










































Below is the uncorrected machine-read text of this chapter, intended to provide our own search engines and external engines with highly rich, chapter-representative searchable text of each book. Because it is UNCORRECTED material, please consider the following text as a useful but insufficient proxy for the authoritative book pages.
14 Nonneoplastic Respiratory Diseases n persons who smoke, the cells that line the bronchi and alveoli come I into direct contact with high concentrations of tobacco toxicants. Not surprisingly, respiratory diseases such as chronic obstructive pulmo- nary disease (COPD) are major health problems in smokers (Murin and Silvestri, 2000). Tobacco-related respiratory diseases predominately affect male smokers, but the prevalence of COPD in women is rising rapidly and it appears to follow the prevalence of smoking by 20-30 years (Tanoue, 2000). Children exposed to environmental tobacco smoke (ETS) are also affected (Joad, 2000). Because low levels of tobacco toxicants from ETS come in direct contact with the lung, it is necessary to consider the health effects of both mainstream and secondary smoke. In evaluating harm reduction strategies for tobacco-related lung dis- eases, three major nonneoplastic respiratory diseases linked to cigarette smoking must be considered: COPD, asthma, and respiratory infections. Numerous other respiratory diseases are strongly related to cigarette smoking as shown in Table 14-1 (Murin et al., 2000). The relative risks of mortality due to smoking-related nonneoplastic respiratory diseases are considerable, and approximately 91,000 Americans died annually of res- piratory diseases attributed to smoking during 1990-1994 (Table 14-1 and 14-2) (Novotny and Giovino, 1998). Cigarette smoking is estimated to contribute to 80-90% of cases of COPD, and the amount and duration of cigarette smoking directly influence the progression of COPD. Asthma and respiratory infections, on the other hand, are not caused by tobacco smoke but are worsened by exposure to cigarette smoke. Of special im- 500
NONNEOPLASTIC RESPIRATORY DISEASES 501 TABLE 14-1 Smoking-Affected Pulmonary Diseases Disease Incidence or Severity Definitely Increased by Smoking Common cold Influenza Bacterial pneumonia Tuberculosis infection Invasive pneumococcal infection Pulmonary hemmorrhage Pulmonary metastatic disease Spontaneous pneumothorax Eosinophillic granuloma Respiratory bronchiolitis-associated interstitial lung disease Idiopathic pulmonary fibrosis Asbestosis Rheumatoid arthritis-associated interstitial lung disease Disease Incidence or Severity Possibly Decreased by Smoking Sarcoidosis Hypersensitivity pneumonitis NOTE: Modified with permission from Murin et al., 2000. Copyright (2000) by W.B. Saunders Company. portance in considering harm reduction strategies is the contribution of ETS to asthma and respiratory infections in children. Abatement strate- gies for susceptible children exposed to environmental tobacco smoke may differ from those used to reduce harm in tobacco smokers. An extensive knowledge base exists describing the contribution of tobacco smoke exposure to nonneoplastic respiratory disease, and key points are described briefly here. The major goal of this chapter is to summarize studies designed to test whether reducing exposure to to- bacco toxicants improves health outcomes for respiratory diseases. As will be described below, there are considerable gaps in information about reducing harm and uncertainties about the quality of the existing knowl- edge base in this regard. Consequently, a research agenda is proposed to guide future studies aimed at reducing the harm from smoking in COPD. BIOMARKERS OF RESPIRATORY DISEASES There are currently no specific biomarkers of respiratory disease due to smoking tobacco products (see Chapter 11). The rare genetic deficiency of α1-antitrypsin is a risk factor for disease, not a biomarker (see below). No unique molecular or genetic defect specific for tobacco-related respi-
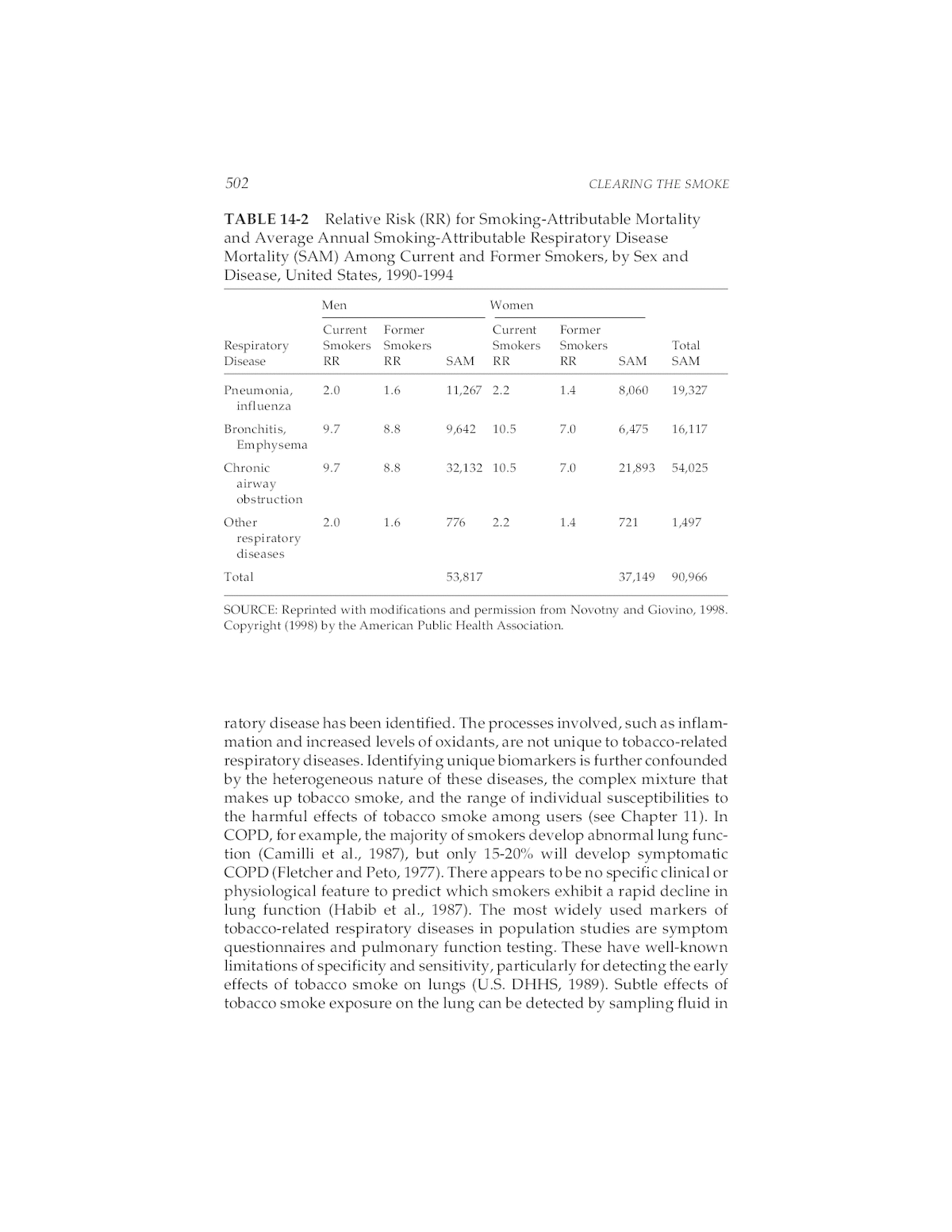
502 CLEARING THE SMOKE TABLE 14-2 Relative Risk (RR) for Smoking-Attributable Mortality and Average Annual Smoking-Attributable Respiratory Disease Mortality (SAM) Among Current and Former Smokers, by Sex and Disease, United States, 1990-1994 Men Women Current Former Current Former Respiratory Smokers Smokers Smokers Smokers Total Disease RR RR SAM RR RR SAM SAM Pneumonia, 2.0 1.6 11,267 2.2 1.4 8,060 19,327 influenza Bronchitis, 9.7 8.8 9,642 10.5 7.0 6,475 16,117 Emphysema Chronic 9.7 8.8 32,132 10.5 7.0 21,893 54,025 airway obstruction Other 2.0 1.6 776 2.2 1.4 721 1,497 respiratory diseases Total 53,817 37,149 90,966 SOURCE: Reprinted with modifications and permission from Novotny and Giovino, 1998. Copyright (1998) by the American Public Health Association. ratory disease has been identified. The processes involved, such as inflam- mation and increased levels of oxidants, are not unique to tobacco-related respiratory diseases. Identifying unique biomarkers is further confounded by the heterogeneous nature of these diseases, the complex mixture that makes up tobacco smoke, and the range of individual susceptibilities to the harmful effects of tobacco smoke among users (see Chapter 11). In COPD, for example, the majority of smokers develop abnormal lung func- tion (Camilli et al., 1987), but only 15-20% will develop symptomatic COPD (Fletcher and Peto, 1977). There appears to be no specific clinical or physiological feature to predict which smokers exhibit a rapid decline in lung function (Habib et al., 1987). The most widely used markers of tobacco-related respiratory diseases in population studies are symptom questionnaires and pulmonary function testing. These have well-known limitations of specificity and sensitivity, particularly for detecting the early effects of tobacco smoke on lungs (U.S. DHHS, 1989). Subtle effects of tobacco smoke exposure on the lung can be detected by sampling fluid in
NONNEOPLASTIC RESPIRATORY DISEASES 503 the lower respiratory tract via a bronchoscope inserted into the airways, but the significance of these changes to clinically important pulmonary disease has not been established. Newer approaches such as sampling the subjectsâ urine (Pratico et al., 1998) or exhaled gas (Ichinose et al., 2000; Paolo et al., 2000) for metabolic products due to tissue injury have the advantage of noninvasive sampling but must be validated. Clearly, the greatest obstacle for developing a specific biomarker is the lack of funda- mental information on mechanisms by which exposure to tobacco smoke causes specific respiratory diseases. Insight into understanding the molecular basis of diseases may come from unraveling the complex inter- actions between genetic makeup and the environment that could evolve from the Human Genome Project or similar molecular studies. CHRONIC OBSTRUCTIVE PULMONARY DISEASE Definition and Epidemiology Chronic obstructive pulmonary disease is an all-inclusive term that encompasses chronic bronchitis (the presence of chronic productive cough) and emphysema (permanent enlargement of the distal airspaces) (Aubry et al., 2000). Usually, chronic bronchitis and emphysema occur in combination. The major clinical features of COPD are chronic cough, ex- pectoration of sputum, breathlessness during exertion, and airflow ob- struction on forced expiration that tends to worsen over time (Barnes, 2000; Piquette et al., 2000). The natural history of COPD and its health care implications have been extensively reviewed (Hensley and Saunders, 1989). The course of COPD is characterized by loss of ventilatory function from the peak at- tained in early adulthood (Figure 14-1). Clinical symptoms of COPD de- velop after substantial loss of ventilatory function, typically in the fourth and fifth decades of life. In longitudinal studies, the average loss of func- tion in nonsmokers begins at about age 30 as assessed by the relationship of change in forced expiratory volume at 1 second (FEV1) with age. In nonsmokers, this amounts to about 20 ml per year. The rate of decline in FEV1 is somewhat greater (40 ml per year) in smokers of 30 cigarettes per day compared to nonsmokers (Camilli et al., 1987; Fletcher and Peto, 1977). A subset of smokers, who will develop COPD, lose function more rapidly (60 ml per year; Habib et al., 1987). However, there is variation in these rates and the rate of decline increases with age (Camilli et al., 1987; Fletcher and Peto, 1977). Smokers who cease smoking do not regain lost function, but the rate of decline of FEV1 slows to that of nonsmokers (Burrows, 1990). Loss of lung function appears to be a risk factor for mortality even among ânever smokersâ (Ashley et al., 1975; Beaty et al.,
504 CLEARING THE SMOKE 4 Non smo ker Me an 30 3 Cig FEV Liters /Da Su y sc ep 1 tib Exâ Sm le oke 30 r Ci 2 g/ Da 1 y DYSPNEA-ACTIVITIES OF DAILY LIVING 0 25 35 45 55 65 75 Age (Years) FIGURE 14-1 FEV1 NOTE: Scheme showing loss of forced expiratory volume at 1 second (FEV1) with age. Nonsmokers lose lung function with age, about 30 ml per year. Smokers of 30 cigarettes/day have a greater rate of loss (dashed line). A small proportion of smokers (10-15%) have a steeper rate of decline, about 60 ml per year (dot and dashed line). This susceptible group of smokers reaches an FEV1 of 0.8 liter (the level at which shortness of breath occurs on activities of daily living) at approxi- mately 60-70 years of age. If a susceptible smoker stops smoking at age 50, the rate of decline follows that of the nonsmoker (dotted line), showing that smoking cessation may prolong onset of symptoms. SOURCE: Reprinted with permission from Piquette, et al., 2000. Copyright (2000) by W.B. Saunders Company.
NONNEOPLASTIC RESPIRATORY DISEASES 505 1985). A loss of lung function may reflect other diseases such as heart disease that decrease life expectancy (Beaty et al., 1985). The annual num- ber of deaths attributable to chronic airway obstruction, bronchitis, and emphysema in the United States during 1990-1994 was 70,000 (Table 14-2). The relative risk of death from COPD in smokers who have symptoms is reduced by cessation, but remains almost as high as that of current smok- ers. Many of those who eventually die of COPD suffer from prolonged disability due to dyspnea, cough, and sputum production. Pathogenesis The role of inflammation and exposure to toxins in cigarette smoke is central to the pathogenesis of COPD (Aubry et al., 2000). There is consid- erable evidence to support the theory that pulmonary emphysema is caused by excessive exposure to elastolytic enzymes in relation to inhibi- tors of these enzymes, the âprotease-antiproteaseâ theory (Barnes, 2000; Piquette et al., 2000). Cigarette smoke promotes injury to the lungs by increasing the proteolytic burden and compromising the antiproteolytic defenses leading to a breakdown in lung structure. The major antiproteo- lytic protein in the lower respiratory tract is α1-antitrypsin (α1-AT), al- though other proteinase inhibitors play a lesser role (Senior and Shapiro, 1998). In chronic bronchitis, an inflammatory airway response caused by chronic exposure to airborne toxins (cigarette smoke, dust, air pollutants) is the central pathogenic mechanism (Barnes, 2000; Piquette et al., 2000). Inflammation leads to edema, cellular infiltration, fibrosis, smooth-muscle hypertrophy, and secretions that narrow the bronchioles. Animal models of emphysema and chronic bronchitis have been used to study the pathophysiology of COPD (Drazen et al., 1999; Shapiro, 2000), including the contribution of tobacco smoke (Witschi et al., 1997) (see Chapter 10). Several etiologic factors in COPD have been investigated with regard to cigarette smoking (Sethi and Rochester, 2000). One factor is airway hyperreactivity, measured by the provocation of airway constriction fol- lowing inhalation of a bronchoconstricting drug or physical condition. Several longitudinal studies have shown that airway hyperreactivity is related to accelerated decline in lung function in cigarette smokers (Frew et al., 1992; OâConner et al., 1995; Postma et al., 1986; Rijcken et al., 1995; Tashkin et al., 1996; Tracey et al., 1995). Hyperresponsiveness in the pres- ence of blood eosinophilia increases the risk of developing respiratory symptoms (Jansen et al., 1999). If airway hyperreactivity plays a role in the progression of COPD, it is possible that regular use of bronchodilators may slow the rate of decline. However, this conclusion was not supported by the Lung Health Study (Anthonisen et al., 1994; see below).
506 CLEARING THE SMOKE Special tests of âsmall-airwayâ function were developed in the late 1960s and early 1970s to detect early changes in small airways (less than 2- mm diameter) that were are not detected on standard tests of pulmonary function (U.S. DHHS, 1984). When applied to prospective studies of popu- lations, tests of small-airway function were not predictive for susceptible subjects who would progress to clinically significant airflow obstruction (U.S. DHHS, 1984). In subjects who exhibit accelerated deterioration of lung function (greater than 60 ml per year), this type of physiological testing was not predictive of the rate of development of clinically signifi- cant airway obstruction, probably because of the heterogeneous nature of COPD (Habib et al., 1987). Mucous hypersecretion and infections have been postulated to play roles in the accelerated decline of pulmonary function in COPD. In adults, prospective studies have failed to show an association between acute chest infections and the rate of decline of FEV1 (Bates, 1973). Chronic mucous hypersecretion in earlier studies (Fletcher and Peto, 1977; Higgins et al., 1982; Kauffmann et al., 1989; Peto et al., 1983) was not shown to be related to a decline in FEV1 in COPD. More recent studies in larger samples of the general population have shown associations between chronic mucous hypersecretion and a decline in FEV1 (Lange at al., 1990; Sherman et al., 1992; Vestbo et al., 1996). In children, however, population studies suggest that childhood respiratory infection is a risk factor for the development of COPD in adults (Gold et al., 1989; Samet et al., 1983). The role of infections is unclear, but it is believed that bacterial colonization/ infection stimulates inflammatory responses that cause local damage and progression of disease. In addition, infections impair host defenses and tissue repair, leading to further infection and perpetuating tissue injury (Murphy and Sethi, 1992). Considerable evidence from human and laboratory studies suggest that oxidant-antioxidant imbalance in favor of oxidants occurs in COPD (Figure 14-2). The evidence is based on a large number of studies showing an increased oxidant burden and markers of oxidative stress in the air- spaces, breath, blood, and urine of smokers and patients with COPD (Koyama and Geddes, 1998; Macnee and Rahman, 1999). Oxidants in pa- tients with COPD are derived from oxygen free radicals in both the gas and tar phases of cigarette smoke (Pryor and Stone, 1993; Zang et al., 1995). Inflammation itself induces oxidant stress in the lungs of smokers as suggested by studies showing greater production of oxygen free radi- cals by leukocytes in smokers compared to nonsmokers (Morrison et al., 1999) and increased iron content, which promotes formation of free radi- cals in alveolar macrophages of smokers (Mateos et al., 1998). Pathologi- cal examination of the alveolar regions of smokersâ lungs has shown increases in the number and percentage of leukocytes compared to non-
NONNEOPLASTIC RESPIRATORY DISEASES 507 FIGURE 14-2 Pathogenesis of emphysema. NOTE: Scheme of smoking-induced pulmonary emphysema. Smoking recruits inflammatory cells to the lower respiratory system via stimulation of alveolar macrophages. Inflammatory cells release enzymes (peroxidase, myeloperoxidase) and oxygen free radicals that degrade the extracellular matrix of the respiratory tissues and interfere with normal repair mechanisms. Tobacco smoke also con- tains oxygen free radicals that, together with products released from inflam- matory cells, inactivate protease inhibitors such as α1-AT. SOURCE: Reprinted with permission from Senior and Shapiro, 1998. Copyright (1998) by McGraw-Hill Companies. smokers (Hunninghake and Crystal, 1983). Recent studies have utilized analysis of exhaled gas in patients with COPD for gaseous products (i.e., reactive nitrogen species, ethane)(Ichinose et al., 2000; Paolo et al., 2000) and urinary products (isoprostane F2-α-III) (Pratico et al., 1998) formed from oxidative stress. It is possible that measurement of products of oxi- dative stress in exhaled gas may be used as surrogate markers of inflam- mation in COPD. A possible mechanism whereby oxidants damage the lung is by inactivating the antielastase α1-AT, thereby decreasing the ca-
508 CLEARING THE SMOKE pacity of α1-antitrypsin to inhibit proteases (Carp et al., 1982; Johnson and Travis, 1979). This mechanism has been expanded to include proteinases other than neutrophil elastase and antiproteinases other than α1-AT (Se- nior and Shapiro, 1998). Dietary deficiency of antioxidants has been pro- posed as a factor accounting for airflow limitation in elderly people (Dow et al., 1996), but dietary supplements of antioxidants have not been shown to modify clinical symptoms of COPD (Macnee and Rahman, 1999; Rautalahti et al., 1997; Traber et al., 2000). Mainstream Smoke Exposure The predominant risk factor for COPD is cigarette smoking, and it is estimated to account for 80-90% of the risk of developing COPD (U.S. DHHS, 1984). Cigarette smoking is associated with a lower FEV1 in cross- sectional studies (Burrows et al., 1977a; Dockery et al., 1988; Knudson et al., 1976) and with an accelerated decline in FEV1 in longitudinal studies (reviewed in Sherman, 1991). The lower FEV1 in cross-sectional studies and accelerated rate of decline in FEV1 exhibit a dose-response relation- ship. Both the duration of smoking and the amount smoked are signifi- cant predictors of lung function impairment (U.S. DHHS, 1989). Individuals who smoke have age-adjusted death rates for COPD that are tenfold higher than those of never smokers (U.S. DHHS, 1989). Mor- tality and morbidity rates for COPD are higher in pipe and cigar smokers than nonsmokers, although the rates are lower than in cigarette smokers. Factors predictive of COPD mortality include age at starting, current smoking status, and total pack-years. For reasons that are not known, only about 15% of smokers develop clinically significant COPD (Fletcher and Peto, 1977). Data suggest that 10-15% of COPD cases are attributable to causes other than smoking, including occupational exposures to coal dust (Marine et al., 1988), grain dust (Zejda et al., 1993), air pollution (Bates, 1973; Buist and Vollmer, 1994; Rokaw et al., 1980), childhood respiratory infections (Burrows et al., 1977b; Shaheen et al., 1995), and airway hyper- responsiveness (Buist and Vollmer, 1994). Genetic factors that are inde- pendent of personal smoking history or environmental exposures also contribute to COPD (Khoury et al., 1985). These include a1-antitrypsin deficiency, which accounts for less than 1% of cases of COPD (Snider, 1989). Additional genetic factors other than α1-AT deficiency that appear to play a role in susceptibility to COPD have not been identified (Khoury et al., 1985). Prospective population studies have shown an additive effect of air pollution on the decline in lung function in smokers (Tashkin et al., 1994;
NONNEOPLASTIC RESPIRATORY DISEASES 509 van der Lende et al., 1981). In a cohort study examining the additive effects of smoking and pollution on lung function decline, the mean ad- justed decrement in FEV1 attributable to smoking more than 24 cigarettes a day was 17.4 ml per year. An adjusted mean annual decrease of 8.9 ml per year was attributed to living in a moderately polluted environment compared to living in a clean environment (van der Lende et al., 1981). The conclusion of this study was that the decline in lung function attribut- able to smoking was twice as great when living in a polluted environ- ment. In a prospective study of persons living in three communities with air pollution, a significant interaction between smoking more than 12 cigarettes a day and area of residence was found for mean decline of adjusted FEV1 in males (Tashkin et al., 1994). These findings suggest an independent adverse effect of air pollution on decline of lung function in smokers. Environmental Tobacco Smoke Exposure Adults Coultas (1998) has reviewed published reports of an association be- tween ETS exposure and COPD. He classified three types of studies: two categories based on âindirectâ measures of COPD (self-reports of symp- toms of COPD; effects on lung function measurements) and a third cat- egory that used âdirectâ measures of COPD (mortality and hospitaliza- tions). The studies focused on environmental tobacco smoke as a risk factor for developing COPD, not as factor that contributed to worsening of symptoms or lung function. In regard to self-reported outcomes, Coultas (1998) summarized the results of three studies (Dayal et al., 1994; Leuenberger et al., 1994; Robbins et al., 1993) published since the Environ- mental Protection Agency (EPA) report (EPA, 1993) that concluded envi- ronmental tobacco smoke âmay increase the frequency of respiratory symptoms in adults.â Results of one population-based survey of self- reported COPD suggest that 3-5% of nonsmokers may be affected (Whittemore et al., 1995). The three published reports combined asthma and COPD. These studies all report similar findings that passive smoking is associated with chronic respiratory symptoms found in adults with COPD. The second type of study examined declines in lung function and passive smoking in the development of COPD. Epidemiological studies have investigated the association between environmental tobacco smoke, respiratory diseases, and reduction in pulmonary function tests (Sherman, 1991; Tredaniel et al., 1994). It is controversial whether ETS exposure is
510 CLEARING THE SMOKE associated with COPD. Some studies have reported greater reduction in pulmonary function in nonsmokers married to smokers and exposed to environmental tobacco smoke in the workplace (Berglund et al., 1999; Hole et al., 1989; Leuenberger et al., 1994; Svendsen et al., 1987; White and Froeb, 1980). However, most of the studies used sensitive indicators of lung function, and the physiological significance of small changes in lung function to COPD is not established. In addition, the effect of bias and confounding factors were not taken into account. No relation between passive smoking and reduced lung function in adults was found in two large cross-sectional studies (Comstock et al., 1981; Schottenfeld, 1984) and one longitudinal study (Jones et al., 1983). Thus, whether environ- mental tobacco smoke causes COPD in adults remains uncertain. Current evidence suggests that the development of COPD in adults results from impaired lung development and growth in childhood, pre- mature onset of declines in lung function, and/or accelerated decline in lung function (Fletcher et al., 1976; Kerstjens et al., 1997; Samet and Lange, 1996). Although passive smoking is a biologically plausible risk factor, the impact of passive smoking during adulthood on the development of COPD remains controversial. In a review of this topic, Tredaniel and associates (1994) summarized the results of 18 relevant publications. Eight reports found no effect of ETS exposure on lung function, and ten demon- strated small decrements. The authors pointed out the methodological limitations of these studies and raised questions about the relevance of small declines in lung function to the development of COPD. Coultas (1998) reviewed the results of three additional studies published since the Tredaniel et al. (1994) review and concluded that the results were incon- sistent (two showing no effect and one showing an effect). In the study showing an effect, a large sample of never-smoking adults in Switzerland (Leuenberger et al., 1994), the association between increased symptoms of chronic bronchitis and environmental tobacco smoke was dose-related. The adjusted odds ratio of chronic bronchitis symptoms increased by years of exposure to environmental tobacco smoke, number of smokers to whom the subject was exposed, and workplace exposures (Leuenberger et al., 1994). An additional report published after Coultasâ review con- cluded that environmental tobacco smoke in adults is associated with small defects in lung function (Carey et al., 1999). Children Exposure to passive smoking in childhood has been associated with reduced rate of growth of the lung as determined by change of ventilatory function with age in children exposed to environmental smoke compared to unexposed children (Berkey et al., 1986; Tager et al., 1979, 1983, 1987)
NONNEOPLASTIC RESPIRATORY DISEASES 511 (see Chapter 15). However, one cohort study failed to find any effect of maternal smoking on lung growth (Lebowitz and Holberg, 1988), possi- bly due to differences in the amount of maternal smoking or differences in indoor environments (i.e., greater air mixing or exchange may decrease the concentration of environmental tobacco smoke). A recent study con- cluded that in utero exposure to maternal smoking is independently asso- ciated with decreased lung function in children of school age, especially for small-airway flows (Gilliland et al., 2000). Studies in neonates that excluded the effect of environmental tobacco smoke by measuring lung function after birth concluded that in utero exposure has an independent effect on reduced lung mechanics (Hanrahan et al., 1992; Stick et al., 1996). In a cross-sectional study by Milner et al. (1999), smoking was associated with a significant reduction in birthweight, but lung volume was not reduced when related to weight. Smoking was associated with a highly significant reduction in static compliance in boys and a small but signifi- cant reduction in respiratory conductance in girls. These authors con- cluded that smoking in pregnancy reduces static compliance in boys and conductance in girls although there was no evidence that maternal smok- ing adversely affected fetal development. Fetal lung hypoplasia has been produced in rats in a model of maternal cigarette smoke exposure (Collins et al., 1985). The weight of the evidence suggests that passive smoking contributes to COPD by causing a slight decrease in airway diameter that increases airway resistance. Latency There are limited data on the latency (Robbins et al., 1993) and dose- response (Dayal et al., 1994) between exposure to environmental tobacco smoke and the development of respiratory symptoms. Additional studies are needed, particularly of latency and dose-response relationships, to establish a causal relationship between passive smoking and COPD. Review of Harm Reduction Strategies Applied to COPD Impact of Smoking Reduction It is plausible that reducing the number of cigarettes smoked per day would reduce disease risk. The presumption is that smoking causes mor- bidity and mortality in a dose-related manner. However, this presump- tion has never been formally tested (Hughes, 2000). One impediment to determining whether reducing smoking reduces risk is that a study would require a large number of subjects over a long time. Hughes (2000) esti- mated that 8,000 subjects would have to be followed for eight years. A
512 CLEARING THE SMOKE large study would be required since there are no adequate surrogate markers of disease. Since a large study of this type would delay imple- mentation of harm reduction measures, one school of thought is that the benefits from smoking reduction can be assumed and we need not wait eight years for the study to be completed (Hughes, 2000). Another school of thought argues that the decline in disease from smoking reduction may not be as great as presumed. This view is based on the following: (1) post hoc corrections for variables associated with smoking reduction may inflate mortality and morbidity among smokers, and (2) the expected reductions from âlow-tarâ cigarettes were not as great as expected (Hughes, 2000). It has been suggested that reducing smoking may undermine attempts at quitting, but based on limited evidence from three trials, Hughes (2000) concluded that there is no evidence to support this concern. It should be noted, however, that these results apply only to people who were self- motivated to reduce smoking, and the effects on quitting may not apply to the general population (Hughes, 2000). There are data from retrospective cross-sectional, multicenter epide- miological studies suggesting that reducing smoking reduces the risk of COPD (Higgins et al., 1984). The risk for cigarette smokers of developing COPD was assessed in 958 men and 1,159 women ages 25 to 64 years over a 10-year period (1967-1969 and retested in 1978-1979). At entry, patients were excluded who already had borderline or definite COPD. Data for the change in daily cigarette consumption were available from only one center. The odds ratio (OR) for developing COPD within ten years pre- dicted from the change in the number of cigarettes per day (number at follow-up minus number at entry into the study) was 1.7 for men and 2.4 for women. These results were interpreted as showing a benefit from stopping or reducing cigarette smoking. The limitations of this study are that it was not designed to show a change in mortality and morbidity for people who reduced smoking, that estimates are based only on survivors, and that it is a cross-sectional population study. Nevertheless, it does provide some evidence that reducing smoking might modify the risk of COPD. The potential benefit to improving survival from COPD by reducing smoking can be analyzed from dose-response relationships between mor- tality and number of cigarettes smoked in a population. Such data are available from two large-scale prospective surveys of smoking and mor- tality among men and women in the United States sponsored by the American Cancer Society. The first survey of approximately one million people covered the period 1959 to 1972 and is referred to as Cancer Pre- vention Study I (Burns et al., 1997; CPS-I). The second survey was con- ducted from 1982 through 1986 on approximately 1.2 million people and is referred to as Cancer Prevention Study II (CPS-II; Thun et al., 1997).
NONNEOPLASTIC RESPIRATORY DISEASES 513 Methodological issues pertaining to calculations of risk-attributable mor- tality in these studies are discussed in the 1989 Surgeon Generalâs report on reducing the health consequences of smoking (U.S. DHHS, 1989). Data are available from CPS-I that illustrate the relationship between cigarettes smoked per day and mortality rates from COPD among white male and female current smokers. Figure 14-3 shows data for two 15-year age groups (65-79 and 50-64 years). For both men and women age 65-79, the results show a monotonic increase in mortality rate from COPD as a function of number of cigarettes smoked per day, with higher rates in men than women. For men and women age 50-64, there is an apparent increase in mortality rate from COPD with number of cigarettes smoked per day, although the slopes are less steep than for the older group. These Men Women 500 200 400 Mortality rates per 100,000 150 Age 65-79 Age 65-79 300 100 200 50 100 Age 50-64 Age 50-64 0 0 1-9 10-19 20 21-39 â¥40 1-9 10-19 20 21-39 â¥40 Cigarettes per day FIGURE 14-3 Mortality rates. NOTE: Graphs showing relationships between mortality rates from COPD among current white smokers by level of cigarette consumption. Plotted are standard- ized mortality rates per 100,000 adjusted within 15-year age-specific rates to 1980 U.S. standard population for men and women. SOURCE: Data for figure gathered from Burns et al., 1997.
514 CLEARING THE SMOKE findings strongly suggest that increased cigarette smoking causes greater mortality due to COPD in current smokers. One cannot infer from these results that reducing smoking will de- crease mortality from COPD. However, the apparent by linear dose- response relationships suggest that decreasing the amount of cigarettes smoked may lead to fewer deaths from COPD. This may be particularly important to heavy smokers (more than 40 cigarettes per day). Caution is needed, however, in interpreting these results too simply. For example, it is unlikely that someone who smoked 40-plus cigarettes per day during adulthood will experience a substantial reduction in the risk of dying from COPD (e.g., comparable to that of a lifelong 20-cigarette per day smoker) if he or she were able to cut down on cigarette consumption. Only prospective studies of mortality in smokers who reduce their expo- sure to cigarettes will provide definitive answers to the question of how much mortality rates are decreased by reducing smoking. Interventional Studies A landmark study in smoking cessation in people at risk for develop- ing symptomatic COPD is the Lung Health Study (Anthonisen et al., 1994). The study population consisted of 5,887 subjects from North America, 35 to 60 years old, who had presymptomatic COPD (no symp- toms but abnormal lung function tests) and included a substantial frac- tion (37%) of women. Participants were moderate to heavy smokers (mean consumption, 31 cigarettes per day) who were motivated to quit smoking. They were randomized into three groups: (1) smoking intervention, (2) smoking intervention plus a brononchodilator, and (3) usual care. Smoking intervention was intensive and involved 12 group meetings in ten weeks. Behavior modification was stressed, and nicotine replacement therapy (NRT) with nicotine gum was used aggressively. Those who ceased smoking entered a maintenance program. Relapses were treated individually. The main outcome was the rate of change in FEV1 measured annually over five years. The cessation rate was among the highest re- ported in any trial. In the intervention group, 35% were abstinent one year and 22% were abstinent for five years (compared to 5% in the non- intervention group). The most important finding of this intent-to-treat analysis was that smoking cessation reduced the rate of decline of FEV1. In a separate analysis of the study population, symptoms of cough and sputum production, and dyspnea improved after smoking cessation (Kanner et al., 1999). The administration of a bronchodilator (ipratropium bromide) resulted in a small improvement in FEV1 at the onset of use. However, this effect reversed when the drug was discontinued at the end
NONNEOPLASTIC RESPIRATORY DISEASES 515 of the study. The major conclusion of the Lung Health Study was that an aggressive intervention program of smoking cessation reduces the age- related rate of decline in FEV1 in middle-aged smokers with mild airway obstruction in the first year. After that time, however, the rate of lung function decline was not different in the two groups. These results pro- vide a measure of the impact of aggressive smoking cessation compared with the usual care in reducing the harm of cigarette smoking. A particularly important audience at which to target harm reduction is young smokers who want to quit. To determine how rapidly lung func- tion returns to normal after smoking is stopped in this group of subjects, Ingram and OâCain (1971) used sensitive tests of lung function (frequency dependence of compliance) in six asymptomatic young smokers. Within eight weeks of smoking cessation, lung function had returned to normal in all subjects, suggesting that structural changes in the peripheral air- ways are reversible in these subjects after cessation of smoking. There is a dearth of information on whether reducing cigarette smok- ing results in improvement in lung function. Two prospective studies in the 1970s and one in 1989 (Buist et al., 1976; Lange et al., 1989; McCarthy et al., 1976) examined the effect of reduced smoking over a one year period using volunteers with small-airway disease and either normal or mild reductions in FEV1. Subjects who were able to reduce smoking con- sumption by 25% or more were considered to have reduced smoking. Tests of small-airway disease improved after smoking reduction, and in one study, FEV1 showed slight improvement at one year (McCarthy et al., 1976). An observational study by Lange and colleagues in (1989) exam- ined the effect of smoking habits on change in FEV1 over a five-year period in more than 7,000 Danish men and women. Among these were a sample of 189 subjects who were grouped as reducing smoking from more than 15 cigarettes per day to less than 15 cigarettes per day between intake and final examination. Regression analysis of change in age- adjusted FEV1 over five years showed that smoking reduction resulted in a beneficial effect in younger subjects (less than 55 years old) but not in older subjects (more than 55 years old). In the younger group, the annual decline in FEV1 unadjusted for age in women who were able to reduce smoking was 14 ml per year versus 30 ml per year in women who contin- ued to smoke more than 15 cigarettes a day. For younger men who were able to cut back on smoking, the reduction was 17 ml per year compared to 42 ml per year in those who continued to smoke more than 15 cigarettes a day. Limitations of this study were that it was an observational study, smoking habits were based on self-reported data, and there were wide interindividual differences in the rate of decline of FEV1. Further limita- tions of these studies are that they were not designed primarily to test
516 CLEARING THE SMOKE whether reducing smoking results in benefit as measured by less decline in lung function, but rather were designed to study the effect of cessation. Although they suggest that reducing smoking (at least in younger sub- jects) may slow the rate of decline in function, more rigorously designed prospective studies are needed to determine whether reducing smoking results in less harm to the lungs. Decreasing Smoking Slows Decline in Lung Function Numerous cross-sectional studies show that the level of smoking is a strong determinate of decline in lung function (U.S. DHHS, 1984, 1989). Multiple regression and analysis has been applied to these cross-sectional studies to determine the quantitative relationships between the rate of decline in FEV1 and pack-years of cigarettes smoked (Burrows et al., 1977a; Dockery et al., 1988). In one study of over 8,000 men and women from U.S. cities (Dockery et al., 1988), it was estimated that the average male smoker lost 7.4 ml of FEV1 for every pack-year of cigarettes smoked and that women lost 4.4 ml of FEV1. FEV1 values in the population were skewed toward lower values for the smokers compared to the nonsmokers. This suggests that the loss in FEV1 was relatively larger in smokers than nonsmokers. It is uncertain whether these data could be used to predict a slower rate of decline in lung function if a smoker reduced the number of cigarettes smoked over a prolonged period of time. There are several uncertainties about using studies of longitudinal change in lung function to predict slowing of lung function after reducing smoking. For example, the observations were made over a limited span of the natural history of COPD and it is unclear whether the measured changes in reduced FEV1 correlated with progression of disease. Function of pack-years smoked remains constant throughout the natural history of the disease. It is pos- sible, for instance, that a more rapid decline in lung function takes place after an event such as an infection or that the rate of decline accelerates with age. Individual factors that contribute to the rate of decline of lung function are poorly understood and also influence progression of disease. Because of uncertainties about the natural history of COPD, it is not pos- sible to use longitudinal studies of decline in lung function as a function of pack-years smoked to estimate the reduction in harm in terms of de- cline in lung function from reducing cigarette smoking. Effect of Short-Term Smoking Reduction on Lung Inflammation The beneficial effect of short-term smoking reduction has been stud- ied in current smokers using a technique that enables inflammation in the lower respiratory tract to be examined directly (ECLIPSE, 2000; Rennard,
NONNEOPLASTIC RESPIRATORY DISEASES 517 2000; Rennard et al., 1990). In current cigarette smokers, three approaches to reduce smoking consumption were used: administration of nicotine medication, switching to a low-tar and nicotine cigarette, and using Eclipse⢠(R.J. Reynolds Tobacco Company), a product that results in less combustion of tobacco and potentially generates relatively less tar than nicotine. All studies used a similar approach. Heavy smokers (at least two packs per day) who had no clinical or physiological evidence of COPD were enrolled. During the reduced smoking period, cigarette smoke ex- posure was monitored by exhaled carbon monoxide (CO) concentration, serum cotinine level, and self-monitoring the number of cigarettes smoked. Lung inflammation was assessed before and two to three months after reduced smoking using bronchoalveolar lavage. Inflammatory cells and protective antiproteinase molecules were measured in the fluid sample, and results were compared to those from nonsmokers. The de- gree of inflammation of the airways was scored by directly visualizing the degree of erythema and edema though the bronchoscope. Reduced smoking by approximately 50% (measured by self-reporting and exhaled CO) was achieved by the use of nicotine medication (Rennard et al., 1990). The amount of visible inflammation decreased but remained greater than that of nonsmokers. In lavage fluid, the number of inflamma- tory cells (particularly the pigment-laden macrophages and neutrophils) and the amount of elastase complexed to its a1-antiproteinase inhibitor decreased after smoking reduction. These results show that reducing smoking from two packs to one pack per day produces a significant de- crease in lung inflammation within two to three months. Whether the reduced inflammation results in less risk of developing COPD remains to be determined. A second study examined the effect of switching to low-tar and nico- tine cigarettes (ECLIPSE, 2000; Rennard, 2000). This study differed from the nicotine replacement study in several ways: the subjects had moder- ate to heavy cigarette smoking (approximately 30 cigarettes per day) at enrollment, subjects chewed either nicotine gum (2 or 4 mg) or a placebo gum, and the duration of the study was six months. The groups that used NRT reduced their cigarette consumption (measured by self-reporting and exhaled CO). However, the group that used the low-tar and nicotine cigarettes did not reduce smoking because they changed their smoking strategy to compensate. The results showed no reduction in the inflam- mation score in the group that used low-tar and nicotine cigarettes, which was attributed to altered smoking strategy. The study using heated tobacco (Eclipseâ¢) was similar in design to the reports cited above and involved 12 smokers studied after a three- month intervention. Exhaled CO was used to monitor cigarette use. Al- though one-third of the study group smoked conventional cigarettes, the
518 CLEARING THE SMOKE number of conventional cigarettes used was small (97% of all products consumed were Eclipseâ¢). In Eclipse⢠users, inflammation scores and number of recovered alveolar macrophages decreased after intervention as did the density of goblet cells (an index of the mucous secreting cells) on bronchial biopsies. (Results of neutrophil counts were confounded by outliers in the control group.) After three months of Eclipseâ¢, inflamma- tion scores and goblet cells density remained above levels found in non- smoking controls. The small number of patients and control subjects limits interpretation of this study. In summary, asymptomatic smokers clearly have inflammation in their lower respiratory tract, and this inflammation is likely to be in- volved in the pathogenesis of COPD. The major conclusion of Rennardâs (Rennard, 2000; ECLIPSE, 2000) studies is that smoking reduction has measurable effects on inflammation. It is plausible that reducing inflam- mation by reduced exposure to cigarettes will mitigate the development of COPD. However, the hypothesis that reduced smoking will lead to less severe COPD needs considerably more study using prospective interven- tion trials in persons susceptible to developing COPD. ASTHMA Definition and Epidemiology Asthma is defined as a âchronic inflammatory disorder of the airways in which many cells and cellular elements play a role, in particular, mast cells, eosinophils, T lymphocytes, neutrophils, and epithelial cells. In sus- ceptible individuals, this inflammation causes recurrent episodes of wheezing, breathlessness, chest tightness, and cough, particularly at night and in the early morning. These episodes are usually associated with widespread but variable airflow obstruction that is often reversible either spontaneously or with treatment. The inflammation also causes an associ- ated increase in the existing airways hyperreactivity to a variety of stimuliâ (NIH, 1997). There is no standardized method for measuring asthma, and the lack of standardization has hampered the investigation of asthma by making it difficult to compare results of different studies (Woolcock, 1994). Asthma is an extremely common disorder in the United States, with approximately 10-15% of boys (Sly, 2000), 7-10% of girls (Sly, 2000), and 5% of adults having signs and symptoms consistent with asthma (Drazen, 2000). Asthma has its origins primarily in childhood, with 50% of all asthma diagnosed by three years of age and 80% by six years of age (Yunginger et al., 1992), although asthma may develop at any time
NONNEOPLASTIC RESPIRATORY DISEASES 519 throughout life. The worldwide incidence of asthma has increased more than 30% since the 1970s (Weiss et al., 1993). A disproportional fraction of these cases have occurred among socioeconomically disadvantaged urban dwellers. The reason for the increased incidence of asthma is not known. In the United States, the overall age-adjusted prevalence of asthma in- creased 54% from 1980 to 1993-1994 (from 31 to 54 per 1,000 population) (NCHS, 1999). Pathogenesis The factors that influence the pathogenesis of asthma include atopy (production of abnormal amounts of immunoglobulin E [IgE]), early res- piratory illness (before age two), and parental history of atopy (Woolcock, 1994). Allergens act as triggers of asthma attacks and provoke inflamma- tion of the airways. Other triggers include exercise and air pollutants such as ozone and sulfur dioxide (Woolcock, 1994). Inflammation of the air- ways with lymphocytes, mast cells, and eosinophils and production of certain interleukins by these cells creates an environment that promotes the production of IgE. The links between infiltration of cells and the pathobiologic processes that account for episodic airway narrowing have not been clearly delineated. The constriction of airway smooth-muscle cells due to the local release of bioactive mediators or neurotransmitters is a widely accepted explanation for acute constriction of the airways, but thickening of airway epithelium and the presence of liquid in the lumen may also contribute. The consequences of airway obstruction are de- creased flow rates throughout expiration. As the asthma attack resolves, airway narrowing reverses and flow rates return toward normal. These physiological and pathological changes account for intermittent symp- toms of shortness of breath, coughing, and wheezing (Drazen, 2000). Mainstream Smoke Exposure Asthmatics have extreme sensitivity of the airways to chemical, physi- cal, and pharmacological stimuli, which can cause pronounced airflow limitation (Drazen, 2000). Cigarette smoke is a complex mixture of irritant compounds. Smoking can potentially lead to amplification of the airway inflammation already present in asthmatics by a number of mechanisms including recruitment of inflammatory cells, enhancement of some cellu- lar function, and release of proinflammatory mediators (Floreani and Rennard, 1999). For example, cigarette smoking stimulates production of cysteinyl leukotrienes (Fauler and Frolich, 1997), a class of mediators that is increased in asthmatics, causes rapid proliferation of cells in small air-
520 CLEARING THE SMOKE ways (Sekhon et al., 1994). In addition to inflammation, cigarette smoking is associated with increased airway wall remodeling (U.S. DHHS, 1990), a change asociated with chronic asthma. Physiological studies have shown that direct cigarette smoking causes acute bronchoconstriction in subjects with asthma (Nadel and Comroe, 1961). Many patients with asthma re- port worsening respiratory symptoms upon active cigarette exposure. In a cross-sectional study of 225 adult asthmatics, current smokers had a higher asthma symptom scores than nonsmokers when adjusted for age, gender, and physician visits (Althuis et al., 1999). However, there are methodological issues that might obscure the associations between active smoking and asthma in cross-sectional studies. One form of selection bias, referred to as the healthy smoker effect (Weiss et al., 1999), refers to self-selection of subjects with less mild asthma as active smokers com- pared to those who remain smokers. The probability of an asthmatic be- coming an active smoker may be less in individuals who have a greater airway hyperreactivity or who become more symptomatic when they smoke. Similarily, adults who had childhood asthma may be more likely to remain nonsmokers than those who did not have asthma in childhood. Thus, cross-sectional studies of smoking and asthma have limitations of interpretation. Nevertheless, observational studies show that asthmatics who are unable to cease smoking have more serious problems with con- trol of asthma and have more frequent hospital admissions than non- smoking asthmatics (Abramson et al., 1995; Floreani and Rennard, 1999). Since the 1960s, it has been suggested that airways hyperresponsive- ness and atopy are risk factors for the development of COPD, the so- called Dutch hypothesis (Orie et al., 1961). Data from several longitudinal studies now clearly show that airways hyperreactivity is a susceptibility factor for accelerated decline of lung function in active cigarette smokers compared to smokers without airways hyperreactivity (Frew et al., 1992; OâConnor et al., 1995; Rijcken et al., 1995; Tashkin et al., 1996; Tracey et al., 1995). Data from the Lung Health Study, a five-year prospective study of subjects with mild COPD, showed that responsiveness to methocholine at the initiation of the study was a strong predictor of change in FEV1 in continuing smokers even after controlling for baseline lung function, num- ber of cigarettes smoked, and demographic variables (Tashkin et al., 1996). Moreover, airway reactivity appeared to be a more important determi- nant of subsequent lung function decline then baseline lung function. In addition, the magnitude of the decline in FEV1 increased with greater levels of airways hyperreactivity. The design of the study did not permit the distinction of whether the risk factor for progression of COPD was airway hyperreactivity itself or an underlying mechanism of reactivity such as inflammation. The finding that the effect of reactivity on lung function decline was greatest in continuing smokers compared to subjects
NONNEOPLASTIC RESPIRATORY DISEASES 521 who quit smoking suggests that the effect is at least partially due to pro- gressive airway damage induced by smoking. Since subjects who reduced smoking were not analyzed, there is insufficient information to determine whether decreasing the number of cigarettes smoked leads to less airways hyperreactivity. Environmental Tobacco Smoke Exposure Adults The three major reviews of the health effects of passive smoking pub- lished between 1986 and 1992 cited no literature directly examining the association between passive smoking and asthma in adults (NRC, 1986; U.S. DHHS, 1986). Coultas (1998) reviewed studies on this topic from 1992 to 1998 and classified them as etiological studies (the association between passive smoking and the diagnosis of asthma) and morbidity studies (the role of passive smoking in causing symptoms or worsening lung function in patients with asthma). Among the papers reviewed was that of Hu and coworkers (1997) who observed in young adults that physician-diagnosed asthma was associated with parental smoking. The odds ratio was 1.6 (95% confidence interval [CI]=1.1, 2.3) for maternal smoking and 1.3 (95% CI=0.9, 1.8) for paternal smoking. Their investiga- tors also found a dose-response relationship with the amount smoked and the number of parents smoking. Greer and associates (1993) studied a large, nonsmoking population over a ten-year period to examine the asso- ciation between workplace exposure to environmental tobacco smoke and the development of asthma. After controlling for confounding factors, workplace ETS exposure was a significant risk for asthma (relative risk [RR]=1.5, 95% CI=1.2, 1.8). In another study of more than 4000 never- smoking adults, Leuenberger and associates (1994) found exposure to environmental tobacco smoke at home or work was associated with physician-diagnosed asthma (OR=1.4; 95% CI=1.0, 1.9). Several studies have examined the role of environmental tobacco smoke as a factor for asthma in adults. There are limitations of present epidemiological studies including potential biases in both subject selec- tion and misclassification of exposure and differing designs. The effects of environmental tobacco smoke on pulmonary function in otherwise healthy adults are likely to be small and unlikely to result in clinically significant chronic respiratory disease (Zeise et al., 1997). Although the results tend to indicate potential effects of environmental tobacco smoke as a cause of asthma in adults, the present epidemiological data are lim- ited and have to be interpreted cautiously. The results of published reports on the effects of ETS exposure on
522 CLEARING THE SMOKE respiratory symptoms and lung function in adults have been summarized by Tredaniel and associates (1994) and Coultas (1998). Six observational studies in patients and six experimental studies of patients with asthma are reviewed. In general, these studies report that environmental tobacco smoke worsens respiratory symptoms and lung function among adult asthmatics. There are no published intervention trials in adults examining whether decreasing exposure reduces asthma symptoms or impairment of lung function. A recent review evaluated the quality of the studies of the relation of environmental tobacco smoke as an exacerbating factor for asthma in adults (Weiss et al., 1999). Although exposure of adult asthmatics to envi- ronmental tobacco smoke was associated with increased symptoms of asthma, these studies are subject to recall and information bias, and no firm conclusions can be made. On the other hand, a number of controlled exposure studies have examined the effect of exposure to environmental smoke in healthy adult volunteers and subjects with asthma (Weiss et al., 1999). In general, the experimental studies show that brief exposure to ETS produces symptoms such as eye and nasopharyngeal irritation with less consistent responses in terms of lung function. Airway hyperreactiv- ity after environmental smoke exposure was assessed by bronchoprovo- cation tests using methacholine. The majority of these studies failed to document an effect on ventilatory function or measures of airway respon- siveness. In general, studies evaluating acute exposure of adult asthmatics to environmental tobacco smoke have generated conflicting data, yet there is evidence that some asthmatics and groups of asthmatics do respond to ETS levels that do not elicit responses in healthy volunteers. However, because of the limited number of studies and potential problems in their design, definitive conclusions cannot be drawn at this time. Children The relationship between environmental tobacco smoke and asthma, lung function, and respiratory symptoms in childhood has been reviewed (Cook et al., 1998; Cook and Strachan, 1997; NRC, 1986; Strachan and Cook, 1998; U.S. DHHS, 1986). Most of the studies support the notion that small but significant differences can be detected in respiratory symptom prevalence and pulmonary function studies comparing children exposed to environmental tobacco smoke and children who are unexposed. The incidence of wheezing illnesses in children whose parents smoke is great- est in early life, whereas the incidence of asthma during school years in less strongly affected by parental smoking. This may be due to a stronger influence of viral-associated wheezing in early childhood or less exposure to maternal smoke among older children.
NONNEOPLASTIC RESPIRATORY DISEASES 523 The results of effects on spirometric indices of passive smoking have been reviewed (Cook et al., 1998; Cook and Strachan, 1999). These analy- ses concluded that passive smoking is associated with a small but signifi- cant reduction in FEV1 and other spirometric indices in school-age chil- dren without asthma. For example, the large Six Cities Study showed a very small but significant effect of maternal smoking on lung growth (minus 3.8 ml per year for FEV1) (Tager et al., 1983). Such subtle reduc- tions are unlikely to impact the rates of development of chronic obstruc- tion in airflow unless data emerge demonstrating that children exposed in early life have more rapid decline of lung function. Unfortunately, none of the longitudinal studies have looked at changes in lung function in relation to change in maternal smoking behavior. Therefore, it is un- known whether their effects are reversible. Similarly, a study in infants demonstrated that a family history of parental smoking contributes to airway hyperresponsiveness at an early age (Young et al., 1991). Whether these normal, healthy infants with airway hyperresponsiveness develop asthma as adults is not known. An important issue in evaluating potential strategies for reducing harm from environmental cigarette smoke among asthmatic children in smoking households is to assess the relationship between biomarkers of cigarette smoke exposure and severity of asthma. If a dose-effect relation- ship exits, these data might indicate the approximate extent of reduction in exposure that may be expected to reduce the severity of asthma in children residing with smokers. Several studies have examined the dose- response relationship in asthmatic children using cross-sectional or case- control designs (Table 14-3). Asthma was assessed by self-reporting, physical examination, or lung function studies, and cotinine was mea- sured in urine or saliva. Two general types of analyses were done: (1) asso- ciations of mean levels of cotinine in asthmatic or control groups (Group A) and (2) relationships between cotinine levels and asthma severity (Group B). Two studies (Ehrlich et al., 1992; Willers et al., 1991) found higher levels of cotinine in urine of asthmatics than controls, and one study found that cotinine levels in saliva were no different in asth- matics and control subjects (Chang et al., 2000). Four studies showed a relationship between urinary cotinine levels and the severity of asthma (Chilmonczyk et al., 1993; Ehrlich et al., 1996; Oddoze et al., 1999; Rylander et al., 1995) (see Table 14-3). There are methodological problems with these studies, however, since most have not controlled for confounding factors such as respiratory ill- nesses or exposure to house mite antigen. It is possible that children of parents who smoke are more likely to become allergic or to have respira- tory infections. Moreover, the effects are applicable only to some age groups or some symptoms of asthma, leading to inconsistencies in the
TABLE 14-3 Relationships Between Cotinine Levels in Asthmatic Children Exposed to Environmental Tobacco 524 Smoke Author Study Type Asthma and Year and Sample Size Population Cotinine Asthma Finding Group AâAssociation of high cotinine levels with asthma Willers et al., Case-control 3-15 yr. old, Urinary Self-reporting Cotinine levels greater in cases 1991 49/77 clinic symptoms than controls (p<0.005) Ehrlich et al., Case-control 3-14 yr. old, Urinary, grouped Clinical exam Cases had higher cotinine levels 1992 72/121 ER and clinic <30 ng/ml or >30 ng/ml than control (OR=1.9, 95% CI 1.0-3.3) Chang et al., Case-control 2 mo.-16 yr. old, Salivary Clinical exam No difference in cotinine levels 2000 165/106 ER between case and control groups Group BâDose-effect relationship between cotinine and asthma severity Chilmonczyk Case-control 8 mo.-13 yr. old, Urinary, grouped, Asthma Monotonic relationship for â et al., 1993 199/199 clinic <10, 10-39, >39 ng/ml exacerbations; exacerbations and â function lung function Rylander Case-control 4 mo.-4 yr. old, Urinary Clinical exam Risk of wheezing increased with et al., 1995 199/351 hospital cotinine concentration Ehrlich et al., Case-control 7-9 yr. old; Urinary Self-reported Positive exposure-response 1996 368/294 heavy symptoms relationship for asthma and household cotinine by quartile (p=0.02) smokers Oddoze Cross sectional 4-14 yr. old, Urinary Bronchial Bronchial responsiveness et al., 1991 90 clinic responsiveness correlated with cotinine level (p<0.03) NOTE: ER=emergency room; OR=odds ratio; CI=confidence interval
NONNEOPLASTIC RESPIRATORY DISEASES 525 reported results. Finally, the clinical significance of the small differences in lung function or symptoms is unknown. Therefore, there is uncertainty about whether a dose-response relationship exists between cigarette smoke exposure as measured by cotinine levels and severity of asthma, and this subject requires more study before harm reduction studies can be planned. Review of Harm Reduction Strategies Applied to Asthma Several studies have examined the effect of reducing tobacco smoke exposure on wheezing symptoms. In one study (OâConnell and Logan, 1974), parents of a subpopulation of children in whom ETS exposure was considered a âsignificant factorâ were given antismoking advice and fol- lowed for 6-24 months. Symptoms improved in 90% of children whose parents stopped smoking and in 27% of children who remained exposed to passive smoking. These results are difficult to interpret because this is an uncontrolled trial. In a second study (Murray and Morrison, 1989), severity of asthma was scored in a group of children with the diagnosis of asthma whose mothers smoked. When the group was scored three years later, there was a highly significant decline in lung function in children of mothers who continued to smoke but an improvement in lung function of children whose mothers no longer smoked. This reduction was attributed to an alteration of maternal smoking habits, but this explanation is based on anecdotal evidence. A randomized, controlled trial to investigate whether brief intervention by advising parents about the impact of pas- sive smoking would reduce salivary cotinine levels in asthmatic children was found to be ineffective (Irvine et al., 1999). A study of the relationship between modifications of parental smoking behavior and nicotine expo- sure found that smoking outside the home was associated with lower urinary cotinine levels only when the parent was the only smoker in the house (Winkelstein et al., 1997). These intervention studies lack assess- ment of confounders and require a more rigorous design. Intervention studies, utilizing dose-response relationships between asthma severity and levels of environmental tobacco smoke, represent a future strategy for evaluating harm reduction from ETS in asthmatic children. RESPIRATORY INFECTIONS Definition and Epidemiology Viral rhinitis, tracheobronchitis, and bacterial bronchitis are acute in- fections of the epithelium of the upper respiratory tract. According to the 1997 National Center for Health Statistics survey (NCHS, 1999), upper
526 CLEARING THE SMOKE respiratory infections are the most common medical reason for school or work absenteeism in the United States. These infections can occur in healthy people during epidemics. Immunocompromised patients are at special risk of developing pneumonia and tuberculosis. More serious in- fections of the lower respiratory tract such as pneumonia are less com- mon in normal hosts. In elderly hospitalized individuals, nosocomial pneumonia is a major health problem and risk to life. Tuberculosis is a disease of the lower respiratory tract present predominately in socially and economically disadvantaged populations and in immunosuppressed patients in the United States, but it is a major health problem worldwide. Respiratory tract infections and pneumonia are particularly impor- tant causes of death in the United States. In 1997, the combined cause-of- death category âpneumonia and influenzaâ ranked sixth among the lead- ing causes of death (CDC, 1999). The age-adjusted mortality rate for pneumonia and influenza was 12.9 deaths per 100,000 population. During the interval from 1977 to 1997, the death rate from pneumonia and influ- enza increased 15.2% (CDC, 1999). The number of deaths in the United States in 1990-1994 due to respiratory infections that were attributed to cigarette smoking was estimated to be 8,000 per year (Table 14-2). Pneu- monia and influenza are particularly important in the elderly, and the death rate reaches 20% in community-acquired pneumonia (Feldman, 1999; Fine et al., 1996). During 1997, in children aged 1-4 years, the com- bined category pneumonia-influenza was the sixth leading cause of death, and it was the seventh leading cause of death in children 5-14 years of age (NCHS, 1999). Pathogenesis The respiratory tract in the normal host is effective in containing mi- crobes present in the environment. The airways below the level of the major bronchi are mostly sterile, and special situations predispose to res- piratory tract infections. These include the introduction of a new species into the environment, such as an antigenic shift in influenza virus. A second situation involves an overwhelming inoculum of organisms, such as exposure to contaminated ventilatory equipment. A third situation results from impairment of part of the host respiratory defense apparatus or systemic immune response. The major components of the respiratory host defenses are the mechanical barriers of the upper respiratory tract, locally secreted immunoglobulins, lymphoid structures of the bronchi, phagocytic macrophages, and a number of immune and nonimmune sub- stances that line the alveolar surfaces (Reynolds, 1997). Components of the host defense system are specially designed to deal with large particu- lates (3-10 µm) in the upper respiratory tract and small particles (0.5-<3
NONNEOPLASTIC RESPIRATORY DISEASES 527 mm) that may reach alveoli. If inflammation occurs in the alveoli, various systemic immune components can enter the airspaces by leakage through the alveolar surface. If local host defenses are insufficient, the host can recruit additional phagocytic cells such as polymorphonuclear leukocytes (PMNs) and create a local inflammatory response. The creation of an inflammatory response considerably increases the repertoire of systemic immune responses that can be recruited to contain the spread of microbes (Delves and Roitt, 2000). If the infection is successfully contained, inflam- mation is terminated and lung tissue is restored to its normal state. If it is not contained, inflammation can spread locally, leading to pneumonia. If severe, the local responses may produce permanent damage such as scar- ring to lung structure. The effects of exposure to cigarette smoke on immune and inflamma- tory responses have been studied extensively in humans (Holt, 1987; Johnson et al., 1990). Alteration of immune and inflammatory processes by cigarette smoke are also relevant to asthma and COPD. Both acute exposure and chronic exposure to cigarette smoke alter the responsive- ness of the immune system. The magnitude and direction of the alteration depend on the quantity and duration of exposure and the particular im- mune functions being studied. In general, low concentrations and short exposures enhance immune response, whereas high concentrations and long-term exposures suppress the responsiveness of the immune system. Many of the effects appear reversible within several weeks or months following cessation of exposure even after long periods (years) of expo- sure. The pulmonary tissues directly in contact with the smoke and the associated regional lymphatics are more affected than the peripheral im- mune system. Smokers have serum immunoglobulin levels that are 10- 20% lower than those of nonsmokers (Holt, 1987; Mili et al., 1991). In addition to impairment of the peripheral immune system, smoking im- pairs mucociliary clearance, enhances bacterial adherence to respiratory epithelium, and disrupts respiratory epithelium (Dye and Adler, 1994; Fainstein and Musher, 1979; Green and Carolin, 1967; Raman et al., 1983). These altered functions may explain the higher rates of nasopharyngeal colonization with meningococcus among active and passive smokers com- pared to nonsmokers (Stuart et al., 1989). Mainstream Smoke Exposure Cigarette smoking is associated with increased susceptibility to cer- tain types of respiratory infections (Aronson et al., 1982; Finklea et al., 1969; Fischer et al., 1997; Haynes et al., 1966; Imrey et al., 1996; Kark et al., 1982; Nuorti et al., 2000; Parnell et al., 1966; Peters and Ferris, 1967; Stanwell-Smith et al., 1994; Straus et al., 1996). The dose-response rela-
528 CLEARING THE SMOKE tionship between smoking and mortality from influenza and pneumonia has been studied in several cohort studies. In the American Cancer Soci- ety CPS-I, the standardized mortality ratio for deaths due to influenza and pneumonia for ever smokers compared to never smokers was 1.9-1.7 for males and 1.3 for females (Hammond, 1965). In the British Physician Study (Doll and Peto, 1976), there was a slight increase in risk for current smokers, and a dose-response relationship was noted by the amount smoked. Similar results were found in the U.S. Veterans Study (Rogot and Murray, 1980) and in a study of Swedish men (Carstensen et al., 1987). Cigarette smoking is an independent risk factor for invasive pneumo- coccal disease among immunocompetent, nonelderly adults (Nuorti et al., 2000). A dose-response relation was found for invasive pneumococcal disease and number of cigarettes smoked daily, pack-years of smoking, and time since quitting. Thus, cigarette smoking increases the risk of cer- tain respiratory diseases and predisposes to increased invasiveness of pneumococcal disease. Smoking has been studied as a possible risk factor for tuberculosis. Although research has been sparse, it has been postulated that smokers are at increased risk of developing active pulmonary tuberculosis. A case- control study by Alcaide and associates (1996) found smoking to be linked to the development of pulmonary tuberculosis in infected subjects who were in close contact with a case of active pulmonary tuberculosis. The risk of active tuberculosis was increased by exposure to environmental tobacco smoke. The significance of the association remained after control- ling for social and economic status, age, and gender, but it became not significant in those exposed only to passive smoking. A dose-response effect was also described in this study, although findings have been in- consistent in other studies. Children exposed to passive smoking were found to have increased risk of developing active pulmonary tuberculosis after having been infected (Altet et al., 1996). This association persisted after controlling for age and social and economic status. Although there are few studies, smoking has been linked to positive skin test conversions (Anderson et al., 1997). The mechanism postulated is that smoking de- creases immune defenses and increases susceptibility to pulmonary tu- berculosis (Plit et al., 1998). Secondary Smoke Exposure Compelling evidence exists for a causal relationship between paren- tal smoking and lower respiratory infections (bronchiolitis, pneumonia) during childhood. Strachan and Cook (1998) have reviewed 40 published series on this topic, which included papers in previous reviews (DiFranza and Lew, 1996; EPA, 1993; U.S. DHHS, 1986). The studies sought to ascer-
NONNEOPLASTIC RESPIRATORY DISEASES 529 tain the relationship between lower respiratory illnesses and parental smoking in children less than three years old. In one group, the occurrence of lower respiratory infections was based on data from a community or ambulatory care setting and included longi- tudinal studies, controlled trials, case-control studies, and retrospective prevalence surveys. The illnesses included unspecified lower respiratory illness, bronchitis, bronchiolitis, and pneumonia. The most widely derived measures of effect related to either parent smoking (compared with neither parent) and the effect of maternal smok- ing (compared with father only or neither parent). Information was sought to determine whether there was a dose-response relationship by relating illness to the amount smoked by either parent. A second series of studies analyzed the association between hospital admissions for lower respira- tory tract infections and parental smoking. Diagnoses included undiffer- entiated chest illness, bronchitis and/or pneumonia, wheezing illness, and bronchiolitis with or without confirmation of respiratory syncytial virus. All 14 studies in this group found an increased risk associated with parental smoking. The results of the community and hospital studies were broadly consistent, with only one study reporting a reduced risk among children of smokers. The pooled odds ratios were 1.6 (95% CI=1.4, 1.9) for smoking by either parent and 1.7 (95% CI=1.6, 1.9) for maternal smoking. The associations for parental smoking were robust to adjustments for confounding factors. In general, the evidence was more convincing for an association with smoking by the mother only than by the father only, probably related to a greater degree of exposure to the mother. Twelve cohort studies presented evidence for dose-response within smoking fami- lies, either to the number of smokers or to the amount smoked in the household. A statistically significant dose-response relationship was found in 10 of the 13 cohort studies (Cook and Strachan, 1997; Cook et al., 1998). In two case-control studies (Reese et al., 1992; Rylander et al., 1995), urinary cotinine levels were measured, but in neither study was it pos- sible to determine if the cotinine levels were related to the risk of respira- tory illnesses. The effect of paternal smoking on respiratory illnesses was most marked in the first year of life (Colley et al., 1974; Fergusson et al., 1981; Fergusson and Horwood, 1985), but the effects of maternal smoking on hospital admissions for respiratory infections were similar at all ages up to five years (Taylor and Wadsworth, 1987). There appears to be no increased risk of respiratory infections in susceptible subgroups (i.e., his- tory of parental allergy, prematurity, low birthweight) (Burr et al., 1989; Chen, 1994; Ehrlich et al., 1996; Lucas et al., 1990). Although the studies are generally consistent with an association be- tween parental smoking and lower respiratory tract infections in chil-
530 CLEARING THE SMOKE dren, the summary odds ratios derived from the pooled studies should be interpreted with caution. Difficulties with the pooled analysis include the process by which the variables were selected, the different types of data collection and analytical approaches applied to each study, and the incon- sistent methods used to account for confounding effects. In addition, the precise nature of the specific diagnosis of lower respiratory tract infec- tions and overlap with childhood asthma are important barriers to the analysis. The quality and sizes of the populations in these studies varied considerably, possibly introducing certain biases. Despite these limita- tions, it is reasonable to conclude that there is a causal relationship be- tween parental smoking and acute lower respiratory illnesses at least during the first two years of life. Latency The effect of active smoking and smoking cessation on age-adjusted mortality from infectious diseases was examined in the American Cancer Society CPS-II study. Male former smoker of less than 15 cigarettes per day have mortality ratios after ten years approaching unity, whereas former smokers of more than 21 cigarettes per day have mortality ratios near unity at 15 years. Female former smokers of any amount have mor- tality ratios from influenza and pneumonia approaching unity within 3-5 years. These results suggest that impaired host defenses are reversible after smoking cessation. Review of Harm Reduction Strategies Applied to Respiratory Infections One controlled intervention study has examined the effect of acute lower respiratory tract infection after an intervention designed to modify postnatal exposure to tobacco smoke (Greenberg et al., 1994). There was a reduced prevalence of lower respiratory symptoms among the interven- tion group of infants of smoking mothers whose education level was high school or less versus the control group (Greenberg et al., 1994). However, there was uncertainty about the effectiveness of the intervention since the mean cotinine levels did not differ between study groups despite a reduc- tion in smoking levels in the intervention group. SUMMARY AND CONCLUSIONS In evaluating the use of potential reduced-exposure products (PREPs) in tobacco-related lung disease, three major nonneoplastic respiratory dis- eases linked to cigarette smoking should be considered: COPD, asthma,
NONNEOPLASTIC RESPIRATORY DISEASES 531 and respiratory infections. Respiratory diseases are major tobacco-related illnesses, and there is a clear need to mitigate the harmful effects of expo- sure to both mainstream and secondary tobacco smoke. It is plausible that decreasing smoking will reduce the severity of chronic lung diseases and the incidence of respiratory infections, but there is no adequate scientific evidence to support this conclusion because the effects of reduced smok- ing on harm reduction have not been extensively studied. Important considerations are determining dose-effect relationships and use of respiratory disease biomarkers. Rational design of studies of harm reduction would rely on dose-effect data, but such data for respira- tory diseases are limited and of uncertain quality. High-quality dose- effect data are required for adequate study design, and such data should be generated. Rational study design would also incorporate biomarkers of disease, and the testing of current and new biomarkers might be done concurrently in the populations studied for dose-effects. The Cancer Pre- vention Studies I and II, large-scale prospective studies, however, do sug- gest a linear dose-effect relationship between number of cigarettes smoked per day and mortality rates from COPD, indicating that decreasing the amount of cigarettes smoked may lead to fewer deaths from COPD. There are currently no specific biomarkers of respiratory disease due to smoking tobacco products. No unique molecular or genetic defect spe- cific for tobacco-related respiratory disease has been identified. The pro- cesses involved, such as inflammation and increased levels of oxidants, are not unique to tobacco-related respiratory diseases. Identifying unique biomarkers is further confounded by the heterogeneous nature of these diseases, the complex mixture of tobacco smoke, and the range of indi- vidual susceptibilities to the harmful effects of tobacco smoke among users. The most widely used markers of tobacco-related respiratory dis- eases in population studies are symptom questionnaires and pulmonary function testing. These have well-known limitations of specificity and sensitivity, particularly for detecting the early effects of tobacco smoke on the lungs (U.S. DHHS, 1989). Subtle effects of tobacco smoke exposure on the lung can be detected by sampling fluid in the lower respiratory tract via a bronchoscope inserted into the airways, but the significance of these changes to clinically important pulmonary disease has not been estab- lished. Newer approaches such as sampling the subjectsâ urine (Pratico et al., 1998) or exhaled gas (Ichinose et al., 2000; Paolo et al., 2000) for meta- bolic products due to tissue injury have the advantage of noninvasive sampling but must be validated. Clearly, the greatest obstacle for devel- oping a specific biomarker is the lack of fundamental information on mechanisms by which exposure to tobacco smoke causes specific respira- tory diseases.
532 CLEARING THE SMOKE The availability of dose-effect data and validated biomarkers may improve the quality of, and provide greater confidence in, the results of contemplated intervention studies. However, the time frame for generat- ing dose-effect data and testing biomarkers is uncertain, and it is unclear whether inclusion of dose-effect considerations and biomarkers will im- prove the quality of clinical trials of harm reduction in respiratory dis- eases. An alternative is to proceed with interventional trials based on cur- rent knowledge if there are uncertainties about the added value of dose- effect data or untested biomarkers to study design. As an example, an intervention study of the effect of smoking reduction on COPD could be considered that is similar in design to the Lung Health Study, a large prospective trial of the effects of smoking cessation on rate of decline of FEV1 in middle-aged smokers with mild COPD (Anthonisen et al., 1994). Another approach is to conduct a trial using a low-tar/moderate-nicotine product from a noncommercial source to avoid product endorsement is- sues. (A more detailed research agenda can be found in the next section.) Design of population studies for harm reduction of major respiratory diseases is challenging because of uncertainties about effectiveness and long-term compliance with harm reduction interventions. Reducing the burden of tobacco-related respiratory diseases through harm reduction strategies should be a major priority of the nationâs public health. RESEARCH AGENDA This section outlines a suggested research agenda for studying harm reduction due to cigarette smoking in respiratory diseases (i.e., COPD, asthma, respiratory infections). Several specific suggestions for research design arise from this re- view. An interventional study of the effect of smoking reduction on COPD could be considered that is similar in design to the Lung Health Study, a large prospective trial of the effects of smoking cessation on rate of de- cline of FEV1 in middle-aged smokers with mild COPD (Anthonisen et al., 1994). In a proposed smoking reduction trial, it might be possible to in- clude an intervention group of smokers who are able to reduce their smoking spontaneously and maintain significant reductions for a long period using behavior intervention and/or nicotine replacement prod- ucts. The goal would be to decrease the number of cigarettes per day to eight to ten, a point below which it is very difficult to reduce the number of cigarettes smoked (Hughes, 2000). The primary end point could be the change in FEV1 over five years, and secondary end points could be ex- haled carbon monoxide concentration, serum cotinine level, survival, and comorbid smoking-related disorders. If successful, this study should be
NONNEOPLASTIC RESPIRATORY DISEASES 533 able to determine whether reduced smoking as measured by change in FEV1 mitigates harm to the lungs. Potential weaknesses are the self- selection of subjects, the large number of subjects required, ethical issues regarding the nonintervention group, and a probable large dropout rate in the intervention group. Another approach is to conduct a trial using a low-tar/moderate- nicotine product from a noncommercial source to avoid product endorse- ment issues. This study might be conducted in two phases. Phase I would be a controlled-exposure study in human smokers to compare the effects of the low-tar/moderate-nicotine product versus a reference cigarette on inflammatory changes in the lower respiratory tract, similar to the obser- vations of Rennard et al. (Rennard, 2000). The objective of Phase I would be to determine if inflammation is reduced by exposure to the low-tar/ moderate-nicotine product. If so, a Phase II intervention trial would com- pare the low-tar/moderate nicotine product to reference cigarettes in co- horts of current smokers. The objectives and design of the Phase II trial would be similar to that described above for intervention using behav- ioral modification or NRT. Potential advantages of this trial are that the low-tar/moderate-nicotine product, if it reduces harm, could be used as a reference product for future regulation of marketed products. Uncertain- ties related to such a study include inference from results of short-term controlled exposures to longer-term studies, controlling use of the inter- vention product, and the large effort that would be needed to complete the study. REFERENCES Abramson M, Kutin J, et al. 1995. Morbidity, medication and trigger factors in a community sample of adults with asthma. Med J Aust 162:78-81. Alcaide J, Altet M, et al. 1996. Cigarette smoking as a risk factor for tuberculosis in young adults: a case-control study. Tubercle and Lung Disease 77:112-116. Altet M, Alcaide J, et al. 1996. Passive smoking and risk of pulmonary tuberculosis in children immediately following infection. A case-control study. Tubercle and Lung Dis- ease 77:537-544. Althuis MD, Sexton M, Prybylski D. 1999. Cigarette smoking and asthma symptom severity among adult asthmatics. J Asthma 36(3):257-264. Anderson R, Sy F, et al. 1997. Cigarette smoking and tuberculin skin test conversion among incarcerated adults. Amer J Prev Med 13:175-181. Anthonisen NR, Connett JE, Kiley JP, et al. 1994. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA 272(19):1497-1505. Aronson MD, Weiss ST, Ben RL, Komaroff AL. 1982. Association between cigarette smok- ing and acute respiratory tract illness in young adults. JAMA 248(2):181-183. Ashley F, Kannel WB, Sorlie PD, Masson R. 1975. Pulmonary function: relation to aging, cigarette habit, and mortality. The Framingham Study. Ann Intern Med 82:739-745.
534 CLEARING THE SMOKE Aubry MC, Wright JL, Myers JL. 2000. The pathology of smoking-related lung diseases. Clin Chest Med 21(1):11-35. Barnes PJ. 2000. Chronic obstructive pulmonary disease. N Engl J Med 343(4):269-280. Bates DV. 1973. The fate of the chronic bronchitic: a report of the ten-year follow-up in the Canadian Department of Veteranâs Affairs coordinated study of chronic bronchitis. The J. Burns Amberson Lecture of the American Thoracic Society. Am Rev Respir Dis 108(5):1043-1065. Beaty TH, Newill CA, Cohen BH, Tockman MS, Bryant SH, Spurgeon HA. 1985. Effects of pulmonary function on mortality. J Chron Dis 38:703-710. Berglund DJ, Abbey DE, Lebowitz MD, Knutsen SF, McDonnell WF. 1999. Respiratory symptoms and pulmonary function in an elderly nonsmoking population. Chest 115(1):49-59. Berkey CS, Ware JH, Dockery DW, Ferris BG Jr, Speizer FE. 1986. Indoor air pollution and pulmonary function growth in preadolescent children. Am J Epidemiol 123(2):250-60. Buist AS, Sexton GJ, Nagy JM, Ross BB. 1976. The effect of smoking cessation and modifica- tion on lung function. Am Rev Respir Dis 114(1):115-122. Buist A, Vollmer W. 1994. Smoking and other risk factors. Murray J, Nadel J, eds. Textbook of Respiratory Medicine. Philadelphia, PA: W.B. Saunders Company. Pp. 1259-1287. Burns DM, Shanks TG, Choi W, Thun MJ, Heath CW, Garfinkel L. 1997. The American Cancer Society Cancer Prevention Study: 12-year follow-up of one million men and women. Changes in Cigarette-Related Diseases Risks and Their Implication for Prevention and Control. Smoking and Tobacco Control Monograph 8. Washington, DC: National Cancer Institute. Pp. 113-304. Burr ML, Miskelly FG, Butland BK, Merrett TG, Vaughan-Williams E. 1989. Environmental factors and symptoms in infants at high risk of allergy. J Epidemiol Community Health 43(2):125-132. Burrows B. 1990. Airways obstructive diseases: pathogenetic mechanisms and natural his- tories of the disorders. Med Clin North Am 74(3):547-559. Burrows B, Knudson RJ, Cline MG, Lebowitz MD. 1977a. Quantitative relationships be- tween cigarette smoking and ventilatory function. Am Rev Respir Dis 115(2):195-205. Burrows B, Knudson RJ, Lebowitz MD. 1977b. The relationship of childhood respiratory illness to adult obstructive airway disease. Am Rev Respir Dis 115(5):751-760. Camilli AE, Burrows B, Knudson RJ, Lyle Sk, Lebowitz MD. 1987. Longitudinal changes in forced expiratory volume in one second in adults. Effects of smoking and smoking cessation. Am Rev Respir Dis 135(4):794-799. Carey IM, Cook DG, Strachan DP. 1999. The effects of environmental tobacco smoke expo- sure on lung function in a longitudinal study of British adults. Epidemiology 10(3):319- 326. Carp H, Miller F, Hoidal JR, Janoff A. 1982. Potential mechanism of emphysema: alpha 1- proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized me- thionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci U S A 79(6):2041-2045. Carstensen JM, Pershagen G, Eklund G. 1987. Mortality in relation to cigarette and pipe smoking: 16 yearsâ observation of 25,000 Swedish men. J Epidemiol Community Health 41(2):166-172. CDC (Centers for Disease Control and Prevention). 1999. Mortality Patterns-United States, 1997. Morbidity and Mortality Weekly Reports 48:664-668. Chang M, Hogan A, et al. 2000. Salivary cotinine levels in children presenting with wheez- ing in an emergency department. Ped Pulm 29:257-263. Chen Y. 1994. Environmental tobacco smoke, low birth weight, and hospitalization for respiratory disease. Am J Respir Crit Care Med 150(1):54-58.
NONNEOPLASTIC RESPIRATORY DISEASES 535 Chilmonczyk B, Salmun L, et al. 1993. Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. N Engl J Med 328:1665-1669. Colley JR, Holland WW, Corkhill RT. 1974. Influence of passive smoking and parental phlegm on pneumonia and bronchitis in early childhood. Lancet 2(7888):1031-1034. Collins M, Moessinger A, et al. 1985. Fetal lung hypoplasia associated with maternal smok- ing: a morphometric analysis. Ped Research 19:408-412. Comstock GW, Meyer MB, Helsing KJ, Tockman MS. 1981. Respiratory effects on house- hold exposures to tobacco smoke and gas cooking. Am Rev Respir Dis 124(2):143-148. Cook DG, Strachan DP. 1997. Health effects of passive smoking. Parental smoking and prevalence of respiratory symptoms and asthma in school age children. Thorax 52(12):1081-1094. Cook DG, Strachan DP. 1999. Health effects of passive smoking. Summary of effects of parental smoking on the respiratory health of children and implications for research. Thorax 54(4):357-366. Cook DG, Strachan DP, Carey IM. 1998. Health effects of passive smoking. Parental smok- ing and spirometric indices in children. Thorax 53(10):884-893. Coultas DB. 1998. Health effects of passive smoking. Passive smoking and risk of adult asthma and COPD: an update. Thorax 53(5):381-387. Dayal HH, Khuder S, Sharrar R, Trieff N. 1994. Passive smoking in obstructive respiratory disease in an industrialized urban population. Environ Res 65(2):161-171. Delves P, Roitt I. 2000. The immune system. NEJM 343:37-49. DiFranza JR, Lew RA. 1996. Morbidity and mortality in children associated with the use of tobacco products by other people. Pediatrics 97(4):560-568. Dockery DW, Speizer FE, Ferris BG Jr, Ware JH, Louis TA, Spiro A 3d. 1988. Cumulative and reversible effects of lifetime smoking on simple tests of lung function in adults. Am Rev Respir Dis 137(2):286-292. Doll R, Peto R. 1976. Mortality in relation to smoking: 20 yearsâ observations on male British doctors. Br Med J 2(6051):1525-1536. Dow L, Tracey M, Villar A, et al. 1996. Does dietary intake of vitamins C and E influence lung function in older people? Am J Respir Crit Care Med 154(5):1401-1404. Drazen J. 2000. Asthma. Goldman L, Bennett J, eds. Cecil Textbook of Medicine. Philadelphia, PA: W.B. Saunders Company. Pp. 387-393. Drazen JM, Takebayashi T, Long NC, De Sanctis GT, Shore SA. 1999. Animal models of asthma and chronic bronchitis. Clin Exp Allergy 9 (Suppl 2):37-47. Dye JA, Adler KB. 1994. Effects of cigarette smoke on epithelial cells of the respiratory tract. Thorax 49(8):825-834. ECLIPSE Expert Panel. 2000. A safer cigarette? A comparative study. A consensus report. Inhalation Toxicology 12 (Suppl.5): 1-48. Ehrlich RI, Du Toit D, Jordaan E, et al. 1996. Risk factors for childhood asthma and wheez- ing. Importance of maternal and household smoking. Am J Respir Crit Care Med 154(3 Pt 1):681-688. Ehrlich R, Kattan M, Godbold J, et al. 1992. Childhood asthma and passive smoking. Uri- nary cotinine as a biomarker of exposure. Am Rev Respir Dis 145(3):594-599. EPA (Environmental Protection Agency). 1993. Respiratory Health Effects of Passive Smoking: Lung Cancer and Other Disorders. NIH Publication No. 93-3605. Washington, DC: U.S. Environmental Protection Agency. Fainstein V, Musher DM. 1979. Bacterial adherence to pharyngeal cells in smokers, non- smokers, and chronic bronchitics. Infect Immun 26(1):178-182. Fauler J, Frolich JC. 1997. Cigarette smoking stimulates cysteinyl leukotriene production in man. Eur J Clin Invest 27(1):43-47. Feldman C. 1999. Pneumonia in the elderly. Clin Chest Med 20(3):563-573.
536 CLEARING THE SMOKE Fergusson DM, Horwood LJ. 1985. Parental smoking and respiratory illness during early childhood: a six-year longitudinal study. Pediatr Pulmonol 1(2):99-106. Fergusson DM, Horwood LJ, Shannon FT, Taylor B. 1981. Parental smoking and lower respiratory illness in the first three years of life. J Epidemiol Community Health 35(3):180- 184. Fine MJ, Smith MA, Carson CA, et al. 1996. Prognosis and outcomes of patients with com- munity-acquired pneumonia. A meta-analysis. JAMA 275(2):134-141. Finklea JF, Sandifer SH, Smith DD. 1969. Cigarette smoking and epidemic influenza. Am J Epidemiol 90(5):390-399. Fischer M, Hedberg K, Cardosi P, et al. 1997. Tobacco smoke as a risk factor for meningo- coccal disease. Pediatr Infect Dis J 16(10):979-983. Fletcher C, Peto R. 1977. The natural history of chronic airflow obstruction. Br Med J 1(6077):1645-1648. Fletcher C, Peto R, et al. 1976. The Natural History of Bronchitis and Emphysema. Oxford, England: Oxford University Press. Floreani AA, Rennard SI. 1999. The role of cigarette smoke in the pathogenesis of asthma and as a trigger for acute symptoms. Curr Opin Pulm Med 5(1):38-46. Frew AJ, Kennedy SM, Chan-Yeung M. 1992. Methacholine responsiveness, smoking, and atopy as risk factors for accelerated FEV1 decline in male working populations. Am Rev Respir Dis 146(4):878-883. Gilliland FD, Berhane K, McConnell R, et al. 2000. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax 55(4):271- 276. Gold DR, Tager IB, Weiss ST, Tosteson TD, Speizer FE. 1989. Acute lower respiratory illness in childhood as a predictor of lung function and chronic respiratory symptoms. Am Rev Respir Dis 140(4):877-884. Green GM, Carolin D. 1967. The depressant effect of cigarette smoke on the in vitro antibac- terial activity of alveolar macrophages. N Engl J Med 276(8):421-427. Greenberg RA, Strecher VJ, Bauman KE, et al. 1994. Evaluation of a home-based interven- tion program to reduce infant passive smoking and lower respiratory illness. J Behav Med 17(3):273-290. Greer JR, Abbey DE, Burchette RJ. 1993. Asthma related to occupational and ambient air pollutants in nonsmokers. J Occup Med 35(9):909-915. Habib MP, Klink ME, Knudson DE, Bloom JW, Kaltenborn WT, Knudson RJ. 1987. Physi- ologic characteristics of subjects exhibiting accelerated deterioration of ventilatory function. Am Rev respir Dis 136:638-645. Hammond E. 1965. Evidence on the effects of giving up cigarette smoking. Am J Public Health 55:682-691. Hanrahan JP, Tager IB, Segal MR, Tosteson TD, Castile RG, Van Vunakis H, Weiss ST, Speizer FE. 1992. The effect of maternal smoking during pregnancy on early infant lung function. Am Rev Respir Dis 145(5):1129-1135. Haynes WF Jr, Krstulovic VJ, Bell AL Jr. 1966. Smoking habit and incidence of respiratory tract infections in a group of adolescent males. Am Rev Respir Dis 93(5):730-735. Hensley M, Saunders N. 1989. Clinical epidemiology of chronic obstructive lung disease. Lung Biology in Health and Disease. New York: Marcel Dekker, Inc. Higgins MW, Keller JB, Becker M, Howatt W, Landis JR, Rotman H, et al. 1982. An index of risk for obstructive airways disease. Am Rev Respir Dis 125:144-151. Higgins MW, Keller JB, Landis JR, et al. 1984. Risk of chronic obstructive pulmonary dis- ease. Collaborative assessment of the validity of the Tecumseh index of risk. Am Rev Respir Dis 130(3):380-385.
NONNEOPLASTIC RESPIRATORY DISEASES 537 Hole DJ, Gillis CR, Chopra C, Hawthorne VM. 1989. Passive smoking and cardiorespiratory health in a general population in the west of Scotland. BMJ 299(6696):423-427. Holt PG. 1987. Immune and inflammatory function in cigarette smokers. Thorax 42(4):241- 249. Hu FB, Persky V, Flay BR, Richardson J. 1997. An epidemiological study of asthma preva- lence and related factors among young adults. J Asthma 34(1):67-76. Hughes J. 2000. Reduced smoking: an introduction and review of the evidence. Addiction 95(Suppl 1):S3-7. Hunninghake GW, Crystal RG. 1983. Cigarette smoking and lung destruction. Accumula- tion of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis 128(5):833-838. Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. 2000. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med 162(2 Pt 1):701-706. Imrey PB, Jackson LA, Ludwinski PH, et al. 1996. Outbreak of serogroup C meningococcal disease associated with campus bar patronage. Am J Epidemiol 143(6):624-630. Ingram RJ, OâCain C. 1971. Frequency dependence of compliance in apparently healthy smokers versus non-smokers. Bull European de Physiopathologie Resp 7:195-212. Irvine L, Crombie I, et al. 1999. Advising parents of asthmatic children on passive smoking: randomized control trial. BMJ 318:1456-1459. Jansen DF, Schouten JP, Vonk JM, Rijcken B, Timens W, Kraan J, Weiss S, Postma DS. 1999. Smoking and airway hyperresponsiveness especially in the presence of blood eosino- philia increase the risk to develop respiratory symptoms: a 25-year follow-up study in the general adult population. Am J Respir Crit Care Med 160(1):259-264. Joad JP. 2000. Smoking and pediatric respiratory health. Clin Chest Med 21(1):37-46. Johnson D, Travis J. 1979. The oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive center. J Biol Chem 254(10):4022-4026. Johnson JD, Houchens DP, Kluwe WM, Craig DK, Fisher GL. 1990. Effects of mainstream and environmental tobacco smoke on the immune system in animals and humans: a review. Crit Rev Toxicol 20(5):369-395. Jones JR, Higgins IT, Higgins MW, Keller JB. 1983. Effects of cooking fuels on lung function in nonsmoking women. Arch Environ Health 38(4):219-222. Kanner RE, Connett JE, Williams DE, Buist AS. 1999. Effects of randomized assignment to a smoking cessation intervention and changes in smoking habits on respiratory symp- toms in smokers with early chronic obstructive pulmonary disease: the Lung Health Study. Am J Med 106(4):410-416. Kark JD, Lebiush M, Rannon L. 1982. Cigarette smoking as a risk factor for epidemic a(h1n1) influenza in young men. N Engl J Med 307(17):1042-1046. Kauffman F, Dockery D, et al. 1989. Respiratory symptoms and lung function in relation to passive smoking: a comparative study of American and French women. Int J Epidemiol 18:334-344. Kerstjens HA, Rijcken B, Schouten JP, Postma DS. 1997. Decline of FEV1 by age and smok- ing status: facts, figures, and fallacies. Thorax 52(9):820-827. Khoury MJ, Beaty TH, Tockman MS, Self SG, Cohen BH. 1985. Familial aggregation in chronic obstructive pulmonary disease: use of the loglinear model to analyze interme- diate environmental and genetic risk factors. Genet Epidemiol 2(2):155-166. Knudson RJ, Burrows B, Lebowitz MD. 1976. The maximal expiratory flow-volume curve: its use in the detection of ventilatory abnormalities in a population study. Am Rev Respir Dis 114(5):871-879. Koyama H, Geddes DM. 1998. Genes, oxidative stress, and the risk of chronic obstructive pulmonary disease. Thorax 53(Suppl 2):S10-14.
538 CLEARING THE SMOKE Lange P, Groth S, Nyboe J, et al. 1989. Effects of smoking and changes in smoking habits on the decline of FEV1. Eur Respir J 2(9):811-816. Lange P, Nyboe J, Appleyard M, Jensen G, Schnohr P. 1990. Relation of ventilatory impair- ment and of chronic mucus hypersecretion to mortality from obstructive lung disease and from all causes. Thorax 45:579-585. Lebowitz MD, Holberg CJ. 1988. Effects of parental smoking and other risk factors on the development of pulmonary function in children and adolescents. Analysis of two lon- gitudinal population studies. Am J Epidemiol 128(3):589-597. Leuenberger P, Schwartz J, Ackermann-Liebrich U, et al. 1994. Passive smoking exposure in adults and chronic respiratory symptoms (SAPALDIA Study). Swiss Study on Air Pollution and Lung Diseases in Adults, SAPALDIA Team. Am J Respir Crit Care Med 150(5 Pt 1):1222-1228. Lucas A, Brooke OG, Cole TJ, Morley R, Bamford MF. 1990. Food and drug reactions, wheezing, and eczema in preterm infants. Arch Dis Child 65(4):411-415. Macnee W, Rahman I. 1999. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 160(5 Pt 2):S58-65. Marine WM, Gurr D, Jacobsen M. 1988. Clinically important respiratory effects of dust exposure and smoking in British coal miners. Am Rev Respir Dis 137(1):106-112. Mateos F, Brock JH, Perez-Arellano JL. 1998. Iron metabolism in the lower respiratory tract. Thorax 53(7):594-600. McCarthy DS, Craig DB, Cherniack RM. 1976. Effect of modification of the smoking habit on lung function. Am Rev Respir Dis 114(1):103-113. Mili F, Flanders WD, Boring JR, Annest JL, Destefano F. 1991. The associations of race, cigarette smoking, and smoking cessation to measures of the immune system in middle-aged men. Clin Immunol Immunopathol 59(2):187-200. Milner AD, Marsh MJ, Ingram DM, Fox GF, Susiva C. 1999. Effects of smoking in preg- nancy on neonatal lung function. Arch Dis Child Fetal Neonatal Ed 80(1):F8-14. Morrison D, Rahman I, Lannan S, MacNee W. 1999. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med 159(2):473- 479. Murin S, Bilello KS, Matthay R. 2000. Other smoking-affected pulmonary diseases. Clin Chest Med 21(1):121-137. Murin S, Silvestri G, eds. 2000. Smoking and pulmonary and cardiovascular diseases. Clinics in Chest Medicine. Philadelphia, PA: W.B. Saunders Co. Murphy TF, Sethi S. 1992. Bacterial infection in chronic obstructive pulmonary disease. Am Rev Respir Dis 146(4):1067-1083. Murray AB, Morrison BJ. 1989. Passive smoking by asthmatics: its greater effect on boys than on girls and on older than on younger children. Pediatrics 84(3):451-459. Nadel J, Comroe J. 1961. Acute effects of inhalation of cigarette smoke on airway conduc- tance. J Applied Physiol 16: 713-716. NCHS (National Center for Health Statistics). 1999. Health, United States 1999 with Health and Aging Chartbook. Hyattsville, MD: NCHS. NIH (National Institutes of Health). 1997. Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: NIH, National Heart, Lung, and Blood Institute. Novotny TE, Giovino GE. 1998. Tobacco Use. Brownson RE, Renington PL, Davis JR, eds. Chronic Disease Epidemiology and Control. 2nd ed. Washington, DC: American Public Health Association Press. Pp.117-148. NRC (National Research Council). 1986. Environmental Tobacco Smoke: Measuring Exposures and Assessing Health Effects. Washington, DC: National Academy Press. Nuorti JP, Butler JC, Farley MM, et al. 2000. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med 342(10):681-689.
NONNEOPLASTIC RESPIRATORY DISEASES 539 OâConnell EJ, Logan GB. 1974. Parental smoking in childhood asthma. Ann Allergy 32(3):142- 145. OâConner G, Sparrow D, et. al. 1995. A prospective longitudinal study of methacholine airway responsiveness as a predictor of pulmonary function decline: Normative Ag- ing Study. American Journal of Respiratory and Critical Care Medicine 152:87-92. Oddoze C, Dunus J, et al. 1999. Urinary cotinine and exposure to parental smoking in a population of children with asthma. Clinical Chemistry 45:505-509. Orie NGM, Sluiter HJ, De Vries K, Tammeling GJ, Witkop J. 1961. The host factor in bron- chitis. Orie NGM, Sluiter HJ, ed. Bronchitis. Assen, Holland: Royal Van Gorcum. Pp. 43-59. Paolo P, Kharitonov S, et al. 2000. Exhaled ethane, a marker of lipid peroxidation, is el- evated in chronic obstructive pulmonary disease. Am J Resp Crit Care Med 162:369-373. Parnell JL, Anderson DO, Kinnis C. 1966. Cigarette smoking and respiratory infections in a class of student nurses. N Engl J Med 274(18):979-984. Peters JM, Ferris BG Jr. 1967. Smoking and morbidity in a college-age group. Am Rev Respir Dis 95(5):783-789. Peto R, Speizer FE, Cochrane AL, et al. 1983. The relevance in adults of air-flow obstruction, but not of mucus hypersecretion, to mortality from chronic lung disease. Results from 20 years of prospective observation. Am Rev Respir Dis 128(3):491-500. Piquette C, Rennard S, et. al. 2000. Chronic bronchitis and emphysema. Murray J, Nadel J, Mason R, Boushey HJ. Textbook of Respiratory Medicine. Philadelphia, PA: W.B. Saunders Company. Pp. 1187-1245. Plit M, Theron A, et al. 1998. Influence of antimicrobial chemotherapy and smoking status on the plasma concentrations of vitamin C, vitamin E, beta-carotene, acute phase reac- tants, iron and lipid peroxides in patients with pulmonary tuberculosis. Inter J Tuber Lung Dis 2(7):590-596. Postma DS, de Vries K, Koeter GH, Sluiter HJ. 1986. Independent influence of reversibility of air-flow obstruction and nonspecific hyperreactivity on the long-term course of lung function in chronic air-flow obstruction. Am Rev Respir Dis 134(2):276-280. Practico D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald GA. 1998. Chronic obstructive pulmonary disease is associated wiht an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stress. Am J Respir Crit Care Med 158(6):1709-1714. Pryor WA, Stone K. 1993. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 686:12-27, Discussion 27-28. Raman AS, Swinburne AJ, Fedullo AJ. 1983. Pneumococcal adherence to the buccal epithe- lial cells of cigarette smokers. Chest 83(1):23-27. Rautalahti M, Virtamo J, Haukka J, et al. 1997. The effect of alpha-tocopherol and beta- carotene supplementation on COPD symptoms. Am J Respir Crit Care Med 156(5):1447- 1452. Reese AC, James IR, Landau LI, Lesouef PN. 1992. Relationship between urinary cotinine level and diagnosis in children admitted to hospital. Am Rev Respir Dis 146(1):66-70. Rennard SI. 2000 Apr 25. Presentation to the IOM Committee to Assess the Science Base for Tobacco Harm Reduction. Washington, DC. Rennard SI, Daughton D, Fujita J, et al. 1990. Short-term smoking reduction is associated with reduction in measures of lower respiratory tract inflammation in heavy smokers. Eur Respir J 3(7):752-759. Reynolds H. 1997. Integrated host defense against infections. Crystal R, West J, Barnes P, Weibel E, eds. The Lung: Scientific Foundations. Vol. 2. Philadelphia, PA: Lippincott- Raven Publishers. Pp. 2353-2365.
540 CLEARING THE SMOKE Rijcken B, Schouten JP, Xu X, Rosner B, Weiss ST. 1995. Airway hyperresponsiveness to histamine associated with accelerated decline in FEV1. Am J Respir Crit Care Med 151(5):1377-1382. Robbins AS, Abbey DE, Lebowitz MD. 1993. Passive smoking and chronic respiratory dis- ease symptoms in non-smoking adults. Int J Epidemiol 22(5):809-817. Rogot E, Murray JL. 1980. Smoking and causes of death among U.S. veterans: 16 years of observation. Public Health Rep 95(3):213-222. Rokaw SN, Detels R, Coulson AH, Sayre JW, Tashkin DP, Allwright SS, Massey FJ. 1980. The UCLA population studies of chronic obstructive respiratory disease. Comparison of pulmonary function in three communities exposed to photochemical oxidants, mul- tiple primary pollutants, or minimal pollutants. Chest 78(2):252-262. Rylander E, Pershagen G, Eriksson M, Bermann G. 1995. Parental smoking, urinary cotinine, and wheezing bronchitis in children. Epidemiology 6(3):289-293. Samet JM, Lange P. 1996. Longitudinal studies of active and passive smoking. Am J Respir Crit Care Med 154(6 Pt 2):S257-265. Samet JM, Tager IB, Speizer FE. 1983. The relationship between respiratory illness in child- hood and chronic air-flow obstruction in adulthood. Am Rev Respir Dis 127(4):508-523. Schottenfeld D. 1984. Epidemiology of cancer of the esophagus. Semin Oncol 11(2):92-100. Sekhon HS, Wright JL, Churg A. 1994. Cigarette smoke causes rapid cell proliferation in small airways and associated pulmonary arteries. Am J Physiol 267(5 Pt 1):L557-563. Senior RM, Shapiro SD. 1998. Chronic obstructive lung disease: epidemiology, pathophysi- ology, and pathogenesis. Fishman A, Elias J, Fishman J, et al., eds. Fishmanâs Pulmonary Diseases and Disorder. 3rd ed. New York: McGraw-Hill. Pp. 680. Sethi JM, Rochester CL. 2000. Smoking and chronic obstructive pulmonary disease. Clin Chest Med 21(1):67-86. Shaheen SO, Barker DJ, Holgate ST. 1995. Do lower respiratory tract infections in early childhood cause chronic obstructive pulmonary disease? Am J Respir Crit Care Med 151(5):1649-51, discussion 1651-1652. Shapiro SD. 2000. Animal models for COPD. Chest 117(5 Suppl 1):223S-227S. Sherman CB. 1991. Health effects of cigarette smoking. Clin Chest Med 12(4):643-658. Sherman CB, Xu X, Speizer FE, Ferris Jr BG, Weiss ST, Dockery DW. 1992. Longitudinal lung function decline in subjects with respiratory symptoms. Am Rev Respir Dis 146:855-859. Sly R. 2000. Asthma. Behrman R, Kliegman R, Jenson H, eds. Nelson Textbook of Pediatrics. Philadelphia: WB Saunders Company. Snider GL. 1989. Pulmonary disease in alpha-1-antitrypsin deficiency. Ann Intern Med 111(12):957-959. Stanwell-Smith RE, Stuart JM, Hughes AO, Robinson P, Griffin MB, Cartwright K. 1994. Smoking, the environment and meningococcal disease: a case control study. Epidemiol Infect 112(2):315-328. Stick SM, Burton PR, Gurrin L, Sly PD, LeSouef PN. 1996. Effects of maternal smoking during pregnancy and a family history of asthma on respiratory function in newborn infants. Lancet 348(9034):1060-1064. Strachan DP, Cook DG. 1998. Health effects of passive smoking. Parental smoking and childhood asthma: longitudinal and case-control studies. Thorax 53(3):204-212. Straus WL, Plouffe JF, File TM Jr, et al. 1996. Risk factors for domestic acquisition of legion- naires disease. Ohio Legionnaires Disease Group. Arch Intern Med 156(15):1685-1692. Stuart J, Cartwright K, et al. 1989. Effect of smoking on meningococcal cariage. Lancet 2:723- 725. Svendsen KH, Kuller LH, Martin MJ, Ockene JK. 1987. Effects of passive smoking in the Multiple Risk Factor Intervention Trial. Am J Epidemiol 126(5):783-795.
NONNEOPLASTIC RESPIRATORY DISEASES 541 Tager IB, Segal MR, Munoz A, Weiss ST, Speizer FE. 1987. The effect of maternal cigarette smoking on the pulmonary function of children and adolescents. Analyses of data from two populations. Am Rev Respir Dis 136(6):1366-1370. Tager IB, Weiss ST, Munoz A, Rosner B, Speizer FE. 1983. Longitudinal study of the effects of maternal smoking on pulmonary function in children. N Engl J Med 309(12):699-703. Tager IB, Weiss ST, Rosner B, Speizer FE. 1979. Effect of parental cigarette smoking on the pulmonary function of children. Am J Epidemiol 110(1):15-26. Tanoue LT. 2000. Cigarette smoking and womenâs respiratory health. Clin Chest Med 21(1):47-65. Tashkin DP, Altose MD, Connett JE, Kanner RE, Lee WW, Wise RA. 1996. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group. Am J Respir Crit Care Med 153(6 Pt 1):1802-1811. Tashkin DP, Detels R, Simmons M, Liu H, Coulson AH, Sayre J, Rokaw S. 1994. The UCLA population studies of chronic obstructive respiratory disease: XI. Impact of air pollu- tion and smoking on annual change in forced expiratory volume in one second. Am J Respir Crit Care Med 149(5):1209-1217. Taylor B, Wadsworth J. 1987. Maternal smoking during pregnancy and lower respiratory tract illness in early life. Arch Dis Child 62(8):786-791. Thun MJ, Myers DG, Day-Lally C, et al. 1997. Age and exposure-response relationships between cigarette smoking and premature death in cancer prevention study II. Na- tional Cancer Institute. Changes in Cigarette-Related Disease Risks and Their Implication for Prevention and Control. NCI Smoking and Tobacco Control Monograph 8. Washington, DC: NCI. Pp. 383-475. Traber MG, van der Vliet A, Reznick AZ, Cross CE. 2000. Tobacco-related diseases. Is there a role for antioxidant micronutrient supplementation? Clin Chest Med 21(1):173-187. Tracey M, Villar A, Dow L, Coggon D, Lampe FC, Holgate ST. 1995. The influence of increased bronchial responsiveness, atopy, and serum IgE on decline in FEV1. A longi- tudinal study in the elderly. Am J Respir Crit Care Med 151(3 Pt 1):656-662. Tredaniel J, Boffetta P, Saracci R, Hirsch A. 1994. Exposure to environmental tobacco smoke and risk of lung cancer: the epidemiological evidence. Eur Respir J 7(10):1877-1888. U.S. DHHS (U.S. Department of Health and Human Services). 1984. The Health Consequences of Smoking: Chronic Obstructive Lung Disease. A Report of the Surgeon General. Washing- ton, DC: U.S. Department of Health and Human Services, Public Health Service, Office of Smoking and Health. U.S. DHHS. 1986. The Health Consequences of Involuntary Smoking: A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. U.S. DHHS. 1989. Reducing the Health Consequences of Smoking; A Report of the Surgeon Gen- eral. Washington, DC: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. U.S. DHHS. 1990. The Health Benefits of Smoking Cessation; A Report of the Surgeon General. Washington, DC: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. van der Lende R, Kok T, et al. 1981. Decreases in VC and FEV1 with time: indicators for effects of smoking and air pollution. Bulletin Europeen de Physiopathologie Respiratoire 17:775-792. Vestbo J, Prescott E, Lange P. 1996. Association of chronic mucus hypersecretion with FEV1 decline and chronic abstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med 153(5):1530-1535.
542 CLEARING THE SMOKE Weiss K, Gergen P, et al. 1993. Breathing better or worse? The changing epidemiology of asthma morbidity and mortality. Ann Rev Public Health 14:491-513. Weiss ST, Utell MJ, Samet JM. 1999. Environmental tobacco smoke exposure and asthma in adults. Environ Health Perspect 107(suppl 6):891-895. White JR, Froeb HF. 1980. Small-airways dysfunction in nonsmokers chronically exposed to tobacco smoke. N Engl J Med 302(13):720-723. Whittemore AS, Perlin SA, DiCiccio Y. 1995. Chronic obstructive pulmonary disease in lifelong nonsmokers: results from NHANES. Am J Public Health 85(5):702-706. Willers S, Svenonius E, Skarping G. 1991. Passive smoking and childhood asthma. Urinary cotinine levels in children with asthma and in referents. Allergy 46(5):330-334. Winkelstein M, Tarzian A, et al. 1997. Paternal smoking behavior and passive smoke expo- sure in children with asthma. Ann Allergy Asthma Immunol 78:419-423. Witschi H, Joad JP, Pinkerton KE. 1997. The toxicology of environmental tobacco smoke. Annu Rev Pharmacol Toxicol 37:29-52. Woolcock A. 1994. Asthma. Murray J, Nadel J, eds. Textbook of Respiratory Medicine. Phila- delphia, PA: W.B. Saunders and Company. Pp. 1288-1330. Young S, Le Souef P, et al. 1991. The influence of a family history of asthma and parental smoking on airway responsiveness in early infancy. N Engl J Med 324:1168-1173. Yunginger JW, Reed CE, OâConnell EJ, Melton LJ 3rd, OâFallon WM, Silverstein MD. 1992. A community-based study of the epidemiology of asthma. Incidence rates, 1964-1983. Am Rev Respir Dis 146(4):888-894. Zang LY, Stone K, Pryor WA. 1995. Detection of free radicals in aqueous extracts of ciga- rette tar by electron spin resonance. Free Radic Biol Med 19(2):161-167. Zeise L, Dunn A, Haroun L, Ting D, Windham G, Golub M, Waller K, Lipsett M, Shusterman D, Mann J, Wu A. 1997. Health Effects of Exposure to Environmental Tobacco Smoke. Office of Health and Environmental Health Hazard Assessment. California: Environmental Pro- tection Agency. Zejda J, McDuffie H, et al. 1993. Respiratory effects of exposure to grain dust. Seminars in Respiratory Medicine 14:20-30.
