as discussed later in this chapter.
Functional Genomics and Microarray Technology
From the outset, it was expected that the completion of sequencing of the human genome would mark but a first step in the HGP. The information about
the sequence and location of genes in the genome will greatly facilitate further studies—not only of human genetic variability but also of functional genomics. As noted above, the latter is the comprehensive analysis of gene expression and gene-product function. In the cases of yeast and C. elegans for which the entire genome sequence is already known, projects are under way to assess systematically the function of every gene product, for example, by knocking out yeast genes (causing a loss of function) one at a time and by associating an identified messenger (m) RNA with every ORF.
Some of this functional analysis can go forward even before a genome is sequenced. In the case of humans, the study of ESTs has been an important step of such analysis. mRNAs can be isolated from the organism and converted to complementary (c) DNAs, by reverse transcription (RT) and the polymerase chain reaction (PCR). The cDNAs are then cloned and sequenced to prepare a large and well-defined library of ESTs. The information is entered in a database. These sequences represent genes expressed in the human. For example, more than 1 million human ESTs are now available, representing greater than 50,000 genes. Each EST reflects an mRNA piece, not a full-length sequence. The most comprehensive libraries are prepared from a wide range of tissues and times of development in an effort to include all expressed genes. (Unfortunately for developmental toxicologists, although the initial sources of RNA included placenta, they were underrepresented in the variety of early embryonic tissue.) These sequences are useful in the course of genome sequencing to identify DNA regions that actually encode proteins (only 5% of the human genome sequence is thought to show up in processed mRNA sequences). New methods have become available to obtain full-length cDNAs from transcripts, and these will be more useful than fragments.
A further step of analysis of genome function is the determination of the time, place, and conditions of expression of each gene. Until recently, this analysis has been done one gene at a time. DNA microarray techniques recently have made possible the description of simultaneous changes of thousands of genes as cells and tissues undergo development or various changes of environmental conditions. In the study of toxicant effects on the organism, the analysis sometimes is called “toxicogenomics” or, in the study of the effects of pharmaceuticals, “pharmacogenomics.” DNA microarray approaches are gaining widespread use (see Nuwaysir et al. 1999 for a discussion of its use in toxicology).
The technology is now suitable for simultaneously comparing the amounts of thousands of kinds of mRNA in two tissues or cell samples (e.g., a normal control tissue versus a tissue treated with a teratogenic agent). To do the comparison, thousands of different DNA sequences (e.g., each an oligomer of at least 25 nucleotides) are robotically spotted onto a microslide, and each sequence is placed on a known spot to make a DNA microarray. The DNA adheres to the glass, and each DNA spot is typically 20 micrometers (m) in diameter. For example, microarrays of 6,200 cDNA sequences, representing all the genes of yeast, have been fitted on a single slide or a few slides, and 8,900 cDNA sequences
representing about 10% of human genes (mostly EST sequences) have been put on a few slides (Iyer et al. 1999). The expense and time of producing such slides are modest enough that a hundred or so can be prepared, each serving for the analysis of one comparison condition (e.g., analysis of several time points and concentration conditions). Each slide is an array of DNA “probes” by which the amounts of thousands of kinds of mRNA in cells, tissues, or embryos can be visualized simultaneously at each time and condition.
In the procedure of Brown and Botstein (1999), the tissue samples for comparison are separately extracted and the mRNAs are labeled with different fluorescent dye molecules, say green for the control and red for the treated tissue (see Figure 5-1). The RNAs are then mixed and added to the microarray slide (in their case, carrying 8,900 human DNA sequences) under conditions suitable for RNA-DNA hybridization, and then washed to remove unbound RNA. The slide is then read in a fluorescence microscope to see if each particular DNA spot has bound more of the green or rRNA. The ratio of red to green tells whether a particular gene is expressed more or less than normal under the treatment condition. Yellow is seen when equal red and green mRNA has hybridized. This technique has been applied recently to human cells cultured in the presence or absence of serum. Indeed, hundreds of genes changed expression, including many genes encoding stress-related proteins seen in wound healing (Iyer et al. 1998), a fact not previously realized in the years of study of the serum response of cultured cells. The technique also has been applied to yeast cells progressing along the sporulation pathway (Chu et al. 1998), yeast cells progressing through the cell cycle (Spellman et al. 1998), and yeast cells in a haploid, diploid, or tetraploid state (Galitski et al. 1999). Recently, it has been used to discover the response of a single kind of cell to two signaling ligands, each acting through a different receptor tyrosine kinase (Fambrough et al. 1999). In all cases, the expression of hundreds of genes changed. Viewing global patterns reveals that each gene does not seem to behave individually; instead, concerted expression of large batteries of genes seems to occur under various conditions. These results give credence to the value of analysis of the global patterns of gene expression. Previous studies of individual genes might have missed large-scale patterns. However, much remains to be done in the interpretation of the manifold changes of gene expression. As mentioned above, hundreds of gene expression changes are observed even with seemingly modest changes in a cell’s circumstances, such as its ploidy.
In the analysis of toxicant effects, it is expected that cells or organisms could be treated with toxicants of unknown mechanism of action, and the changes of gene expression could be profiled by the DNA microarray method. If enough were known about the function and interaction of proteins encoded by the genes undergoing changes of expression, sound deductions might be made about the mechanism of action of the toxicant. In the future, it is expected that DNA microarray methods will allow rapid and detailed characterization of a cell or organism’s response to a toxicant. As more information is collected, different
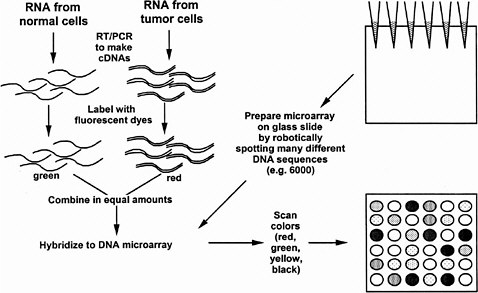
FIGURE 5-1 DNA microarrays as a means to determine simultaneously the amounts of thousands of kinds of mRNA in a cell or tissue. As shown in the upper left, mRNA is prepared separately from two kinds of cells, such as normal and tumorous, and each mRNA is converted to cDNA by reverse transcription and the polymerase chain reaction (RT-PCR) (see text). The separate cDNA samples are reacted with fluorescent dyes (e.g., green for the normal cDNA sample and red for the tumorous cDNA sample). The two are mixed in equal amounts. To prepare the DNA microarray, thousands of different DNA sequences are spotted robotically on a glass slide, each sequence in a known spot. The DNA adheres to the glass. The fluorescent cDNA mix is spread over the slide under hybridization conditions so that cDNAs stick to specific spots based on their complementary base sequence. Unbound cDNA is washed away and the slide is scanned in a fluorescence microscope to determine the green-to-red ratio of each spot. Pure red means that the RNA is present only in normal cells. Pure green means that the mRNA is present only in tumorous cells. Yellow means that the mRNA is present in equal amounts in both cells, and more red or green reflects more or less of the mRNA in the normal or tumorous cell. Black means that there is no mRNA in either kind of cell. Source: Adapted from the National Human Genome Research Institute’s Glossary of Genetic Terms. Available via the Internet at <http://www.nhgri.nih.gov/DIR/VIP/Glossary/Illustration/microarray_technology.htm>.
toxicants can be grouped by their similarities of effect, and the analysis of toxicant action can be pursued on a more systematic basis. The DNA microarray method is already in use to compare normal cells and cancer cells. Many of the questions of interpretation of gene expression differences are being explored in that case as well.
Although it is preferrable to have a large set of cloned and sequenced DNAs (representing different identified genes) for the microarray, as is the case for yeast,
it is not necessary. ESTs have been useful already for human and mouse studies, as in the serum study mentioned above (Iyer et al. 1999). If expression of a particular sequence, known only by its EST, is found to change greatly in the test condition, it might then qualify as interesting enough to deserve full-length cloning, sequencing, and further analysis of function.
Vast amounts of data accumulate in such comparisons (e.g., when 6,200 yeast genes (the entire yeast genome) or 8,900 human sequences are expressed differently under several conditions at several time points). The multidimensional data sets have challenged applied mathematicians to find means to express them in ways useful to biologists (e.g., see the methods of two-dimensional clustering analysis in Eisen et al. 1998; Alon et al. 1999; Tamayo et al. 1999). Yet, larger data sets loom on the horizon (e.g., the expression of perhaps 100,000 mouse or human genes at all times and places in development, not to mention with different toxicant exposures). As described below, the demand is great for managers and analysts of these data sets.
Although the various microarray techniques promise to reveal exciting new information about where, when, and under what conditions the genes of the genome are transcribed, this approach will not provide information concerning the translation and post-translational modification of proteins encoded by these mRNAs—that is, information about when and where the proteins are present and active. Protein function is almost always the immediate cause of cell function. To provide such functional information is the goal of proteomics. Proteomics has been defined (Anderson and Anderson 1998) as “the use of quantitative protein-level measurements of gene expression to characterize biological processes (e.g., disease processes and drug effects) and decipher the mechanisms of gene expression control.” At the core of proteomics is the Human Protein Index—that is, the systematic identification of all human proteins (Anderson and Anderson 1982) using high-throughput, high-resolution, two-dimensional (2D)-gel electrophoresis to generate a gel with as many as 1,000 separate protein spots on it. A large amount of 2D-gel information is stored in the Proteomics database (see Appendix B for the Internet address). Plans have been made to identify every protein spot on a 2D gel, because the nanogram amount of protein in a spot is sufficient to determine a partial amino acid sequence by tandem mass spectrometry (Yates 1998). The partial sequence can then be looked up in the genome database and the protein identified. When proteins are modified by phosphorylation, acylation, glycosylation, farnesylation, limited proteolysis, or any of the other 30 or so covalent post-translational alterations, their migration on a 2D gel changes, thus allowing a correlation to be made with their activity or inactivity in the tissue. Such activity information cannot be gained from DNA microarray measurements of mRNA amounts.
Finally, many proteins are required to associate with other proteins in order to achieve activity, and there are various nondenaturing gel-electrophoresis methods to detect such associations. Current and future efforts can be expected to
produce new technological advances for the analysis of proteins and their functions. In developmental toxicology, the combination of genomics and proteomics offers the possibility of assessing developmental toxicants not only for their capacity to alter gene expression but also for their capacity to alter protein function.
Applications of Genomic Technologies
Researchers have recently made use of the genomic technologies to identify and sequence genes with a role in disease etiology. It is probable that these genomic technologies will be applied to the study of the effects of chemicals on development in the near future. Two models already exist that demonstrate the application of new genomic technologies: the Cancer Genome Anatomy Project (CGAP), which began in 1997 and is administered by the National Cancer Institute, and the Environmental Genome Project (EGP), which began in 1998 and is administered by the National Institute of Environmental Health Sciences.
The goal of CGAP is to provide a complete catalogue of all genes whose expression changes in cancer cells relative to normal cells for all types of cancer (Pennisi 1997). In the past, a major barrier to the analysis of cancer cells has been the mixture of cell types (normal and cancerous) present in a typical tumor. It is difficult to get an accurate picture of gene expression in cancer cells if the RNA extracted from the whole tumor includes sequences from many different cell types. Using recently developed methods, CGAP researchers minimize that problem by microscopically selecting a small homogeneous group of cells, either normal, precancerous, or malignant (Emmert-Buck et al. 1996; Simone et al. 1998), which are then isolated by adhering them to a special laser-sensitive film. RNA sequences then are extracted from the isolated cells and amplified by RT-PCR to make cDNA libraries (Peterson et al. 1998). About 5,000 cells of homogeneous morphology have been required to obtain a wide mRNA representation. As few as 500 cells are needed to obtain cDNAs of abundant mRNA species. Such libraries will be important tools for describing the progression of cancer and providing diagnostic markers of the disease. They also provide sequences for functional analysis by way of DNA microarrays. As data are obtained, they are entered on the CGAP Web site for ready access and use by other investigators and for integration with other data in the large repository.
New technologies are expected to reduce the minimum number of cells needed to 1,000 or fewer and still get wide RNA representation. The lower the minimum number of cells required, the better for future developmental toxicological studies, because many developing cell types are present in small numbers during embryogenesis and fetal development.
The goal of EGP is to study the interaction between genetic susceptibility, environmental exposures, and disease. Specifically, researchers working on this 5-year project are attempting to identify allelic variants (i.e., polymorphisms) of 200 genes important in environmental diseases, to develop a centralized database
of polymorphisms for those genes, and to foster epidemiological studies of gene-environment interactions in disease etiology. As discussed later in this chapter, evidence is particularly strong for increased chemical susceptibilities of individuals with polymorphisms of genes encoding drug-metabolizing enzymes (DMEs). EGP is using genomic technologies, such as high-throughput sequence analysis developed for the HGP, which will facilitate epidemiological studies of gene-environment interactions in disease.
The type of genomic technologies used in CGAP and EGP, and many of the data themselves, are anticipated to have an enormous impact on future research in developmental toxicology. Ideally, a developmental toxicology counterpart to such programs as CGAP and EGP would focus on obtaining data on gene expression at all times and in all tissues during normal development. Comparisons then could be made between embryos and fetuses from pregnant control (normal) animals and pregnant treated animals to look for differences in gene expression. Changes could be identified in the expression of genes encoding proteins known to function in cell signaling, transcriptional regulation, cell division, cell motility, cell adhesion, apoptosis, differentiation, metabolism, repair, electrolyte balance, homeostasis, or transport. Such studies will help to elucidate mechanisms by which extrinsic chemicals (potential developmental toxicants) act as agonists or antagonists to receptor- and enzyme-mediated subcellular processes during embryogenesis and fetal development. Such research will be a major force in merging the fields of developmental biology, genomics, and developmental toxicology.
Management of Genome Sequence and Functional Genomics Data
The explosion of molecular biology in the past two decades has led to enormous advances in DNA sequencing, which in turn has led to the increasingly rapid identification of genes as ORFs and as EST sites and the identification of the function of gene products by sequence motifs (e.g., homeodomains, zinc-finger domains, kinase domains, and SH2 and SH3 domains). There are four major nucleotide sequence databases: GenBank and the Genome Sequence Database (GSDB) in the United States, the European Molecular Biology Library (EMBL), and the DNA Data Bank of Japan (DDBJ). All groups exchange new and updated sequences electronically and usually on the same day of submission.
There is a two-decade history of these databases. The goal of the Los Alamos Sequence Library in 1979 at the Department of Energy (DOE)-sponsored Los Alamos National Laboratory (LANL) was to store DNA sequence data in electronic form. Within the same year, a similar database was also established at the EMBL in Heidelberg. In 1982, it was agreed that any data submitted or entered by one group would be forwarded immediately to the other, thereby avoiding duplication of effort. In 1982, the LANL database became GenBank when Bolt, Beranek and Newman (BBN) became the primary contractor for distribution of
data-and-user support, and LANL was changed to a subcontractor of BBN. Sequence data activities at BBN and LANL were sponsored by the National Institute of General Medical Sciences (NIGMS), as well as DOE and other agencies. At the end of the first 5-year contract, IntelliGenetics became the primary contractor, and LANL again became the subcontractor in charge of designing and building the database. In October 1992, at the end of the second 5-year contract, NIGMS transferred GenBank to the National Center for Biotechnology Information (NCBI) at the National Library of Medicine. In August 1993, LANL and NCBI database resources became independent of one another. GenBank remained at NCBI, and LANL took the new name of Genome Sequence Database (GSDB) and moved to the National Center for Genome Resources (NCGR) in Santa Fe, New Mexico. The EMBL Nucleotide Sequence Database, which originated in 1982, is now maintained by the European Bioinformatics Institute (EBI), located near Cambridge, England—which also oversees the SWISS-PROT Protein Sequence Database and more than 30 other specialty databases. DDBJ, created in 1984 and sponsored by the Japanese Ministry of Education, Science, and Culture since 1986, accumulates nucleotide sequence data, mostly from Japanese scientists, and through electronic transfer makes more than a dozen other databases available.
Within a decade the genomes of at least 200 organisms, from numerous bacterial species to humans, will have been sequenced. By then, expression assays using high-throughput microarrays of DNA or cDNA or protein microarrays will be commonplace and provide us with overwhelming amounts of new information on the time and location (i.e., tissue and cell type) of expression of various genes and on the changes of gene expression in the organism’s development and response to different exposure conditions (Reichhardt 1999). There will be a tremendous need for departments, or divisions, of bioinformatics in universities and industries to keep track of the data and to analyze it with respect to interesting questions about genome organization and function (see commentary by Reichhardt 1999). Additional information is likely to arise from the comparison of genomes of different organisms. The need to train large numbers of people in the new field of bioinformatics will be great. Information readily available on the Internet should facilitate the integration of the fields of developmental toxicology, human genetics, genomics, and developmental biology. In the future, developmental toxicologists will certainly benefit in many ways from ready and immediate access to this new information, but it should be appreciated that training will be required before the vast amounts of information can be used effectively. It is also unspecified at present how best to organize the data so that those involved in risk assessment can obtain what is most relevant.
RECENT DEVELOPMENTS IN MOLECULAR EPIDEMIOLOGY
Molecular tools have been used recently to identify interactions between genetic and environmental factors in the causation of complex diseases such as
developmental defects. The majority of adverse pregnancy outcomes in humans are of unknown etiology and are viewed as complex traits in which exogenous agents might interact with particular combinations of allelic variants of genes controlling development and differentiation to produce adverse pregnancy outcomes. Studies of gene-environment interactions during embryogenesis and fetal development have become commonplace and increasingly appreciated. Categories of disease etiology can be viewed as spanning the range from totally genetic in causation to totally environmental in causation. These categories include single gene causation, chromosomal causation, multifactorial causation with high heritability, multifactorial causation with low heritability, infectious causes, and environmental causes (Khoury et al. 1993a).
Many single-gene disorders are characterized by a low frequency of the disease allele in the general population (allelic frequency, 1% or less) and high penetrance (a high proportion of individuals with the disease allele develop the disorder). Susceptibility genes for these single-gene disorders typically demonstrate Mendelian patterns of inheritance and are associated with high disease risk. Although individually rare, single-gene Mendelian disorders contribute significantly to infant morbidity and mortality. Approximately 4-7% of pediatric hospital admissions are made for recognized Mendelian diseases (Khoury et al. 1993b). From more than 2,000 likely single-gene developmental-defect syndromes in humans, the gene has been isolated and mapped for 100 of these syndromes and mapped but not yet isolated for another 100 (Winter 1996). In the mouse, for comparison, there are approximately 500 spontaneously occurring single-gene defects associated with developmental defects. Approximately 75 of these genes have been isolated, and greater than 400 have been mapped (Winter 1996). Furthermore, more than 1,000 mouse mutants have been prepared with known gene defects (many in components of signaling pathways), and many of these have phenotypes that fully qualify them as mouse single-gene developmental defects. Despite the availability of mouse mutants and the identification of genes important in development of C. elegans, Drosophila, and zebrafish (see Chapters 6 and 7), the study of single-gene defects contributing to developmental defects in humans has not yet received much experimental attention. Genes identified as important for development in C. elegans, Drosophila, and zebrafish, however, provide a rich source of information for identification of potential susceptibility genes in humans. That strikes this committee as an underutilized resource.
Multifactorial disorders, or complex diseases, are characterized by genetic complexity and probable gene-environment interactions (Ellsworth et al. 1997). These diseases tend to aggregate within families but are not inherited in simple Mendelian fashion. They are typically found in a higher proportion within affected families than expected in the general population. In contrast to single-gene disorders, susceptibility genes for complex disorders tend to be common in the population (allelic frequency more than 1%) and can be considered polymorphisms. Susceptibility genes are usually associated with low risk to the indi-
vidual but high attributable risk in the population. In cases of high heritability, the disease trait might have alleles of several major genes and of several modifier genes contributing to its penetrance and expressivity. In those with low heritability, there might be alleles of several genes as well as specific environmental factors that together increase the risk of disease in a population.
Recent examples in which gene-environment interactions have been sucessfully elucidated for developmental defects are the following:
-
Transforming growth factor (TGFα) polymorphisms and oral clefts: Evidence for an association between maternal smoking and oral clefts has been equivocal (Hwang et al. 1995). In this study, there was not an overall significant association between maternal smoking and oral clefts in the newborn. However, if the newborn had a variant allele (TAQL C2) of the TGFα gene, the odds ratio for oral clefts in infants of smoking mothers (more than 10 cigarettes per day) was 8.7, a 10-fold increase compared with infants of smoking mothers who did not have this variant allele. The variant allele alone was not associated with increased risk for oral clefts. TGFα is a ligand of a tyrosine kinase receptor.
-
Homeobox gene MSX1, limb deficiencies, and smoking: Frequencies of rare alleles at the MSX1 locus are slightly higher in infants with limb deficiencies compared with infants having other types of developmental defects (odds ratio 2.4). Infants carrying the rare alleles had a 2-fold increased risk of a limb deficiency when the mother smoked during pregnancy (odds ratio 4.8) compared with infants harboring the rare allele whose mothers did not smoke. Smoking alone was not associated with increased risk for limb deficiencies in this study (Hwang et al. 1998). MSX1 is a transcription factor whose activity often depends on BMP2,4 signals.
-
Variable human susceptibility to developmental defects due to diphenylhydantoin (DPH or phenytoin): 10-20% of the offspring of epileptic women taking phenytoin during pregnancy have the fetal hydantoin syndrome (Hanson et al. 1976; van Dyke et al. 1988). Phenytoin is thought to be converted to a reactive intermediate to have teratogenic effects (Martz et al. 1977; for a review, see Finnell et al. 1997a). The population variability in response to phenytoin possibly reflects a heterogeneity of DME genotypes. In a pair of twin births in which only one twin had dysmorphologies of the hydantoin syndrome, the mother and the affected twin had decreased activity of the enzyme epoxide hydroxylase compared with the unaffected twin (Buehler 1984). Buehler et al. (1990) subsequently showed that children with the hydantoin syndrome indeed have lower activity of epoxide hydrolase. Epoxide hydrolase would serve to detoxify an arene oxide intermediate of phenytoin, and it has been suggested that reduced activity of this enzyme is responsible for increased susceptibility to phenytoin. Hassett et al. (1994) reported a polymorphism in the epoxide hydrolase gene that markedly decreases enzyme activity. Conversely, the DPH parent compound might be teratogenic, and genetic defects in CYP2C9 and CYP2C19 (the two
-
enzymes that metabolize DPH) could result in an accumulation of the DPH teratogen. The frequencies of CYP2C9 and CYP2C19 poor metabolizing polymorphisms range between 10% and 25% of individuals in different populations—very similar to the percent of women taking DHP who have children with the fetal hydantoin syndrome.
-
Aldehyde dehydrogenase 2, alcohol dehydrogenase 2, and the susceptibility to fetal alcohol syndrome: In the metabolism of ethyl alcohol, alcohol dehydrogenase (ADH) catalyzes the conversion of alcohol to acetaldyhyde, and acetylaldehyde dehydrogenase (ALDH) oxidizes the conversion of this product to acetic acid. Alcohol and acetaldehyde, but not acetic acid, are thought to have the potential for deleterious effects. Humans possess at least seven ADH genes and 13 ALDH genes. Crabb (1990) pointed out that the single base mutation in ALDH2 (the mitochondrial as opposed to the cytosolic ALDH), which is responsible for acute alcohol-flushing reaction and alcohol intolerance mostly in Asians, is the best-characterized genetic factor influencing alcohol drinking behavior (lower activity correlating with intolerance). He raised the possibility that polymorphisms in the several alcohol dehydrogenase genes might be related to risk of fetal alcohol syndrome (FAS). A genetic influence in fetal alcohol syndrome is suggested by twin studies: Streissguth and Dehaene (1993) established that the rate of concordance for the diagnosis of fetal alcohol syndrome was 5 out of 5 for monozygotic and 7 out of 11 for dizygotic twins. In two dizygotic pairs, one twin had FAS, and the other had fetal alcohol effects (FAE). In two other dizygotic pairs, one twin had no evident abnormality, and the other had FAE. Intelligence Quotient scores were most similar within pairs of monozygotic twins and least similar within pairs of dizygotic twins discordant for diagnosis. Johnson et al. (1996) documented the central nervous system (CNS) anomalies of FAS by magnetic resonance imaging. CNS and craniofacial abnormalities were predominantly symmetric and central or midline. The authors stated that the association emphasized the concept of the midline as a special developmental field. The CNS is vulnerable to adverse factors during embryogenesis and fetal growth and development.
As those four examples indicate, further investigation of gene-environment interactions using the tools of molecular epidemiology is likely to yield important new information on multifactorial causes of developmental defects. Two of the above-cited examples concern polymorphisms of genes encoding enzymes involved in the metabolism of an agent, namely, phenytoin or alcohol, and two of the examples concern polymorphisms of genes encoding protein intermediates of signal transduction pathways and genetic regulatory circuits (TGFα or MSX1), which are components of developmental processes.
The examination of gene-environment interactions is particularly advanced for disease conditions related to the DMEs, and this area of study is called ecogenetics or pharmacogenetics. There are phase I and phase II metabolizing
enzymes. Phase I enzymes catalyze a conversion of the exogenous agent to a modified form, often an oxidized form. In some cases, the exogenous agent is toxic and the intermediate is not, but in other cases, the agent is nontoxic and the intermediate is toxic, an example of metabolic potentiation or activation. There probably are several hundred kinds of phase I enzymes (and genes encoding them) in mammals (including humans). The majority of them are members of the large cytochrome P450 monooxygenase family. Three or four kinds of P450 enzymes are thought to metabolize 70-80% of the prescription drugs taken by patients, and defects in phase I enzymes correlate with drug sensitivities and hazardous side effects. Phase II enzymes subsequently catalyze the conjugation of the modified intermediate to an endogenous harmless metabolite, such as a sugar or amino acid, and the conjugated form, which is usually nontoxic, is then excreted. In several well-analyzed cases, patients with high levels of phase I enzyme (hence, producing high amounts of a toxic intermediate) and low levels of phase II enzyme (hence, unable to get rid of that intermediate) were found to be particularly at risk from chemical exposures. Thus, human variants with altered levels of enzymes of one group or the other, or both, can have abnormal drug responses, as much as a 20- or 30-fold increase in drug sensitivity.
At least 60 ecogenetic or pharmacogenetic differences are now known; many are listed in Table 5-1. In this research, epidemiological methods and genomic methods are complementary, and progress in the near future seems assured. It seems likely that the fetus is at increased risk of developmental defects, because either the mother or the fetus cannot metabolize chemicals as well as others can or because they metabolize them better.
The other large area to investigate for the correlation of polymorphisms with developmental defects is that of the components of the developmental processes themselves, namely, key components of developmental processes, such as those of signal transduction pathways and genetic regulatory circuits. These components are the targets of exogenous agents that elude detoxification or are potentiated by phase I enzymes. The examples of TGFα with smoking and MSX1 with limb defects are two that have been clarified. At this time, however, there are few good examples, perhaps simply because information about developmental processes has not been available until recently. Because the developmental components are conserved across phyla and have been well described in Drosophila, C. elegans, and now the mouse, the means are available to obtain related human sequences and search for polymorphisms. This research will be further discussed in Chapters 8 and 9. The phenotypes of mouse null mutants generated by the embryonic stem-cell-(ES) knockout technology have already contributed substantially to our knowledge about how alterations in those genes and pathways impact development. This kind of research is progressing rapidly. Complete deletion of some components of a variety of pathways fundamental to development results in embryo lethality, but in other cases for which there is a gene redundancy for the component, the deletion of the component leads to mice born with developmental
TABLE 5-1 Classification of a Partial List of Human Pharmacogenetic or Ecogenetic Differencesa
|
Less enzyme or a defective protein |
|
N-acetylation polymorphisms (NAT2, NAT1) Increased susceptibility to chemical-induced hemolysis (G6PD deficiency) (G6PD) Hereditary methemoglobinemias; hemoglobinopathies P450 monooxygenase polymorphisms (oxidation deficiencies). Debrisoquine (CYP2D6), S-mephenytoin (CYP2C19 & 2C9), phenytoin (CYP2C9 & 2C19), nifedipine (CYP3A4), coumarin and nicotine (CYP2A6), theophylline (CYP1A2), acetaminophen (CYP2E1) Null mutants of glutathione transferase, mu class (GSTM1); theta class (GSTT1) Thiopurine methyltransferase (TPMT) Paraoxonase deficiency, sarinase (PON1) UDP glucuronosyltransferase (Gilbert’s disease, UGT1A1; (S)-oxazepam, UGT2B7) NAD(P)H:quinone oxidoreductase (NQO1) Epoxide hydrolase (HYL1) Atypical alcohol dehydrogenase (ADH) Atypical or absent aldehyde dehydrogenase (ALDH2) Defect in converting aldophosphamide to carboxyphosphamide α1-antitrypsin (PI) α1-antichymotrypsin (ACT) Angiotensin-converting enzyme (DCP1, ACE) Acatalesemia (CAT) Dihydropyrimidine dehydrogenase (DPD) Succinyl sensitivity, atypical or absent serum cholinesterase (CHE1) Cholesteryl ester transfer protein (CETP) Butyrylcholinesterase (BCE1) Fish odor syndrome (FMO3) Glucocorticoid-remediable aldosteronism (CYP11B1, CYP11B2) Dubin-Johnson syndrome; multispecific organic anion transporter (MOAT, MRP) Altered serotonin transporter (5HHT) Altered dopamine transporter (DAT) Dopamine receptors (D2DR, D4DR) Defective drug transporters (e.g., MDR1), resistance to chemotherapeutic agents Licorice-induced pseudoaldosteronism (HSD11B1) Mineralocorticoid excess with hypertension (HSD11B2) Pyridoxine (vitamin B6)-responsive anemia (ALAS2) |
|
Increased resistance to chemicals |
|
Inability to taste phenylthiourea Coumarin anticoagulant resistance Androgen resistance Estrogen resistance Cushing syndrome from low doses of dexamethasone Insulin resistance Rhodopsin variants; dominant form of retinitis pigmentosa Vasopressin resistance (AVPR2) Increased metabolism—Atypical liver alcohol dehydrogenase (ADH) Defective receptor—Malignant hyperthermia / general anesthesia (Ca2+-release channel ryanodine receptor) (RYR1, MHS1) |
|
Change in response due to altered enzyme induction |
|
Porphyrias Aryl hydrocarbon receptor (AHR) polymorphism (CYP1A1 and CYP1A2 inducibility polymorphism) correlated with cancer, immunosuppression, birth defects, chloracne, porphyria, and, possibly, eye toxicity and ovarian toxicity |
|
Abnormal metal distribution |
|
Iron (hemochromatosis, HFE), copper (Wilson disease, Menkes disease), and, possibly, lead, cadmium, and others |
|
Disorders of unknown etiology (known to run in families) |
|
Corticosteroid (eye drops)-induced glaucoma Halothane-induced hepatitis Chloramphenicol-induced aplastic anemia Aminoglycoside antibiotic-induced deafness Beryllium-induced lung disease Hepatitis B vaccine resistance Susceptibility to human immunodeficiency virus infection (polymorphism of CCR5 coreceptors) Long-QT syndrome Retinoic acid resistance and acute promyelocytic leukemia Thombophilia (activated protein C resistance) Lactose intolerance Fructose intolerance Beeturia; red urine after eating beets Malodorous urine after eating asparagus Reproductive disadvantage in F508 cystic fibrosis heterozygotes who smoke cigarettes (CFTR) High risk of cerebral vein thrombosis in defective prothrombin (F2) heterozygotes High risk of cerebral vein thrombosis in users of oral contraceptives |
|
aModified from Nebert (1999). See also refs. 11 and 18 of Nebert (1999). All of these are pharmacogenetic or ecogenetic in the sense that health risk correlates not only with the polymorphic state of the individual, but also exposure of that individual to a particular chemical (drug or environmental agent). Many of these are searchable in the online Mendelian Inheritance in Man (OMIM) database at http://www.ncbi.nlm.nih.gov/omim/. Not all of these are correlated with developmental defects. |
defects resembling human defects (see examples in Chapter 6). The work on key developmental components in animals can greatly benefit the search for human variants of developmental components.
The final step, though, will be to evaluate how specific toxicants interact with those altered pathways to produce abnormal development. Relevant susceptibility genes of development can then be examined in human populations, and interactions between alleles of those genes and toxicant exposures can be identified. Whether allelic variants of genes controlling development, such as those encoding components of the major signal transduction pathways, will be more important, as important, or less important than those controlling DMEs remains to be determined and should be given high priority for future research.
SUMMARY
The sequencing of the human genome and a variety of animal genomes will provide fundamental information about genome organization, genome evolution, gene sequence variety, and genetic polymorphisms. Sequencing will also provide a platform for global systematic analysis of gene function and gene expression. Developmental toxicology and risk assessment are expected to benefit in major ways from the new methodologies and information, namely, in the analysis of gene-environment interactions in human development defects and in the analysis of toxicant action on developmental processes.
A quarter to a half of the human developmental defects are believed to be attributed to interactions of the genotype and environment—that is, the exposure of individuals of a particular genetic composition to particular environmental conditions to which they are more sensitive than are others. Complex gene-environment interactions present a great challenge to developmental toxicology. The best epidemiological methods, the most discriminating molecular assessments of exposure and effect, and the most detailed analysis of genetic differences will be needed to make progress in understanding gene-environment interactions. Recent improvements in high-throughput sequencing of the human genome and in the identification of polymorphic markers conveniently spaced along each chromosome increase the chances for progress in this direction. Within this new area of molecular epidemiology, recent insights into human differences in activity levels of various DMEs and the genetic basis for those differences, offer great promise. Other kinds of gene products that might be important in susceptibility but are less well known, include components of developmental processes, particularly the components of signal transduction pathways and genetic regulatory circuits. These components will be discussed in later chapters.
Methods now are available to describe patterns of simultaneous expression of thousands of genes of developing cells and tissues and, in principle, to describe the changes of expression in the embryos of normal experimental animals and those following testing with toxicants. The use of such methods is expected to improve the categorization and analysis of toxicant-induced developmental defects.
The amount of data generated by modern genomic methods is prodigious. For the full benefit of the data, departments or divisions of bioinformatics in universities and industries will be needed to keep track of the data and to analyze it with respect to questions about genome organization and function.




















