
6
Chemical Theory and Computer Modeling: From Computational Chemistry to Process Systems Engineering1
|
Some Challenges for Chemists and Chemical Engineers
|
|
The phenomenal increase in speed and computational power of computers— as well as their dramatic reduction in cost—has continued at an astonishing pace over the last decade. In terms of CPU speed, Moore’s law implies that computing power should double every year or two,2 a pattern that is expected to continue for at least 10 years into the future. At the same time, key advances in areas such as ultraviolet lithography techniques, nonleaking complementary metal oxide semiconductor (CMOS) transistors, and multiple instruction, multiple data (MIMD) computer architecture are already in place to support clock-speeds at the 10-GHz plateau with power requirements at 1 V or lower. In addition, the technology and software infrastructure now exist for researchers to routinely build large parallel supercomputing clusters using off-the-shelf commodity computers and networking components, thus increasing the impact of Moore’s law by orders of magnitude. Finally, 10 years ago, few could envision how the dimension of communications—via the Internet—would enhance computing in what may have been the most revolutionary development in the late 20th century.
Chemistry and chemical engineering, like many other disciplines, are being profoundly influenced by increased computing power. This has happened in part by enhancing many existing computational procedures, providing a new impetus to quantum mechanical and molecular simulations at the atomic level, and optimizing processes and supply chain management at the macrosystem level. Furthermore, these computational tools have helped test new conceptual approaches to understanding matter and molecules. While we expect all these developments
|
2 |
Moore originally stated that “The complexity for minimum component costs has increased at a rate of roughly a factor of two per year,” Moore, G.E., Electronics, 38 (8), 1965. This has been restated as “Moore’s Law, the doubling of transistors every couple of years,” (http://www.intel.com/research/silicon/mooreslaw.htm). There are two related laws: (1) The cost of a fabrication plant to make each generation of processors doubles in price from the previous one. (2) Since the discovery of the giant magnetoresistance (GMR) effect, the density of disk storage increases by a factor of 2 every 12 months. See also: Gelsinger, P., Gargini, P., Parker, G., Yu, A., IEEE Spectrum, October 1989. |
to continue, the important challenge is likely to arise in dramatic growth of new computing needs. These needs are driven by the increasing shift in the chemical industry toward biotechnology products and pharmaceuticals, the emergence of industrially relevant nanotechnologies, the requirement to optimize existing large-scale commodity chemicals plants, and the increased size and complexity of many new problems and systems. Instruments used by chemists and chemical engineers are already substantially controlled by on-board computer systems, the complexity of which will increase with that of the purpose and use of the instruments.
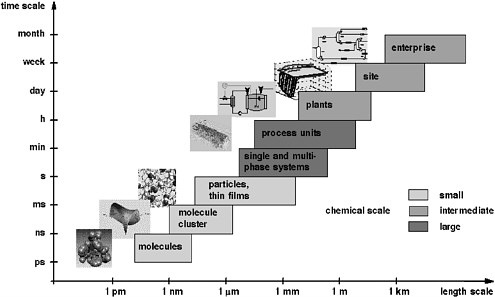
Computational chemistry and process systems engineering play a major role in providing new understanding and development of computational procedures for the simulation, design, and operation of systems ranging from atoms and molecules to industrial-scale processes. The enormous span of scales of space and time that range from computational chemistry to process systems engineering can be visualized with the “chemical supply chain” shown in Figure 6-1. The supply chain starts with the set of chemicals that industry must synthesize and characterize at the molecular level. Subsequent steps aggregate these molecules into clusters, particles, and films—as single and multiphase systems that finally take the form of macroscopic mixtures. At the process engineering scale, the figure illustrates the design and analysis of production units that are integrated into a chemical process, which in turn is part of a site with multiple processes. Finally, this site is part of the commercial enterprise that is driven by business considerations. The multiple scales of this chain are a fact of life in chemical sciences and technology. To date, the field has neither sufficient tools nor enough trained people to pursue computational chemistry and chemical engineering across all these scales. The field will qualitatively change—in new insights, in what experiments are done and how chemical products and processes are designed—when this is achievable.
Advances in computing have facilitated major progress in computational chemistry and biochemistry, computational materials design, computational fluid dynamics, process synthesis, planning and scheduling, model-based process control, fault diagnosis, and real-time process optimization. This progress has been enabled by parallel advances in fundamental chemical physics theory, mathematics, operations research, and computer science—including computational techniques for simulation and optimization of chemical systems. This chapter shows how areas that span computational chemistry, from the atomic level to process systems engineering at the macrosystem level, are full of exciting computational challenges that await solutions from bright minds.3,4
GOALS
Science proceeds by both experiment and theory. Simple experimental facts without a theory to interpret them do not satisfy our need for understanding. Indeed, many experimental measurements cannot be interpreted without theory, and as experiments probe phenomena and structures at ever smaller spatial and temporal scales, the role of theory in interpreting experiment increases. However, theory without experiment can lead to unrealistic dreams.The birth of modern science came when it was realized that truth is obtained from facts, not just from speculation. Theory validated by experiment runs through all of chemistry, and almost all its branches now use computers. Structures are determined by computer treatments of x-ray data, potential new drugs are analyzed by computer modeling, and even synthetic strategies to make a desired target molecule are developed using computer programs created for the purpose.
Quantum Mechanics
The chemical sciences are built on a set of fundamental mathematical theories that have increasing utility as computational hardware and software become more powerful. As the basis for calculating the electronic structure of molecules, quantum mechanics permits calculations, often based on rational approximations, of structure and properties of molecules, and of reactivity and reaction mechanisms. The Schrödinger wave equation can in principle predict the electronic states of any chemical species, but in practice it can be applied only approximately to molecules of any significant size. A continuing, important goal is to devise better and more accurate ways to obtain predictions of molecular structures, bond energies, molecular properties, transition state structures, and energies for systems of increasing size.
Good approximate quantum calculations usually can be done reliably only for isolated molecules. Another important objective in this field is to develop methodologies for solvated molecules and molecules that are parts of membranes or other organized biological systems. Engineers are attempting to use quantum mechanical calculations to predict practical phenomena—for example, making and breaking of bonds in adhesion and fracture—that are based on electronic interactions. Additional goals are to learn how to accurately include heavier elements and to calculate the properties of molecules as a function of time, when they are interacting with other species.
Quantum calculations are the starting point for another objective of theoretical and computational chemical science, multiscale calculations. The overall objective is to understand and predict large-scale phenomena, such as deformation in solids or transport in porous media, beginning with fundamental calculation of electronic structure and interactions, then using the results of that calculation as input to the next level of a more coarse-grained approximation.
An important goal is to improve or supplement quantum mechanical methods in order to calculate reliably the energy and geometry of a transition state. This is one piece of information that can lead to the larger objective of predicting the rates of unmeasured reactions, but a second goal must also be met to achieve this. The accurate prediction of reaction rates also depends on our knowledge of dynamical properties of molecules and the dynamics of their sampling of accessible electronic, rotational, and vibrational states. Another goal is to predict the course of excited-state reactions, often initiated by adsorption of light. Such molecules do not traverse the lowest energy paths, and they usually form products that are different from those produced in ground-state reactions. Molecules that absorb light are transformed into various different excited states, depending on the energy of the light that is absorbed. Each excited state can in principle undergo a unique photochemical transformation. The development of successful theoretical treatments for such complex phenomena presents a substantial challenge. This goal also extends to the calculation of magnetic, optical, electrical, and mechanical properties of molecular and extended solids in both the ground and excited states.
Yet another related goal is to be able to predict the catalytic activity of a given surface for a particular reaction. Using computational quantum chemistry, it is becoming possible to predict with reasonable accuracy the energy barriers and transition states of molecules reacting on catalytic surfaces, thus leading to insights into reaction rates. This is enabling a new field of rational catalyst design, which offers the promise of designing and optimizing new catalysts computationally so that synthetic efforts can be focused on high-priority candidates.
Molecular Mechanics
One tool for working toward this objective is molecular mechanics. In this approach, the bonds in a molecule are treated as classical objects, with continuous interaction potentials (sometimes called force fields) that can be developed empirically or calculated by quantum theory. This is a powerful method that allows the application of predictive theory to much larger systems if sufficiently accurate and robust force fields can be developed. Predicting the structures of proteins and polymers is an important objective, but at present this often requires prohibitively large calculations. Molecular mechanics with classical interaction potentials has been the principal tool in the development of molecular models of polymer dynamics. The ability to model isolated polymer molecules (in dilute solution) is well developed, but fundamental molecular mechanics models of dense systems of entangled polymers remains an important goal.
A particular goal of chemical theory is to predict protein structure from the amino acid sequence—to calculate how polypeptides fold into the compact geometries of proteins. One strategy is to develop methods (often based on bioinformatics) for predicting structures approximately and then refining the structures
using atomic-level molecular modeling methods. Molecular mechanics is also the theoretical approach employed in calculating how a proposed drug might bind into a protein.
Modeling and Simulation
Modeling and simulation are extremely important tools in the chemical sciences. The understanding and engineering of complex chemical processes, such as combustion or atmospheric chemistry and transport, generally rely heavily and increasingly on modeling and computation. Recent advances in computing not only have enabled more accurate and reliable calculations, but they have also provided new tools for interpreting the output of the calculations. Modern computer graphics—including molecular graphics, simulations, and animations— have greatly enhanced the ability of scientists and engineers to understand and utilize the results of their computations.
Yet modeling can be no better than its assumptions. It often suffers from the problem that we cannot follow any computed process for a long duration (many time steps)—primarily because the computer time needed per time step is significant, but also because of the cumulative propagation of round-off errors. The typical time step is on the order of 1 femtosecond (i.e., 10−15 s) of real time for an atomically detailed molecular simulation. Consequently, modeling phenomena on the femtosecond time scale would require about 103 time steps, which is not difficult, and modeling on the picosecond time scale (106 time steps) is fairly routine. However, many phenomena of interest (e.g., the time to fold a protein) are on the millisecond or larger time scale and would require 1015 time steps or more. Another goal is to learn how to improve the calculations by overcoming these problems. One approach is to use stochastic approaches based on cleverly chosen Monte Carlo methods; another is to reduce the level of detail in the models for the molecules (so-called coarse grained models).
Statistical Mechanics and Fluid Mechanics
Sometimes the theoretical or computational approach to description of molecular structure, properties, and reactivity cannot be based on deterministic equations that can be solved by analytical or computational methods. The properties of a molecule or assembly of molecules may be known or describable only in a statistical sense. Molecules and assemblies of molecules exist in distributions of configuration, composition, momentum, and energy. Sometimes, this statistical character is best captured and studied by computer experiments: molecular dynamics, Brownian dynamics, Stokesian dynamics, and Monte Carlo methods. Interaction potentials based on quantum mechanics, classical particle mechanics, continuum mechanics, or empiricism are specified and the evolution of the system is then followed in time by simulation of motions resulting from these direct
interparticle, stochastic, or fluid mechanical forces. The larger the size of the computation, the better the statistical representation of the system.
Statistical mechanics is the science that deals with average properties of the molecules, atoms, or elementary particles in random motion in a system of many such particles and relates these properties to the thermodynamic and other macroscopic properties of the system.5 Use of statistical mechanics reflects the view that broad features of a system may be captured best as a description of the statistical distribution of the elements within a population rather than as a precise description of the elements themselves. One advantage of statistical mechanics is that a good model often reveals some underlying generality governing the system, thereby permitting analogies to be made between properties or behaviors that superficially are quite different. Simulations and statistical mechanics are key tools for physical chemists and engineers working on understanding rheological behavior, mass transport, modeling of microfluidic devices, flow of granular media, and behavior of dense particle suspensions.
At the next higher level of coarse-graining, fluid mechanics and other continuum mechanics methods are active arenas of theory and computation. For example, electrorheology and magnetorheology have provided tremendous impetus in the last decade. Efforts to determine the dependence of basic scaling on field strength and particle volume fraction quickly require answers to questions about material properties. The links between the thinking of chemical engineers studying transport and chemists designing and producing materials are crucial for progress. Granular media is another area where the clear interplay between science and technology is facilitating progress in solving sophisticated scientific questions that have immediate impact on technology and practice. From the viewpoint of computation and simulation, engineers working in granular media are addressing problems of flow and mixing (such as those arising in the processing of powdered pharmaceuticals); discrete computational approaches encompass particle dynamics, Monte Carlo, and cellular automata calculations.
Spanning Length and Time Scales
Many of the problems cited above highlight the need for being able to bridge calculations across several length and time scales. It is thus worthwhile to consider this in some additional detail. For example, reactions involve changes in the molecules, and hence are inherently quantum mechanical in nature. But a reaction taking place in a solution at finite temperature implies that the reaction is influenced by a dynamic environment more pragmatically described by classical molecular simulation methods. Hence, a scale-bridging method is needed to allow the dynamics of the solvent around the reactants to influence the electronic
structure of the reactants in various conformations, as well as the reactants influencing the motion of the solvent molecules around them. One solution is to perform molecular simulations with force fields calculated “on the fly” by quantum mechanical methods at each time step. Such “first principles” or ab initio molecular simulation methods are presently limited to fairly small numbers of atoms (around 100 to 1000) for rather short simulations (10 ps is a typical value). Other hybrid methods, which treat just the environment around reactants quantum mechanically, are less accurate in principle but allow much longer time scales and much larger spatial scales to be accessed, since the vast majority of the molecules are treated by classical molecular simulation.
The problem of spanning scales goes well beyond spanning from the electronic structure scale to the molecular (which might be thought of as the Ångströmor nanoscale). Chemical processes at the commercial scale ultimately involve spatial scales on the order of meters, and time scales (corresponding to processing times in reactors and separations equipment) ranging from seconds to hours and, in the case of many bioengineering processes involving fermentations, days or weeks. How do we connect phenomena at the electronic and molecular scale to the commercial chemical process scale?
In some cases, the connection is trivial: for low-molecular-weight molecular systems, properties calculated at the electronic structure level (e.g., reaction rates, and free energies of formation and reaction) or at the molecular simulation level (e.g., the so-called critical constants, and transport properties such as viscosity) can be used directly at the process scale as input data for process models. However, for complex molecules (such as polymers and proteins) the properties at the molecular level are not decoupled from the process level. For example, flow at process level can result in changes in the conformation of the molecules, which in turn changes the properties of the molecules. In such cases, the problem of developing accurate molecular modeling methods that will span the scales from electronic to molecular to mesoscopic to macroscopic (the process scale) cannot be avoided. Since such complex molecules and their processing are the focus of the modern chemical, pharmaceutical, and materials industries, it is imperative that the problem of developing effective multiscale modeling methods be solved. It is currently one of the most active research focuses of the molecular modeling community. It is also a major focus of nanotechnology modeling efforts, since in such systems the need to connect nanoscale structure to macroscale functionality is even more apparent.
Making the “scale-up” connection between the electronic and molecular structure of molecules and their macroscopic properties, and the design of processes to manufacture them, is one half of the story. The other half is making the connection “scale-down”—from specification of a process-level manufacturing need (e.g., a solvent with better properties) to the design of a molecule that meets the need. Harnessing these yet-to-be-developed methods and combining them with state-of-the-art process modeling and optimization tools will result in inte-
grated process and product design: the ability to computationally design and optimize products (e.g., chemicals, drugs, materials) as well as the manufacturing processes to make them. The molecular modeling and process design communities recognize that, with the continued rapid pace of computer hardware, software, and algorithmic advances, this goal is now accessible in the next decade or two. Hence, conferences are beginning to spring up whose main goal is to bring together members of these communities to learn from each other. Cross-fertilization between the fields has led to computational chemistry methods (e.g., simulated annealing) crossing over to the process design community, just as techniques from the process design community (e.g., mathematical programming methods) have crossed over to the protein folding community.
|
The Problem of Time Scales in Molecular Simulation Brute-force classical molecular dynamics simulation will not span the time scales present in complex molecular systems, even with fully efficient implementation on the world’s fastest parallel supercomputers. This time scale problem was captured well by Ken Dill in 1993.6 Although this scheme did not fully take into account the impact of massively parallel computers (by which the current generation of ~10 teraflops computers theoretically increase the level of computing 1,000- to 10,000-fold over workstations), the essential problem of simulating over long periods of time relative to the shortest time scale motion is not fundamentally changed even by recent advances in parallel supercomputing. While parallel supercomputing makes it possible to span larger spatial scales rather easily, spanning larger time scales remains a fundamental difficulty. To be more concrete, there are about 30 million seconds in a year, and the maximum speed at which messages can be passed between any two processors on a parallel supercomputer is roughly 10,000 per second. Hence, even if the time to compute forces could be reduced by sufficient parallelization to essentially zero, a molecular dynamics simulation could not execute more than 3 x 1011 time steps per year; in practice, it will be considerably less. When a calculation is limited by the time required to pass messages, it is referred to as communications-limited. Thus, with a time step of 10−15 s, in a year of computing the most one could hope to achieve is around 10−4 s of real time. This conclusion highlights the need to develop theoretically sound methods for spanning the many time scales present in the molecular simulation of complex systems (such as proteins and polymers). |
|
How well has Dill’s prediction held up? In 2000, the first ever microsecond-long molecular dynamics simulation of protein folding was reported. It required 750,000 node hours (equal to the product of the number of hours times the number of processors) of computer time on a Cray T3 supercomputer. According to Dill’s prediction, this length of simulation was not to be expected until around 2010. However, as noted above, Dill’s analysis does not take into account large-scale parallelization—which, unless the computation is communications-limited, will effectively increase the speed of a computation in proportion to the number of processors available. 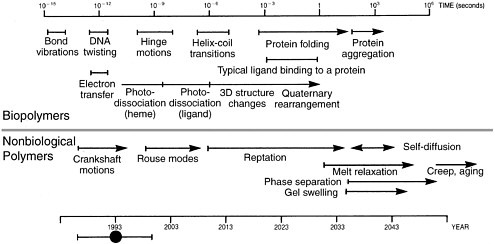 Time scales for various motions within biopolymers (upper) and nonbiological polymers (lower). The year scale at the bottom shows estimates of when each such process might be accessible to brute force molecular simulation on supercomputers, assuming that parallel processing capability on supercomputers increases by about a factor of 1,000 every 10 years (i.e., one order of magnitude more than Moore’s law) and neglecting new approaches or breakthroughs. Reprinted with permission from H.S. Chan and K. A. Dill. Physics Today, 46, 2, 24, (1993). |
PROGRESS TO DATE
Important insights have been developed using approximate methods that were not highly precise quantitatively, and excellent high-level methods for solving the Schrödinger equation have been developed, but the methods still have used approximations. A Nobel Prize in 1998 went to John Pople and Walter Kohn for their different successful approaches to this problem. Earlier methods used many
different parameters derived from experiment, but current so-called ab initio methods as pioneered by Pople are essentially free of such experimentally derived parameters. With the current methods, systems of 5 to 20 common atoms are handled well. Density-functional theory (DFT) as developed by Kohn is also moderately successful with larger systems, and with more unusual atoms.
In molecular mechanics, force fields and sampling methods used in the calculations are being improved continually, but they are not yet good enough that atomic-level refinement of protein structures is feasible on a robust and routine basis. It is unclear what the relative contribution of force field and sampling errors are to the problem. Similar comments apply to the prediction of protein-ligand binding. The protein-folding problem is thus currently being approached mainly by analogy, using data from existing protein structures to derive various folding rules. The field of ab initio theoretical protein folding—prediction of full three-dimensional protein structure using a combination of an optimization algorithm to minimize energy with an explicit force-field representation of the functional groups in the protein—is a computationally intensive approach that has recorded some successes in protein structure prediction.7Ab initio protein-folding approaches are the only methods capable of predicting fundamentally new structures in proteins (i.e., substructures not previously found in any experimentally measured protein structures). One forefront area of research at the interface of polymers and biology is the synthesis (by genetic engineering) of synthetic proteins/biopolymers containing building blocks other than the usual 20 amino acids, including inorganic species such as silicon. Ab initio protein-folding approaches offer routes to predicting the properties of such biopolymers.
Initially, most theoretical methods calculated the properties of molecules in the gas phase as isolated species, but chemical reactions are most often carried out in solution. Biochemical reactions normally take place in water. Consequently, there is increasing interest in methods for including solvents in the calculations. In the simplest approach, solvents are treated as a continuum, whose average properties are included in the calculation. Explicit inclusion of solvent molecules in the calculation greatly expands the size of the problem, but newer approaches do this for at least those solvent molecules next to the dissolved species of interest. The detailed structures and properties of these solvent molecules affect their direct interaction with the dissolved species. Reactions at catalytic surfaces present an additional challenge, as the theoretical techniques must be able to handle the reactants and the atoms in the surface, as well as possible solvent species. The first concrete examples of computationally based rational catalyst design have begun to appear in publications and to have impact in industry.
In the area of collective properties, simulation methods have been developed over the past decade and a half that make it possible to predict the phase equilibria of systems directly from force fields, dramatically widening the applicability of molecular simulation for process-engineering calculations. Phase equilibria of various kinds (vapor-liquid, liquid-liquid, fluid-solid) lie at the center of most of the separation techniques (e.g., distillation and crystallization) commonly used in the chemical industry. The last decade has also seen dramatic improvements in molecular simulation methodologies—largely based on stochastic methods—for overcoming the long relaxation times inherent in the equilibration of polymers and similar complex materials. A recent international comparative study of the industrial application of molecular modeling in the chemical, pharmaceutical, and materials industries in the United States, Europe, and Japan gathered information from over 75 sites; the report documents some of the remarkably diverse ways in which molecular modeling methods are successfully impacting industry today.8 Given the limitations of today’s tools, the future for integrated molecular-based product design and process-level design and optimization is very bright indeed.
Goals in Process Systems Engineering, and Progress to Date
Process systems engineering is the study of the processes used in manufacturing. It encompasses all aspects of the design, construction, control, and operation of the chemical processes used in chemical manufacturing. The goal of process systems engineering remains the understanding and development of tools for the advantaged synthesis, analysis, optimization, evaluation, design, control, scheduling, and operation of chemical process systems consistent with new societal and economic objectives and constraints. This is true for all processes—no matter what the chemistry or biochemistry, whether isolated or in supply chain networks, dedicated or general purpose, batch or continuous, and whether microscale or macroscale. This goal requires the further creation and exploitation of a science base that includes novel representations of the underlying chemical and physical phenomena, computationally efficient formulation and solution methods, and the expansion of and incorporation of advanced engineering expertise and judgment. As discussed below, two major objectives in process systems engineering are the creation or invention of new processes (process synthesis), and the systematization of decision-making with prediction of systems performance (modeling and optimization).
|
8 |
The report is available on the Internet at http://itri.loyola.edu/molmodel. |
Process Synthesis
Creation of new processes for building new plants or retrofitting existing facilities is a key area for addressing the goals of process systems engineering cited above. The major reason is that process synthesis deals with the systematic generation and search of alternatives that can potentially lead to major structural discoveries and modifications. Significant progress has been made in the development of systematic techniques for the synthesis of energy recovery networks, distillation systems (including azeotropic separations), and reactor networks. For synthesizing process flow sheets that integrate these subsystems, hierarchical decomposition and superstructure optimization have emerged as two major approaches. In the design of multiproduct batch plants, progress has been made with mathematical programming models that integrate scheduling as part of the design decisions.
In the future we will see the application of synthesis techniques to new processes that involve greater use of more abundant or renewable raw materials, and greater reuse of materials such as carbon dioxide, salts, tars, and sludges, which are currently generally disposed of. Exploiting some of these alternative raw materials and chemistries may involve greater energy input than required for raw materials presently in use, and the source and impact of any such increased energy requirement must also be carefully considered. At the same time, pressures to increase reaction selectivity will continue unabated.
Process synthesis is the invention of chemistry-implementing processing concepts. It is a creative open-ended activity characterized by a combinatorially large number of feasible alternatives. Process synthesis defines the overall structure of the manufacturing process and identifies and designs the processing equipment to be employed and specifies how they are to be interconnected. The number of potential feasible process designs is in general very large, and selecting the right process structure generally determines most of the economic potential of a new or retrofitted chemical process. In recent industrial experience, energy reductions of 50% and overall cost reductions of 35% using systematic process synthesis techniques have been achieved. Nevertheless, a significant need remains for even better approaches.
Some success has been achieved in developing systematic process synthesis methods based on recursive hierarchical means-ends analysis architectures adapted from artificial intelligence research. These have been greatly aided by the recent development of new representations of the underlying physical science and thermodynamic knowledge that define the forces generally engaged in chemical reaction and separation processes. Exploiting these representations—problem-solving strategies that focus primarily on the processing tasks to be accomplished rather than the equipment to be employed—has proven to be particularly effective in creating novel superior designs.
The chemical process synthesis problem might also be formulated as an optimization over all feasible process structures. In principle, if we employed all different possible approaches to implement some desired chemistry—including all possible different reactor configurations, all possible different separation and purification schemes, techniques, and equipment, and all interconnections among these potential units in one gigantic tentative process flow sheet—and if we then subjected such a superstructure to economic optimization constrained by environmental, safety, and other criteria (during which process inferior equipment and interconnections were eliminated), the best manufacturing process in terms of both structure and the design of each surviving piece of equipment would emerge. This, of course, is a massive and extremely difficult optimization problem involving a mixture of continuous, integer, and logical variables and relationships. However, new mathematical techniques such as generalized disjunctive programming and global optimization, combined with the tremendous increases in available computing power made possible through large clusters of fast independent processors, give hope that this superstructure optimization approach to chemical process synthesis may be practical in the near future.
Modeling and Optimization
The chemical industry can largely be viewed as being composed of two major segments. One is the “value preservation” industry that is largely based on the large-scale production of commodity chemicals. The other is the “value growth” industry that is based on the small-scale production of specialty chemicals, biotechnology products, and pharmaceuticals. For the chemical industry to remain strong, it is essential that these two segments be competitive and economically strong. The “value preservation” industry must be able to reduce costs, operate efficiently, and continuously improve product quality, thereby making process simulation and process optimization its key technologies. The “value growth” industry must be agile and quick to market new products, making supply chain management one of its key technologies. In both cases, major challenges over the next two decades will be to gain a better understanding of the structure and information flows underlying the chemical supply chain of Figure 6-1, and to develop novel mathematical models and methods for its simulation and optimization.
In the past, most of the modeling and optimization activity has taken place in isolated parts of the chemical supply chain. In chemical engineering it has been mostly at the level of process units and plants, but more recently it has been moving in two opposing directions—the molecular and the enterprise levels. In computational chemistry, the modeling research has been directed at the molecular level and is increasingly moving toward the atomic and quantum scale, as discussed in the previous sections. Developments in planning and scheduling are being increasingly directed to address the optimization of the supply chain at the
enterprise level. Major difficulties in developing mathematical models that integrate the various parts of the chemical supply chain result from the huge differences in length and time scales in the supply chain and the number of chemical species considered at each level. Furthermore, at the longer scales, new situations can arise that may not be reliably predicted from the basic models.
|
Process Simulation: A Revolution Process simulation, which emerged in the 1960s, has become one of the great success stories in the use of computing in the chemical industry. For instance, steady-state simulation has largely replaced experimentation and pilot plant testing in process development for commodity chemicals, except in the case of reactions having new mechanisms or requiring new separation technologies. Tools for steady-state process simulation are nowadays universally available to aid in the decisions for design, operation, and debottlenecking; they are part of every process engineer’s toolkit. Their accuracy and predictive ability for decision-making is widely accepted to make routine plant trials and most experimental scale-up obsolete in the commodity chemicals industry. Dynamic simulation plays a dominant role in the design of training simulators for operators. As with aircraft simulation, these programs allow operators to deal with disturbances, start-ups, and upsets that go beyond conventional regulatory control. For hazardous processes, they are a requirement for operator certification on distributed control systems. Environmental, safety and hazard, and operability studies are often handled through steady-state and dynamic simulation tools. In short, process simulation is the engine that drives decision-making in process engineering. It is the accepted “virtual reality” of the process industries. Some recent applications have benefited from advances in computing and computational techniques. Steady-state simulation is being used off-line for process analysis, design, and retrofit; process simulators can model flow sheets with up to about a million equations by employing nested procedures. Other applications have resulted in great economic benefits; these include on-line real-time optimization models for data reconciliation and parameter estimation followed by optimal adjustment of operating conditions. Models of up to 500,000 variables have been used on a refinery-wide basis. |
|
Off-line analysis, controller design, and optimization are now performed in the area of dynamics. The largest dynamic simulation has been about 100,000 differential algebraic equations (DAEs) for analysis of control systems. Simulations formulated with process models having over 10,000 DAEs are considered frequently. Also, detailed training simulators have models with over 10,000 DAEs. On-line model predictive control (MPC) and nonlinear MPC using first-principle models are seeing a number of industrial applications, particularly in polymeric reactions and processes. At this point, systems with over 100 DAEs have been implemented for on-line dynamic optimization and control. While the developments described above are currently implemented and available, significant remaining issues are being investigated to further improve the capability of process simulators. One example is the use of object-oriented approaches to integrating models from different sources and vendors in a single simulation. Another challenge is combining partial differential equation models with DAE and algebraic process models, which will require parallel computations to solve models with millions of variables. Finally, nonlinear optimization algorithms are needed that can handle very large models as well as discrete variables for performing topology optimization in process flow sheets. |
To tackle these problems successfully, new concepts will be required for developing systematic modeling techniques that can describe parts of the chemical supply chain at different levels of abstraction. A specific example is the integration of molecular thermodynamics in process simulation computations. This would fulfill the objective of predicting the properties of new chemical products when designing a new manufacturing plant. However, such computations remain unachievable at the present time and probably will remain so for the next decade. The challenge is how to abstract the details and description of a complex system into a reduced dimensional space.
Another important aspect in the modeling and optimization of the chemical supply chain is the description of the dynamics of the information and material flow through the chain. This will require a better understanding of the integration of R&D, process design, process operation, and business logistics. The challenge will be to develop quantitative models that can be used to better coordinate and optimize the chemical enterprise. Progress will be facilitated by new advances in information technology, particularly through advances in the Internet and by new methods and mathematical concepts. Advances in computer technology will play a central role. Fulfilling the goal of effectively integrating the various functions
(R&D, design, production, logistics) in the chemical supply chain will help to better meet customer demands, and effectively adapt in the new world of e-commerce. Concepts related to planning, scheduling, and control that have not been widely adopted by chemical engineers should play a prominent role in the modeling part of this problem. Concepts and tools of computer science and operations research will play an even greater role in terms of impacting the implementation of solutions for this problem.
|
Supply-Chain Management Increases Profitability of the Chemical Industry Through Computer Tools New optimization tools, information management systems, and the Internet are revolutionizing supply-chain management in chemical companies. The objective of these tools is not only to help manage more effectively the flow of materials from their production facilities to their distribution centers, but also to incorporate the suppliers and the customers. Instead of running businesses from the inside out, forward thinking companies are integrating their business processes to run the business the way their customers demand: from the outside in. The ultimate winners in this new world will be the ones who manage their supply chains more efficiently than their competitors, harness the customer loyalty factor, and find ways to facilitate business transactions. Making the customer and the other business partners more profitable is the new way to compete in the chemical industry, where many products rapidly become commodities. From simple ways to reduce the cost of each transaction, to working together to reduce overall inventories, to true collaboration of business processes such as product design, logistics management, manufacturing execution, product marketing, and overall management, the use of advanced computer and optimization tools and process models deployed through the Internet will give those companies a significant competitive advantage. Supply-chain management can be applied at three major levels: strategic planning, tactical planning, and production scheduling. Furthermore, its integration can lead to new business practices such as improved “capability to promise” in the sales of products. An example of strategic planning is the integration of a single product through the entire supply chain, from raw materials delivery, through manufacturing, to delivery to customers. Major decisions at this level include rationalizing the distribution network, building new plants, or evaluating new markets. This is in contrast to tactical planning, which involves developing an operational plan, meeting capacity constraints, and developing inventory and overtime strategies to increase the efficiency in the supply chain. BASF per- |
|
formed a corporate network optimization of packaged finished goods in North America. There were 17 operating divisions with multiple, heterogeneous systems: 25,000 stockkeeping units (SKUs), 134 shipping points, 15,000 ship-to locations, 956 million pounds shipped direct to customers, and 696 million pounds shipped to customers through distribution centers. By using optimization tools BASF reduced transportation and facility costs by 10%, next-day volume delivery increased from 77% to 96%, the number of distribution centers was reduced from 86 to 15, generating $10 million per year savings in operating costs (personal communication, Dr. Vladimir Mahlaec, Aspen Technology). An example of production scheduling involves scheduling a plant in continuous time to optimize sequencing, while providing an executable production schedule. This problem is complicated by the fact that it must take into account forecast and customer orders, inventories of raw material, intermediates and finished goods, facility status, manufacturing information, rates, facility preferences, and recipes. Pharmacia-Upjohn was faced with such a problem in the scheduling of a fermentation process. Using modern software, schedules were generated for 6 days of production. The major benefit was an effective and flexible computer tool for rescheduling that can readily accommodate changes in customer orders. Finally, an interesting trend in supply-chain management is improving business practices by providing capabilities to improve service for customers. One such concept is “capability to promise.” The idea is not just to ensure that the product is available, but that the supply chain is capable of delivering the product. This involves, for instance, capability to rapidly change the schedule to accommodate the customer order and interact with the active plant schedule. Such a system was developed at Celanese Fibers, which is integrated with a Customer Order System and enabled via a Web browser. |
CHALLENGES AND OPPORTUNITIES FOR THE FUTURE
Chemical Theory and Computation
Faster and cheaper computers will extend the range of high-level methods. Improvements in DFT functionals will improve the accuracy of DFT calculations. One specific challenge will be to derive density functional theory from fundamental theory in a way that reveals how to incorporate successive approximations for the exchange and correlation terms. Some work on high-level methods with better scaling may allow more immediate extension of high-accuracy calculations to larger systems. Combining quantum mechanical and molecular
mechanics methods will allow very large systems to be treated using quantum methods in the reactive core and force fields for the remainder of the systems, for example in enzymatic chemistry. Many specific needs can be identified:
-
methods to simulate quantum molecular dynamics in condensed systems without approximating the system as a quantum system coupled to a classical bath. This is not now possible although there has been some progress toward this end in the treatment of simple physical models. This is a development that will benefit greatly from the development of practical quantum computers.
-
development of methods to bypass the problem of multiple time scales in molecular dynamics. This difficulty is particularly egregious in the protein-folding problem.
-
methods for the efficient sampling of rough energy landscapes such as those found in proteins. Because of high energy barriers in such systems, most of the time is spent sampling energy basins near the starting configuration. The development of efficient methods is required for the determination of structural and thermodynamic properties as well as for efficient refinement of protein structure.
-
accurate polarizable force fields for peptides, water, etc. Improved force fields explicitly incorporating polarization are being developed. Until rapid ab initio molecular dynamics methods exist, such force fields are required for the simulation of chemical systems with chemical accuracy.
-
to understand the kinetics of protein folding from a mechanistic point of view. Abstract schemes have been proposed, but there is much yet to do before this goal is realized.
-
to develop methods for understanding and predicting energy transfer, electron transport, and the entirely new quantum effects involving coherence that arise in nanoscale devices.
-
to develop the statistical mechanics of fluids and fluid mixtures—for example, to obtain improved understanding of associating fluids, hydrophobicity, and ionic systems.
-
to develop computational tools for solid-state problems, including calculation of magnetic, optical, electrical, and mechanical properties of molecular and extended solids in both ground and excited states.
-
to correlate theoretical predictions with experimental results, designing experiments specifically to test various theoretical predictions. As this is done, and is successful, the role of mathematical theory in chemistry will increase in value.
Process Systems Engineering
A number of major challenges exist in process systems engineering in which computing will play a major role. These can be grouped by major areas of application:
-
Process and Product Design. The traditional process design will be expanded to include product design as an integral part of this area. Within the commodity chemicals industry major challenges that will be addressed include process intensification for novel unit operations, and design of environmentally benign processes. Areas that are likely to receive increased attention due to the growth in new industries include molecular design, synthesis of microchips, smart materials, bioprocess systems, bioinformatics, and design and analysis of metabolic networks.
-
Process Control. The traditional process control will be expanded toward new applications such as nonlinear process control of biosystems. However, in the commodity chemicals industry there will be increased need for synthesizing plantwide control systems, as well as integrating dynamics, discrete events, and safety functions, which will be achieved through new mathematical and computer science developments in hybrid systems.
-
R&D and Process Operations. The traditional area of process operations will expand upstream and downstream in terms of integrating R&D as well as logistics and distribution functions. Areas that are likely to receive increased attention include logistics for new product development, planning and supply chain management, real-time scheduling, and synthesis of operating procedures (safety).
-
Integration. As is also described below, the integration of several parts of the chemical supply chain will give rise to a number of challenges, such as multiscale modeling for molecular dynamics, integration of planning, scheduling and control (including Internet based), and integration of measurements, control, and information systems.
Progress in these areas will require a number of new supporting tools that can effectively handle and solve a variety of mathematical models involving thousands and millions of variables. These supporting tools in turn will require that chemical engineers become acquainted with new advances in numerical analysis, mathematical programming, and local search techniques.
-
There is a need for large-scale differential-algebraic methods for simulating systems at multiple scales (e.g., fluid mechanics and molecular dynamics), a capability that is still at a very early stage.
-
There is a need for methods for simulating and optimizing models whose parameters are described by probability distribution functions, a capability that is in its infancy.
-
There is a need for advanced discrete-continuous optimization tools that can handle mixed-integer, discrete-logic, and quantitative-qualitative equations to model synthesis and planning and scheduling problems.
-
There is a need for methods that can determine global optima for arbitrary nonlinear functions, and that can handle extremely large nonlinear models for real-time optimization (on the order of millions of variables).
-
There is a need for improved modeling tools to accommodate heuristic and qualitative reasoning.
-
There is a need for hierarchical computations for conceptual design, which will require knowledge of new developments in computer science.
-
There is a need for information-modeling tools, which will become increasingly important for supporting integration problems as well as teamwork.
Finally, there are new potential applications from software and Internet-based computing. While the former is likely to be most relevant for a few specialists, the demand for software development may increase as chemistry and chemical engineering move to new areas in which there are no standard software packages. For Internet-based computing an exciting possibility will be to share more readily new software developments directly from the developers, bypassing the commercial software vendors. Another area of sharing leading to powerful new computational opportunities in the chemical sciences is the use of peer-to-peer computing in the form of sharing unused cycles on small computers. We can do very large-scale computations on networks of personal computers, as is being done in studies of protein folding9 and molecular docking.10 The potential here is to tackle computational problems of unprecedented size and complexity, with a relatively low investment in the actual computational resources.
Scientific Computing
Advances in scientific computing will help to address some of the challenges in computational chemistry and process systems engineering, particularly computational tasks that scale exponentially with size. While single-threaded execution speed is important and needed, coordination of multiple instruction multiple data (MIMD) computer systems is rapidly becoming the major challenge in scientific computing. The optimal parallel organization is application dependent: synchronous systems execute multiple elementary tasks per clock cycle while asynchronous models use clusters, vector units, or hypercubes. Although work in automatic task parallelization has made significant progress, the major issues in scientific high-performance computing are likely to be identification of suitable hardware architectures, algorithms to reformulate problems into a sequence of parallelizable subtasks with the “weakest possible” couplings, and nondeterministic procedures with statistical properties. On the positive side, parallel computing is becoming almost routine as individual researchers, groups, universities,
|
9 |
http://www.stanford.edu/group/pandegroup/Cosm; V.S. Pande and D.S. Rokhsar, Proceedings of the National Academy of Sciences, U.S.A. 96, 9062-9067, 1999; V. S. Pande, A. Y. Grosberg, T. Tanaka, and D. S. Rokhsar, Current Opinions in Structural Biology, 8:68-79, 1997. |
|
10 |
http://www.chem.ox.ac.uk/curecancer.html; Robinson, Daniel D.; Lyne, Paul D.; Richards, W. Graham, J. Chem. Inf. Comput. Sci., 40(2), 503-512, 2000. |
and companies embrace low-cost cluster parallel computers made from commodity off-the-shelf processors and network interconnects. From a computational science point of view, however, this multiplies the complexity of delivering higher performance computational tools to practicing researchers. Even when the number of parallel supercomputer vendors peaked in the mid-1990s, the number of manufacturers, processors, and network architectures was limited to a handful of such systems; by contrast, the number of possible cluster configurations is enormous.
Beyond parallelization, the major thrust of high-performance computing today is computational grids, modeled after the electrical power grid, in which the computational resources of a large number of sites are combined through wide area networks into a computational metaresource. Significant scientific and technical obstacles associated with the grid computational environment must be overcome, since grids embody all the complexity of parallel computers with additional difficulties such as distributed ownership of resources, multiple and wide-ranging network latencies, and heterogeneous architectures, to name just three. Resolution of these obstacles is one of the major thrusts of computational science research today. Nevertheless, assuming these obstacles can be overcome, grid computing offers the possibility of solving the largest scale problems confronting the computational chemistry and process engineering communities in the most cost-effective way. They can also facilitate collaboration between groups with complementary expertise that can share their capabilities while maintaining control over them. One can foresee the day when integrated process/product design/optimization (IPPDO) is performed on a wide area grid in which dedicated servers perform needed parts of the overall calculation, with the most compute-intensive computations being executed on the highest performance nodes of the grid.
As the sophistication of computation approaches increases, and the desire to integrate product and process design and optimization becomes reality, we anticipate that large-scale computations will be required that may last for days or weeks (even on the fastest computational resources) and that these computations will require intervention and redirection (known as computational steering) by the experts managing the computation. Computational steering is a relatively new field that is being applied in such areas as global climate modeling. Enabling computational steering requires the development of appropriate visualization tools that communicate the relevant information to the users, and interfaces to the computation that enable quantities and methods to be changed. Tools specific to the integrated IPPDO application will need to be developed.
Another new capability whose impact is still somewhat difficult to predict is wireless computing. One area that is likely to benefit is in the integration of measurements, control, and information systems. Wireless computing may also play an important role in computational steering of long computations described above, since examining the state of a computation, consulting with colleagues, and steer
ing the computation can all be envisaged as taking place on an appropriately enabled handheld device. Many of these tools exist in a disconnected and primitive form today: Much of the challenge lies in integrating the tools and adapting them for the IPPDO application.
WHY ALL THIS IS IMPORTANT
In the final analysis, basic understanding of chemistry will require successful theoretical approaches. For example, in our picture of the exact pathways involved in a chemical reaction there is no current hope that we can directly observe it in full molecular detail on the fast and microscopic scale on which it occurs. As discussed in Chapter 4, our ability to make a detailed picture of every aspect of a chemical reaction will come most readily from theories in which those aspects can be calculated, but theories whose predictions have been validated by particular incisive experiments.
When we have the information from the sequencing of the human genome, and want to understand the properties of those proteins that are coded by some of the genes but not yet known experimentally, we need to solve the protein-folding problem. Then we can translate the gene sequence—which specifies the sequence of amino acids—into the three-dimensional structure of the unknown protein.
For practical applications, good effective theories and computational tools are invaluable. We want to calculate the properties of molecules that have not yet been made, to select a likely medicine for synthesis. We want to be able to calculate what catalyst would best speed a particular reaction with selectivity, so that catalyst can be created and used in manufacturing. We want to calculate the properties of organized chemical systems, nanometer-sized particles, and aggregates whose properties can be valuable in computers and in other electronic devices. We need to develop new and powerful computational methods that span from the atomic and molecular level to the chemical-process and chemical-enterprise level in order to allow their effective integration for multiscale simulation and optimization. We want to synthesize energy-efficient and environmentally benign processes that are cost effective. We want to manage networks of plants that eliminate inventories and can be operated in a safe and responsive manner. With increasingly powerful computers and better software, these goals seem within reach in the future, and they will greatly enhance our capabilities—both in basic and applied chemistry and in chemical engineering.
As discussed above, simulation and modeling are central components of process engineering. Improvements in these techniques will permit the design of much more efficient processes and facilities. Integrating the current and future capabilities of computational chemistry and process engineering will result in improved materials, chemicals, and pharmaceuticals; yield more efficient environmentally benign processes to manufacture them; and accomplish this while providing greater financial return.
























