
required to answer the study question of interest to the ISIS group was 10,000 patients. Using unpaid clinical investigators and a one-page case report form the ISIS group randomized the 10,000 patients necessary to answer the relevant clinical question. The ISIS trial changed the study of ACS in that future trials sought larger sample sizes to appropriately answer the main outcome questions of interest in cardiovascular disease. However, the fact that the ISIS trial did not pay investigators makes its duplication in any country today impossible, according to Califf. Another critical change in the conduct of STEMI trials came when the U.S. Food and Drug Administration (FDA) moved toward requiring outcome trials to evaluate critical clinical endpoints related to the safety and effectiveness of a drug. Califf explained that the new FDA standard for STEMI trials changed this field of research and improved the clinical relevance of trials conducted by industry and academia.
Recognizing a practical, clinical inception time for the spectrum of ACS improved the ability of practitioners to identify a study population. The primary medical action in ACS occurs in the first 24 hours of a patient’s hospital stay. In the past, diagnosing a type of ACS often required a patient to be admitted, receive an ECG, and have a physician evaluate enzyme levels 24 hours later to determine whether the patient had experienced a heart attack, and then for the physician to refer to the ECG results to determine whether it was STEMI or non-STEMI. Elliott Antman, Senior Investigator in the TIMI Study Group, developed a system for classifying patients at the time of the ECG (Figure 4-1). Califf explained that creating an easily identifiable marker by which to classify patients in the emergency room made it less expensive and time-consuming for busy practitioners to become involved in locating patients for a clinical trial. This practitioner-oriented classification system is especially helpful in facilitating enrollment in cardiovascular disease trials, which typically seek to recruit a large number of patients per trial. Box 4-1 presents a case study illustrating the importance of developing a practical disease inception point.
THE THROMBOLYSIS IN MYOCARDIAL INFARCTION STUDY GROUP
As an example of a disease-focused research network, Marc Sabatine, investigator in the TIMI Study Group, discussed the group’s research work and structure. Headquartered at Brigham and Women’s Hospital and Harvard Medical School, TIMI is an academic research organization (ARO) conducting clinical trials to improve health outcomes in patients with cardiovascular disease. Conducting studies from phase I to IV, TIMI has completed 45 clinical trials to date, and has 6 ongoing trials and 7 in the planning stages. Trial sizes range from 30 to 25,000 subjects. Sabatine
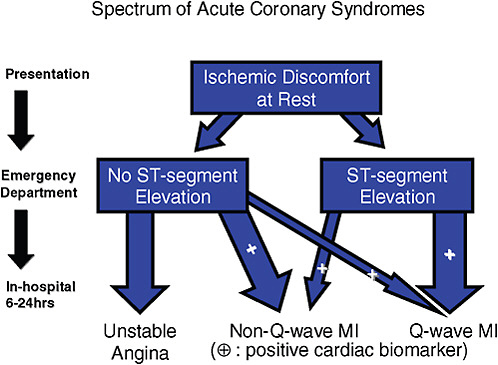
FIGURE 4-1 Classification system for the spectrum of acute coronary syndromes that helps practitioners identify a study population more easily.
NOTE: MI = myocardial infarction.
SOURCE: Califf, 2009. (Adapted from 2007 American Heart Association/American College of Cardiology Guidelines.) Reprinted with permission from Robert Califf 2010.
|
BOX 4-1 Case Study: Acute Decompensated Heart Failure and Acute Myocardial Infarction The importance of developing a practical inception point for a disease is highlighted by comparing two diseases—acute decompensated heart failure and acute myocardial infarction (MI). The two diseases affect the same number of people and have roughly the same mortality and readmission risks. As illustrated in the table below, in 2006 acute decompensated heart failure lacked evidence-based guidelines, whereas acute MI had a robust base of scientific evidence based on large randomized trials. |
noted that the academic leadership at the core of TIMI’s work participates in each stage of the clinical trial process, including:
-
Reviewing the compound—reviewing the pharmacokinetic (PK), pharmacodynamic (PD), animal model, and phase I data for a compound being considered for further study.
-
Refining the scientific question—asking whether a new compound addresses an unmet clinical need in cardiovascular medicine, as well as what utility the compound has and how it relates to current and evolving concomitant treatments.
-
Initiating the study—helping to plan and initiate both investigator-and industry-initiated studies.
-
Developing study design—helping to determine the study population, the timing of the intervention, the appropriate control arm and background therapy, the endpoints and timing of ascertainments, and the statistical analysis plan.
-
Developing key trial documents—collaborating on the development of the trial protocol, case report form, Clinical Events Committee (CEC) charter, and Data Safety Monitoring Board (DSMB) charter.
-
Study startup—considering the country and site for a trial, the applicability of trial results in the United States, the acceptability of the trial to other countries, and the cost of conducting the trial.
-
Monitoring study progress—monitoring patient enrollment, any changes in the medical landscape, aggregate event rates (efficacy and safety), and the retention of patients.
-
Leading study analysis—conducting data analysis on a database separate from that received by trial sponsors and moving rapidly to present the data at scientific meetings, as well as drafting primary manuscript of the trial results and engaging in subsequent data analyses.
When Eugene Braunwald founded the group in 1984, TIMI trials were funded by the National Heart, Lung, and Blood Institute (NHLBI). Today the trials are funded by industry, with some NIH grant support for ancillary studies. In response to a question from Califf, Sabatine described TIMI’s studies as being relatively complex, having case report forms that are more than one or two pages, and seeking to answer a variety of questions. Strong academic support for the trial activities listed above is important to the group as a whole and would be very difficult to maintain through government funding alone.
Sabatine stated that one of the strengths of the TIMI group is the relative leanness of the organization. The physician staff includes the study chairman, 12 staff cardiologists, three senior cardiology fellows, and a
rotating staff of research residents from Brigham and Women’s Hospital. Operational staff includes a director, eight project directors and managers, and research assistants. Core biostatistics staffs, including a director and multiple programmers, round out the TIMI group. Sabatine described an intense working relationship between physician staff and operational staff as important to ensuring that the trials are conducted in the best possible way. For a typical TIMI trial, two physicians and multiple operational staff work closely on planning and execution. Physician and operational staff offices are located next to one another, which facilitates daily conversations to guide a trial from inception to completion.
Investigator- and Industry-Initiated TIMI Studies
TIMI trials generally fall into two broad categories: (1) investigator-initiated studies, in which an academic investigator who is interested in the field of cardiovascular medicine is the driving force behind the study, and (2) industry-initiated studies, in which the TIMI group is approached by a company to address a particular question. Both Sabatine and Christopher Cannon, senior investigator, TIMI Study Group, noted that TIMI involves practicing cardiologists and researchers in the development of scientific questions to be addressed and encourages them to consider where they see the field moving in terms of current and evolving therapy for cardiovascular disease.
Sabatine presented case examples of both investigator- and industry-initiated studies. In each example, the flexibility of the TIMI group is highlighted, as well as its trademark of core academic leadership.
Investigator-Initiated TIMI Study
At the suggestion of TIMI investigator Cannon, the study group approached the makers of an antiplatelet drug, clopidogrel, which had been approved for patients with less severe types of heart attacks. Cannon wanted to test whether clopidogrel would also benefit patients with more severe STEMI. As a result, the phase III CLARITY TIMI 28 trial involving nearly 3,500 patients was launched. The trial showed that administering clopidogrel to patients receiving a thrombolytic therapy for their heart attack decreased the odds of their having a blocked artery and translated to a decreased rate of clinical events. Simultaneous to the CLARITY TIMI 28 study, a very large, simple trial of the drug in China in which patients received a non-Western form of care (e.g., without the use of angiography) yielded similar results. Taken together, the results of CLARITY TIMI 28 and the Chinese trial provided a compelling case for including clopidogrel
in the care of STEMI patients. As a result, in 2007 guidelines adopted this change and made STEMI a Class I indication for the drug.
The TIMI work with clopidogrel continued when data were released indicating that there was some variability in response to the drug. Certain individuals were found to be “clopidogrel-resistant,” meaning the drug did not result in substantial platelet inhibition. Observational studies linked the clopidogrel resistance to worse clinical outcomes. Eli Lilly subsequently approached TIMI with a new drug, prasugrel, which is in the same class as clopidogrel. The academic leadership at TIMI examined data for prasugrel and clopidogrel and determined that the potential increased platelet inhibition of prasugrel made it a promising compound to study. TIMI conducted a phase II efficacy trial, which found that prasugrel showed promise compared with clopidogrel for decreasing the risk of heart attack. These phase II results led to a very large international, multicenter phase III double-blind RCT of prasugrel and clopidogrel involving more than 13,000 patients. The TRITON TIMI 38 trial showed a benefit of prasugrel over clopidogrel with respect to cardiovascular death, nonfatal heart attack, and nonfatal stroke. However, the increased platelet inhibition that brought these heart benefits also translated into an increased risk of bleeding in patients taking prasugrel. The end result of this series of trials was FDA approval of Eli Lilly’s prasugrel in July 2009. In this case, the investigator-initiated small phase II trial led to a more robust clinical trial that ultimately resulted in a new FDA-approved drug to treat cardiovascular disease.
Industry-Initiated TIMI Study
TIMI is frequently contacted by companies with new compounds for study and potential further development. Sabatine shared an example of an industry-initiated study run by TIMI that yielded a positive result for the sponsoring company’s competitor drug. PROVE-IT TIMI 22 was sponsored by Bristol-Myers Squibb to compare its drug, pravastatin, with a new drug, atorvastatin, which had been shown in earlier trials to be effective in lowering LDL cholesterol. According to Sabatine, the company was confident of the positive results of a number of RCTs comparing pravastatin with placebo and believed pravastatin offered patients additional benefits that atorvastatin lacked. However, PROVE-IT TIMI 22 showed that atorvastatin outperformed pravastatin in helping patients achieve lower LDL. The results of this trial and others led to an update in clinical practice guidelines for targeting LDL.
U.S. Enrollment in TIMI Trials
Sabatine discussed the competing factors that influence the decision of TIMI leaders to study a drug in the United States or internationally. He noted the importance of conducting trials in the United States so as to understand the drug’s applicability to the U.S. patient population. But, he noted, there are also reasons to ensure that clinical trials have a global presence. For example, in a global market, regulatory bodies in other countries will insist that a trial be conducted in their countries as well. When TIMI conducts phase II trials, involvement in multiple countries is desirable in anticipation of an international phase III trial. The lower cost associated with international trial sites is also a consideration in determining trial locations (see Chapter 3). TIMI created a Steering Committee of National Lead Investigators comprising key opinion leaders from various countries. This infrastructure allows TIMI to maintain an international presence throughout the clinical trial process, from site selection to patient follow-up and retention.
As Michael Lauer, Director of the Division of Cardiovascular Diseases, NHLBI, indicated in his presentation (see below), medical practice in the United States recently shifted from thrombolytic, or clot-busting, medications to primary angioplasty. As the manual opening of arteries became more prevalent in the United States, it became more difficult to find centers that were using medications, or lytic therapy, to break up clots. Worldwide, thrombolytic medications are still the most common form of reperfusion therapy, according to Sabatine. Thus, for trials that involve testing lytic therapy, international sites can provide patients more readily than U.S. sites.
Sabatine noted that not having adequate U.S. representation in clinical trials can be dangerous: when the proportion of U.S. patients decreases, the risk of spurious subgroup findings increases. In the recent PLATO trial comparing clopidogrel and ticagrelor (platelet inhibitors), the subgroup of patients in North America showed no benefit of ticagrelor, despite a finding that the 29 other patient subgroups experienced lower rates of clinical events due to the drug. This lack of effect in the U.S. population is likely a spurious finding, according to Sabatine, considering that subgroups in Europe, where practice patterns are similar to those in the United States, saw a large benefit from the drug.
THE OCCLUDED ARTERY TRIAL
Lauer discussed OAT as a case study that highlights a number of common obstacles encountered in conducting a large clinical trial with significant ramifications for clinical practice.
Acute MI (heart attack) is caused by the occlusion of coronary arteries
that supply blood to the heart. A cardiologist treating such a case can perform a percutaneous intervention (PCI), or cardiac catheterization, in which a stent is inserted to open the affected arteries. In the 1990s, observational studies showed a marked improvement in mortality rates for those patients with open arteries. As a result, there was a widespread increase in the use of cardiac catheterization to open the arteries of heart attack patients, and, despite the lack of robust scientific evidence supporting PCI, it became conventional medical wisdom to use this procedure for patients with chronically occluded arteries after a heart attack. OAT, an NHLBI-supported experimental trial, eventually revealed that health outcomes were worse among those subjects randomized to receive stents, dispelling the belief that opening arteries through PCI is always desirable. OAT illustrates the importance of basing medical decision making on well-controlled clinical experiments as opposed to observational correlations. According to Lauer, the trial is also an example of how the federal government can provide real value in clinical research, particularly when focusing on existing clinical practices for which evidence is lacking.
Lauer also explained that a number of logistical and administrative obstacles were encountered in running OAT. Each obstacle highlights the extent to which the incentives and interests of stakeholders, as well as regulatory burdens, can hinder a trial. As indicated in Figure 4-2, OAT included 2,166 patients and took 10 years to complete. Along the way, investigators encountered delays in grant review. Investigators also faced an unexpected requirement to conduct a pilot study, for which they received no funding, to improve their chances of ultimately receiving NIH funding. After final grant approval, of the 926 sites approached to participate in the study, only one-third agreed to do so. The investigators believe this unwillingness to participate was due partly to a pro-PCI bias on the part of physicians. Under this theory, because some physicians believed PCI was clearly the appropriate course of treatment, they saw no value in participating in a clinical trial to test this assumption. Some physicians claimed it would be unethical to assign patients to a control group that would receive the best medicine for the condition but not PCI. Califf suggested that the lack of physician interest in participating in this trial was also associated with the fact that PCI is a profitable procedure for physicians, and participation in the trial would randomize only a portion of their patients to receive the procedure, thus eliminating the opportunity to administer it to every patient.
Recruiting patients to participate in the trial also created delays. Of those sites that did participate, only two-thirds enrolled at least one patient. One-third of the sites filled out all required paperwork and received Institutional Review Board (IRB) approval, but never enrolled a patient. The overall enrollment rate was 0.25 patients per site per month. Ultimately, only 488 of the 2,166 patients were enrolled from 85 U.S. sites; the vast majority of subjects had to be enrolled abroad.
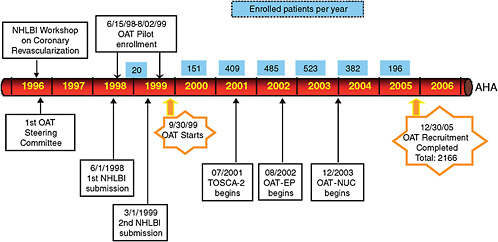
FIGURE 4-2 Timeline for the NIH-sponsored Occluded Artery Trial (OAT).
NOTE: NHLBI = National Heart, Lung, and Blood Institute; OAT = Occluded Artery Trial; OAT-NUC = Occluded Artery Trial Nuclear Study; TOSCA-2 = Total Occlusion Study of Canada-2.
SOURCE: Lauer, 2009. Reprinted with permission from Michael Lauer and Judith Hochman 2009.
According to Lauer, the OAT experience highlights a number of important points:
-
Relying on the gold standard—RCTs remain the gold standard for proving the effectiveness of a drug or therapy. Observational data and unproven associations are susceptible to confounding and can be misinterpreted.
-
Federally sponsored research—The federal government plays an important role in funding and conducting large clinical trials to test the effectiveness of clinical practices that are in widespread use but do not have an evidence base.
-
Biases—Physicians and patients often have strong preferences for and biases toward certain procedures or levels of care they believe to be appropriate, regardless of the evidence supporting them.
-
Misaligned financial incentives—Financial incentives for physicians are focused on performing a high volume of procedures, which creates a disincentive for them to refer patients to a clinical trial outside of their practice.
-
Lengthy timeline to conduct a large, multicenter trial—Navigating regulatory barriers and overcoming difficulties in recruiting the right group of patients to participate in a study can lead to substantial delays in conducting a trial (see Chapter 3). In the case of OAT, the timeline was 10 years.










