
5
Health Considerations Related to Chemical Contaminants and Physical Factors
The 1986 National Research Council (NRC) report on commercial airliner cabin air quality notes that information regarding the environmental characteristics (e.g., relative humidity and air pressure) and contaminants identified in surveys of airline cabin air “suggests a diverse set of adverse health effects that could arise from exposure to the cabin environment—from acute effects…to long-term effects.”
Any consideration of health effects in the context of airline cabin air must distinguish between effects of exposures that result from the ambient environment encountered during boarding, waiting at the gate with the aircraft door open, and normal operation of the aircraft and effects of exposures that result from incidents during flight. Examples of the two categories of exposures are listed in Table 5–1. The myriad health complaints registered by flight crews and passengers are broad and nonspecific, and that makes it difficult to define or discern a precise illness or syndrome.
Among the many plausible explanations of the complaints are the flight environment (e.g., partial pressure of oxygen (PO2) and relative humidity), chemical or biological contaminants, psychological and physiological stressors, and exacerbation of pre-existing medical conditions. (Biological agents are discussed in Chapter 4).
TABLE 5–1 Exposure Sources Relevant to Aircraft Cabin Air Quality
|
Exposures Related to Normal Operations of the Aircraft |
Exposures Related to Incidents |
|
Ozone Carbon dioxide Temperature Relative humidity Off-gassing from interior material and cleaning agents Bioeffluents Personal-care products Allergens Infectious or inflammatory agents Ambient airport air Cabin pressure/partial pressure of oxygen Pesticides Jet exhaust fumes (runway) Alcohol |
Carbon monoxide Smoke, fumes, mists, vapors from leaks of engine oils, hydraulic fluids, and deicing fluids and their combustion products |
FLIGHT ENVIRONMENT
Cabin Pressure
As discussed in Chapter 2, at cruise altitude, the aircraft cabin is typically pressurized to the equivalent of an altitude of 6,000–8,000 ft (1,829–2,438 m), with a corresponding barometric pressure of 609–564 mm Hg and an ambient PO2 of 128–118 mm Hg (see Table 5–2). As specified in the Federal Aviation Regulation (FAR) 25.841, aircraft “cabin pressure altitude” must not exceed 8,000 ft at the aircraft’s highest operating altitude (14 CFR 1986).
The reduced pressure in the cabin environment results in several physiological changes in the passengers and crew. Specifically, the reduced ambient air pressure will cause the gas in body cavities (e.g., middle ear, sinuses, and gastrointestinal tract) to expand in volume by as much as 25%. In the lungs, the lower PO2 in ambient air will reduce the oxygen (O2) pressure in the alveoli from the normal value of 105 mm Hg. That decrease will lower systemic arterial PO2. In healthy people, the arterial PO2 is usually about 5–10 mm Hg lower than the alveolar PO2 (see Table 5–2). It is the arterial PO2 that determines the amount of O2 that is carried by the hemoglobin in the blood, expressed as the percent hemoglobin saturation.
TABLE 5–2 Barometric Pressure and PO2
|
Altitude, ft (m) above sea level |
Barometric Pressure, mm Hg (kPa) |
Ambient PO2, mm Hg (kPa) |
Alveolar PO2 (for healthy person at rest), mm Hg |
Arterial PO2 (for healthy person at rest), mm Hg |
|
0 |
760 (101.3) |
160 (21.3) |
105 |
95–100 |
|
6,000 (1,828) |
609 (81.2) |
128 (17.1) |
76 |
66–71 |
|
8,000 (2,438) |
564 (75.2) |
118 (15.7) |
72 |
62–67 |
|
Source: Adapted from Slonim and Hamilton (1971). |
||||
Table 5–3 shows the arterial O2 saturation at various altitudes without supplemental O2. At an altitude of 7,500 ft (2,287 m), the PO2 begins to approach the steep-slope portion of the O2-hemoglobin dissociation curve (Figure 5–1 ). Further small changes in PO2 in the cabin can lead to large changes in the O2 content of the blood.
The relationship between arterial PO2 and hemoglobin saturation is an Sshaped curve. As shown in Figure 5–1, at an arterial PO2 greater than 60 mm Hg, hemoglobin is more than 90% saturated with O2.
For healthy adults at sea level, hemoglobin is 95–97% saturated. Even at an altitude of 8,000 ft (2,439 m), the O2 saturation of hemoglobin remains at least 90% for healthy adults at rest because their arterial PO2 is above 60 mm Hg. Below arterial PO2 of 60 mm Hg, there is a steep decline in the curve, over which a slight change in arterial PO2 can lead to large changes in hemoglobin saturation. The significance of this curve with respect to the aircraft
TABLE 5–3 Hypobaric Pressure and Arterial O2 Saturation
|
Pressure Altitude, ft |
Atmospheric Pressure, mm Hg |
PO2, mm Hga |
Arterial O2 Saturation Without Supplemental O2, % |
|
0 |
760 |
160 |
96 |
|
2,500 |
694 |
147 |
95 |
|
5,000 |
632 |
133 |
95 |
|
7,500 |
575 |
121 |
93 |
|
10,000 |
523 |
110 |
89 |
|
a21% of atmospheric pressure. Source: NRC (1986). |
|||
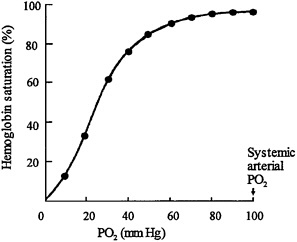
FIGURE 5–1 O2-hemoglobin dissociation curve shows relationship between PO2 in blood (X-axis) and amount of O2 held by blood hemoglobin (Y-axis). Important feature of curve is sharp drop in O2 content of hemoglobin when PO2 falls below 60 mm Hg.
environment is that at cruise altitudes (reduced air pressure), alveolar and arterial PO2 are reduced. However, the hemoglobin saturation decreases only slightly unless arterial PO2 falls below 60 mm Hg. A drop in arterial PO2 from 100 to 60 mm Hg will result in only a 10% drop in hemoglobin saturation.
In persons with chronic obstructive pulmonary disease (COPD) or asthma, there may be inadequate O2 exchange between the air in the lungs and the blood. In this case, even at sea level, arterial PO2 may be considerably lower than alveolar PO2 (i.e., some affected persons may have less than 90% hemoglobin saturation when at rest; many more will experience this effect on exertion). The situation is worsened at higher altitudes or with exercise; under these conditions, hemoglobin saturation will be substantially lower than in healthy persons at sea level or in sedentary persons, respectively. For these people, lowering alveolar PO2 by lowering ambient air pressure may also decrease the arterial PO2 and substantially lower the amount of O2 carried by the blood
The 1986 NRC report on airliner cabin environment summarized the effects of altitude on PO2 and recommended that passengers with heart or lung disease be educated about the risks posed by flight. It also recommended that passengers with middle ear problems be told about the effects of cabin pressure in general. However, that committee had few direct data on the hemoglo-
bin saturation that might be expected in passengers and cabin crew under normal flight conditions of commercial aircraft. Some studies have since examined the effects of cabin pressurization on PO2 in humans. Real-time continuous monitoring of hemoglobin saturation in flight has indicated that considerable changes can occur in a given person and that there are considerable differences among people.
The effects of hypoxia have been studied in a number of situations, including combat flights and passenger transport (Ernsting 1978). Studies conducted during the 1940s suggested that the maximal acceptable degree of hypoxia in passenger aircraft corresponded to a cabin altitude of 8,000 ft, but it was recommended that under routine operating conditions cabin pressure altitude should not exceed 5,000–6,000 ft. The altitude of 8,000 ft was a compromise between the aircraft design and operation requirements and the human performance impairments. Studies by McFarland and Evans (1939), McFarland (1946), and others (Ernsting et al. 1962; Denison et al. 1966; Ledwith 1970) showed that mild hypoxia, as is found in subjects at 8,000 ft, might impair the learning of new tasks and the performance of complex tasks. Most of the studies were conducted on young, healthy men, primarily in the military, who were engaged in vigilance tasks. McFarland and Evans (1939) found an increase in the absolute brightness threshold of the dark-adapted eye at hypoxia equivalent to 7,400 ft but concluded that the change was so small that it was of no practical significance. Ernsting (1978) noted that a “reduction of the cabin altitude from 8,000 to 6,000 ft is associated with a lower incidence of otiotic barotrauma and disturbances in passengers with cardiorespiratory disease.” Studies on performance deficit under hypoxic conditions—86% arterial oxyhemoglobin equivalent to 8,900 ft—showed no effect on performance, including night vision; the threshold for effects appeared to be 82% oxyhemoglobin (9,750 ft) (Fowler et al. 1987).
Cottrell et al. (1995) used continuously reading pulse oximeters to measure O2 saturation in 38 pilots on 21 flights of about 4 h each. Pressure altitudes in the aircraft cockpits during the cruise portion of the flight were 6,000–9,000 ft (average, 7,610 ft). Maximal and minimal O2 saturations were 95–99% (mean, 97%±1.1%) and 80–93% (mean, 88.6%±2.9%), respectively. Of the 38 subjects, 20 (53%) developed an O2 saturation of less than 90% at some time during the flight (duration of time below 90% not given). Baseline O2 saturation was 95–99%; at 6,000–7,000 ft, saturation was 87–92%; and at 8,000 ft or more, it was 80–91%. No symptoms were reported, and no attempt was made to measure performance decrement. There was no correlation between the minimal oxyhemoglobin saturation and age, height, weight, length of flight,
maximal saturation, smoking history, peak cabin altitude, or whether the flight was during the day or night.
In a study of the effects of hypoxia on healthy infants, 34 babies (1–6 mo old; average 3.1 mo) were exposed to 15% O2 in nitrogen for a mean duration of 6.3 h±2.9 h (Parkins et al. 1998). During exposure, there was a significant increase in the infants’ heart rate and time spent in periodic apnea and a decrease in the amount of time spent in regular breathing; respiratory rates did not change significantly. Baseline O2 saturation decreased from a median of 97.6% (range, 94–100%) in ambient air to 92.8% (range, 84.7–100%) in 15% O2. Four of the infants had to be removed from the hypoxic conditions early because their O2 saturation fell below 80% for more than 1 min. The authors suggest that exposure to reduced PO2, like that encountered on high-altitude flights can result in hypoxia in some infants.
A newborn infant’s red blood cells contain up to 77% fetal hemoglobin (Delivoria-Papadopoulos and Wagerle 1990). At 6 mo, the red cells still contain up to 4.7% fetal hemoglobin. Fetal hemoglobin binds O2 with greater affinity than does adult hemoglobin (see Figure 5–2). That results in greater saturation for any given partial pressure, but it also means that at any given partial pressure fetal hemoglobin holds O2 more than adult hemoglobin. The net effect is that less O2 is available for tissue metabolic needs. Thus, infants who experience substantial falls in arterial O2 saturation may be at greater risk for tissue hypoxia than adults at the same O2 saturation.
With decreasing cabin pressure, air in the middle ear and sinuses expands and escapes via nostrils and ostia to the outside via the nasopharynx. On descent, increasing ambient pressure requires gas to re-enter the middle ear and sinuses through the same pathways. Both movements can happen only if air can move freely in either direction. If there is blockage due, for example, to an upper respiratory infection, allergy, or tumor, the free flow of air may be impeded, and earache, a feeling of sinus fullness, or dizziness can ensue, particularly on descent. In severe cases, the pressure differential can cause extreme pain, bleeding, or rupture of a tympanic membrane. Likewise, gas in the respiratory tract and gastrointestinal tract expands and contracts with decreasing and increasing cabin pressure. Although gas readily escapes from those parts of the body, minor stomach cramping or bloating might still occur.
At cabin altitudes during cruise, all crew and passengers are in a decreased-O2 environment. It is clear that under ordinary conditions of commercial flight, PO2 can be reduced substantially during rest or in situations of minimal exertion. As stated above, this relatively small decrement usually causes
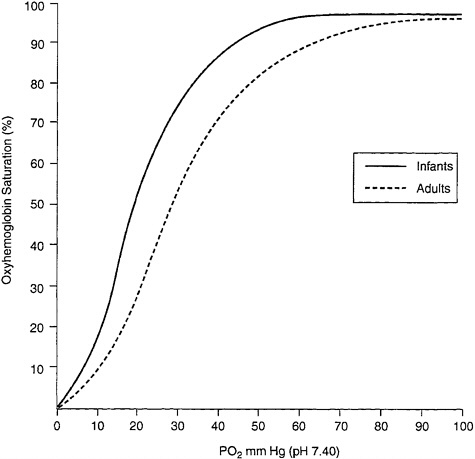
FIGURE 5–2 Oxyhemoglobin equilibrium curves of blood from infants and adults. Source: Adapted from Delivoria-Papadopoulos and Wagerle (1990).
no symptoms in healthy people because hemoglobin remains well saturated with O2 at altitude. Nevertheless, some people might be sensitive to a lowering of PO2 and experience various symptoms, including headache, dizziness, fatigue, numbness, and tingling (Sheffield and Heimbach 1996). The array of possible symptoms varies from person to person or even in the same person on different days. The PO2 values are such that substantial reductions in arterial O2 content occur and could pose a definite health risk for persons with underlying pulmonary or cardiac disease or untreated or partially treated anemia.
Relative Humidity
As noted in Chapters 1 and 2, the relative humidity in most aircraft cabins is low, typically from 10 to 20% (see Table 1–2) with an average of 15–19%, depending on the aircraft (Nagda et al. 2000). Although, low relative humidity is not an air contaminant, it can affect passenger and crew comfort and health (Rayman 1997). Low relative humidity may cause drying of the skin, mucous membranes, and conjunctivae in the latter case adding a risk for conjunctivitis with its symptoms of tearing and pain, especially in those wearing contact lenses (Eng 1979). Studies have indicated that passengers and cabin crews find the air in the aircraft cabin to be too dry and to lead to such symptoms as dry, itchy, or irritated eyes; dry or stuffy noses; and skin dryness or irritation (Lee et al. 2000). Stuffy, dry nose was the primary complaint in another study of 3,630 passengers in all cabin classes on standard and wide-body aircraft. Although symptoms could not be correlated with aircraft type (comfort rating, 5.34 of 7 for first-class versus 4.46 for coach and no difference in passenger comfort ratings for relative humidity between standard-body and wide-body aircraft), they did reflect flight length: longer flights resulted in more symptoms (Rankin et al. 2000). Low relative humidity has also been associated with fatigue, headaches, and nosebleeds (Space et al. 2000). Low humidity can have a greater effect on passengers who have respiratory infections, asthma, or tracheotomy (Rayman 1997). It has been found that in dry environments, mucus can concentrate; this can reduce ciliary clearance and phagocytic activities in the respiratory tract (Berglund 1998).
In a study of the effects of low humidity on the human eye, Laviana et al. (1988, as cited in Nagda and Hodgson 2001) found that at a relative humidity of either 10% or 30%, eye pain (described as scratchiness, pain, or burning) increased over time up to the fourth hour of exposure (total exposure duration of 10 h) for both a naked eye and an eye covered with a soft contact lens. However, there was no difference in the severity of responses at either 10% or 30% relative humidity and the humidity level did not significantly affect acuity, refractive error or cornea curvature in either eye. In aircraft cabins, symptoms of low humidity, such as eye and nasal irritation, seem to occur within 2 h after exposure begins (Eng 1979); skin symptoms may require at least 4 h to occur (Carleton and Welch 1971, as cited in Nagda and Hodgson 2001). Furthermore, all symptoms increase in severity with time, and an adaptive response to low-humidity environments is not evident (Nagda and Hodgson 2001). At reduced barometric pressures (259–700 mm Hg), symptoms associ-
ated with low humidity develop more quickly and are more severe (Carleton and Welch 1971, as cited in Nagda and Hodgson 2001). In a review of the studies that examined potential health effects of exposure to low relative humidity, Nagda and Hodgson (2001) indicated that the study subjects in general were relatively young and that an older study population might have been more likely to perceive changes in relative humidity and also might be more susceptible to health effects of such exposures. They concluded that a modest increase in the relative humidity in an aircraft cabin (e.g., from the current average of 14–19% to about 22–24%) might have beneficial effects similar to those seen in building studies in which a 10% increase in humidity alleviated many of the symptoms of “sick-building syndrome.” Such humidities are below the values that may affect the safety of an aircraft—that is, that might cause condensation in and corrosion of the aircraft shell—or that would result in increased microbial growth.
Although cabin relative humidity is well below the preferred values of 30– 60% suggested in American Society of Heating, Refrigerating and Air-Conditioning Engineers Standard 62–1999, it is questionable whether low humidity has substantial short- or long-term health effects. One investigator reported that in a room with 0% humidity, the degree of dehydration in undressed subjects was insignificant over a period of 7 h (Nicholson 1996). It might be expected that the adverse effects of low relative humidity experienced by crew and passengers will be temporary and will be alleviated when they leave the aircraft. The time required for rehydration will depend on individual physiology and the ambient environment.
Humidity influences the perception of air quality. In one study, as relative humidity increased from 24% to 79%, the air was considered to be less fresh (Berglund 1998). Acceptability of air-quality decreases with increasing temperature and relative humidity (Fang et al. 1998a,b).
CHEMICAL CONTAMINANTS OF CONCERN
During flight, passengers and crew can be exposed to a variety of air contaminants. Some of them are naturally occurring chemicals, such as ozone (O3), which is found in greater concentrations in the upper troposphere and lower stratosphere; others result from incidents that suggest equipment failure; and still others result from preventive measures, such as the use of pesticides. The toxicity of these contaminants is discussed below.
Ozone
Although ground-level O3 is a major contributor to photochemical air pollution in urban air, the presence of O3 in the upper troposphere and lower stratosphere provides a necessary health benefit to humans by screening out harmful ultraviolet radiation. As discussed in Chapter 3, many commercial flight paths are at altitudes where O3 concentration might be greater than those typically found at ground level. O3 in the cabin is required not to exceed 0.25 ppm (250 ppb) during a flight (FAR 25.8321). Mean O3 measured on aircraft has ranged from 22 ppb (Nagda et al. 1989) to 200 ppb (Waters 2001). The health effects of ground-level O3 have been well studied (EPA 2001). Those effects are relevant to travelers who might be exposed to increased O3 during high-altitude flights if the aircraft is not equipped with an O3 converter or the equipment is not operating properly. The ground-level national ambient air-quality standard for O3, established by the Environmental Protection Agency (EPA) in 1997, is 0.12 ppm by volume for a 1-h exposure and 0.08 ppm by volume for 8-h exposures (EPA 1997).
O3 can cause acute respiratory problems, aggravate asthma, and impair the body’s immune system making people more susceptible to respiratory illnesses, including bronchitis and pneumonia (EPA 1996). Exposures to O3 as low as approximately 0.08–0.10 ppm for about 6 h have resulted in impairments of the immune system (EPA 1996). Inflammatory responses occurred within 1 h after a 1-h exposure to O3 at 0.3 ppm or more. Exposures for up to 7 h to O3 as low as 0.08 ppm caused small decrements in lung function and increases in respiratory symptoms (EPA 1996). Those effects are exacerbated by exercise. In a study of healthy adults exposed during exercise to ambient O3 at 21–124 ppb, there was a statistically significant decrement in lung function (Spektor et al. 1988a). Similar lung function decrements were seen in healthy children (at a summer camp) exposed to O3 at the ambient air standard of 120 ppb, where average forced vital capacity (FEV), forced expiratory volume in the first second (FEV1), peak expiratory flow rate (PEFR), and forced expiratory flow (FEF25–75) decrements were 4.9%, 7.7%, 17%, and 11%, respectively (Spektor et al. 1988b).
|
1 |
FAR Section 25.832 Cabin ozone concentration: “(a) The airplane cabin ozone concentration during flight must be shown not to exceed: (1) 0.25 parts per million by volume, sea level equivalent, at any time above flight level 320, and (2) 0.1 parts per million by volume, sea level equivalent, time-weighted average during any 3-hour interval above flight level 270.” |
Asthmatics, particularly children, show an increase in respiratory symptoms and decrements in peak expiratory flow rate with increasing O3 (EPA 1996). Studies on children with moderate-to-severe asthma found that increasing O3 from 84 to 160 ppb for 1 h resulted in increased medication use and an increase in the number of chest symptoms (Thurston et al. 1997). The long-term sequelae of inhalation exposure are unknown, but continued exposure O3 at low concentrations could result in chronic effects in humans (EPA 1996).
The committee was able to identify only one study that examined the possible effects of O3 on flight attendants (Tashkin et al. 1983). The authors attempted to determine, on the basis of symptoms reported on a questionnaire distributed to flight attendants on high-altitude flights of Boeing 747SP aircraft, whether the symptoms were consistent with possible exposure to increased O3. Although the authors concluded that the reported symptoms were consistent with exposure to toxic concentrations of O3 (0.4–1.09 ppm, measured onboard other 747SP aircraft), no O3 measurements were made on flights where the questionnaire was used. There were substantial limitations in the study, as discussed in Chapter 6.
Engine Oils and Hydraulic Fluids
Engine lubricating oils and hydraulic fluids are complex mixtures of primarily organic compounds (see Table 3–12 for major components). Among the major additives to the petroleum base of these mixtures are organophosphate compounds, of which those organophosphates, of greatest toxicological concern are tricresyl phosphate (TCP) derivatives and the generation of trimethylolpropane (TMPP). Other organophosphates that can be present in the oils and fluids are tributyl phosphate, dibutyl phenyl phosphate, and triphenyl phosphate. The general toxicity of these agents is discussed below.
Engine lubricating oils and hydraulic fluids have been reported to enter the passenger cabin of aircraft through the environmental control system (ECS) as discussed in Chapter 3. The fluids (and their possible pyrolysis products) can result in mists, fumes, vapors, and smoke in the cabin. The major pyrolysis products of jet engine oils and hydraulic fluids, at less than very high temperatures, will probably be volatile organic compounds (VOCs) and carbon monoxide (CO). Although there is considerable toxicological information on the major constituents of engine oils and hydraulic fluids, there are few data on the toxicity of the formulated oils and fluids themselves. Only a couple of studies were found in the published literature, and they are discussed below.
In an early study to determine whether the introduction of engine oils or hydraulic fluids or their combustion products into the ECS would impair aircraft crew, rats were exposed via inhalation to synthetic engine lubricating oils (Exxon turbo oil 2380 or Mobil II jet oil) or one synthetic hydraulic fluid (Skydrol 550B) (Crane et al. 1983). The authors concluded that for combustion of the engine oils at 400°C, the major toxic component was CO, whether or not combustion produced flames or only smoke. The hydraulic fluid Skydrol, under flaming conditions, was more toxic (the animals became incapacitated and died more quickly) than the engine oils, but it was not toxic under non-flaming (smoking) conditions.
In another study, the hydraulic fluid Skydrol 500B-4 (manufactured by Monsanto), containing primarily tributyl phosphate and dibutyl phenyl phosphate (80–90% phosphate esters) was tested with rats via inhalation. The animals were exposed for 6 h/d, 5 d/wk, to very high Skydrol concentrations (up to 300 mg/m3, with about 95% of particles less than 10 μm in diameter) for up to 13 wk. Rats exhibited reddish nasal discharge and salivation, suggesting irritation. No pathological changes were seen in animals exposed at 100 mg/m3 (the no-observed-adverse-effect level). Pathological changes at 300 mg/m3 included increased liver weight with hepatocellular hypertrophy, decreased hematocrit in both males and females, and decreased plasma cholinesterase in females only (Healy et al. 1992). In other animal studies with organophosphate ester hydraulic fluids, rabbits exposed for less than 4 h/d, 5 d/wk, for 11 or 22 d, to aerosols of Cellulube 200 hydraulic fluid at 2,000 mg/m3 died with severe dyspnea and mild diarrhea (Carpenter et al. 1959, as cited in ATSDR 1997).
Tricresyl Phosphate Esters
As noted in Table 3–12, engine oils and hydraulic fluids contain several phosphate esters as antiwear additives. Hydraulic fluids contain tributyl phosphate (at up to 80%), dibutyl phenyl phosphate, and butyl diphenyl phosphate and can also contain up to 1% TCP (Hewstone 1994). Engine lubricating oils contain TCP at 1–5% (typically 3%). The commercial TCP found in engine oils and hydraulic fluids is actually a mixture of aryl phosphates, including the ortho, meta, and para isomers and other o-cresol compounds. Although older TCP contained up to about 25–40% o-cresyl residues, currently manufactured TCP contains less than 0.3% o-cresyl residues (some reports indicate that o-cresyl isomers are present in TCP at 0.05–0.13%) (Goode 2000), and some
TCP mixtures contain almost no o-cresyls or o-xylenyls (Craig and Barth 1999).
TCP is a toxic mixture that can cause a wide array of transitory or permanent neurological dysfunction—including convulsions, flaccid paralysis, and polyneuropathy—although these serious sequelae are almost always secondary to ingestion. The committee did not identify any reports of human neuropathy following inhalation of TCP. Episodes of penetration of engine oil mists and fumes into the aircraft cabin might lead to dermal and inhalation exposure of passengers and crew members. Although the committee was unable to find any objective information that would substantiate and document frank neurotoxicity in persons exposed to such mists and fumes on aircraft, such a possibility cannot be a priori discounted if airborne concentrations of TCP are substantial and exposures long enough.
One of the most toxic components of TCP is tri-o-cresyl phosphate (TOCP). The primary consequence of TOCP ingestion is delayed neuropathy, characterized by degenerative changes in the axons (“dying-back neuropathy”), also known as OPIDN (organophosphate-induced delayed neurotoxicity or neuropathy), that generally occurs 1–2 wk after exposure (Baron 1981). Acute toxicity may be manifest immediately as nausea, vomiting, diarrhea, and abdominal pain and be followed by a long asymptomatic period (8–35 d), after which there is a bilaterally symmetrical degeneration of sensory and motor axons in peripheral nerves and spinal-cord tracts; fibers that are longest, and have the largest diameter tend to be the most affected. Signs and symptoms are paresthesias in the extremities (numbing and tingling), pain in the calves and legs, and absence of reflexes, mostly in the feet and legs; in severe cases of poisoning, all four limbs can be affected in the form of a “glove and stocking” distribution. Development of uncoordinated movements (ataxia) may occur at the same time and progress to flaccid paralysis. Recovery is usually poor; in time, flaccidity may be replaced with spasticity, reflecting some regeneration of the peripheral nerves, but with residual damage in the spinal cord.
Studies with experimental animals (primarily hens and cats) have shown that modern lubricating oils have comparatively low toxicity via ingestion and inhalation. An acute oral 5-g/kg dose of engine lubricating oil containing 3% TCP did not result in any clinical signs of delayed neurotoxicity in hens, the animals most sensitive to the neurotoxic action of TCP and related phosphate esters, after a 3-wk followup period; a repeat oral dose also produced no toxic or histopathological changes (Daughtrey et al 1990). Groups of 17–20 hens received a daily oral 1-g/kg body weight dose of engine lubricating oils containing either 3% TCP (the commercial TCP contained less than 1% TOCP),
triphenyl phosphorothionate, or butylated triphenyl phosphate for 5 d/wk for 13 wk. The animals did not exhibit any inhibition of neuropathic target esterase (NTE) activity in the brain and spinal cord at 6 wk; at 13 wk, NTE was inhibited by 23–34% compared with the result in saline-treated hens; no clinical or neuropathological signs of OPIDN were evident at either 6 or 13 wk. Hens that received TOCP daily at 7.5 mg/kg (plus an oral dose of 500 mg/kg 12 d before the end of treatment) exhibited clinical impairment and lesions indicative of OPIDN at 6 and 13 wk (Daughtrey et al. 1996). The authors concluded that TCP should not pose a hazard to humans under exposure conditions that can realistically be expected in the handling of these materials.
Only one set of animal studies of the inhalation toxicity of TOCP was found by the committee (Siegel et al. 1965). One of four hens with continuous (23 h/d), whole-body exposure to hydraulic fluid mist containing not more than 1.5% TOCP (with trixylenyl phosphates and other trialkylphenyl esters) at to 23 mg/m3 exhibited signs of neurotoxicity at day 58. At hydraulic fluid concentrations of 102–110 mg/m3, early signs of neurotoxicity were evident in 19 of 20 hens between days 22 and 29. Four of 12 squirrel monkeys exposed continuously to hydraulic fluid mists at 4.3 or 4.4 mg/m3 (six animals per concentration) died within the 108-day exposure period although no signs of neurotoxicity were seen in the surviving animals (four of the 10 control animals also died) (Siegel et al. 1965). For intermittent exposures, groups of six chickens were exposed to hydraulic fluid mist containing 1.5% TOCP at 25 or 50 mg/m3 for 8 h/d, 5 d/wk, for a total of 30 exposures. Hens in the lower concentration group showed no signs of neurotoxicity although there was an increase in weight; at the higher concentration, three of the hens developed signs of paralysis by day 29 (Siegel et al. 1965).
In humans, exposures to TOCP that are not likely to produce toxic effects are estimated to be 2.5 mg/kg for a single oral dose (175 mg for a 70-kg person) and 0.13 mg/kg per day for repeated exposures (9 mg/d for a 70-kg person) on the basis of the assumption that there was no significant difference in TOCP sensitivity among cats, hens, and humans. A safety factor of 10 was used to extrapolate from animals to humans (Craig and Barth 1999). Because of the lack of data on TOCP toxicity in humans, the committee is unable to assess whether the assumption of equivalent sensitivity in humans, cats, and hens is appropriate but notes that a safety factor of 100 is typically used, which includes a safety factor of 10 for extrapolation from animals to humans and a second safety factor of 10 to account for interhuman variability (Faustman and Omenn 2001).
A further assessment of the inhalation toxicity of TOCP was conducted
by comparing the amount of TOCP in an oil mist with worst-case occupational-exposure scenarios (Craig and Barth 1999). For the purposes of comparing risks, the authors assumed that inhaled and ingested doses of TOCP were of equivalent toxic potential (but do not justify the assumption). If the TOCP concentration in the oil mist were 3%, a sedentary worker (ventilation rate, 0.5 m3/h) with an 8-h workday would inhale TOCP at 0.6 mg/d, and a worker engaged in heavy activity (ventilation rate, 3.6 m3/h) with a 12-h workday would inhale TOCP at 6.5 mg/d. Those values do not exceed the estimated safe dose for a 70-kg person of 9 mg/d. The authors note that although a lubricating oil or hydraulic fluid is unlikely to contain 3% pure TOCP, if the other o-aryl phosphates present approach this concentration, it is the atmospheric concentrations of the neurotoxic components that might be of concern because they could exceed the American Conference of Governmental Industrial Hygienists (ACGIH) 8-h threshold limit value of 0.1 mg/m3 time-weighted average for pure TOCP (Craig and Barth 1999; ACGIH 2001).
Some reports suggest that although the “conventional” type of jet engine oils contains substantial concentrations of TOCP (2–3% o-cresyl isomers in the TCP), a newer “low-toxicity” jet engine oil contains very little of the ortho isomer and instead contains primarily the less-toxic meta and para isomers. It was estimated that oral doses of jet engine oil of 5.7–33.3 g/kg containing 3% “conventional” TCP would be required to inhibit brain NTE in the hen (the most sensitive species) by 70%; an oral dose of 87–330 g/kg would be required to elicit the same effect if jet engine oil containing 3% “low-toxicity” TCP were used (Craig and Barth 1999; Mackerer et al. 1999).
A recent risk assessment to estimate the potential of conventional and low-toxicity jet engine oils containing TCP to cause OPIDN found that ingestion of jet engine oil containing 3% TCP at approximately 9–10 g/d would be a minimal toxic dose for a 70-kg person; for conventional jet engine oils, the minimal toxic dose was estimated to be 280 mg/kg per day (Mackerer et al. 1999). Studies by Henschler (1958a,b) on older TCP that contained up to 30% o-cresyl isomers showed that the asymmetrical mono-o-cresyl-substituted mixed esters were about 10 times as toxic as pure TOCP, with the di-o-cresyl isomers in between (Henschler 1959). It has been estimated that the conventional TCP can contain the mono-o-cresyl isomers at 3,070 ppm and the di-o-cresyl isomers at 6 ppm, compared with less than 0.005 ppm for TOCP; however, the newer TCP has been calculated to contain TOCP at less than 0.001 ppm, mono-o-cresyl isomers at approximately 1,760 ppm, and di-o-cresyl isomers at 1.1 ppm (Mackerer and Ladov 2000).
Other Phosphate Esters
Hydraulic fluids contain phosphate esters in addition to the TCP mixture. Among them are tributyl phosphate, dibutyl phenyl phosphate, butyl diphenyl phosphate, and triisobutyl phosphate. Toxicity information on those phosphate esters in the published literature is relatively sparse and is summarized below.
The use of synthetic engine lubricants raises the possibility that at extremely high temperatures (250–700°C) a very potent neurotoxicant can be formed (Centers 1992; Wright 1996). If the lubricant contains trimethylolpropane esters as well as TCP, they may thermally degrade to form TMPP at rates of 7–10 mg/mL of lubricant at those very high temperatures (Centers 1992). Detection of TMPP after a fire (on a ship that used a lubricant containing the compounds) confirmed that the generation of this extremely toxic compound can occur under actual fire conditions (Wyman et al. 1993). The intraperitoneal dose of TMPP required to kill 50% of mice was 1.0 mg/kg; 50% mortality was achieved with dermal exposures of 50–100 mg/kg (Centers 1992).
Dibutyl phenyl phosphate and di-tert-butylphenyl phosphate manifest many of the health effects typical of organophosphate compounds in that they are expected to produce neurotoxic symptoms after ingestion. Irritation of the mucous membranes of the eyes, nose, and upper respiratory tract with coughing and wheezing follow inhalation exposure to their vapors or aerosolized formulations. Repeated dermal contact with dibutyl phenyl phosphate has resulted in drying and cracking of exposed skin (Hazardous Substances Data Bank [HSDB] 2000). Specific toxicity data were not identified for di-tert-butylphenyl phosphate.
Tributyl phosphate (TBP) appears to be much less toxic than TCP. Vapors of TBP can cause irritation of the mucous membranes in the eyes, nose, and throat. Workers exposed to TBP at 15 mg/m3 complained of headache and nausea. Prolonged exposure may result in paralysis; however, neurotoxic effects are seen only at very high doses and then primarily via ingestion. TBP has only weak cholinesterase-inhibition activity and only via ingestion of large quantities (e.g., 100 mL). It has been estimated that the probable oral lethal dose for a 70-kg person is 0.5–5 g/kg (HSDB 2000). Skin contact may result in irritation.
In summary, the presence of phosphate esters in engine lubricating oils and hydraulic fluids may constitute a potential neurotoxic hazard. Practically all known cases of human toxicity of these compounds involve ingestion of sub-
stantial amounts. It should be noted that no TCP isomers, including TOCP, and no TMPP were detected on several flights of different aircraft (Nagda et al. 2001); however, TCP vapors were detected in air when engine lubricating oils and hydraulic fluids were pyrolized under laboratory conditions (van Netten 2000, van Netten and Leung 2000, van Netten and Leung 2001). Measurement of the airborne concentrations of these compounds or modeling of possible exposure scenarios in aircraft cabins, with objective demonstration of neurotoxicity in experimental animals exposed to these compounds by inhalation, is needed for a proper assessment of risk.
Carbon Monoxide
CO is a colorless, odorless gas produced by incomplete combustion of carbonaceous material. EPA has established standards for ambient air concentrations of CO of 9 ppm (10 mg/m3) for an 8-h exposure and 35 ppm (40 mg/m3) for a 1-h exposure (EPA 2000). It has also established “significant harm” concentrations of 50 ppm (8-h average), 75 ppm (4-h average) and 125 ppm (1-h average) as conditions in which exposure could result in carboxyhemoglobin (COHb) concentrations of 5–10%, which could cause significant health effects in sensitive people (EPA 2000). CO in cabin air is regulated by FAR 25.831.2 Concentrations measured in aircraft under normal operating conditions are typically below 1.0 ppm (Nagda et al. 2001) and in a recent survey did not exceed 0.87 ppm (Waters 2001). The presence of CO in the aircraft cabin is important for health because it has an affinity for hemoglobin about 250 times that of O2, forming COHb, reducing the O2-carrying capacity of the blood, and possibly causing hypoxia. Formation of COHb depends on CO concentrations in the ambient air and on respiratory minute volume. Under normal physiological conditions, the brain can increase the blood flow or tissue O2 extraction to compensate for the hypoxia. When ambient CO is high—for example, in a fire or in the presence of incomplete combustion—blood CO can reach lethal concentrations within minutes and are not necessarily preceded by such symptoms as headaches and dizziness. At
|
2 |
FAR Section 25.831 “Ventilation: 1. Carbon monoxide concentrations in excess of 1 part in 20,000 parts of air (50 ppm) are considered hazardous. For test purposes, any acceptable carbon monoxide detection method may be used.” |
COHb of at least 2.3 and at least 4.3%, maximal exercise duration and performance, respectively, are slightly reduced in healthy people; no effects on submaximal exercise were observed when COHb levels was increased to 15– 20% (EPA 2000). A large clinical study of the cardiovascular effects of CO on patients with angina was sponsored by the Health Effects Institute (Allred et al. 1991). Exposure to CO concentrations during exercise sufficient to raise the blood COHb concentration to 2%, resulted in a reduced time to angina symptoms, 4% COHb resulted in a further reduction. These CO exposures also produced depressions of one temporal segment of the cardiograms.
Smokers are more susceptible to the effects of CO because they have COHb at about 5%; nonsmokers typically have COHb at close to zero. Early symptoms of CO poisoning in humans are headaches and lightheadedness. At COHb of 15%, hypoxia begins. When blood COHb reaches 30–40%, severe headache, weakness, dimness of vision, confusion, dizziness, nausea, and vomiting may occur; consciousness can be lost sometimes for hours before death. At higher concentrations—usually more than 40–50% blood saturation with CO—severe ataxia, increasing confusion, hallucinations, and accelerated respiration occur. At 50–60% saturation, convulsions, tachycardia, and coma are evident; death occurs at COHb of greater than 70% (Gosselin et al. 1984, as cited in HSDB 2000). Fatal COHb concentrations would probably be lower at higher altitudes, with reduced PO2, than at sea level.
Although CO is known to have fetotoxic effects in laboratory animals, effects on human fetuses are less evident, although of concern. In animals, COHb concentrations of less than 10% do not appear to affect fetal development adversely until possibly later in gestation, and it has been postulated that the same response may be expected in human pregnancy (Robkin 1997; as cited in EPA 2000). One prospective study of 40 cases of acute CO poisoning of pregnant women (Koren et al. 1991, as cited in EPA 2000) found that even hypoxemia resulting in mild to moderate COHb saturations, up to 18%, did not impair fetal growth; this suggests that most pregnant women are not increasing fetal risk through ordinary ambient exposure. In another study, maternal exposures to CO at 150 to 200 ppm, resulting in 15–25% COHb, did result in lower birth weights, heart abnormalities, delays in behavioral development, and disruption of cognitive function in several laboratory animal species (EPA 2000). Exposures to CO at levels as low as 60–65 ppm CO (6–11% COHb) throughout gestation produced many of the same developmental effects (EPA 2000).
Healthy people have shown central nervous effects after 1-h peak CO exposure which resulted in 5–20% COHb. Effects included decrements in
hand-eye coordination (driving or tracking), attention, and vigilance (detection of infrequent events) (EPA 2000).
People who have suffered and recovered from acute CO poisoning occasionally develop neuronal loss in the cortex and necrosis in the basal ganglia, which result in parkinsonism-like signs (Smith, 1996; Steinmetz 1998).
Formaldehyde
Another cabin air contaminant that may have adverse effects is formaldehyde. Formaldehyde is a colorless gas with a distinct odor that is noticeable at approximately 0.5–1.0 ppm. It is a ubiquitous chemical in ambient air at very low concentrations. However, excessive concentrations could be found in aircraft cabins as a result of the thermal decomposition of engine oils or hydraulic fluids or the reaction of O3 with cabin surfaces. Chapter 3 contains more information on the generation of formaldehyde in cabin air. Under normal operating conditions, measured concentrations on aircraft have not exceeded 26 ppb (13 μg/m3) (Nagda et al. 2001)
Formaldehyde is a known irritant of the mucous membranes of the eyes, nose, and respiratory tract at concentrations of approximately 0.4–3 ppm (ATSDR, 1999). Paustenbach et al. (1997) reported that for most people, eye irritation does not occur until at least 1 ppm; more severe eye, throat, and lung irritation occurs at 2–3 ppm; and acclimation occurs (Paustenbach et al. 1997). On the basis of its evaluation of the literature, the Industrial Health Panel recommended an 8-h time-weighted average occupational exposure limit of 0.3 ppm for formaldehyde with a ceiling value of 1.0 ppm. The panel did not identify any hypersensitive subgroups, such as asthmatics, and found no evidence of sensitization (Paustenbach et al. 1997).
Chronic inhalation exposures (for 6.8 yr; range, 2–19 yr) to formaldehyde at 0.39 ppm resulted in abnormalities of the nasal epithelium in plywood-factory workers (Ballarin et al., 1992, as cited in ATSDR 1999). In 70 chemical workers who produced formaldehyde for an average of 7.3 yr (range, 1–36 yr), expousre to it at approximately 0.24 ppm also resulted in nasal epithelium abnormalities (Holmstrom et al. 1989, as cited in ATSDR 1999); the chronic daily human exposure that is estimated to be without risk of adverse effects is 0.008 ppm, on the basis of this study. Concentrations of greater than 20 ppm are life-threatening. Formaldehyde has been designated by EPA as a probable human carcinogen (Integrated Risk Information System [IRIS] 2001).
Deicing Fluids
Glycols (ethylene glycol and propylene glycol) are the main constituents of deicing fluids used for aircraft. Exposure to deicing fluids is expected to be season- and climate-dependent. Deicing fluid is typically applied hot. As noted in Chapter 3, there are no published studies on the potential for deicing fluids to enter the cabin air via bleed air.
Ethylene glycol can be extremely toxic, particularly if ingested at high doses or if high concentrations of heated vapors are inhaled. Most known cases of ethylene glycol toxicity resulted from ingestion; ethylene glycol has a low vapor pressure. Ingestion of small amounts can cause drowsiness and slurred speech. Ingestion of antifreeze fluids containing ethylene glycol, often accidentally or suicidally, causes serious metabolic acidosis and renal toxicity (tubular necrosis) in humans—because of the toxic metabolite oxalic acid—and CNS depression, cardiopulmonary disturbances, pulmonary edema, and congestive heart failure (Snyder and Andrews 1996). The minimal lethal oral dose of ethylene glycol for adults is 1.4 mL/kg (1,330 mg/kg of body weight) (ATSDR 1997). Because of its poor absorption through the skin, systemic effects through skin contact are unlikely.
There are few data on inhalation exposures to ethylene or propylene glycol and no reports of death following exposure by this route (ATSDR 1997). After inhalation exposure to ethylene glycol aerosol at 3–67 mg/m3 for 20–22 h/d for about 4 wk, human volunteers complained of upper respiratory tract irritation and occasionally of slight headache and low backache. Ethylene glycol at above 140 mg/m3 was highly irritating and above 200 mg/m3 was intolerable. No ethylene glycol or its metabolites were found in the blood or urine of the subjects (Wills et al. 1974).
Propylene glycol has almost no intrinsic oral toxicity and is widely used in foods, cosmetics, and pharmaceuticals. Its low toxicity is explained by its metabolic conversion to lactic acid and pyruvic acid, normal body constituents (Snyder and Andrews 1996). Both propylene glycol and ethylene glycol are rapidly metabolized by humans and cannot be detected in tissues 48 h after exposure. Repeated, short-term exposures to propylene glycol may result in some irritation of the eyes, skin, and nasal and oral mucosa; some skin sensitization is possible (ATSDR 1997). Over the last 5 yr, the market share of propylene glycol used in deicing fluids has grown from 10% (124 million pounds) to 70% (140 million pounds), primarily at the expense of ethylene glycol (Ritter 2001). The increased use of propylene glycol in deicing fluids
has reduced the potential for toxic effects after accidental exposure to these compounds.
Pyrethroid Pesticides
Today, only a few foreign countries require the use of insecticides on aircraft coming from other countries, including aircraft from the United States. Consequently, the interiors of U.S. aircraft destined for those countries must be disinsected, a practice the United States discontinued in 1979 (see Chapter 3). The pesticides are usually synthetic pyrethroids, primarily phenothrin and permethrin. Chapter 3 describes the application methods for these pesticides.
The disinsection practices of some countries are expected to result in the exposure of cabin crews and passengers to pesticides. Although pyrethroid pesticides have very low toxicity in humans, they can cause adverse effects in some people and are recognized as neurotoxicants at very high doses. The acute oral dose that is lethal to 50% of rats is 0.5–5 g/kg; the dose that is lethal to 50% of animals in 4-h is 2,280 mg/kg; and the concentration that is lethal to 50% of animals via inhalation with intermittent exposure is 500 mg/m3 (NRC 1994). The pesticides act by slowing inactivation of the neuronal sodium channels, thus causing hyperexcitability of the nerves. That hyperexcitability produces signs of CNS and peripheral nerve disturbances. CNS toxicity is manifested as coarse tremors, involuntary muscle movements, and excessive lacrimation. Although their lethality is low, the pyrethroids are recognized as dermal irritants and, when they are in fume or vapor form, as respiratory and ocular irritants (Ecobichon and Joy 1994).
Systemic poisoning is less likely from dermal exposure than from oral exposure oral because absorption through the skin is considerably less than absorption through the gastrointestinal tract. In general, dermal exposure to pyrethroids results in a localized tingling sensation of the skin (paresthesia) without loss of normal sensation within 0.5–5 h and persists for up to 3 d (WHO 1995). Because skin appears to bind pyrethroids, localized effects can be expected to last for long periods, and the skin might act as a reservoir; this could result in symptoms that last for several weeks in acute cases of pyrethroid poisoning (He et al. 1989), although thorough cleaning of the skin may prevent or minimize this effect. In a study with 184 human volunteers, a 21 -d repeat patch test with a 40% solution of permethrin did not result in any skin sensitization, although there were some reports of transient burning, stinging,
and itching (NRC 1994). Patch tests with permethrin have shown no allergic response (Naumann and McLachlan 1999).
Areas on the body where skin is thin are affected first, because the agent can interact more readily with nerve endings. The average threshold dose for producing paresthesia is 0.2 mg/cm2 of skin in humans (WHO 1995). About 2% of permethrin applied directly to human skin is expected to be absorbed (NRC 1994). In a scabies-control program involving about 1,000 people, skin application of a cream containing 5% pyrethroids resulted in no complaints of paresthesia (Yonkosky et al. 1990). There appear to be individual susceptibilities to the effect; this may be related to the variation in the concentrations of carboxyesterase enzymes in the skin (Leng et al. 1999). Thus, lymphocyte carboxyesterase activity might serve as a biomarker of the effect (Leng et al. 1999). It has been suggested that persons with pre-existing disease (including skin disease and lowered immunity), infants, and children can be more sensitive than healthy adults to the effects of permethrin (Naumann and McLachlan 1999). Phenothrin is used to control headlice in children and has been reported to have relatively few adverse health effects (Naumann and McLachlan 1999).
Occupational exposure to or inappropriate handling of synthetic pyrethroids has been found to produce dizziness, burning, itching, or tingling of the skin. More severe contact, such as spilling of the pesticides onto the skin, resulted in lacrimation, photophobia, and eye irritation. Accidental swallowing may cause headaches, dizziness, vomiting, fatigue, anorexia, chest tightness, blurred vision, paresthesias, and, in severe cases, convulsive attacks. However, the signs and symptoms of pyrethrin toxicity are usually fully reversible, and no long-term human toxicity has been reported after single or repeated exposure (He et al. 1989; Ecobichon and Joy 1994; Harp 1998; Leng et al. 1999; Vandenplas et al. 2000). It is possible that after exposure sensitive people will develop a rash or allergy-like symptoms such as wheezing, cough, and shortness of breath. That might happen particularly in people who have atopy or bronchial asthma, although the relationship between asthma and pyrethroid exposure is not clear (WHO 1995). Aerosolized pesticides can trigger a “nonspecific” asthmatic response—bronchoconstriction and respiratory symptoms—although it is has been difficult to link sensitization in nonasthmatics to exposure to aerosolized pyrethroid insecticides (WHO 1995).
Aside from the obvious inhalation and dermal exposures that occur when passengers and crew are actively sprayed, typically with phenothrin, residual exposures can occur when permethrin is used to treat unoccupied cabins.
Residual exposures are likely to be dermal and oral. See Chapter 3 for a more detailed description of possible exposures resulting from disinsection practices.
Pyrethroid pesticides are rapidly destroyed by carboxyesterase enzymes in the liver and to a lesser extent in skin, muscle, kidney, brain, and serum (Leng et al. 1999). Organophosphates are also destroyed by carboxyesterases and so can compete with pyrethroids for the enzymes, the competition can result in a synergism that increases overall toxicity. The enzyme cholinesterase, which is also inhibited by organophosphates, does not appear to be important in pyrethroid metabolism (Leng et al. 1999). There is little information on long-term exposure to permethrin and none such on exposure of humans (Naumann and McLachlan 1999).
Most flight attendants’ reports of pesticide incidents describe lung, eye, throat, or skin irritation as the primary adverse effect. Sensitization, or enhanced responsiveness with successive pesticide exposures, may be a problem faced by cabin attendants because they are intermittently exposed; such an exposure regimen is ideal for inducing sensitization or magnified responses to the same exposures (Weiss and Santelli 1978; Gilbert 1995; Jones et al. 1996).
If people are exposed to both pyrethroid and some enzyme inhibitors, there is a potential for synergistic effects. Pyrethroids are lipophilic and are rapidly absorbed and metabolized after ingestion by mammals. However, they do not accumulate in mammalian tissues. In mammals and insects, pyrethroids are generally metabolized by carboxylases and the mixed-function oxidase (MFO) system of the microsomes (Casida et al. 1983). Piperonyl butoxide, a potent inhibitor of the MFO system, is often added to the pesticide formulations to increase the toxicity by a factor of 10–300 (Casida et al. 1983). Carboxyesterase inhibitors may also act synergistically to enhance the toxicity of pyrethroids. The synergism is an asset for insect control, but it can have potentially serious implications for humans in the rare event that a leak of engine oils or hydraulic fluids causes these compounds to enter the cabin and causes simultaneous exposure to carboxyesterase inhibitors. No incidents of such interactions have been documented in humans, but engine-oil seal failures and hydraulic-fluid leaks into the ECS of aircraft have been reported (Parliament of Commonwealth of Australia 2000; van Netten 2000; van Netten and Leung 2000, 2001). See Chapter 3 for a more detailed discussion of engine-oil and hydraulic fluid leaks.
Two other features of possible pyrethroid toxicity may warrant further research and discussion: their potential as developmental neurotoxicants (Ericsson, 1997; Landrigan et al, 1999) and their potential as endocrine disruptors in vivo, which so far has been demonstrated only in vitro (Go et al. 1999).
Aldehydes
Aldehydes, such as acetaldehyde and acrolein (an unsaturated aldehyde), could be found in cabin air in the case of leaking or pyrolysis of engine oil or hydraulic fluids. Concentrations of aldehydes (and ketones) measured in aircraft cabins are “lower than those encountered in ground level buildings” (Nagda et al. 2001).
Maximal concentrations of acetaldehyde were 26.4–30.7 μg/m3 in bleed air and 20.8–70.2 μg/in3 in cabin air (see Chapter 3). Some sensitive people might suffer adverse effects, and there could be repeated exposures. Exposure to other aldehydes results in decreasing toxicity as the chain length of the aldehyde increases in the order of acetaldehyde, propionaldehyde, isobutyraldehyde, n-butyraldehyde, valeraldehyde, and isovaleraldehyde (HSDB 2000). Effects of inhalation exposure to acetaldehyde at high concentrations (about 100–200) ppm include irritation of the mucous membranes (200 ppm for 15 min), irritation of the respiratory tract (134 ppm for 30 min), and lung edema. Prolonged inhalation exposure to acetaldehyde results in symptoms that mimic alcohol intoxication. Repeated dermal or ocular exposures may cause dermatitis or conjuncitivitis, respectively (HSDB 2000). Irritation of the eye may occur at acetaldehyde vapor concentrations as low as 25 ppm after 15 min. Acetaldehyde may facilitate the uptake of other air contaminants by humans because of its ciliotoxic and mucus-coagulating effects (HSDB 2000). Acetaldehyde has been classified as a possible human carcinogen.
The unsaturated aldehyde acrolein may also cause adverse effects when inhaled. However, acrolein was not detected in bleed air or cabin air of several aircraft under normal operating conditions (Nagda et al. 2001). It is an irritant of the eyes and respiratory tract. The odor of acrolein can be perceived at 0.7 mg/m3. The threshold concentrations of acrolein for irritation and health effects are 0.13 mg/m3 for eye irritation, 0.3 mg/m3 for nasal irritation and blinking, and 0.7 mg/m3 for decreased respiratory rate. Exposure to acrolein vapor in air at 1 ppm (2.3 mg/m3) causes lacrimation and marked eye, nose, and throat irritation within 5 min. It irritates the conjunctiva and mucous membranes of the upper respiratory tract. Above 3 mg/m3, it causes injury to the lungs, and respiratory insufficiency may persist for 18 mo or more. A10-min exposure at 350 mg/m3 was lethal (HSDB 2000). Acrolein has been classified as a possible human carcinogen on the basis of an increased incidence of adrenal cortical adenomas in female rats (IRIS 2001). EPA established a
reference concentration3 of 0.0002 mg/m3 for acrolein on the basis of squamous metaplasia and neutrophilic infiltration of nasal epithelium in subchronic inhalation studies in rats (Kutzman 1981, as cited in IRIS 2001).
Carbon Dioxide
Carbon dioxide (CO2) in the aircraft cabin is primarily a “bioeffluent” in that it is released in the exhaled breath of the occupants and is generated by normal metabolism (see also the section on sources inside the cabin in Chapter 3). CO2 inhaled at 2% in air increases pulmonary ventilation by 50% (Thienes and Haley 1972, as cited in HSDB 2000). Asphyxiant effects may occur as the concentration exceeds 6%, causing shortness of breath, dizziness, tingling, and slowed mentation (Maresh et al. 1997). Adding 1% CO2 to air increased pulmonary ventilation rate by 37% at sea level. Under a pressure that simulated an altitude of 5,000 m (16,400 ft), pulmonary ventilation rate increased by 7%. CO2 concentrations of 0.5% or 1% stimulated hyperventilation to a degree that prevented a decrease in psychomotor performance at a simulated altitude of 5,800 m (19,000 ft) but not at 5,000 m (16,400 ft) (Vieillefond et al. 1981, as cited in HSDB 2000). Repeated daily exposures to 0.5–1.5% inspired CO2 at 1 atm are well tolerated by healthy people (ACGIH 1980).
FAR 25.831 has a set limit for CO2 in the cabin at 0.5% (5,000 ppm).4 Cabin pressure altitude is limited to 2,440 m (8,000 ft) and that limit appears to protect occupants from adverse effects of exposure to CO2. The highest CO2 concentrations measured in aircraft cabins and reported in the literature are below the limit (see Table 1–2, Chapter 1). Typical values reported are in the range of 1,100–1,700 ppm, although concentrations as high as 4,200 ppm have been reported (Waters 2001). Backman and Highighat (2000) reported CO2 concentrations of 455 ppm, 586 ppm, and 706 ppm on an Airbus 320 (altitude, 39,000 ft; 86 passengers), a Boeing 767 (39,000 ft; 70 passengers), and a DC-9 (35,000 ft; 75 passengers), respectively.
|
3 |
The reference concentration is an estimate of a daily exposure to the human population (including sensitive subgroups) that is likely to be without an appreciable risk of deleterious effects during a lifetime. |
|
4 |
FAR Section 25.831: “Ventilation: 2. CO2 concentration during flight must be shown not to exceed 0.5 percent by volume (sea level equivalent) in compartments normally occupied by passengers or crew members.” |
CO2 is used to assess the adequacy of ventilation. Normally, CO2 produced as the metabolic byproduct of occupants—respiration—is the primary source of CO2 generation in an occupied space. Although the CO2 generated by respiration is unlikely to reach the point at which adverse effects would be expected, other bioeffluents are generated more or less in proportion to CO2. These other bioeffluents may be responsible for complaints about odors, stale air, lack of fresh air, and stuffiness.
Particulate Matter
Airborne particles in the passenger cabin may comprise coarse particles (such as powders, dusts, dirt, and hair with aerodynamic diameters over 2.5 (μm) and fine and ultrafine particles. Fine particles (aerodynamic diameter, less than 2.5 μm) and ultrafine particles (aerodynamic diameter, less than 0.1 μm) are the products of combustion of materials such as fuels, engine oil, and hydraulic and deicing fluids. Particulate matter (PM), generally with an aerodynamic diameter of less than 10 μm (PM10) has been measured aboard aircraft during normal operations with concentration means ranging from less than 10 μg/m3 (Nagda et al. 2001) to 176 μg/m3 (CSS 1994). Typical indoor air PM concentrations are 3–35 μg/m3 (Burton et al. 2000). Coarse particles, because of their size, are deposited in the upper respiratory airways and can lead to coughing, sneezing, and nasal irritation. Chapter 4 discusses exposure to and health effects of particles of biological origin.
PM10 or smaller may be deposited in the lower (thoracic) regions of the human respiratory tract, including PM2.5, which deposit primarily in deep lung airways. Exposure to fine and ultrafine PM has been associated with heart rate variability (Peters et al. 2000, as cited in EPA 2001) and may be related to cough. Asthmatics have small decrements in lung function and increases in coughing, phlegm, breathing difficulty, and bronchodilator use after exposure to PM10 and PM2.5 (EPA 2001). Fine and ultrafine particles, which are deposited in the small airways, may contain toxic constituents that have been shown to cause cardiopulmonary effects. However, few data are available on the acute effects of particle-associated organic carbon constituents (EPA 2001). See Chapter 4 for a more detailed discussion of the possible health effects associated with exposure to biological agents.
OTHER HEALTH CONSIDERATIONS
Physiological Stressors
In addition to the environmental and chemical stressors discussed above, other stressors may play a role in the perception of and susceptibility to air quality or changes in it.
Fatigue is a common complaint among flight attendants. Work schedules can be erratic, with unplanned layovers and long hours. Furthermore, jet lag can pose a problem. Sleeping accommodations are not always conducive to good rest, and meals can be erratic. Fatigue can also be accentuated by increased CO2 in the air: other factors contributing to fatigue are the effects of vibration, turbulence, and noise; however, these are beyond the scope of this report and will not be discussed further here.
Multiple and Interactive Factors
As discussed earlier in this chapter, cabin occupants (whether flight crew or passengers) may be subjected to numerous air contaminants and other physiological stessors in some situations. Space et al. (2000) argue that cabin air quality generally is better than that in most indoor environments, according to standard criteria. Although that assessment may describe the experience of passengers under most circumstances, problems do occur. Engine oil and hydraulic fluids, and their pyrolysis products, as noted in other chapters, may contaminate the cabin environment, releasing organic compounds, including cresyl phosphates, some of which are neurotoxic. Because cabin pressurization is limited to a virtual altitude of 8,000 ft, PO2 declines; this a potential hazard for passengers with cardiovascular impairment. Elevated CO2 concentrations in that cabin may not present a serious health threat, but in office buildings they are reported to be associated with respiratory symptoms (Apte et al. 2000). Some countries expose passengers to insecticides as part of a program designed to prevent the introduction of nonnative insects.
Although individual factors in isolation might each contribute only a small diminution in well-being, in combination they might be additive, synergistic, or even antagonistic, with their combined influence on the health status of crew and passengers virtually unknown.
Such factors as cramped seating also influence passenger comfort. How-
ever, physical activity is more likely to be a potentiating than an additive factor. Unlike the relatively sedentary passengers and cockpit crew, cabin attendants tend to have sustained periods of moderate activity during the course of a flight. As shown for O3 earlier, exercise could enhance the effects of exposure to many possible cabin air contaminants, including CO and CO2.
The interactions of CO and high altitude have been examined, and results suggest that the effects are additive (EPA 2000). By binding to hemoglobin, CO may increase the hypoxic effects of high altitude in the elderly and those with coronary arterial disease. At high altitudes on earth (e.g., 15,000 ft), the body adjusts to these hypoxic effects within a few days, but the adjustments do not occur over the duration of even a long-distance flight. In a review of the long-term effects of CO at high altitude, EPA found few effects at CO concentrations of less than 100 ppm and altitudes below 15,000 ft (4,573 m) (EPA 2000).
Passengers and crew members frequently report fatigue, dizziness, headaches, sinus and ear problems, dry eyes, and sore throats during and after air travel. Such symptoms are common to many conditions and can be mistaken for infections (IEH 2001). Other characteristics of air travel, however, can make people more susceptible to infection. For example, travelers are subject to stresses (unrelated to their time on an aircraft) that may reduce their resistance to infectious diseases, such as long hours of waiting, disruption of eating habits, and changes in climate and time zone (Rayman 1997; CDC 2001). Psychological stress has been associated with increased susceptibility to the common cold (Cohen et al., 1991) and may play a role in other infectious diseases. Drying of the nasal mucosa during air travel (due to the low moisture content of the air) may result in nosebleeds and is thought by some to increase susceptibility to respiratory infections, although this is uncertain (BRE 2001). Rose et al. (1999) hypothesized that the reduced-O2 environment in aircraft leads to mild hypoxia and suppression of the cellular immune system, which in turn contributes to increased susceptibility to infection. If that is borne out, this pathway could explain in part why some frequent flyers complain of infections after air travel.
Other gases, such as those associated with human body odor, are present in the cabin air and may contribute to the discomfort of passengers. Emissions vary widely and depend on individual health, diet, activity, and personal hygiene, such as bathing habits, frequency of clothing change, and use of deodorant (Berg-Munch et al. 1986). Personal-hygiene products and fragrances contain various plant and animal components, such as aldehydes and sterols,
that may be in the cabin air. Additional sources of disagreeable biological odors on aircraft are the toilets, waste-storage areas in galleys, and bacterial and fungal growth in the cabin. Evidence of adverse health effects (as opposed to annoyance) of body odors, cosmetics and colognes, or VOCs of microbial origin remains controversial. Respiratory irritation is one condition that could result from exposure to odors on aircraft (Batterman 1995; Ammann 1999; Korpi et al. 1999; Rose et al. 2000).
Susceptibility Factors
Although most members of the traveling public are healthy, given the large number of people who fly each year it is to be expected that many of them will have some level of compromised health. At cabin altitudes during cruise, all crew and passengers are in an environment with decreased O2. Under ordinary conditions of commercial flight, relatively small O2 decrements usually cause no symptoms in healthy people, because hemoglobin remains well saturated with O2 at altitude. Nevertheless, some persons with underlying pulmonary or cardiac disease or significant untreated or partially treated anemia might be sensitive to a lowered PO2 and might experience any number of symptoms, including headache, lightheadedness, dizziness, fatigue, numbness, and tingling (Sheffield and Heimbach 1996). The health implications of commercial flight for passengers with pre-existing illness are described in greater detail in Medical Guidelines for Airline Travel (Aerospace Medical Association 1997). Air crew and passengers with particular preexisting medical conditions may not tolerate the cabin environment described above as well as would a healthy traveler. People with underlying cardiovascular or respiratory diseases, particularly adults and children with asthma, may also be at increased risk because of the increased O3 exposures that occur in high-altitude flights. Data are lacking on the relationship between asthma and O3 in aircraft cabins at high altitudes. People with COPD or emphysema have lung-tissue damage with an abnormal capability for transporting O2 across the lung tissue into the bloodstream. Consequently, they have less O2 in their blood than a normal person, whether at sea level or, worse, at altitude. Such passengers may become symptomatic and have shortness of breath, wheezing, and coughing. Schwartz et al. (1984) studied subjects with severe COPD (average resting arterial PO2, 68.0±7.3 mm Hg) during flights at altitudes of 1,650–2,250 m (about 5,400–7,400 ft) in unpressurized aircraft cabins. PO2 decreased to an
average of 51.0±9.1 mm Hg at an altitude of 1,650 m; there was little further change at 2,250 m.
Several simulation studies have been carried out in hypobaric chambers with COPD patients and healthy subjects (Dillard et al. 1989; Naughton et al. 1995). Declines in PO2 were observed in all patients at rest; values fell to below 50 mm Hg in many healthy and COPD subjects and the declines were made worse by light exercise (Figure 5–3). Of the 100 patients with COPD, 44 reported traveling by air in the 2 years before the interview (Dillard et al. 1991). Eight of the 44 patients reported increased symptoms during flight (no direct physiological measurements were available). Five of the 8 patients experienced shortness of breath when walking in the cabin, and two requested supplemental O2 for their symptoms. Variables to consider are clinical evaluation, pulmonary-function tests, and blood-gas analysis. The measurement of arterial blood gas is most useful because it is considered the best predictor of arterial PO2 at altitude. A ground-level value greater than 9.3 kPa (70 mm Hg) is considered adequate in most cases (Gong et al. 1984; Cottrell 1998). If it is lower, consideration must be given to prescribing inflight medical O2 or postponing air travel depending on the clinical circumstances. It cannot be overemphasized that the clinical judgment of a physician is essential for a person with COPD in deciding whether to fly. The committee was unable to locate any data to suggest that most people with COPD cannot fly safely.
People with significant coronary arterial disease may also be at increased risk for cardiac symptoms because of reduced PO2 at altitude. The committee was unable to locate any published studies of air-travel experiences of passengers with coronary arterial disease. Because of the lack of data, the broad spectrum of disease severity, and individual variability, the committee cannot put forward any recommendations other than to note that, as with COPD, a decision on a traveler’s fitness to fly should be made by a physician.
A number of characteristics may affect a person’s risk of becoming infected if exposed and affect the severity of disease that could result from infection (see Chapter 4 for more information on infectious agents on aircraft). Those characteristics include age, nutritional condition, alcoholism, coexisting (particularly chronic) diseases (such as those discussed above), immune status, and abnormalities in the skin or respiratory tract that allow the entry of infectious agents (Macher and Rosenberg 1999). Many persons with altered immunocompetence are able to travel, but they should consult their physicians before traveling.
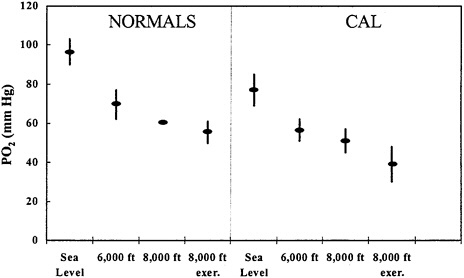
FIGURE 5–3 PO2 in six healthy subjects and nine subjects with chronic air-flow limitation (CAL; FEV1/FVC≤70%) during hypobaric high-attitude simulation. Data at 8,000 ft are at rest and after 2 min of “light exercise” (approximate work rate, 200 kJ/min) to approximate a short walk on aircraft. Source: Naughton et al. 1995. Redrawn from the American Journal of Respiratory and Critical Care Medicine; copyright 1995, American Thoracic Society.
People with medical conditions that may require treatment during travel (e.g., COPD, asthma, and autoimmune diseases) should carry letters describing their condition and treatment. Various types of medical alert tags and bracelets are available and should be worn to inform emergency-care providers of appropriate treatments or precautions for various medical conditions and how to reach travelers’ health-care providers.
Populations that may be particularly sensitive to CO poisoning are anemic persons who have low hemoglobin concentrations, children who have higher metabolic rates that would exacerbate the adverse effects of CO, persons with a history of coronary heart disease or respiratory disease, and the elderly. Smokers typically have increased COHb and may have an adaptive response to elevated COHb (EPA 2000). People with coronary arterial disease and reproducible exercise-induced angina have decreased exercise tolerance at COHb concentrations of 3–6% (EPA 2000). Pregnant women may be at particular risk. The fetus may be very susceptible to the effects of CO be
cause it readily crosses the placenta and might result in neurological damage to the infant (EPA 2000). Asthmatics may be especially sensitive to formaldehyde.
Cabin crews as a cohort population are relatively healthy. Air-transport pilots are required by FAA to undergo a flight physical examination every 6 mo, and must be granted an exception for significant illness or some classes of medications. Air-transport pilots can no longer fly as pilots once they are 60 yr old. However, flight attendants are required to undergo only an entry physical examination. There is no requirement for periodic medical examinations as there is for pilots. There are no health requirements for passengers.
CONCLUSIONS
-
The flight environment, with its lowered barometric pressure, may result in passenger and crew discomfort and in susceptible people, in health effects. Infants may also be at greater risk for hypoxia under conditions of reduced PO2.
-
Although low relative humidity in the aircraft cabin can result in temporary discomfort as a result of the drying of mucous membranes and eye, nose, and respiratory tract irritation, symptoms are expected to subside after exposure is discontinued. There is no information on the potential for long- or short-term adverse effects associated with exposure to low relative humidity.
-
High-altitude flights might result in increased O3 levels in an aircraft. Elevated O3 concentrations have been associated with increased respiratory symptoms, such as coughing, wheezing, and asthma.
-
Phosphate esters, formaldehyde, other aldehydes, and CO—found in engine oil, hydraulic fluids, and their pyrolysis products—may cause respiratory and neurological effects, particularly at high concentrations. However, more data are needed to establish an association between the presence and concentrations of cabin contaminants and potential health effects in passengers and crew.
-
Although pyrethroid pesticides, used for disinsection on some aircraft, have very low toxicity in humans, they can cause adverse effects and are recognized as neurotoxins.
-
In passengers and cabin crew who have pre-existing illness—such as anemia, asthma, COPD, and coronary arterial disease—the stresses of flight could exacerbate symptoms.
RECOMMENDATIONS
-
Potential synergistic and interactive effects of exposure in the aircraft cabin to reduced barometric pressure, low humidity, O3, other chemical contaminants, and pesticides should be examined.
-
If future research, such as that described in Chapter 8, indicates that some cabin air contaminants or other environmental characteristics, such as relative humidity, pose hazards to the health of passengers or crew, FAA should work with other organizations—such as the Occupational Safety and Health Administration, EPA, and ACGIH—to establish standards or guidelines to regulate them.
REFERENCES
ACGIH (American Conference of Governmental Industrial Hygienists). 1980. Documentation of the Threshold Limit Values, 4th Ed. Cincinnati, OH: ACGIH.
ACGIH (American Conference of Governmental Industrial Hygienists). 2001. 2001 TLVs and BEIs: Threshold Limit Values for Chemical Substances and Physical Agents: Biological Exposure Indices. Cincinnati, OH: ACGIH.
Aerospace Medical Association. 1997. Medical Guidelines for Airline Travel. Alexandria, VA: Aerospace Medical Association.
Allred, E.N., E.R.Bleecker, B.R.Chaitman, T.E.Dahms, S.O.Gottlieb, J.D.Hackney, M. Pagano, R.H.Selvester, S.M.Walden, and J.Warren. 1991. Effects of carbon monoxide on myocardial ischemia. Environ. Health Perspect. 91:89–132.
Ammann, H.M. 1999. Microbial volatile organic compounds. Pp. 26.1–26.17 in Bioaerosols: Assessment and Control, J.M.Macher, H.M.Ammann, H.A.Burge, D.K.Milton, and P.R.Morey, eds. Cincinnati, OH: American Conference of Government Industrial Hygienists.
Apte, M.G., W.J.Fisk, and J.M.Daisey. 2000. Associations between indoor CO2 concentrations and sick building syndrome symptoms in U.S. office buildings: An analysis of the 1994–1996 BASE study data. Indoor Air 10(4):246–257.
ATSDR (Agency for Toxic Substances and Disease Registry). 1997. Toxicological Profile for Ethylene Glycol and Propylene Glycol. U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA.
ATSDR (Agency for Toxic Substances and Disease Registry). 1999. Toxicological Profile for Formaldehyde. U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA.
Backman, H., and F.Haghighat. 2000. Air quality and ocular discomfort aboard commercial aircraft. Optometry 71(10):653–656.
Ballarin, C., F.Sarto, L.Giacomelli, G.B.Bartolucci, and E.Clonfero. 1992. Micronucle-
ated cells in nasal mucosa of formaldehyde-exposed workers. Mutat. Res. 280(1):1–7.
Baron, R.L. 1981. Delayed neurotoxicity and other consequences of organophosphate esters. Annu. Rev. Entomol. 26:29–48.
Batterman, S.A. 1995. Sampling and analysis of biological volatile organic compounds. Pp. 249–268 in Bioaerosols, H.A.Burge, ed. Boca Raton: Lewis.
Berglund, L.G. 1998. Comfort and humidity. ASHRAE J. 40(10):35–41.
Berg-Munch, B., G.Clausen, and P.O.Fanger. 1986. Ventilation requirements for the control of body odor inspaces occupied by women. Environ. Int. 12(1–4):195–199.
BRE (British Research Establishment). 2001. Study of Possible Effects on Health of Aircraft Cabin Environments- Stage 2. British Research Establishment, Environment Division, Garston, Watford, UK. [Online]. Available: http://www.aviation.dtlr.gov.uk/healthcab/aircab/index.htm [September 2001].
Burton, L.E., J.G.Girman, and S.E.Womble. 2000. Airborne particulate matter within 100 randomly selected office buildings in the United States (BASE). Pp. 157–162 in Healthy Buildings 2000: Exposure, Human Responses, and Building Investigations, Proceedings, Vol. 1, O.Seppänen, and J.Säteri, eds. Helsinki, Finland: SIY Indoor Air Information.
Carleton, W.M., and B.Welch. 1971. Fluid Balance in Artificial Environment. U.S. Air Force School of Aerospace Medicine. NASA Report CR-114977.
Carpenter, H.M., D.J.Jenden, N.R.Shulman, and J.R.Tureman. 1959. Toxicology of a triaryl phosphate oil: I. Experimental toxicology. AMA Arch. Ind. Health. 20(3):234–252.
Casida, J.E., D.W.Gammon, A.H.Glickman, and L.J.Lawrence. 1983. Mechanisms of selective action of pyrethroid insecticides. Annu. Rev. Pharmacol. Toxicol. 23:413– 438.
CDC (Centers for Disease Control and Prevention). 2001. Health Information for International Travel 2001–2002. Atlanta, GA: National Center for Infectious Disease, Division of Quarantine, Centers for Disease Control and Prevention.
Centers, P. 1992. Potential neurotoxin formation in thermally degraded synthetic ester turbine lubricants. Arch. Toxicol. 66(9):679–680.
Cohen, S., D.A.Tyrrell, and A.P.Smith. 1991. Psychological stress and susceptibility to the common cold. N. Engl. J. Med. 325(9):606–612.
Commonwealth of Australia Senate. 1999. Air Safety-BAe 146 Air Quality. The Proof and Official Hansard transcripts of Senate Rural and Regional Affairs and Transport References Committee, Canberra, Australia. November 1, 1999 .
Cottrell, J.J. 1998. Altitude exposures during aircraft flight. Flying higher. Chest 93(1):81–84.
Cottrell, J.J., B.L.Lebovitz, R.G.Fennell, and G.M.Kohn. 1995. Inflight arterial saturation: Continuous monitoring by pulse oximetry. Aviat. Space Environ. Med. 66(2):126–130.
Craig, P.H., and M.L.Barth. 1999. Evaluation of the hazards of industrial exposure to tricresyl phosphate: A review and interpretation of the literature. J. Toxicol. Environ. Health B Crit. Rev. 2(4):281–300.
Crane, C.R., D.C.Sanders, B.R.Endecott, and J.K.Abbot. 1983. Inhalation Toxicology: III. Evaluation of Thermal Degradation Products From Aircraft and Automobile Engine Oils, Aircraft Hydraulic Fluid, and Mineral Oil. FAA-AM-83–12. Washington, DC: Federal Aviation Administration.
CSS (Consolidated Safety Services). 1994. Airline Cabin Air Quality Study. Prepared for Air Transport Association of America, Washington, DC. April 1994.
Daughtrey, W.C., R.A.Scala, L.N.Curcio, and J.O.Kuhn. 1990. Evaluation of the Acute Delayed Neurotoxic Potential of a Commercial Tricresylphosphate-Containing Turbo Oil. Toxicologist 10: Abstracts of the 29th Annual Meeting No. 343.
Daughtrey, W., R.Biles, B.Jortner, and M.Ehrich. 1996. Subchronic delayed neurotoxicity evaluation of jet engine lubricants containing phosphorus additives. Fundam. Appl. Toxicol. 32(2):244–249.
Delivoria-Papadopoulos, M., and L.S.Wagerle. 1990. Oxygen diffusion and transport. Pp. 281–305 in Pulmonary Physiology: Fetus, Newborn, Child and Adolescent, 2nd Ed., E.M.Scarpelli, ed. Philadelphia: Lea and Febiger.
Denison, D.M., F.Ledwith, and E.C.Poulton. 1966. Complex reaction times at simulated cabin altitudes of 5,000 feet and 8,000 feet. Aerosp. Med. 37(10):1010–1013.
Dillard, T.A., B.W.Berg, K.R.Rajagopal, J.W.Dooley, and W.J.Mehm. 1989. Hypoxemia during air travel in patients with chronic obstructive pulmonary disease. Ann. Inter. Med. 111(5):362–367.
Dillard, T.A., W.A.Beninati, and B.W.Berg. 1991. Air travel in patients with chronic obstructive pulmonary disease. Arch. Intern. Med. 151(9):1793–1795.
Ecobichon, D.J., and R.M.Joy. 1994. Pesticides and Neurological Diseases, 2nd Ed. Boca Raton, FL: CRC.
Eng, W.G. 1979. Survey on eye comfort in aircraft: I. Flight attendants. Aviat. Space Environ. Med. 50(4):401–404.
EPA (U.S. Environmental Protection Agency). 1996. Air Quality Criteria for Ozone and Related Photochemical Oxidants. EPA/600/P-93/004aF, Vol. 1; EPA/600/P-93/004bF, Vol. 2; EPA/600/P-93/004cF, Vol. 3. National Center for Environmental Assessment, Office of Research and Development, U.S. Environmental Protection Agency, Research Triangle Park, NC. [Online]. Available: www.epa.gov/ncea/ozone.htm. [July 11, 2001].
EPA (U.S. Environmental Protection Agency). 1997. National Ambient Air Quality Standards (NAAQS). Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency. [Online]. Available: http://www.epa.gov/airs/criteria.html [Oct. 24, 2001].
EPA (U.S. Environmental Protection Agency). 2000. Air Quality Criteria for Carbon Monoxide. EPA/600/P-99/001 F. National Center for Environmental Assessment, Office of Research and Development, U.S. Environmental Protection Agency, Research Triangle Park, NC.
EPA (U.S. Environmental Protection Agency). 2001. Air Quality Criteria for Particulate Matter, Vol. 2, 2nd External Review Draft. EPA/600/P-99/002bB. National Center for Environmental Assessment, Office of Research and Development, U.S. Environmental Protection Agency, Research Triangle Park, NC. [Online]. Available:
http://www.epa.gov/NCEA/pdfs/partmatt/VOL_II_AQCD_PM_2nd_Review_Draft.pdf [July 12, 2001].
Eriksson, P. 1997. Developmental neurotoxicity of environmental agents in the neonate. Neurotoxicology 18(3):719–726.
Ernsting, J. 1978. Prevention of hypoxia—Acceptable compromises. Aviat. Space Environ. Med. 49(3):495–502.
Ernsting, J., J.L.Gedye, and G.J.R.McHardy. 1962. Anoxia subsequent to rapid decompression. Pp. 359–368 in Human Problems of Supersonic and Hypersonic Flight, A.Buchanan-Barbour, and H.E.Whittingham, eds. Oxford, England: Pergamon Press.
Fang, L., G.Clausen, and P.O.Fanger. 1998a. Impact of temperature and humidity on the perception of indoor air quality. Indoor Air 8(2):80–90.
Fang, L., G.Clausen, and P.O.Fanger. 1998b. Impact of temperature and humidity on perception of indoor air quality during immediate and longer whole-body exposures. Indoor Air 8(4):276–284.
Faustman, E.M., and G.S.Omenn. 2001. Risk assessment. Pp. 83–104 in Casarett and Doull’s Toxicology, The Basis Science of Poisons, 6th Ed., C.D.Klaassen, ed. New York: McGraw-Hill.
Fowler, B., D.D.Elcombe, B.Kelso, and G.Porlier. 1987. The threshold for hypoxia effects on perceptual-motor performance. Human Factors 29(1):61–66.
Gilbert, M.E. 1995. Repeated exposure to lindane leads to behavioral sensitization and facilitates electrical kindling . Neurotoxicol. Teratol. 17(2):131–141.
Go, V., J.Garey, M.S.Wolff, and B.G.Pogo. 1999. Estrogenic potential of certain pyrethroid compounds in the MCF-7 human breast carcinoma cell line. Environ. Health Perspect. 107(3):173–177.
Gong Jr., H., D.P.Tashkin, E.Y.Lee, and M.S.Simmons. 1984. Hypoxia-altitude simulation test. Evaluation of patients with chronic airway obstruction. Am. Rev. Respir. Dis. 130(6):980–986.
Goode, M. 2000. Chemistry and History of TCP Usage in Aviation Lubricating. Health Safety and Environmental Overview. Presentation at Chemical Exxon Mobile Conference, Great Lakes, December 2000.
Gosselin, R.E., R.P.Smith, and H.C.Hodge. 1984. Pp. 111–98 in Clinical Toxicology of Commercial Products, 5th Ed. Baltimore: Williams and Wilkins.
Harp, P.R. 1998. Pyrethrin/pyrethroids. Pp. 610–611 in Encyclopedia of Toxicology, Vol. 2., P.Wexler and S.C. Gad, eds. San Diego: Academic Press.
He, F., S.Wang, L.Liu, S.Chen, Z.Zhang, and J.Sun. 1989. Clinical manifestations and diagnosis of acute pyrethroid poisoning. Arch. Toxicol. 63(1):54–58.
Healy, C.E., R.S.Nair, W.E.Ribelin, and C.L.Bechtel. 1992. Subchronic rat inhalation study with Skydrol 500B-4 fire resistant hydraulic fluid. Am. Ind. Hyg. Assoc. J. 53(3):175–180.
Henschler, D. 1958a. Hazards in use of modem tricresyl phosphates [in German]. Zbl. Arbeitsmed. 8(11):265–267.
Henschler, D. 1958b. Tricresyl phosphate poisoning. Experimental clarification of
problems of etiology and pathogenesis [in German]. Klinische Wochenschrift 36(14):663–674.
Henschler, D. 1959. Relationships between the chemical structure and the paralyzing action of triaryl phosphates [in German]. Naunyn-Schmiedeberg’s Arch. Exp. Path. U. Pharmak. 237:459–472.
Hewstone, R.K. 1994. Environmental health aspects of lubricant additives. Sci. Total Environ. 156(3):243–254.
Holmstrom, M., B.Wilhelmsson, H.Hellquist, and G.Rosen. 1989. Histological changes in the nasal mucosa in persons occupationally exposed to formaldehyde alone and in combination with wood dust. Acta Otolaryngol. 107(1–2):120–129.
HSDB [Hazardous Substances Data Bank]. 2000. Toxicology Data Network, National Library of Medicine’s (NLM). [Online]. Available: http://toxnet.nlm.nih.gov/
IEH (Institute for Environment and Health). 2001. Consultation on the Possible Effects on Health, Comfort and Safety on Aircraft Cabin Environments. IEH Web Report W5. Leicester, UK: Institute for Environment and Health. [Online]. Available: http://www.le.ac.uk/ieh/webpub.html [March 2001].
IPCS (International Programme on Chemical Safety). 1999. Carbon monoxide. Environmental Health Criteria 213, 2nd Ed. Geneva: World Health Organization.
IRIS. 2001. Integrated Risk Information System. U.S. Environmental Protection Agency. [Online]. Available: http://www.epa.gov/iriswebp/iris/subst/0419.htm [October 19, 2001].
Jones, S.R., T.H.Lee, R.M.Wightman, and E.H.Ellinwood. 1996. Effects of intermittent and continuous cocaine administration on dopamine release and uptake regulation in the striatum: In vitro voltammetric assessment. Psychopharmacology 126(4):331–338.
Koren, G., T.Sharay, A.Pastuszak, L.K.Garrettson, K.Hill, I.Samson, M.Rorem, A. King , and J.E.Dolgin. 1991. A multicenter, prospective study of fetal outcome following accidental carbon monoxide poisoning in pregnancy. Reprod. Toxicol. 5(5):397–403.
Korpi, A., J.P.Kasanen, Y.Alarie, V.M.Kosma, and A.L.Pasanen. 1999. Sensory irritating potency of some microbial volatile organic compounds (MVOCs) and a mixture of five MVOCs. Arch. Environ. Health 54(5):347–352.
Kutzman, R.S. 1981. A Subchronic Inhalation Study of Fischer 344 Rats Exposed to 0, 0.4, 1.4, or 4.0 ppm Acrolein. Brookhaven National Laboratory, Upton, NY. National Toxicology Program: Interagency Agreement No. 222-Y01-ES-9–0043.
Landrigan, P.J., L.Claudio, S.B.Markowitz, G.S.Berkowitz, B.L.Brenner, H.Romero, J.G.Wetmur, T.D.Matte, A.C.Gore, J.H.Godbold, and M.S.Wolff. 1999. Pesticides and inner-city children: Exposures, risks, and prevention. Environ. Health Perspect. 107 (Suppl. 3):431–437.
Laviana, J.E., F.H.Rohles Jr., and P.E.Bullock. 1988. Humidity, comfort and contact lenses. ASHRAE Transactions 94:3–11.
Ledwith, F. 1970. The effects of hypoxia on choice reaction time and movement time. Ergonomics 13(4):465–482.
Lee, S.C., C.S.Poon, X.D.Li, F.Luk, M.Chang, and S.Lam. 2000. Air quality measurements on sixteen commercial aircraft. Pp. 45–58 in Air Quality and Comfort in Airliner Cabins, N.L.Nagda, ed. West Conshohocken, PA: American Society for Testing and Materials.
Leng, G., J.Lewalter, B.Roehrig, and H.Idel. 1999. The influence of individual susceptibility in pyrethroid exposure. Toxicol. Lett. 107(1–3):123–130.
Macher, J.M., and J.Rosenberg. 1999. Evaluation and management of exposure to infectious agents. Pp. 287–371 in Handbook of Occupational Safety and Health, 2nd Ed., L.J.DiBerardinis, ed. New York: John Wiley & Sons.
Mackerer, C.R, and E.N.Ladov. 2000. Submission 13A to the Senate References Committee Rural and Regional Affairs and Transport on The Inquiry into Air Safety- BAe 146 Cabin Air Quality. Pp. 44–54 in Air Safety and Cabin Air Quality in the BAe 146 Aircraft, Vol. 3. Report by the Senate Rural and Regional Affairs and Transport References Committee, Parliament of the Commonwealth of Australia, Parliament House, Canberra. October 2000 .
Mackerer, C.R., M.L.Barth, A.J.Krueger, B.Chawla, and T.A.Roy. 1999. Comparison of neurotoxic effects and potential risks from oral administration or ingestion of tricresyl phosphate and jet engine oil containing tricresyl phosphate. J. Toxicol. Environ. Health A. 57(5):293–328.
Maresh, C.M., L.E.Armstrong, S.A.Kavouras, G.J.Allen, D.J.Casa, M.Whittlesey, and K.E.LaGasse. 1997. Physiological and psychological effects associated with high carbon dioxide levels in healthy men. Aviat. Space Environ. Med. 68(1): 41–45.
McFarland, R.A. 1946. Pp. 69–101 in Human Factors in Air Transport Design, 1 st Ed. New York: McGraw-Hill.
McFarland, R.A., and J.N.Evans. 1939. Alterations to dark adaptions under reduced oxygen tensions. Am. J. Physiol. 127:37–50.
Nagda, N.L, ed. 2000. Air Quality and Comfort in Airliner Cabins. West Conshohocken, PA: American Society for Testing and Materials.
Nagda, N.L., and M.Hodgson. 2001. Low relative humidity and aircraft cabin air quality. [Review]. Indoor Air 11(3):200–214.
Nagda, N.L., M.D.Fortmann, M.D.Koontz, S.R.Baker, and M.E.Ginevan. 1989. Airliner Cabin Environment: Contaminant Measurements, Health Risks, and Mitigation Options. DOT-P-15–89–5. NTIS/PB91–159384. Prepared by GEOMET Technologies, Germantown, MD, for the U.S. Department of Transportation, Washington DC.
Nagda, N.L., H.E.Rector, Z.Li, and D.R.Space. 2000. Aircraft cabin air quality: A critical review of past monitoring studies. Pp. 215–235 in Air Quality and Comfort in Airliner Cabins, N.L.Nagda, ed. West Conshohocken, PA: American Society for Testing and Materials.
Nagda, N.L., H.E.Rector, Z.Li, and E.H.Hunt. 2001. Determine Aircraft Supply Air Contaminants in the Engine Bleed Air Supply System on Commercial Aircraft. ENERGEN Report AS20151. Prepared for American Society of Heating, Refrigerat-
ing, and Air-Conditioning Engineers, Atlanta, GA, by ENERGEN Consulting, Inc., Germantown, MD. March 2001.
Naughton, M.T., P.D.Rochford, J.J.Pretto, R.J.Pierce, N.F.Cain, and L.B.Irving. 1995. Is normobaric simulation of hypobaric hypoxia accurate in chronic airflow limitation? Am. J. Respir. Crit. Care Med. 152(6 Pt 1):1956–1960.
Naumann, I.D., and K.McLachlan. 1999. Aircraft Disinsection. Report commissioned by the Australian Quarantine and Inspection Service. June 1999.
Nicholson, A.N. 1996. Low Humidity: Dehydration, Dipsosis or Just Dryness? Royal Air Force School of Aviation Medicine Report No 01/96, Hampshire, UK. May 1996.
NRC (National Research Council). 1986. The Airliner Cabin Environment: Air Quality and Safety. Washington, DC: National Academy Press.
NRC (National Research Council. 1994. Health Effects of Permethrin-Impregnated Army Battle-Dress Uniforms. Washington, DC: National Academy Press.
Parkins, K.J., C.F.Poets, L.M.O’Brien, V.A.Stebbens, and D.P.Southall. 1998. Effect of exposure to 15% oxygen on breathing patterns and oxygen saturation in infants: Interventional study. BMJ. 316(7135):887–894.
Parliament of the Commonwealth of Australia. 2000. Air Safety and Cabin Air Quality in the BAe 146 Aircraft. Report by the Senate Rural and Regional Affairs and Transport References Committee, Parliament House, Canberra. October 2000.
Paustenbach, D., Y.Alarie, T.Kulle, N.Schachter, R.Smith, J.Swenberg, H.Witschi, and S.B.Horowitz. 1997. A recommended occupational exposure limit for formaldehyde based on irritation. J. Toxicol. Environ. Health 50(3):217–263.
Peters, A., E.Liu, R.L.Verrier, J.Schwartz, D.R.Gold, M.Mittleman, J.Baliff, J.A.Oh, G.Allen, K.Monahan, and D.W.Dockery. 2000. Air pollution and incidence of cardiac arrhythmia. Epidemiology 11(1):11–17.
Rankin, W.L., D.R.Space, and N.L.Nagda. 2000. Passenger comfort and the effect of air quality. Pp. 269–290 in Air Quality and Comfort in Airline Cabins, N.L. Nagda, ed. West Conshohocken: American Society for Testing and Materials.
Rayman, R.B. 1997. Passenger safety, health, and comfort: A review. Aviat. Space Environ. Med. 68(5):432–440.
Ritter, S. 2001. Aircraft deicers. Chem. Eng. News 79(1):30.
Rose, D.M., D.Jung, D.Parera, and J.Konietzko. 1999. Time zone shift and the immune system during long-distance flights, [in German]. Z. Arztl. Fortbild. Qualitatssich. 93(7):481–484.
Rose, L.J., R.B.Simmons, S.A.Crow, and D.G.Ahearn. 2000. Volatile organic compounds associated with microbial growth in automobile air conditioning systems. Curr. Microbiol. 41(3):206–209.
Robkin, M.A. 1997. Carbon monoxide and the embryo. Int. J. Dev. Biol. 41(2):283–289.
Schwartz, J.S., H.Z.Bencowitz, and K.M.Moser. 1984. Air travel hypoxemia with chronic obstructive pulmonary disease. Ann. Intern. Med. 100(4):473–477.
Sheffield, P.J., and R.D.Heimbach. 1996. Respiratory physiology. Pp. 68–108 in Fundamentals of Aerospace Medicine, 2nd Ed., R.DeHart, ed. Baltimore: Williams and Wilkins.
Siegel, J., H.S.Rudolph, A.J.Getzkin, and R.A.Jones. 1965. Effects on experimental animals of long-term continuous inhalation of a triaryl phosphate hydraulic fluid. Toxicol. Appl. Pharmacol. 7(4):543–549.
Slonim, N.B., and L.H.Hamilton. 1971. Respiratory Physiology, 2nd Ed. St. Louis, MO: Mosby.
Smith, R.P. 1996. Toxic responses of the blood. Pp. 335–354 in Casarett and Doull’s Toxicology, The Basis Science of Poisons, 5th Ed., C.D.Klaassen, ed. New York: McGraw-Hill.
Snyder, R., and L.S.Andrews. 1996. Toxic effects of solvents and vapors. Pp. 737–772 in Casarett and Doull’s Toxicology, The Basis Science of Poisons, 5th Ed., C.D. Klaassen, ed. New York: McGraw-Hill.
Space, D.R., R.A.Johnson, W.L.Rankin, and N.L.Nagda. 2000. The airplane cabin environment: Past, present and future research. Pp. 189–210 in Air Quality and Comfort in Airliner Cabins, N.L.Nagda, ed. West Conshohocken, PA: American Society for Testing and Materials.
Spektor, D.M., M.Lippmann, G.D.Thurston, P.J.Lioy, J.Stecko, G.O’Connor, E. Garshick, F.E.Speizer, and C.Hayes. 1988a. Effects of ambient ozone on respiratory function in healthy adults exercising outdoors. Am. Rev. Respir. Dis. 136(4):821–828.
Spektor, D.M., M.Lippmann, G.D.Thurston, P.J.Lioy, K.Citak, D.J.James, N.Bock, F.E.Speizer, and C.Hayes. 1988b. Effects of ambient ozone on respiratory function in active, normal children. Am. Rev. Respir. Dis. 137(2):313–320.
Steinmetz, D. 1998. Carbon monoxide. Pp. 224–226 in Encyclopedia of Toxicology, Vol. 1., P.Wexler, and S.C.Gad, eds. San Diego: Academic Press.
Tashkin, D.P., A.H.Coulson, M.S.Simmons, and G.H.Spivey. 1983. Respiratory symptoms of flight attendants during high-altitude flight: Possible relation to cabin ozone exposure. Int. Arch. Occup. Environ. Health. 52(2):117–137.
Thienes, C., and T.J.Haley. 1972. Pp. 57 in Clinical Toxicology, 5th Ed. Philadelphia: Lea and Febiger.
Thurston, G.D., M.Lippmann, M.B.Scott, and J.M.Fine. 1997. Summertime haze air pollution and children with asthma. Am. J. Respir. Crit. Care Med. 155(2):654–660.
Vandenplas, O., J.P.Dewiche, J.Auverdin, J.M.Caroyer, and F.Binard-Van Cangh. 2000. Asthma to tetramethrin. Allergy 55(4):418–419.
Van Netten, C. 1998. Air quality and health effects associated with the operation of BAe 246–200 aircraft. Appl. Occup. Environ. Hyg. 13(10):733–739.
Van Netten, C. 2000. Analysis of two jet engine lubricating oils and a hydraulic fluid: Their pyrolytic breakdown products and their implication on aircraft air quality. Pp. 61–75 in Air Quality and Comfort in Airliner Cabins, N.L.Nagda, ed. West Conshohocken, PA: American Society for Testing and Materials.
Van Netten, C., and V.Leung. 2000. Comparison of the constituents of two jet engine lubricating oils and their volatile pyrolytic degradation products. Appl. Occup. Environ. Hyg. 15(3):277–283.
Van Netten, C., and V.Leung. 2001. Hydraulic fluids and jet engine oil: Pyrolysis and aircraft air quality. Arch. Environ. Health 56(2):181–186.
Vieillefond, H., J.L.Poirier, and H.Marotte. 1981. Influence de l’ altitude sur la toxicite des oxydes de carbone. Pp. B11.1-B11.4 in Toxic Hazards in Aviation: Papers presented at the Aerospace Medical Panel Specialists’ Meeting held in Toronto, Canada, 15–19 September 1980. AGARD-Conference Proceedings No. 309. Neuilly sur Seine, France: AGARD, Advisory Group for Aerospace Research & Development.
Waters, M., T.Bloom, and B.Grajewski. 2001. Cabin Air Quality Exposure Assessment. National Institute for Occupational Safety and Health, Cincinnati, OH. Federal Aviation Administration Civil Aeromedical Institute. Presented to the NRC Committee on Air Quality in Passenger Cabins of Commercial Aircraft, January 3, 2001, Washington, DC.
Weiss, B., and S.Santelli. 1978. Dyskinesias evoked in monkeys by weekly administration of haloperidol. Science 200(4343):799–801.
WHO (World Health Organization). 1995. Report of the Informal Consultation on Aircraft Disinsection, WHO/HQ, Geneva, 6–10 November 1995, International Programme on Chemical Safety. Geneva, Switzerland: World Health Organization.
Wills, J.H., F.Coulston, E.S.Harris, E.W.McChesney, J.C.Russell, and D.M.Serrone. 1974. Inhalation of aerosolized ethylene glycol by man. Clin. Toxicol. 7(5):463–476.
Wright, R.L. 1996. Formation of the neurotoxin TMPP from TMPE-Phosphate Formulations. Tribology Trans. 39(4):827–834.
Wyman, J., E.Pitzer, F.Williams, J.Rivera, A.Durkin, J.Gehringer, P.Serve, D. von Minden , and D.Macys. 1993. Evaluation of shipboard formation of a neurotoxicant (trimethyolpropane phosphate) from thermal decomposition of synthetic aircraft engine lubricant. Am. Ind. Hyg. Assoc. J. 54(10):584–592.
Yonkosky, D., L. Ladia, L.Gackenheimer, and M.W.Schultz. 1990. Scabies in nursing homes: An eradication program with permethrin 5% cream. J. Am. Acad. Dermatol. 23(6 Pt.1):1133–1136.









































