
Lyme Disease: A Growing Threat to Urban Populations
ALLEN C. STEERE
Lyme disease was recognized as a separate entity in 1975–1976 because of geographic clustering of children in Lyme, Connecticut, who were thought to have juvenile rheumatoid arthritis (1). The rural setting of the case clusters, the usual onset of illness in the summer and early fall, the onset in members of the same family in different years, and the recognition that erythema migrans was a feature of the illness suggested that the disorder was transmitted by an arthropod. Epidemiologic studies of patients with erythema migrans implicated certain ixodes ticks as vectors of the disease (2, 3). Prospective studies of these patients showed that the illness could affect multiple systems over a period of years, including the skin, nervous system, heart, or joints (4).
Erythema migrans also linked Lyme disease in the United States with certain syndromes described previously in Europe. Early in this century, Afzelius in Sweden (5) and Lipschutz in Austria (6) described a characteristic expanding skin lesion, which they called erythema migrans or erythema chronicum migrans. Many years later, it was recognized that erythema migrans could be followed by a chronic skin disease, acrodermatitis chronica atrophicans, which had already been described as a separate entity (7). In the 1940s, Bannwarth (8) defined a syndrome that consisted of radicular pain followed by
Allen C. Steere is chief of rheumatology and immunology at New England Medical Center and professor of medicine at the Tufts University School of Medicine.
chronic lymphocytic meningitis and sometimes cranial or peripheral neuritis. In a few cases, the neurologic syndrome was preceded by an erythema.
These various syndromes were brought together conclusively in 1982 when a previously unrecognized spirochete, now called Borrelia burgdorferi, was recovered from Ixodes dammini ticks (also called Ixodes scapularis) (9). The spirochete was then isolated from patients with Lyme disease in the United States (10, 11) and from those with erythema migrans, Bannwarth's syndrome, or acrodermatitis in Europe (12–14). In addition, the immune responses of patients were linked conclusively with infection with this organism.
Although the basic outlines of the illness are similar worldwide, regional variations have been noted (15). Lymphocytoma, acrodermatitis, and encephalomyelitis have been seen primarily in Europe (16, 17), whereas widely disseminated early infection, secondary annular skin lesions, and arthritis have been found more commonly in America (18, 19). Recent work in the classification of B. burgdorferi has begun to clarify the issue of geographic differences in Lyme disease. Using a variety of methods, three genomic groups of B. burgdorferi have now been identified (20, 21). To date, all North American strains have belonged to the first group, B. burgdorferi sensu stricto. Although all three groups have been found in Europe, most isolates have been group 2 or 3 strains. Group 2 strains have been renamed Borrelia garinii (21), and group 3 strains have been renamed Borrelia afzelii (81).
VECTOR AND ANIMAL HOSTS
Lyme disease is transmitted in all of these locations by several closely related ixodid ticks that are part of the Ixodes ricinus complex. These include I. dammini (also named I. scapularis) in the northeastern and midwestern United States (3, 22), Ixodes pacificus in the western United States (82), I. ricinus in Europe (23), and Ixodes persulcatus in Asia (24). The endemic cycle of B. burgdorferi varies among geographic locations. In the northeastern and midwestern United States, the preferred host for both the larval and nymphal stages of I. dammini is the white-footed mouse Peromyscus leucopus (22). It is critical that this rodent host is tolerant to infection with B. burgdorferi and that both of the tick's immature stages feed upon this host, since the life cycle of the spirochete depends upon horizontal transmission between immature ticks and mice (25). The preferred host for the adult stage of I. dammini is the white-tailed deer Odocoileus virginianus (26). Although deer are not involved in the life cycle of the spirochete, they seem to be critical for
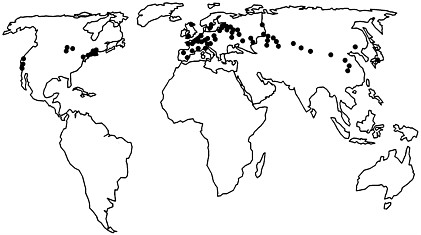
FIGURE 1 Worldwide locations of Lyme disease: Areas in North America, Europe, and Asia that are affected by Lyme disease. These areas are along the terminal moraine of the glaciers 15,000 years ago.
survival of the ticks (27). The endemic cycle of B. burgdorferi is different in the western United States. There, the spirochete is maintained in nature in a horizontal cycle between the dusky-footed woodrat Neotoma fuscipes and Ixodes neotomae, a tick that does not feed on humans (28). Only the relatively few larval and nymphal I. pacificus that feed upon infected woodrats, rather than lizards, are then responsible for transmitting the spirochete to humans. For this reason, only about 1% of nymphal I. pacificus are infected with B. burgdorferi (28), whereas infection rates in nymphal I. dammini in the northeast range from 20% to 50% (10, 29).
EVOLUTION OF ENVIRONMENT
The primary areas now affected by Lyme disease in the United States, Europe, and Asia are sites near the terminal moraine of the glaciers 15,000 years ago (Figure 1) (30, 31). At that time, the northeastern and upper midwestern parts of what is now the United States were covered by tundra. As the glaciers retreated, forests grew in these areas, which eventually became populated with large numbers of deer. Early descriptions of colonial New England include comments about the abundance of deer and annoying ticks (32). During the 18th and 19th centuries, forests were destroyed in New England
to make farms, and deer were hunted practically to extinction. Deer are thought to have survived only in a few isolated locations, such as Naushon Island near Cape Cod and on the eastern end of Long Island, New York (31). Analysis of museum tick collections by polymerase chain reaction (PCR) has documented the presence of B. burgdorferi -infected I. scapularis on Montauk Point, Long Island 50 years ago (33).
The emergence of Lyme disease in the United States in the last several decades is thought to have occurred primarily because of ecological conditions favorable for deer (22, 34). As farmland in the northeast reverted to woodland, the habitat for deer improved: their natural predators were gone, the number of deer increased dramatically, they migrated to new areas, and federal programs protected them. With the advent of the automobile and superhighways, rural and suburban areas, where deer now lived, became populated with large numbers of susceptible suburbanites who had never been exposed to the spirochete. Lyme disease and the deer tick do not occur in all areas where deer are found. Other factors such as temperature, vegetation, and rodent populations must also play an important role in the ecology of this disease.
EPIDEMIOLOGY OF LYME DISEASE
Lyme disease is now the most common vector-borne infection in the United States (35). The infection is usually acquired when nymphal ticks feed between May and July, but adult ticks occasionally transmit the
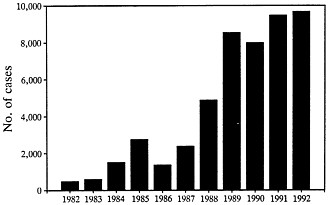
FIGURE 2 Cases of Lyme disease reported to the Centers for Disease Control from 1982 through 1992 (data from ref. 36).
disease when they feed in autumn. People of all ages and both sexes are affected. From 1982 through 1991, 40,195 cases occurring in 47 states were reported to the Centers for Disease Control (Figure 2) (35). In 1992, 45 states reported a provisional total of 9677 cases, a 19-fold increase over the number of cases reported in 1982. However, enzootic cycles of B. burgdorferi have been identified in only 19 states, and 94% of the cases reported in the last 2 years have come from these states (35). Three distinct areas are most affected: the northeast from Massachusetts to Maryland, the upper midwest in Wisconsin and Minnesota, and the west in northern California and Oregon (3, 35). As would be anticipated from the infection rate in ticks, the frequency of the disease is much greater in the northeast and midwest than in the west. The infection also occurs in most European countries, particularly in northern parts of the continent, including Germany, Austria, and Sweden (36). In the former Soviet Union, a central area is affected from the Baltic Sea to the Pacific Ocean (24). Cases have also been reported in China (37), Japan (38), and Australia (39).
During the last several decades, Lyme disease has spread and has caused focal epidemics, particularly in the northeastern United States. In 1980, in Ipswich, Massachusetts, a community close to Boston, I. dammini became established in a nature preserve that had many deer (40). During the next 7 years, clinical symptoms of Lyme disease developed in 35% of the 190 residents of the area adjacent to the preserve. In other outbreaks, 16% of the 162 permanent residents of Great Island, Massachusetts, had the illness, in most instances between 1972 and 1979 (41); and 7.5% of the 200 people who participated in a study on Fire Island, New York, had the disorder during a 5-year period (42). Between 1985 and 1989, the number of counties in New York State with documented I. dammini ticks increased from 4 to 22, and the number of counties endemic for Lyme disease increased from 4 to 8 (43). In addition to the eastern end of Long Island, Westchester County, a heavily populated area close to New York City, became a hyperendemic site of the disease. In recent years, suburban communities in Morris and Monmouth Counties in New Jersey, which are also close to New York City, have become heavily affected (44), and suburban areas near Philadelphia are now endemic for the disease. I. dammini continues to invade new areas, and the ultimate limit of this spread is not known.
The fear of Lyme disease in these suburban communities has become marked (45). Residents in these areas regularly see deer in their yards. Protective measures, such as long clothing, sprays, and tick checks, are difficult to keep up throughout the summer. Because of the small size of nymphal I. dammini, tick bites often go unnoticed.
TABLE 1 Stages of Lyme disease
|
Early infection |
|
Stage 1 (localized infection) Erythema migrans |
|
Stage 2 (disseminated) Skin Musculoskeletal Nervous system Lymphadenopathy Heart Eyes Liver Respiratory Genitourinary |
|
Late infection |
|
Stage 3 (persistent infection) Chronic arthritis Late neurologic involvement Acrodermatitis |
CLINICAL PICTURE
Lyme disease or Lyme borreliosis is most like syphilis in its multisystem involvement, occurrence in stages, and mimicry of other diseases (15). Early infection (stage 1) consists of localized erythema migrans (Table 1). Within days to weeks (stage 2), the spirochete often disseminates to multiple sites, manifested most commonly by secondary annular skin lesions (18), meningitis, facial palsy, radiculoneuritis (46), atrioventricular nodal block (47), or migratory pain in joints, tendons, bursae, muscle, or bone (19). Even in untreated patients, these early manifestations usually resolve or improve within weeks or months. Late or persistent infection (stage 3) usually begins months to years later and typically consists of intermittent or chronic arthritis (19), chronic neurologic involvement (48–51), or acrodermatitis chronica atrophicans (52).
Late neurologic abnormalities of Lyme disease are still being defined, and diagnosis of this manifestation of the disorder has created the most difficulty. From months to years after disease onset, sometimes following long periods of latent infection, patients may develop a subtle encephalopathy, primarily manifested as memory deficit, irritability, or somnolence (49). Neuropsychological tests of memory are often abnormal, and, in some cases, white matter lesions are found on magnetic resonance imaging scans of the brain. Cerebrospinal fluid may show elevated total protein, intrathecal antibody production to B. burgdorferi (53), or a positive PCR test result for borrelial DNA sequences (54, 55).
Most patients with subacute encephalopathy also have an axonal polyneuropathy manifested by spinal radicular pain or by numbness and tingling in the hands or feet (50, 51). Electromyography generally shows extensive abnormalities of proximal and distal nerve segments (51). Leukoencephalitis, a rare manifestation of Lyme disease, is a severe neurologic picture that may include spastic paraparesis, upper motor neuron bladder dysfunction, and lesions in the periventricular white matter (16, 49). These patients have been distinguished from those with multiple sclerosis on the basis of selective concentration of antibody to B. burgdorferi in cerebrospinal fluid. Since patients with late neurologic abnormalities of Lyme disease usually respond to antibiotic therapy, active spirochetal infection is thought to be important in the pathogenesis of these syndromes.
CHRONIC FATIGUE OR FIBROMYALGIA FOLLOWING LYME DISEASE
Chronic fatigue syndrome or fibromyalgia, which may be variants of the same disorder, share certain symptoms with the encephalopathy or polyneuropathy of Lyme disease. The distinction among these entities has been further confused by the fact that a small percentage of patients have developed chronic fatigue or fibromyalgia in association with or soon after Lyme disease (56), suggesting that B. burgdorferi is one of the infectious agents or stressful events that may trigger these syndromes. Compared with Lyme disease, chronic fatigue syndrome or fibromyalgia tends to produce more generalized and disabling symptoms. They include marked fatigue, severe headache, diffuse musculoskeletal pain, multiple symmetric tender points in characteristic locations, pain and stiffness in many joints, diffuse dysesthesias, difficulty with concentration, or sleep disturbance. These patients lack evidence of joint inflammation; they have normal neurologic test results; and they usually have a greater degree of anxiety and depression (57). In the author's experience, the symptoms of fibromyalgia, even when triggered by infection with B. burgdorferi, continue to wax and wane for years, despite treatment with antibiotic therapy. Therefore, chronic fatigue syndrome or fibromyalgia associated with Lyme disease would not appear to require the presence of a live spirochete for continued symptoms of the illness.
MISDIAGNOSIS OF CHRONIC LYME DISEASE
A recent phenomenon is that a number of poorly understood conditions, such as fibromyalgia or chronic fatigue syndrome, are misdiagnosed as ''chronic Lyme disease." Of the 788 patients seen in the
author's Lyme disease clinic during a 4.5-year period, 180 (23%) had active Lyme disease, usually arthritis, encephalopathy, or polyneuropathy (58). One hundred and fifty-six patients (20%) had previous Lyme disease and another current illness, most commonly chronic fatigue or fibromyalgia; and in 49 patients, these symptoms began soon after objective manifestations of Lyme disease. The remaining 452 patients (57%) did not have Lyme disease. The majority of these patients also had the chronic fatigue syndrome or fibromyalgia; the others usually had rheumatic or neurologic diseases. Prior to referral, 409 of the 788 patients had been treated with antibiotic therapy. In 322 of these patients (79%), the reason for lack of response was incorrect diagnosis. Thus, only a minority of the patients referred to the clinic met diagnostic criteria for Lyme disease, and the most common reason for lack of response to antibiotic therapy was misdiagnosis.
DIAGNOSTIC TESTS
Part of the reason for misdiagnosis is due to problems associated with diagnostic tests. In most instances, culture has yielded positive results only from biopsy samples of erythema migrans skin lesions (59, 60). Therefore, serologic testing is currently the only practical laboratory aid in diagnosis. Serologic testing for Lyme disease can be done with a high degree of sensitivity and specificity, and after the first weeks of infection, almost all patients have an elevated antibody response to B. burgdorferi (61). However, B. burgdorferi contains a number of cross-reactive antigens, the test is insensitive early in the infection, and test procedures are not standardized. For these reasons, false-negative and, more commonly, false-positive results have been a considerable problem (62, 63). In the author's laboratory, antibody determinations are performed by ELISA, using sonicated whole spirochetal lysates as the antigen preparation (61). The results are divided into three groups: negative, indeterminate, or positive (Table 2). In patients with indeterminate responses by ELISA, Western blotting is done to determine whether the response is positive or negative. In the future, serologic tests that employ several recombinant antigens will probably replace the current antigen preparations, which use whole cell lysates.
Regardless of the antigen preparation, serologic tests do not distinguish active from inactive infection. Patients who have had late manifestations of Lyme disease usually remain seropositive for years after treatment. If these patients develop other illnesses, particularly those with joint or neurologic symptoms, the positive test for Lyme disease may cause diagnostic confusion. It has been hoped that the PCR might have a role in the diagnosis of active Lyme disease equivalent to culture
TABLE 2 Criteria for positive serologic tests for Lyme disease*
in common bacterial infections. In a recent study, B. burgdorferi DNA was detected in the joint fluid of 75 of 88 patients with Lyme arthritis (85%) and in none of 64 control patients (83). Borrelial DNA sequences have also been detected in blood, cerebrospinal fluid, urine, or skin of patients with Lyme disease (54, 55, 64, 65), but the value of PCR as a reliable diagnostic test is still being researched.
TREATMENT
The various manifestations of Lyme disease can usually be treated successfully with oral antibiotic therapy, except for objective neurologic abnormalities, which seem to require intravenous therapy (15, 66). For early Lyme disease, doxycycline, 100 mg twice a day, or amoxicillin, 500 mg four times a day, is effective therapy (67, 68). For patients with infection localized to the skin, 10 days of therapy is generally sufficient, but for patients with disseminated infection, longer courses of 20–30 days are usually needed. For objective neurologic abnormalities, with the possible exception of facial palsy alone, intravenous ceftriaxone, 2 g/day, is most commonly used (49, 69, 70), but intravenous cefotaxime, 2 g three times a day (70), or intravenous sodium penicillin G, 5 million units four times a day, may also be effective (71). Treatment failures have occurred with any of these regimens, and the course of antibiotic therapy may need to be repeated.
PREVENTION
Most vector-borne diseases are prevented through vector control, but this has proved difficult with tick-borne diseases. In one study, eradication of deer on a small island greatly reduced the number of deer ticks during a 5-year period (26). Aerial application of carbaryl during the fall has been reported to be successful at reducing the number of ticks through the following spring (72). Habitat destruction by burning has long been considered an effective alternative to synthetic insecticides as a means of reducing tick populations (73). Another method involves the distribution of permethrin-treated cotton balls, intended as rodent nesting material, around individual residences during the summer and fall (74, 75). However, with each of these methods, fear of environmental consequences or lack of efficacy has limited their use.
Consequently, reduction of the risk of Lyme disease has been limited primarily to personal protection measures. The risk of tick bites can be reduced by wearing long clothing and by checking for ticks after exposure in wooded areas. Insecticides containing N,N-diethylmetatoluamide (DEET) or permethrin effectively deter ticks (76), but permethrin can only be applied on clothing, and DEET may cause serious side effects when excessive amounts are applied directly to the skin (77). Should patients with tick bites receive prophylactic antibiotic therapy? In one recent study, the risk of acquiring Lyme disease from a recognized tick bite was only 1.2%, perhaps because 24–48 hours of tick attachment is often required for transmission of the spirochete (78). Although amoxicillin or doxycycline therapy for 10 days will probably prevent the occurrence of Lyme disease in these patients, many patients must be treated to prevent one case. Thus, personal protection measures may help the city dweller who takes a long walk in the woods, but such measures are of limited benefit in areas where people have constant exposure to large numbers of ticks.
It is hoped that a vaccine can be developed for Lyme disease to provide protection in high-risk areas. Although reinfection may occur in patients treated early in the course of the illness, those with an expanded antibody response to the spirochete appear to have protective immunity. In an animal model of Lyme disease, mice vaccinated with recombinant outer-surface protein A (OspA) have been shown to be protected from infection with B. burgdorferi, both by antibody-mediated killing of the spirochete within the host and by destruction of the organism within the tick prior to disease transmission (79, 80). Trials are now under way to determine the efficacy and safety of OspA immunization in human subjects.
SUMMARY
Lyme disease or Lyme borreliosis, which is caused by three groups of the spirochete Borrelia burgdorferi, is transmitted in North America, Europe, and Asia by ticks of the Ixodes ricinus complex. The primary areas around the world that are now affected by Lyme disease are near the terminal moraine of the glaciers 15,000 years ago. The emergence of Lyme disease in the United States in this century is thought to have occurred because of ecological conditions favorable for deer. From 1982 through 1991, 40,195 cases occurring in 47 states were reported to the Centers for Disease Control, but enzootic cycles of B. burgdorferi have been identified in only 19 states. During the last several decades, the disease has spread to new areas and has caused focal outbreaks, including locations near Boston, New York, and Philadelphia. Lyme disease is like syphilis in its multisystem involvement, occurrence in stages, and mimicry of other diseases. Diagnosis of late neurologic abnormalities of the disorder has created the most difficulty. A recent phenomenon is that a number of poorly understood conditions, such as chronic fatigue syndrome or fibromyalgia, are misdiagnosed as "chronic Lyme disease." Part of the reason for misdiagnosis is due to problems associated with diagnostic tests. The various manifestations of Lyme disease can usually be treated successfully with oral doxycycline or amoxicillin, except for objective neurologic manifestations, which seem to require intravenous therapy. Vector control of tick-borne diseases has been difficult and, therefore, reduction of the risk of infection has been limited primarily to personal protection measures.
This work was supported in part by Grant AR-20358 from the National Institutes of Health.
REFERENCES
1. Steere, A. C., Malawista, S. E., Snydman, D. R., Shope, R. E., Andiman, W. A., Ross, M. R. & Steele, F. M. (1977) Arthritis Rheum. 20, 7–17.
2. Steere, A. C., Broderick, T. F. & Malawista, S. E. (1978) Am. J. Epidemiol. 108, 312–321.
3. Steere, A. C. & Malawista, S. E. (1979) Ann. Intern. Med. 91, 730–733.
4. Steere, A. C., Malawista, S. E., Hardin, J. A., Ruddy, S., Askenase, P. W. & Andiman, W. A. (1977) Ann. Intern. Med. 86, 685–698.
5. Afzelius, A. (1921) Acta Derm. Venereol. 2, 120–125.
6. Lipschutz, B. (1923) Arch. Dermatol. Syph. 143, 365–374.
7. Herxheimer, K. & Hartmann, K. (1902) Arch. Dermatol. Syph. 61, 57–76, 255–300.
8. Bannwarth, A. (1944) Arch. Psychiatr. Nervenkrankh. 117, 161–185.
9. Burgdorfer, W., Barbour, A. G., Hayes, S. F., Benach, J. L., Grunwaldt, E. & Davis, J. P. (1982) Science 216, 1317–1319.
10. Steere, A. C., Grodzicki, R. L., Kornblatt, A. N., Craft, J. E., Barbour, A. G., Burgdorfer, W., Schmid, G. P., Johnson, E. & Malawista, S. E. (1983) N. Engl. J. Med. 308, 733–740.
11. Benach, J. L., Bosler, E. M. Hanrahan, J. P., Coleman, J. L., Habicht, G. S., Bast, T. F., Cameron, D. J., Ziegler, J. L., Barbour, A. G., Burgdorfer, W., Edelman, R. & Kaslow, R. A. (1983) N. Engl. J. Med. 308, 740–742.
12. Ackermann, R., Kabatzki, J., Boisten, H. P., Steere, A. C., Hartung, S. & Runne, U. (1984) Dtsch. Med. Wochenschr. 109, 92–97.
13. Preac-Mursic, V., Wilske, B., Schierz, G., Pfister, H. W. & Einhaupl, K. (1984) Eur. J. Clin. Microbiol. 3, 564–565.
14. Asbrink, E., Brehmer-Andersson, E. & Hovmark, A. (1986) Am. J. Dermatopathol. 8, 209–219.
15. Steere, A. C. (1989) N. Engl. J. Med. 321, 586–596.
16. Ackermann, R., Rehse-Kupper, B., Gollmer, E. & Schmitt, R. (1988) Ann. N.Y. Acad. Sci. 539, 16–23.
17. Weber, K., Schierz, G., Wilske, B. & Preac-Mursic, V. (1984) Yale J. Biol. Med. 57, 13–21.
18. Steere, A. C., Bartenhagen, N. H., Craft, J. E., Hutchinson, G. J., Newman, J. H., Rahn, D. W., Sigal, L. H., Spieler, P. H., Stenn, K. S. & Malawista, S. E. (1983) Ann. Intern. Med. 99, 76–82.
19. Steere, A. C., Schoen, R. T. & Taylor, E. (1987) Ann. Intern. Med. 107, 725–731.
20. Marconi, R. T., Lubke, L., Hauglum, W. & Garon, C. F. (1992) J. Clin. Microbiol. 30, 628–632.
21. Baranton, G., Postic, D., Saint Girons, I., Borelin, P., Piffaretti, J. C., Assous, M. & Grimont, P. A. (1992) Int. J. Syst. Bacteriol. 42, 378–383.
22. Levine, J. F., Wilson, M. L. & Spielman, A. (1985) Am. J. Trop. Med. Hyg. 34, 355–360.
23. Krampitz, H. E. (1986) Zentralbl. Bakteriol. Mikrobiol. Hyg. 263, 21–28.
24. Dekonenko, E. J., Steere, A. C., Berardi, V. P. & Kravchuk, L. N. (1988) J. Infect. Dis. 158, 748–753.
25. Matuschka, F. R. & Spielman, A. (1986) Exp. Appl. Acarol. 2, 337–353.
26. Wilson, M. L., Adler, G. H. & Spielman, A. (1986) Ann. Entomol. Soc. Am. 78, 172–176.
27. Wilson, M. L., Telford, S. R., III, Piesman, J. & Spielman, A. (1988) J. Med. Entomol. 25, 224–228.
28. Brown, R. N. & Lane, R. S. (1992) Science 256, 1439–1442.
29. Bosler, E. M., Ormiston, B. G., Coleman, J. L., Hanrahan, J. P. & Benach, J. L. (1984) Yale J. Biol. Med. 57, 651–659.
30. Davis, M. B. (1983) in Late Quaternary Environments of the U.S., ed. Wright, H. E., Jr. (Univ. of Minn. Press, Minneapolis), Vol. 2, pp. 166–179.
31. Spielman, A., Telford, S. R., III, & Pollack, R. J. (1993) in Ecology and Environmental Management of Lyme Disease, ed. Ginsberg, H. S. (Rutgers Univ. Press, New Brunswick, NJ), pp. 83–96.
32. Severinghaus, C. W. & Brown, C. Y. (1983) in Changes in the Land: Indians, Colonists, and the Ecology of New England, ed. Cronon, W. (Hill & Wang, New York), p. 241.
33. Persing, D. H., Telford, S. R., Rys, P. N., Dodge, D. E., White, T. J., Malawista, S. E. & Spielman, A. (1990) Science 249, 1420–1423.
34. Mather, T. N., Ribiero, J. M. C. & Spielman, A. (1987) Am. J. Trop. Med. Hyg. 36, 609–614.
35. Centers for Disease Control (1993) Morbid. Mortal. Wkly. Rep. 42, 345–350.
36. Stanek, G., Pletschette, M., Flamm, H., Hirschl, A. M., Aberer, E., Kristoferitsch, W. & Schmutzhard, E. (1988) Ann. N.Y. Acad. Sci. 539, 274–282.
37. Ai, C., Hu, R., Hyland, K. E., Wen, Y., Zhang, Y., Qui, Q., Liu, X., Shi, Z., Zhao, J. & Cheng, D. (1990) Int. J. Epidemiol. 19, 1061–1065.
38. Kawabata, M., Baba, S., Iguchi, K., Yamaguti, N. & Russell, H. J. (1987) J. Infect. Dis. 156, 854.
39. Steward, A., Glass, J., Patel, A., Watt, G., Cripps, A. & Clancy, R. (1982) Med. J. Aust. 1, 139.
40. Lastavica, C. C., Wilson, M. L., Berardi, V. P., Spielman, A. & Deblinger, R. D. (1989) N. Engl. J. Med. 320, 133–137.
41. Steere, A. C., Taylor, E., Wilson, M. L., Levine, J. L. & Spielman, A. (1986) J. Infect. Dis. 154, 295–300.
42. Hanrahan, J. P., Benach, J. L., Coleman, J. L., Bosler, E. M., Morse, D. L., Cameron, D. J., Edelman, R. & Kaslow, R. A. (1984) J. Infect. Dis. 150, 489–496.
43. White, D. J., Chang, H. G., Benach, J. L., Bosler, E. M., Meldrum, S. C., Means, R. G., Debbie, J. G., Birkhead, G. S. & Morse, D. L. (1991) J. Am. Med. Assoc. 266, 1230–1236.
44. Goldstein, M. D., Schwartz, B. S., Friedmann, C., Maccarillo, B., Borbi, M. & Tuccillo, R. (1990) Am. J. Public Health 80, 1225–1229.
45. Barbour, A. G. & Fish, D. (1993) Science 260, 1610–1616.
46. Pachner, A. R. & Steere, A. C. (1985) Neurology 35, 47–53.
47. Steere, A. C., Batsford, W. P., Weinberg, M., Alexander, J., Berger, H. J., Wolfson, S. & Malawista, S. E. (1980) Ann. Intern. Med. 93, 8–16.
48. Halperin, J. J., Luft, B. J., Anand, A. K., Roque, M. D., Alvarez, O., Volkman, D. J. & Dattwyler, R. J. (1989) Neurology 39, 753–759.
49. Logigian, E. L., Kaplan, R. F. & Steere, A. C. (1990) N. Engl. J. Med. 323, 1438–1444.
50. Halperin, J. J., Little, B. W., Coyle, P. K. & Dattwyler, R. J. (1987) Neurology 37, 1700–1706.
51. Logigian, E. L. & Steere, A. C. (1992) Neurology 42, 303–311.
52. Asbrink, E. & Hovmark, A. (1988) Ann. N.Y. Acad. Sci. 539, 4–15.
53. Steere, A. C., Berardi, V. P., Weeks, K. E., Logigian, E. L. & Ackermann, R. (1990) J. Infect. Dis. 161, 1203–1209.
54. Lebech, A. M. & Hansen, K. (1992) J. Clin. Microbiol. 30, 1646–1653.
55. Keller, T. L., Halperin, J. J. & Whitman, M. (1992) Neurology 32, 32–42.
56. Dinerman, H. & Steere, A. C. (1992) Ann. Intern. Med. 117, 281–285.
57. Kaplan, R. F., Meadows, M. E., Vincent, L. C., Logigian, E. L. & Steere, A. C. (1992) Neurology 42, 1263–1267.
58. Steere, A. C., Taylor, E., McHugh, G. L. & Logigian, E. L. (1993) J. Am. Med. Assoc. 269, 1812–1816.
59. Berger, B. W., Johnson, R. C., Kodner, C. & Coleman, L. (1992) J. Clin. Microbiol. 30, 359–361.
60. Melski, J. W., Reed, K. D., Mitchell, P. D. & Barth, G. D. (1993) Arch. Dermatol. 129, 709–716.
61. Dressler, F., Whalen, J. A., Reinhardt, B. N. & Steere, A. C. (1993) J. Infect. Dis. 167, 392–400.
62. Bakken, L. L., Case, K. L., Callister, S. M., Bourdeau, N. J. & Schell, R. F. (1992) J. Am. Med. Assoc. 268, 891–895.
63. Schwartz, B. S., Goldstein, M. D., Ribeiro, J. M. C., Schulze, T. L. & Shahied, S. I. (1989) J. Am. Med. Assoc. 262, 3431–3434.
64. Goodman, J. L., Jurkovich, P., Kramber, J. M. & Johnson, R. C. (1991) Infect. Immun. 59, 269–278.
65. Liebling, M. R., Nishio, M. J., Rodriguez, A., Sigal, L. H., Jin, T. & Louis, J. S. (1993) Arthritis Rheum. 36, 665–675.
66. Rahn, D. W. & Malawista, S. E. (1991) Ann. Intern. Med. 114, 472–481.
67. Dattwyler, R. J., Volkman, D. J., Conaty, S. M., Platkin, S. P. & Luft, B. J. (1990) Lancet 336, 1404–1406.
68. Massarotti, E. M., Luger, S. W., Rahn, D. W., Messner, R. P., Wong, J. B., Johnson, R. C. & Steere, A. C. (1992) Am. J. Med. 92, 396–403.
69. Dattwyler, R. J., Volkman, D. J., Conaty, S. M., Platkin, S. P. & Luft, B. J. (1990) Lancet 336, 1404–1406.
70. Pfister, H. W., Preac-Mursic, V., Wilske, B., Schielke, E., Sorgel, F. & Einhaupl, K. M. (1991) J. Infect. Dis. 163, 311–318.
71. Steere, A. C., Pachner, A. R. & Malawista, S. E. (1983) Ann. Intern. Med. 99, 767–772.
72. Schulze, T. L., Taylor, G. C., Vasvary, L. M., Simmons, W. & Jordan, R. A. (1992) J. Med. Entomol. 29, 544–547.
73. Mather, T. N., Duffy, D. C. & Campbell, S. R. (1993) J. Med. Entomol. 30, 642–645.
74. Mather, T. N., Ribeiro, J. M. C. & Spielman, A. (1987) Am. J. Trop. Med. Hyg. 36, 609–614.
75. Stafford, K. C., III (1991) J. Med. Entomol. 28, 611–617.
76. Schreck, C. E., Snoddy, E. L. & Spielman, A. (1986) J. Med. Entomol. 23, 396–399.
77. Oransky, S., Roseman, B., Fish, D., Gentile, T., Melius, J., Cartter, M. L. & Hadler, J. L. (1989) Morbid. Mortal. Wkly. Rep. 38, 678–680.
78. Shapiro, E. D., Gerber, M. A., Holabird, N. B., Berg, A. T., Feder, H. M., Jr., Bell, G. L., Rys, P. N. & Persing, D. H. (1992) N. Engl. J. Med. 327, 1769–1773.
79. Fikrig, E., Barthold, S. W., Kantor, F. S. & Flavel, R. A. (1990) Science 250, 553–556.
80. Fikrig, E., Telford, S. R., III, Barthold, S. W., Kantor, F. S., Spielman, A. & Flavell, R. A. (1992) Proc. Natl. Acad. Sci. USA 89, 5418–5421.
81. Canica, M. M., Nato, F., duMerle, L., Mazic, J. C., Baranton, G. & Postic, D. (1993) Scand. J. Infect. Dis. 25, 441–448.
82. Burgdorfer, W., Lane, R. S., Barbour, A. G., Gresbrink, R. A. & Anderson, J. R. (1985) Am. J. Trop. Med. Hyg. 34, 925–930.
83. Nocton, J. J., Dressler, F., Rutledge, B. J., Rys, P. N., Persing, D. H. & Steere, A. C. (1994) N. Engl. J. Med. 330, 229–234.














