
2
Endemic Infectious Diseases Linked to Chronic Diseases: Implications for Developing Countries
OVERVIEW
Successful disease control efforts in some economically developing countries have increased life expectancy and resulted in changes in demographics from predominantly youthful populations to older and aging ones. Consequently, during the next 20 years, chronic diseases are expected to become increasingly important in economically developing regions and to encompass chronic conditions currently attributed to industrialized nations. Not only will changing economics, demographic shifts with lower childhood mortality, and changing lifestyles affect this trend, but migration from rural to urban areas and into previously uninhabited ecosystems may expose populations to new infectious agents that underlie chronic disease. Both newly identified and well-recognized infectious etiologies of chronic disease, including infections known to enter a chronic state, such as tuberculosis and malaria, will acquire increasing importance to domestic and global health. As such, countries with limited research capacities and health care services will face increasing burdens from both infectious and chronic disease.
Richard Guerrant illustrated the wide-ranging nature of the threats from chronic diseases caused by infections, using as an example the long-term consequences of early childhood enteric and parasitic infections. The chronic impact of repeated malnourishing diarrheal illnesses is greater than that of acute deaths from enteric illness, which claims more than 6,000 children each day. Early diarrheal illnesses have significant long-term effects not only on physical fitness, but on growth, cognition, and school performance. Diarrhea appears to be a cofactor with malnutrition in that it reduces nutritional absorption.
Josemir Sander detailed the relationship between epilepsy, the most common serious neurological condition worldwide, and a number of parasites. Epilepsy is a symptom complex, so diagnosis relies on clinical history rather than a specific test. Incidence is higher in developing countries than in the industrialized world, and appears to be higher in rural areas than in urban areas. Furthermore, endemic infections may be responsible for the increased incidence in low-income countries.
Maureen Durkin discussed ostensibly preventable or controllable infections that are important causes of childhood cognitive disability, paralysis, epilepsy, blindness, and deafness in developing countries. These infections include congenital disorders, such as syphilis, rubella, and cytomegalovirus, as well as infections occurring during infancy and childhood, such as malaria, meningitis, Japanese viral encephalitis, measles, poliomyelitis, and trachoma.
Eduardo Gotuzzo described clinical experience with HTLV-1, a retrovirus that causes adult T-cell leukemia and is endemic in much of Latin America.The virus produces 3 different clinical patterns: cancer, autoimmune disease, and immunosuppression disease. In developing countries, 80 percent of lymphomas are non-Hodgkins lymphoma, and 10 pecent of the non-Hodgkins lymphomas seen by the Peruvian national cancer center are associated with HTLV-1. A second clinical presentation is tropical dysplastic paraparesia (TSP). The third clinical pattern associated with the infection is immunosuppression.
Sanaa Kamal described chronic hepatitis C infection with and without schistosomiasis. Patients typically present in their thirties or forties with gastrointestinal bleeding, usually massive, and compromised liver function and status. These patients progress rapidly to end stage disease, usually dying in their forties. Coinfected individuals have significantly higher fibrosis levels and are unable to achieve spontaneous viral clearance.
Altaf Lal described interactions between the human immunodeficiency virus (HIV) and malaria to illustrate how different pathogens interact with each other and how they modulate the disease process. Infant mortality is higher in babies born to mothers who are infected with placental malaria and HIV-1, and these infants have lower levels of acquired passive immunity. Concurrent infections also promote pathogen diversity. The interactions, however, are extremely complex. For example, acute measles suppresses HIV replication significantly.
POTENTIAL LONG-TERM CONSEQUENCES OF EARLY CHILDHOOD ENTERIC AND PARASITIC INFECTIONS*
Richard L. Guerrant, M.D.; Aldo A.M. Lima, M.D., Ph.D.; Sean R. Moore, M.S.; Breyette Lorntz, M.S.; and Peter Patrick, Ph.D.
Center for Global Health, University of Virginia School of Medicine; and Federal University of Ceara, Fortaleza, Brazil
The assessment of the global burden of diseases is increasingly important in recognizing and analyzing their importance as well as the priority of economic investments in their amelioration. In this perspective we recognize the quality of life or years lived with varying degrees of disability in addition to the quantity of life lost to premature mortality, as important outcomes or consequences of all diseases or conditions. Recognizing disability or quality of life is especially important, as mortality from a growing list of acute diseases is reduced, and chronic diseases or long-term consequences of diseases or conditions are now being appreciated. Only such a global view can begin to capture the full human and economic costs of diseases, injuries, or other conditions. Only as these true costs are appreciated, can we affect the necessary investments in their alleviation (Guerrant, 2001; Guerrant and Blackwood, 1999).
Importance of Measuring Morbidity as well as Mortality
Major advances have been made in understanding the quality and quantity elements of health outcomes and the global burdens of disease. Two of these “quality of life” measures are Quality-Adjusted Life Years (QALYs) and Disability-Adjusted Life Years (DALYs). QALYs have been devised by economists to capture both quality and quantity elements of a health care outcome in a single measure, and have been used primarily in assessing the effectiveness of specific interventions to improve health. However, QALYs suffer problems of subjective value assignments that vary considerably with who makes the choices, and they do not capture wider benefits (externalities) that may accrue to society, family, or friends.
DALYs involve not only calculating age-specific mortality (as years of potential life lost [YPLL] to fatal conditions) but also taking into account the quality of life affected by disabilities (by formulating years lost to disability [YLD] with nonfatal conditions, injuries, and diseases) (Murray et al., 1994; Murray and Lopez, 1997). In calculating DALYs, perfect health is weighted as 0 disability with disability weights progressing to 1, the equivalent of death. DALYs have the
advantages that they can also help assess effectiveness of interventions as well as the burden of disease and are standardized to permit age weighting and comparability across studies.
All conditions affecting health as well as interventions that prevent or reverse the adverse effects of these conditions are measured in economic as well as human terms. These include, in addition to the causes of death and the YPLL due to premature mortality, the morbidity costs or YLD from conditions that impair the ability of individuals to reach their full human and economic potential or productivity. As causes of premature mortality are brought under control worldwide, the morbidity costs are becoming increasingly recognized and their quantitation is increasingly important. Thus, in addition to diseases or conditions like meningitis, AIDS, or automobile accidents that are often fatal at young ages and are thus responsible for disproportionately greater years of life lost, we must also weigh the burden of chronic diseases, like arthritis or depression, that often disable much more than they kill. Both YPLL and YLD are included in the DALYs that are being used to assess the burdens of all diseases or conditions that threaten healthy life worldwide, as well as the “cost-effectiveness” of interventions designed for their amelioration. Both mortality (YPLL) and morbidity (YLD) pose profound economic costs, whether a young, productive working parent dies with AIDS or violence, or whether a child with repeated bouts of diarrhea, parasitic infection, or malnutrition fails to develop normally to meet his or her full human and economic potential.
It is just such an analysis that has brought appropriate attention to conditions like neuropsychiatric diseases or depression that kill few but disable many. Likewise, from placebo-controlled prospective studies of albendazole treatment of helminthic infections in Kenyan and Jamaican schoolchildren, intestinal helminths have been found to impair growth, fitness, and even cognitive function (Adams et al., 1994; Nokes et al., 1992a,b; Nokes and Bundy, 1992; Stephenson et al., 1993). Such studies have enabled Chan and Bundy to suggest potential recalculation of the long-term impact of childhood helminthic infections on DALYs to essentially double their previous values (Chan et al., 1994; Guerrant and Blackwood, 1999).
Indeed, the disability component of the DALY calculations for malnutrition and the “tropical cluster” (trypanosomiasis, Chagas’ disease, schistosomiasis, and leishmaniasis), like neuropsychiatric conditions, chronic obstructive lung disease, and rheumatoid arthritis, outweigh their mortality components (Guerrant and Blackwood, 1999; Murray and Lopez, 1997). However, the initially calculated DALY for diarrheal diseases, from a 1997 assessment (Murray and Lopez, 1997), initially comprised 95 percent mortality (YPLL) and only 5 percent disability (YLD, from the transient 10 percent incapacitation during just the overt diarrheal illness [i.e., liquid stools] itself). No long-term disability from repeated dehydrating and malnourishing diarrheal illnesses in the critical formative developmental
first 2 years of life is considered, largely because there had been no data to suggest such long-term effects (Guerrant and Blackwood, 1999).
Potential Long-Term Morbidity from Diarrheal Disease
The challenge is to obtain data implicating specific diseases or conditions with long-term impaired outcomes. Best studied perhaps are nutritional effects that may even involve genetic “imprinting” from the regulation of critical developmental genes at pivotal times by DNA methylation, that might further extend the developmental impact of early childhood illnesses perhaps even beyond 2–3 generations (Golden, 1994). In addition, iron deficiency has a well recognized impact on cognitive development (Basta et al., 1979; Soewondo et al., 1989). Nevertheless, despite the lack of a specific single drug (like albendazole for intestinal helminths) to control diarrheal diseases, long-term cohort studies are now enabling associations to be made of heavy early childhood disease burdens with later functional as well as nutritional outcomes. The growing evidence for lasting disability consequences of early childhood diarrhea and specific parasitic infections (including cryptosporidiosis and intestinal helminthic infections in the first 6–24 months of life) is presented in Table 2-1.
Perhaps one of the greatest of all overlooked costs of the diseases of poverty, such as diarrhea and intestinal parasitic infections, are the increasingly recognized, long-term developmental impact of early childhood illnesses, so common in developing areas. For example, we are now learning that the 4–8 dehydrating, malnourishing diarrheal illnesses that often occur each year in the critically formative first two years of life may have profound, lasting consequences for impaired fitness, growth, cognitive development, and school performance several years later. Initial studies in Northeast Brazil show reduced fitness 4 to 6 years later associated with early childhood diarrhea, and specifically with cryptosporidial infections in the first 2 years of life, independent of respiratory illnesses, anthropometry, anemia, and intestinal helminths (Guerrant and Blackwood, 1999). The fitness deficits alone that associate with the median diarrhea burdens in the first 2 years of life in these studies in Northeast Brazil are comparable to that associated with a 17 percent decrement in work productivity in Zimbabwe sugarcane workers (Guerrant et al., 1999; Ndamba et al., 1993).
Furthermore, these early childhood diarrheal illnesses and intestinal helminthic infections in the first 2 years of life independently and additively associate with substantial long-term linear growth shortfalls that continue beyond six years of age (totaling an average of 8.2 cm [3 1/4 inches] growth shortfall at 7 years old, 3.6 cm with diarrhea alone after controlling for early childhood intestinal helminthic infections) (Moore et al., 2001). In addition, longitudinal studies in Peru (Checkley et al., 1997, 1998) have also shown that cryptosporidial infections (even without overt diarrhea) in young or stunted children predispose to an average 1 cm growth shortfall 1 year after infection.
TABLE 2-1 Evidence for Lasting Disability Effects from Early Childhood Diarrhea
|
Disease |
Outcome |
References |
|
Growth shortfalls |
||
|
Cryptosporidial infections and persistent diarrhea |
Increased diarrhea morbidity and nutritional shortfalls for up to 18 months |
Agnew et al., 1998 Lima et al., 2000 Newman et al., 1999 |
|
Cryptosporidial infections at < 6 months of age and in stunted children |
0.95–1.05 cm growth deficits at 1 year later |
Checkley et al., 1998 |
|
Early childhood diarrhea (0–2 y.o.) |
Lasting growth shortfalls, persisting at 3.6 cm at 7 y.o. (additive to 8.2 cm with intestinal helminths at 0–2 y.o.) |
Moore et al., 2001 |
|
Fitness impairment |
||
|
Early childhood diarrhea (0–2 y.o.) |
Impaired fitness scores (assessed by the Harvard Step Test, HST) 4–7 years later (by 4–8.2 percent for median and high diarrhea burdens, respectively; for comparison, fitness scores improved 6.9 percent 4 months after albendazole treatment of schoolboys in Kenya and a 4.3 percent increase in HST scores correlated with a 16.6 percent increase in work productivity in sugarcane cutters in Zimbabwe |
Stephenson et al., 1993 Guerrant et al., 1999 Ndamba et al., 1993 |
|
Cognitive impairment Early childhood diarrhea (0–2 y.o.) |
Impaired cognitive function at 6–9 y.o. by McCarthy Draw-A-Design (p = 0.017 when controlling for early childhood helminthic infections), and WISC coding and reverse digit span testing (p = 0.045) |
Guerrant et al., 1999 |
We also find significant associations of early childhood diarrhea with long-term cognitive deficits (by standard “Test of Nonverbal Intelligence” [TONI]) even when controlling for maternal education, breast feeding duration, and early helminthic infections (Niehaus et al., 2002). Furthermore, WISC (Wechsler Intelligence Scale for Children; The Psychological Corp, San Antonio, TX) coding and digit span scores were lower in children with persistent diarrheal illnesses in their first 2 years of life, even when controlling for helminths and maternal education (Niehaus et al., 2002). And these effects are seen in a “best case” scenario in which we have documented substantial improvements in disease rates and in nutritional status over the several years in which we have conducted close, long-term surveillance of this population (Moore et al., 2000), effects that we have subsequently not found in other nearby shantytown communities that had not
|
Disease |
Outcome |
References |
|
Early childhood diarrhea (0–2 y.o.) |
Impaired Test of Nonverbal Intelligence (TONI-III) scores at 6–10 y.o., when controlling for maternal education, breast feeding duration, and early helminthic infections; and WISC coding and digit span scores were lower in children who had one or more persistent diarrheal illnesses in their first 2 years of life. |
Niehaus et al., 2002 |
|
School performance (increased age at starting school and age-for-grade) |
||
|
Early childhood diarrhea |
Delayed age at starting school and older age-for-grade, independent of maternal education, socioeconomic status, other illnesses and of also significant effects (of ECD) on height for age Z scores (i.e., stunting) at 0, 2, or 7 years of age (p < 0.02, N = 77). Late starters also are 2-fold more likely to have experienced cryptosporidial infections in their first 2 years of life. |
Lorntz et al., 2000 |
been under such intensive surveillance (Lima, Guerrant et al., unpublished observations).
We are now finding that these correlations of early childhood diarrhea are also extending to school performance, with significant associations of diarrhea in the first 2 years of life with delayed age at starting school and age for grade that remain even after controlling for maternal education and (also affected) stature. Late starters are also two-fold more likely to have experienced cryptosporidial infections (Lorntz et al., 2000).
A recent report describes the significant associations of stunting in the first 2 years of life and multiple episodes of Giardia infection with impaired intelligence quotients on the WISC-R test among children in Peru (Berkman et al., 2002). This is the setting in which diarrhea is also associated with reduced WISC-R scores albeit not independently of its association with stunting. This is also the
setting in which cryptosporidial infections are associated with persistent stunting as well (Checkley et al., 1997, 1998).
Most recently we have launched studies of sensitive measures of higher order frontal lobe development and critical “executive” functioning that predict functional recovery from brain injury in children. We conducted semantic and phonetic fluency testing among 74 children who have now reached 6–12 years old from our prospective surveillance population (with their diarrheal illnesses recorded from birth). Early childhood diarrhea, whether measured by total numbers of episodes or as days of diarrhea in the first 2 years of life was a highly significant predictor of total fluency scores at 6–12 years of age (i.e., 4–10 years later). Impressively, early childhood diarrhea remained a significant predictor of fluency even when controlling for maternal education and for household income (p = 0.02; beta = –0.31)1 or when controlling for birth size (p = 0.007; beta = –0.325) or height-for-age Z score (HAZ) at 6.5 years old. Since early childhood diarrhea has such profound effects on TONI III scores and on HAZ at age 2 years old, its association with fluency was not significantly independent of TONI III or HAZ at 2 years old. The persistence of strong associations of early diarrhea with fluency to 6–12 years old and its independence of HAZ at birth and at 6.5 years old (despite persistent associations of diarrhea with HAZ to 6–7 years old) suggests that despite the growth effects recovering in part, the lasting impact of early childhood diarrhea does not recover and is even greater on functional verbal fluency than on growth. We conclude that the higher frontal lobe executive functioning impairment seen at 6–12 years old associated with diarrhea in the first 2 years of life, especially with impaired schooling, growth and cognition, suggest that early childhood diarrhea results in critical neurodevelopmental impairment that greatly magnifies the importance of ameliorating these diarrheal illnesses and their long-term consequences.
These potential consequences of early childhood malnourishing and dehydrating diarrheal illnesses should not be a great surprise when one considers the importance of early childhood years in human brain development (Dobbing, 1985; 1990; Dobbing and Sands, 1985; Niehaus et al., 2002). Unlike other species such as monkeys, sheep or opossums, which have most of their brain development in utero, it is during the first 2 years of life in humans that the major brain growth and synapse formation occurs. Furthermore, if impaired at this formative stage, it is apparently difficult if not impossible to compensate or build these synapses later in life. Add to this the recognized potential for genetic imprinting noted above, and the duration of impact of early childhood illnesses may well be lifelong and even extend even to the next generation(s).
Thus the disability impact and ultimate societal costs of these early child-
hood diarrheal illnesses of poverty is potentially far greater and more critical a global investment than is generally appreciated, i.e., a global “tax” that is paid for the impaired work productivity in the global economy because these largely preventable illnesses continue unabated. Thus, beyond their obvious human toll, the diseases of poverty may well require an economic investment (as they are readily prevented) that we cannot afford not to make.
Persistent High Diarrhea Morbidity Despite Improving Mortality
The importance of an accurate assessment of the YLD, years lost to disability from early childhood illnesses like diarrheal diseases is further accentuated by the striking relative shift from mortality to morbidity seen over recent decades. Despite clear reductions in diarrhea mortality (from 4.2 to 3.3 to 2.5 million) from 1955 to the present (Bern et al., 1992; Kosek et al., 2003; Snyder and Merson, 1982), the morbidity rates from a third 10-year update review (Kosek et al., 2003) have not decreased; instead, with the fastest growing populations occurring in the poorest areas with the highest disease rates, the total global morbidity from diarrhea has actually substantially increased. The potential impact of these still common early childhood diarrheal illnesses on long-term development or disability only further adds to their morbidity costs.
Refining DALYs for Diarrheal Disease
As shown in the first row of Table 2-2, following the standard formulas with age-weighting and discounting at 3 percent, and all disability falling into the lowest class (weight of 0.096), the DALY calculations for diarrheal diseases are presented.
The morbidity in 0–4 year olds is presented in 5 different scenarios as follows:
-
Scenario 1 applies the original assumptions by Murray and Lopez of 2.27 million attacks of 1 week duration, in which the 1.3 million DALYs from morbidity in 0–4 year olds represents 1 percent of the total of 100.9 million global diarrhea DALYs.
-
Scenario 2 assumes that 17 percent of 0–4 year olds (or 33 percent at half the 9.6 percent disability weight) are at risk of at least 1 diarrheal attack (or a diarrhea burden) which could have life-long disability (with a life expectancy of 81.25 years as used by Murray and Lopez).
-
Scenario 3 assumes that 25 percent of 0–4 year olds (or 50 percent at half the 9.6 percent disability weight) are at life-long risk.
-
Scenario 4 assumes that 10 percent of 0–4 year olds (or 50 percent at 20 percent of the 9.6 percent disability weight, i.e., half experience a 2 percent lifelong disability).
TABLE 2-2 Revised Calculations of Disability-Adjusted Life Years (DALYs) for Diarrheal Diseases
|
Scenario |
Attack rate/year |
Proportion disabled |
Duration of disability |
DALYS for morbidity in 0–4 year olds (millions) [percentage of total] |
Total DALYs (millions) |
|
|
1 |
3.6 |
1 |
0.02 |
1.3 |
[1] |
100.9 |
|
2 |
1 |
0.17 |
81.25 |
351.7 |
[78] |
451.3 |
|
3 |
1 |
0.25 |
81.25 |
517.2 |
[84] |
616.8 |
|
4 |
1 |
0.10 |
81.25 |
215.2 |
[68] |
314.8 |
|
5 |
1 |
0.05 |
81.25 |
107.6 |
[52] |
207.2 |
-
Scenario 5 assumes that only 5 percent of 0–4 year olds (or half experience a 1 percent life-long disability).
Thus, a 1 to 4.8 percent disability affecting one-third to one-half of 0–4-year-old children would increase the total global diarrhea DALYs to 2 to 6-fold the current estimates. Considered differently, for every 5 percent of children affected lifelong, DALYs increase by about 100 million; 25 percent of children affected would increase current DALY estimates by over six-fold; only 5 percent affected lifelong (or 10 percent affected for only 25 years) would more than double the total global diarrhea DALYs (Guerrant et al., 2002a).
Add to this the concept that even subclinical enteric infections that may alter critical absorptive function without necessarily producing overt symptoms of liquid stools, like those with Cryptosporidium or enteroaggregative E. coli may impair growth (Checkley et al., 1997, 1998; Steiner et al., 1998), or impede the absorption of (and potentially thus enhance resistance to) key anti-HIV or antituberculosis drugs (Lima et al., 1997; Brantley et al., 2003), and the potential cost of these diseases of poverty, inadequate water, and inadequate sanitation become increasingly unacceptable.
Conclusions
Critical to understanding and making this case for investing adequate resources in the presentation or amelioration of the diseases of poverty like diarrhea is obtaining solid information about the potential long-term correlates with illness rates and even subclinical infections, controlling to the extent possible the numerous confounding variables, and careful studies of potential interventions that could alter these adverse outcomes. Only improved data and careful, accurate analyses will direct adequate attention to alleviation of these diseases of poverty that are so potentially costly to human and societal development for us all.
REFERENCES
Adams EJ, Stephenson LS, Latham MC, Kinoti SN. 1994. Physical activity and growth of Kenyan school children with hookworm, Trichuris trichiura and Ascaris lumbricoides infections are improved after treatment with albendazole. Journal of Nutrition 124:1199–1206.
Agnew DG, Lima AA, Newman RD, Wuhib T, Moore RD, Guerrant RL, Sears CL. 1998. Cryptosporidiosis in northeastern Brazilian children: association with increased diarrhea morbidity. Journal of Infectious Diseases 177:754–760.
Basta SS, Soerkirman, D Karyadi, NS Scrimshaw. 1979. Iron deficiency anaemia and the productivity of males in Indonesia. The American Journal of Clinical Nutrition 32: 916–925.
Berkman DS, Lescano AG, Gilman RH, Lopez SL, Black MM. 2002. Effects of stunting, diarrhoeal disease, and parasitic infection during infancy on cognition in late childhood: a follow-up study. Lancet 359:564–571.
Bern C, Martines J, de Zoysa I, Glass RI. 1992. The magnitude of the global problem of diarrhoeal disease: a ten-year update. Bulletin of the World Health Organization 70:705–714.
Brantley RK, Williams KR, Silva TM, Sistrom M, Thielman NM, Ward H, Lima AA, Guerrant RL. 2003. AIDS-associated diarrhea and wasting in Northeast Brazil is associated with subtherapeutic plasma levels of antiretroviral medications and with both bovine and human subtypes of Cryptosporidium parvum. Brazilian Journal of Infectious Diseases 7:16–22.
Chan MS, Medley GF, Jamison D, Bundy DA. 1994. The evaluation of potential global morbidity attributable to intestinal nematode infections. Parasitology 109:373–387.
Checkley W, Gilman RH, Epstein LD, Suarez M, Diaz JF, Cabrera L, Black RE, Sterling CR. 1997. Asymptomatic and symptomatic cryptosporidiosis: their acute effect on weight gain in Peruvian children. American Journal of Epidemiology 145:156–163.
Checkley W, Epstein LD, Gilman RH, Black RE, Cabrera L, Sterling CR. 1998. Effects of Cryptosporidium parvum infection in Peruvian children: growth faltering and subsequent catchup growth. American Journal of Epidemiology 148:497–506.
Dobbing J. 1985. Infant nutrition and later achievement. The American Journal of Clinical Nutrition 41:477–484.
Dobbing J. 1990. Boyd Orr memorial lecture. Early nutrition and later achievement. Proceedings of the Nutrition Society 49:103–118.
Dobbing J and Sands J. 1985. Cell size and cell number in tissue growth and development. An old hypothesis reconsidered. Archives Francaises de Pediatrie 42:199–203.
Golden MH. 1994. Is complete catch-up possible for stunted malnourished children? European Journal of Clinical Nutrition 48:S58–S70.
Guerrant DI, Moore SR, Lima AA, Patrick PD, Schorling JB, Guerrant RL. 1999. Association of early childhood diarrhea and cryptosporidiosis with impaired physical fitness and cognitive function four-seven years later in a poor urban community in northeast Brazil. The American Journal of Tropical Medicine and Hygiene 61:707–713.
Guerrant RL. 2001. The unacceptable costs of the diseases of poverty. Current Infectious Disease Reports 3:1–3.
Guerrant RL and Blackwood BL. 1999. Threats to global health and survival: the growing crises of tropical infectious diseases—our “unfinished agenda.” Clinical Infectious Diseases 28:966–986.
Guerrant RL, Kosek M, Lima AA, Lorntz B, Guyatt HL. 2002a. Updating the DALYs for diarrhoeal disease. Trends in Parasitology 18:191–193.
Guerrant RL, Kosek M, Moore S, Lorntz B, Brantley R, Lima AA. 2002b. Magnitude and impact of diarrheal diseases. Archives of Medical Research 33:351–355.
Kosek M, Bern C, Guerrant RL. 2003. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bulletin of the World Health Organization 81:197–204.
Lima AA, Silva TM, Gifoni AM, Barrett LJ, McAuliffe IT, Bao Y, Fox JW, Fedorko DP, Guerrant RL. 1997. Mucosal injury and disruption of intestinal barrier function in HIV-infected individuals with and without diarrhea and cryptosporidiosis in northeast Brazil. American Journal of Gastroenterology 92:1861–1866.
Lima AA, Moore SR, Barboza MS, Soares AM, Schleupner MA, Newman RD, Sears CL, Nataro JP, Fedorko DP, Wuhib T, Schorling JB, Guerrant RL. 2000. Persistent diarrhea signals a critical period of increased diarrhea burdens and nutritional shortfalls: A prospective cohort study among children in northeastern Brazil. Journal of Infectious Diseases 181:1643–1651.
Lorntz et al. 2000. Presentation at the ASTMH.
Moore SR, Lima AA, Schorling JB, Barboza MS Jr., Soares AM, Guerrant RL. 2000. Changes over time in the epidemiology of diarrhea and malnutrition among children in an urban Brazilian shantytown, 1989 to 1996. International Journal of Infectious Diseases 4:179–186.
Moore SR, Lima AA, Conaway MR, Schorling JB, Soares AM, Guerrant RL. 2001. Early childhood diarrhoea and helminthiases associate with long-term linear growth faltering. International Journal of Epidemiology 30:1457–1464.
Murray CJ and Lopez AD, eds. 1997. The Global Burden of Disease: A Comprehensive Assessment of Mortality and Disability from Diseases, Injuries, and Risk Factors in 1900 and Projected to 2020. Cambridge, MA: Harvard University Press.
Murray CJ, Lopez AD, Jamison DT. 1994. The global burden of disease in 1990: summary results, sensitivity analysis and future directions. Bulletin of the World Health Organization 72:495–509.
Ndamba J, Makaza N, Munjoma M, Gomo E, Kaondera KC. 1993. The physical fitness and work performance of agricultural workers infected with Schistosoma mansoni in Zimbabwe. Annals of Tropical Medicine & Parasitology 87:553–561.
Newman RD, Sears CL, Moore SR, Nataro JP, Wuhib T, Agnew DA, Guerrant RL, Lima AA. 1999. Longitudinal study of Cryptosporidium infection in children in northeastern Brazil. Journal of Infectious Diseases 180:167–175.
Niehaus MD, Moore SR, Patrick PD, Derr LL, Lorntz B, Lima AA, Guerrant RL. 2002. Early childhood diarrhea is associated with diminished cognitive function 4 to 7 years later in children in a northeast Brazilian shantytown. The American Journal of Tropical Medicine and Hygiene 66:590–593.
Nokes C and Bundy DA. 1992. Trichuris trichiura infection and mental development in children. Lancet 339:500.
Nokes C, Grantham-McGregor SM, Sawyer AW, Cooper ES, Bundy DA. 1992a. Parasitic helminth infection and cognitive function in school children. Proceedings of the Royal Society of London. Series B Biological Sciences 247:77–81.
Nokes C, Grantham-McGregor SM, Sawyer AW, Cooper ES, Robinson BA, Bundy DA. 1992b. Moderate to heavy infections of Trichuris trichiura affect cognitive function in Jamaican school children. Parasitology 104:539–547.
Snyder JD and Merson MH. 1982. The magnitude of the global problem of acute diarrhoeal disease: a review of active surveillance data. Bulletin of the World Health Organization 60:605–613.
Soewondo S, Husaini M, Pollitt E. 1989. Effects of iron deficiency on attention and learning processes in preschool children: Bandung, Indonesia. The American Journal of Clinical Nutrition 50:667–673.
Steiner TS, Lima AA, Nataro JP, Guerrant RL. 1998. Enteroaggregative Escherichia coli produce intestinal inflammation and growth impairment and cause interleukin-8 release from intestinal epithelial cells. Journal of Infectious Diseases 177:88–96.
Stephenson LS, Latham MC, Adams EJ, Kinoti SN, Pertet A. 1993. Physical fitness, growth and appetite of Kenyan school boys with hookworm, Trichuris trichiura and Ascaris lumbricoides infections are improved four months after a single dose of albendazole. Journal of Nutrition 123:1036–1046.
INFECTIOUS AGENTS AND EPILEPSY
Josemir W. Sander, M.D., Ph.D., M.R.C.P.
Department of Clinical and Experimental Epilepsy
University College London Institute of Neurology, and WHO Collaborative Centre for Research and Training in Neurosciences London, United Kingdom
Epilepsy is the tendency to have unprovoked epileptic seizures. Anything causing structural or functional derangement of the cortical physiology may lead to seizures and different conditions may express themselves solely by recurrent seizures and thus be labelled “epilepsy.” The semiology of seizures and the consequences for the sufferers are, however, similar and therefore epilepsy could be better described as a symptom complex or a condition rather than a disease on its own right (Sander, 2003).
Throughout the world, epilepsy is the most common serious neurological condition (Bergen, 1998). In high-income economies its incidence is around 50 per 100,000/year (range 40 to 70 per 100,000/year) and socioeconomically deprived people are at higher risk (Heaney et al., 2002). In low income countries incidence is generally quoted as between 100 and 190 cases per 100,000/year (Sander, 2003). Most large-scale studies have reported prevalence rates for active epilepsy between 4 and 10/1,000; many of these studies, particularly in low-income countries, have reported different rates for urban and rural areas, usually with higher rates in the latter (Sander and Shorvon, 1996). No clear explanation has been advanced for these differences. It is estimated that worldwide there are at least 50 million sufferers from epilepsy, the great majority of whom are in low-income countries (Scott et al., 2001). The overall prognosis for seizure control is quite good if epilepsy is treated. Epilepsy does, however, carry an increased mortality, particularly if untreated (Cockerell et al., 1994; Sander, 2003).
The range of risk factors for the development of epilepsy varies with age and geographic location (Sander, 2003). Congenital, developmental and genetic conditions are mostly associated with the development of epilepsy in childhood, adolescence and early adulthood. Head trauma, infections of the central nervous system and tumours may occur at any age and may lead to the development of epilepsy. Infections of the central nervous system have one of the highest risks for causing epilepsy (Hauser and Annegers, 1991; Annegers et al., 1996; Bittencourt et al., 1999). For instance, over three-quarters of the survivors of cerebral abscess develop severe epilepsy and survivors of viral encephalitis have an odds ratio of 16.2 for the development of epilepsy (Annegers et al., 1996). In the elderly, cerebrovascular disease is the commonest risk factor and accounts for over half the cases of epilepsy in this age group (Sander, 2003). The presence of a family history of epilepsy seems to enhance other risk factors and this suggests that the aetiology of epilepsy is multifactorial, with genetic predisposition play-
ing a role (Johnson and Sander, 2001). It might be difficult, however, to say whether individuals share predisposition or are exposed to the same environmental sources. In epilepsy due to infections, it could also be argued that the interaction between infective agents and social, genetic, and environmental factors determines the extent of the risk (Bittencourt et al., 1999).
Endemic infections such as malaria, neurocysticercosis and paragonomiasis are associated with epilepsy in certain environments particularly in low-income countries (Sander, 2003). Neurocysticercosis, for instance, is the commonest cause of newly diagnosed epilepsy in large areas of the tropical belt, and malaria is the commonest cause of fever in febrile convulsions in endemic areas (Medina et al., 1990; Waruiru et al., 1996; Carpio, 2002). These infections are probably responsible for the higher incidence of epilepsy in low-income economies and this makes epilepsy one of the world’s most preventable non-communicable conditions (Commission on Tropical Diseases of the International League Against Epilepsy, 1994; Bittencourt et al., 1999; Bergen and Silberberg, 2002). This paper briefly reviews central nervous infections and infestations that may lead to chronic epilepsy. The contribution of social and geographic factors and the putative pathophysiology are discussed in general terms and the natural history of the commonest infections is reviewed. Seizures that occur during the acute phase of an infection are termed acute symptomatic seizures and do not constitute epilepsy even if repeated, and are not covered here.
Social and Geographical Factors
The fact that the incidence of epilepsy seems to be higher in low-income countries is often attributed to social problems in these countries (Commission on Tropical Diseases of the International League Against Epilepsy, 1994; Sander and Shorvon, 1996; Bittencourt et al., 1999). Indeed, poor sanitation and malnutrition are risk factors for infections and these are common in low-income countries. In the past, malaria, schistosomiasis and neurocysticercosis were problems in parts of the high-income countries but improvements in social conditions and basic sanitation have resolved this. In most low-income countries there are inadequate health delivery systems, which results in late or no diagnosis and treatment for infective conditions that would carry a low risk if prompt action were instituted. As a result, neurological disabilities, including seizures, may be higher in survivors of CNS infection in low-income countries than in more developed economies (Bittencourt et al., 1999).
The tropics provide the ideal environment for a number of organisms that may occasionally invade the CNS; most of them would not thrive in colder or temperate climates. Other factors may also play a role: malaria, highly prevalent in endemic coastal areas and lowlands, is non-existent at higher altitudes. Some fungi are restricted to small ecological niches. Other agents exhibit seasonal variation in their infectability. The interaction between infective organism and social,
geographic and environmental factors determine the extent of infection (Bittencourt et al., 1999). There is, however, no objective information on the relative distribution of risk factors or attributable risk for the epilepsies in the community in most of the world and this is an area that requires urgent research (Sander and Shorvon, 1996).
Pathophysiology
Seizures in the aftermath of CNS infectious diseases are usually partial or focal in nature, i.e., they start in the epileptic focus, a localised area of (usually damaged) cortex (Bittencourt et al., 1999). The route of entry of infective agents to the CNS may be arterial—(through the blood-brain barrier or the choroid plexus), by passive venous transport through the spinal plexuses, by direct invasion through trauma or from cranial sinuses. Viruses may enter the CNS by the haematogenous route or via neuronal routes (Eeg-Olofsson, 2003). The infectious agent needs to reach and damage the cerebral cortex for seizures to develop, and this may be achieved through various mechanisms (Bittencourt et al., 1999). Fungal infections are often dependent on the immunological status of the person, and are therefore more prevalent in immunocompromised subjects. Cortical damage will not invariably lead to epilepsy but is a major risk factor affected by the location, severity and individual predisposition, which is likely to be genetically determined (Sander and Shorvon, 1996). There may be months, or even years, between the insult and the onset of epilepsy and the reasons for this are not well understood. The existence of critical modulators, which can turn damaged cortical tissue into an epileptic focus, has been postulated (Walker et al., 2002).
Arteritis, ischaemia and infarction are the main pathological outcome of severe viral or bacterial CNS disease and if this affects the cortical ribbon it may be the substrate for an epileptic focus (Bittencourt et al., 1999). Cerebral malaria may lead to capillary thrombosis, which is probably caused by intravascular aggregates of parasitised erythrocytes in cerebral tissues, particularly in white matter (Molyneux, 2000). Astroglial reaction results in the formation of granulomata and infarcts affecting the cortical ribbon and leading to seizures. Most other protozoan and helminthic infestations of the CNS lead to formation of granulomata, which, if located in cortical tissues, may lead to partial seizures (Bittencourt et al., 1999).
Viral Infections
Among the many viruses that have been associated with the development of encephalitis are arboviruses, coxsackie, rubella, measles, herpes simplex, flavivirus (Japanese encephalitis), and cytomegalovirus. Patients may present with seizures during the acute encephalitic process but more often develop neurological disability, including epilepsy, as a long-term complication (Eeg-Olofsson, 2003).
Herpes simplex virus is the commonest and most severe viral encephalitis in immunocompetent subjects and epilepsy as its aftermath is particularly devastating (Marks et al., 1992).
HIV infections may be complicated by a subacute cortical and subcortical encephalopathy with progressive dementia, myoclunus and tonic-clonic seizures (Modi et al., 2000). Partial seizures in patients with HIV are usually the result of secondary infections with cytomegalovirus, cryptococcus or toxoplasmosis.
Bacterial Infections
Bacterial infections of the CNS usually involve the meninges or cerebral parenchyma and present as either meningitis or cerebral abscess. Acute bacterial meningitis is usually caused by H. influenzae, N. meningitidis, S. pneumoniae or streptococcus infections. Although it may occur in any age group, children are the group more likely to contract bacterial meningitis. Five to ten percent of survivors of acute bacterial meningitis will develop chronic epilepsy and this is usually associated with learning deficits and other neurological disabilities (Marks et al., 1992; Bittencourt et al., 1999; Oostenbrink et al., 2002).
Cerebral abscesses and intracranial empyemas are usually associated with a clear port of entry like sinusitis, otitis media, dental abscess or cardiac valvopathies (Bittencourt et al., 1999). In the majority of cases anaerobic organisms are involved. Epilepsy in the aftermath of a cortical abscess is the rule, and it is usually highly refractory to treatment and often associated with other neurological disabilities. Tuberculosis of the central nervous system may involve the meninges and cerebral parenchyma and is associated with neurological disabilities in a large number of survivors (Bittencourt et al., 1999). Many of these will have partial epilepsy that is often refractory to treatment.
Fungal Infections
Fungal infections of the CNS are rare in immunocompetent subjects, particularly in high-income economies. The fungi are acquired by inhalation of spores that lodge initially in the lungs or paranasal sinuses and may seed to any organ, although with certain topographic preferences depending on the organism (Bittencourt et al., 1999). C. neoformans, C. immitis, H. capsulatum, C. albicans, A. fumigatus and A. flavus, and Mucoraceae sp. are the fungal species most likely to be involved and all of them may eventually provoke seizures.
Protozoal Infections
Plasmodium falciparum and Toxoplasma gondii are associated with epilepsy, although the former is by far the bigger culprit. Cerebral malaria may develop abruptly or subacutely, during systemic uncomplicated, as well as during severe,
falciparum malaria and may have severe consequences. Survivors are at high risk of neurological disabilities including epilepsy (Waruiru et al., 1996; Molyneux, 2000; Versteeg et al., 2003). It is likely that this is responsible for the higher prevalence of epilepsy in endemic area. Intrauterine T. gondii infections are associated with a severe congenital encephalopathy with epilepsy as one of the symptoms. It may also cause seizures in immunocompromised patients. Recently, a suggestion has been made that it may be responsible for many cases of cryptogenic partial epilepsy but this has not been fully elucidated (Stommel et al., 2001).
Helminthic Infestations
A number of helminthic infestations can occasionally reach the CNS and lead to seizures. Taenia solium is probably the commonest of these helminthic infestations but Paragonomiasis westermani, Echinoccocus granulosis, Spargonomiasis mansonoides and Schistosoma japonicum and S. mansoni have also been implicated (Pal et al., 2000; Bittencourt et al., 1999). Recently, suggestions have been made that Toxocara canis could be a major culprit for the higher prevalence of epilepsy in low-income economies (Nicoletti et al., 2002).
Taeniasis and cysticercosis are caused by Taenia solium (Carpio, 2002). They are closely related to poor sanitation, and the coexistence of humans and pigs is a major factor. Humans are the final host for Taenia solium while hogs are the intermediate host. Eating uncooked pork contaminated with taenia cysts will lead to intestinal taeniasis. When humans, instead of pigs, ingest taenia eggs they may become the intermediate host and this may lead to neurocysticercosis. In pigs the cysts tend to lodge in subcutaneous and muscle tissues but in humans there is an attraction for the brain, particularly well irrigated areas like the cortex and the choroidal plexus, Here infestation may lead to epilepsy and other neurological symptoms. Indeed, neurological problems resulting from neurocysticercosis are very common in vast areas of South America, West Africa and Asia (Medina et al., 1990; Bergen, 1998; Sander, 2003). Neurocysticercosis is probably the most preventable form of epilepsy worldwide.
Cerebral hydatidosis is caused by Echinococcus granulosus and occurs in sheep-raising areas. It is acquired mainly by eating food contaminated with dog feces. Epilepsy is a rare complication of this condition (Bittencourt et al., 1999).
Paragonimiasis is a parasitic disease caused by Paragonimiasis westermanii and is common in some endemic areas in the Far East. Like neurocysticercosis, it may be associated with epilepsy when humans become the intermediate host (usually a fish). It is acquired by eating undercooked or raw crab or crayfish (Bittencourt et al., 1999).
A recent report has suggested the possibility of Toxocara canis being the culprit for partial epilepsy in low-income countries (Nicoletti et al., 2002). An odds ratio of 18.2 for the development of late onset epilepsy has been reported in association with positive serology for Toxocara canis. This same study in Bolivia
found an odds ratio for positive serology for Taenia solium of 3.6. This is interesting as, over 30 years ago, Woodruff claimed that dog ownership was a major risk factor for epilepsy, but this was never taken forward (Woodruff et al., 1966). Further studies are urgently needed to clarify this issue.
Conclusion
Much of the existing evidence indicates that epilepsy resulting from infections is a major cause of neurological disability in low-income countries. Indeed, it is probably responsible for the higher incidence of epilepsy in these areas and is the commonest preventable cause of epilepsy worldwide. Improvement in basic sanitation is likely to be crucial to decrease the global burden of epilepsy. Much remains to be done in this area. Studies are urgently needed to elucidate the whole spectrum of attributable risk factors for epilepsy. More research is also needed to understand the molecular basis of all epilepsies particularly the ones caused by infectious agents.
REFERENCES
Annegers JF, Rocca WA, Hauser WA. 1996. Causes of epilepsy: contributions of the Rochester epidemiology project. Mayo Clinic Proceedings 71:570–575.
Bergen DC. 1998. Preventable neurological diseases worldwide. Neuroepidemiology 17:67–73.
Bergen DC and Silberberg D. 2002. Nervous system disorders: a global epidemic. Archives of Neurology 59:1194–1196.
Bittencourt PR, Sander JW, Mazer S. 1999. Viral, bacterial, fungal and parasitic infections associated with seizure disorders. Pp. 145–174 in Handbook of Clinical Neurology, Vol. 72: The Epilepsies, H.Meinardi, ed. Amsterdam: Elsevier Sciences.
Carpio A. 2002. Neurocysticercosis: an update. Lancet Infectious Diseases 2:751–762.
Cockerell OC, Johnson AL, Sander JW, Hart YM, Goodridge DM, Shorvon SD. 1994. Mortality from epilepsy: results from a prospective population-based study. Lancet 344:918–921.
Commission on Tropical Diseases of the International League Against Epilepsy. 1994. Relationship between epilepsy and tropical diseases. Epilepsia 35:89–93.
Eeg-Olofsson O. 2003. Virological and immunological aspects of seizure disorders. Brain and Development 25:9–13.
Hauser WA and Annegers JF. 1991. Risk factors for epilepsy. Epilepsy Research Supplement 4:45–52.
Heaney DC, MacDonald BK, Everitt A, Stevenson S, Leonardi GS, Wilkinson P, Sander JW. 2002. Socioeconomic variation in incidence of epilepsy: prospective community based study in south east England. British Medical Journal 325:1013–1016.
Johnson MR and Sander JW. 2001. The clinical impact of epilepsy genetics. Journal of Neurology, Neurosurgery, and Psychiatry 70:428–430.
Marks DA, Kim J, Spencer DD, Spencer SS. 1992. Characteristics of intractable seizures following meningitis and encephalitis. Neurology 42:1513–1518.
Medina MT, Rosas E, Rubio-Donnadieu F, Sotelo J. 1990. Neurocysticercosis as the main cause of late-onset epilepsy in Mexico. Archives of Internal Medicine 150:325–327.
Modi G, Modi M, Martinus I, Saffer D. 2000. New-onset seizures associated with HIV infection. Neurology 55:1558–1561.
Molyneux ME. 2000. Impact of malaria on the brain and its prevention. Lancet 355:671–672.
Nicoletti A, Bartoloni A, Reggio A, Bartalesi F, Roselli M, Sofia V, Rosado Chavez J, Gamboa Barahona H, Paradisi F, Cancrini G, Tsang VC, Hall AJ. 2002. Epilepsy, cysticercosis, and toxocariasis: a population-based case-control study in rural Bolivia. Neurology 58:1256–1261.
Oostenbrink R, Moons KG, Derksen-Lubsen G, Grobbee DE, Moll HA. 2002. Early prediction of neurological sequelae or death after bacterial meningitis. Acta Paediatrica 91:391–398.
Pal DK, Carpio A, Sander JW. 2000. Neurocysticercosis and epilepsy in developing countries. Journal of Neurology, Neurosurgery, and Psychiatry 68:137–143.
Sander JW. 2003. The epidemiology of epilepsy revisited. Current Opinion in Neurology 16:165–170.
Sander JW and Shorvon SD. 1996. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, and Psychiatry 61:433–443.
Scott RA, Lhatoo SD, Sander JW. 2001. The treatment of epilepsy in developing countries: where do we go from here? Bulletin of the World Health Organization 79:344–351.
Stommel EW, Seguin R, Thadani VM, Schwartzman JD, Gilbert K, Ryan KA, Tosteson TD, Kasper LH. 2001. Cryptogenic epilepsy: an infectious etiology? Epilepsia 42:436–438.
Versteeg AC, Carter JA, Dzombo J, Neville BG, Newton CR. 2003. Seizure disorders among relatives of Kenyan children with severe falciparum malaria. Tropical Medicine and International Health 8:12–16.
Walker MC, White HS, Sander JW. 2002. Disease modification in partial epilepsy. Brain 125:1937–1950.
Waruiru CM, Newton CR, Forster D, New L, Winstanley P, Mwangi I, Marsh V, Winstanley M, Snow RW, Marsh K. 1996. Epileptic seizures and malaria in Kenyan children. Transactions of the Royal Society of Tropical Medicine and Hygiene 90:152–155.
Woodruff AW, Bisseru B, Bowe JC. 1966. Infection with animal helminths as a factor in causing poliomyelitis and epilepsy. British Medical Journal 5503:1576–1579.
CONTROL OF INFECTIOUS CAUSES OF CHILDHOOD DISABILITY IN DEVELOPING COUNTRIES
Maureen Durkin, Ph.D., Dr.P.H.
Department of Population Health Sciences and Waisman Center University of Wisconsin-Madison, Madison, WI
Too often, infectious diseases have been distinguished from chronic diseases, as though these are mutually exclusive categories competing for recognition as a leading public health priority. Nowhere is this view less sustainable than in the field of childhood disability, particularly in developing countries. Worldwide, infections are among the leading causes of chronic, developmental disabilities in children, along with and sometimes interacting with genetic and nutritional causes (Institute of Medicine, 2001). In developing countries today, infections that are ostensibly preventable or controllable continue to be important causes of damage to the developing nervous system resulting in early and long-term cognitive, motor, seizure, hearing, vision, and behavioral disabilities. Infectious causes of developmental disabilities thus take a major and potentially unavoidable toll on the population health and economies of low-income countries today. This paper reviews some of the major infectious causes of develop-
mental disabilities in low-income countries and discusses strategies and inputs needed for their prevention.
Congenital Infections
Numerous prenatal infections can damage the developing nervous system or senses, causing long-term disabilities in children (Levine et al., 2001). The occurrence, nature, and severity of effects vary not only with the type of organism but also often with the timing of the exposure. For example, first or second trimester exposure to toxoplasmosis, cytomegalovirus, and varicella infections may result in a range of impairments recognizable at birth, including microcephaly, hydrocephaly, growth retardation, blindness, seizures, and skin disorders (Remington et al., 1995; Dunn et al., 1999), whereas exposure late in pregnancy or during delivery may result in unapparent infection at birth and onset of developmental delay during infancy or childhood (Koppe et al., 1986).
Congenital Syphilis
The first congenital disability to be linked to an infectious cause (the spirochete Treponema pallidum), congenital syphilis is preventable through routine antenatal screening and treatment with penicillin. As a result, it is now a relatively rare occurrence in developed countries, but in some low-resource settings where routine antenatal care is lacking or where cost barriers prevent access to treatment, recent studies have reported that 4 to 11 percent of births occur to women with positive syphilis tests at delivery (Southwick et al., 2001; Frank and Duke, 2000; Walker and Walker, 2002). The outcomes of congenital syphilis range from fetal and infant death to premature birth, and survival with or without neurological manifestations, which can include deafness, interstitial keratitis, and mental retardation. The most severe outcomes generally result when conception occurs during the early stages of maternal syphilis infection. Outcomes are less severe when conception occurs during the latent state of maternal infection, and clinical manifestations of congenital syphilis are thought to be least severe when onset of maternal infection occurs during the third trimester of pregnancy (Wicher and Wicher, 2001). Animal studies suggest that outcomes may also be modulated by the genetic background of the conceptus (Wicher et al., 1994). Prevention of congenital syphilis requires interventions to reduce the risk of sexual transmission to women of childbearing age, and expansion of antenatal screening and access to treatment. Although the effectiveness and cost-effectiveness of these interventions have been established in developed countries, a recent study in South Africa identified logistical difficulties that prevent timely diagnostic results and access to treatment even when antenatal screening can be accomplished (Beksinska et al., 2002). These difficulties include late presentation for antenatal care, transportation delays that delay access to accurate laboratory results, and
lack of record keeping, tracking mechanisms, and counseling services. Considerations such as these have led some to recommend antibiotic treatment of all pregnant women in selected high risk populations (Walker and Walker, 2002).
Congenital Toxoplasmosis
Congenital toxoplasmosis results from transplacental transmission of infection with the protozoan parasite Toxoplasma gondii following an acute episode of maternal infection during pregnancy. The clinical manifestations can include chorioretinitis, intracranial calcification, necrotizing encephalopathy, microcephaly, cranial nerve palsies, spastic hemi- or quadriparesis, seizures, cognitive disability, and death. Clinical signs may be absent at birth, but infants with congenital toxoplasmosis may develop cognitive and vision disabilities by late childhood. While the risk of transplacental transmission has been found to increase with increasing gestational age, approaching 90 percent during the third trimester, the severity of clinical manifestations appears to decrease with increasing gestational age (Jones et al., 2001). Those previously uninfected are susceptible to acute toxoplasmosis infection through ingestion of raw or inadequately cooked infected meat, contaminated unwashed fruits and vegetables, or oocytes from the feces of infected cats. Although little is known about the frequency of congenital toxoplasmosis in low- and middle-income countries generally, a recent study from Brazil reported a prevalence of 1 per 3,000 live births (Neto et al., 2000), more than three times the rate reported in developed countries (Jara et al., 2001). Evidence of the cost-effectiveness and safety of early detection (via prenatal or newborn screening) and treatment of acute infection with antiparasitics is not consistent or conclusive at this time (Jones et al., 2001; Roizen et al., 1995). Thus, prevention of congenital toxoplasmosis in low-income countries requires further research and perhaps more emphasis on educational programs regarding the risks and specific hygienic precautions that can prevent acute infections during pregnancy.
Congenital Rubella
Congenial rubella leads to a range of adverse pregnancy outcomes or birth defects but only when maternal rubella virus infection occurs within the first 18 weeks of pregnancy. Outcomes include fetal death, spontaneous abortion, stillbirth, premature birth and, among surviving infants, sensorineural deafness, cataracts and other visual impairments, mental retardation, autistic features, cardiac defects, and increased susceptibility to juvenile diabetes and other chronic conditions (Peckham and Newell, 2001). The earlier in gestation that the fetus becomes infected, the greater the likelihood of multiple defects. Although congenital rubella has been nearly eliminated in successfully vaccinated populations and with a very high benefit-to-cost ratio (Plotkin et al., 1999), epidemics continue to oc-
cur in many developing countries (Lawn et al., 2000). Cutts and Vynnycky have concluded from an extensive review of evidence that “Congenital rubella syndrome is an under-recognized public health problem in many developing countries. There is an urgent need for collection of appropriate data to estimate the cost-effectiveness of a potential global rubella control program” (Cutts and Vynnycky, 1999). A difficulty facing developing countries is that vaccination can prevent congenital rubella only if high coverage is maintained. Incomplete vaccine coverage may actually increase the risk of congenital rubella infection by reducing opportunities for natural immunity and increasing the mean age of infection, thus increasing the susceptibility to infection of women of childbearing age (Panagiotopoulos et al., 1999). The availability of a combined measles and rubella vaccine may increase the feasibility of achieving adequate rubella vaccination and improve opportunities to prevent congenital rubella throughout the world (Banatvala, 1998).
Mother-to-Child Transmission of HIV and Herpes Viruses
This is an emerging cause of developmental disabilities in populations where high HIV prevalence among childbearing women is combined with lack of access to antenatal antiretroviral therapy and cesarean delivery, which in combination are highly effective in preventing vertical transmission of HIV (European Mode of Delivery Collaboration, 1999; International Perinatal HIV Group, 1999). The neurodevelopmental effects of pediatric AIDS include microcephaly and significant delays in cognitive and motor development (Belman, 1990; Macmillan et al., 2001). These effects may be greater when transmission of the virus from mother to child occurs in utero or early in gestation versus during parturition (Smith et al., 2000). In developed countries, improvements in postnatal treatment and survival of children with HIV may be associated with a reduction in adverse neurodevelopmental outcomes. One study of HIV-infected children in the US found no detriment in verbal or performance IQ when compared to controls matched on ethnicity and prenatal drug exposure (Fishkin et al., 2000). Estimates are not available of the prevalence of pediatric HIV-associated neurodevelopmental disorders from low-income countries where few infected children have access to antiretroviral therapy. In addition to direct effects of AIDS on the developing nervous system, the AIDS epidemic may increase children’s exposure to social, emotional, and economic deprivation during critical periods of development. Cost-effective and accessible methods of prevention and treatment of HIV in developing countries are needed.
Perinatal transmission of herpes viruses, including cytomegalovirus and Herpes simplex can also result in severe neurodevelopmental disorders (Levine et al., 2001; Peckham et al., 1983), but little is known about their occurrence in developing countries.
Infections Contributing to Perinatal Complications
In addition to infections known to directly damage the developing nervous system or senses, other prenatal and perinatal infections associated with perinatal complications may contribute to developmental disabilities either directly or indirectly (Breslau et al., 1994). Perinatal complications that occur more frequently in the presence of maternal and fetal infections include premature birth, low birth weight, intrauterine growth restriction and asphyxia. For example, maternal malaria infection may result in placental parasitemia and intrauterine growth restriction, as well as maternal anemia and death. Infants born with perinatal complications are often at increased risk for brain and sensory abnormalities and disabilities. For example, retinopathy of prematurity is a leading cause of childhood blindness worldwide (WHO, 2000a), and prematurity is an important risk factor for cerebral palsy and cognitive disabilities in childhood. Yet the role and timing of infections in these disorders are not fully understood (Donders et al., 1993; O’Shea and Dammann, 2000). Many factors may contribute to the elevated frequency of perinatal complications in low-income countries, including the scarcity of resources for obstetrical care and management of complications of labor and delivery, nutritional deficiencies, and increased risk of maternal infections. Research is urgently needed on the role of maternal infections in the etiology of adverse perinatal outcomes; on the causal role of infections in developmental disabilities; and on the impact of infection treatment and control on the prevalence of neurodevelopmental disabilities in low-income countries.
Infections During Infancy and Childhood
Infections acquired during infancy and childhood that continue to cause developmental disabilities among children in low-income countries, where access to prophylaxis and treatment is often limited and delayed, include neonatal infections (Wolf et al., 1999; Durkin et al., 2000) as well as bacterial meningitis, viral encephalitis, measles, poliomyelitis, trachoma, and parasitic conditions such as malaria, neurocysticercosis, and other helminth conditions (Institute of Medicine, 2001).
Malaria and Helminthic Diseases
Malaria is a public health problem in many countries and is estimated to cause hundreds of millions of cases and approximately one million deaths in children each year (WHO, 1998). Repeated episodes of malaria are responsible for poor school attendance and childhood anemia. Cerebral malaria occurs in a percentage of affected children, with major clinical manifestations, including convulsions and coma. Measures to prevent malaria infection include use of protective clothing, insect repellents, insecticide-treated bednets, and environmental
management to control mosquito vectors. Once infection has occurred, chemoprophylaxis is effective against the development of disease. The cost-effectiveness of malaria prophylaxis and treatment programs is well established in populations where malaria is endemic, even without accounting for the potential for long-term neurological deficits in children who survive cerebral malaria. Other parasitic diseases, such as intestinal helminthic diseases, also affect a large proportion of the world’s child population and may adversely affect school performance and cognitive development (Dickson et al., 2000).
Meningitis
Meningitis from major bacterial agents probably occurs more commonly in the developing than in developed countries, though specific data are lacking. Children under age 5 and the elderly are at highest risk. In developing countries, pneumonia is the most common presentation of Haemophilus influenzae Type b meningitis; it has been estimated that this cause of meningitis in developing countries has a case fatality rate of 30 percent and results in permanent nervous system impairment in 20 percent of survivors (WHO, 2001a). Meningococcal meningitis occurs sporadically in developed countries, but major epidemics of the disease occur every several years in sub-Saharan Africa and South America. Case fatality exceeds 50 percent in the absence of early and adequate treatment, and it is estimated that 15 to 20 percent of survivors are left with deafness, seizures, and mental retardation (Levine et al., 1998). Primary prevention of Haemophilus influenzae Type b meningitis can be achieved by means of vaccination of all infants or by chemoprophylaxis following close contact with an affected child. Vaccination is the only practical method of preventing infection on a population level. In developed countries where immunization against this disease during infancy is routine, the incidence of Haemophilus influenzae Type b meningitis has dropped dramatically (Levine et al., 1998). It has been argued that vaccination against Haemophilus influenzae Type b infection is cost-effective in developing countries as well (Levine et al., 1998), but information on the frequency of the disease and its sequelae in developing countries is needed to guide the implementation of control strategies. Epidemics of meningococcal meningitis can be controlled effectively by means of mass immunization campaigns resulting in over 80 percent coverage, while infection in endemic situations can be prevented by chemoprophylaxis administered to close contacts of patients (Levine et al., 1998). Information on the cost-effectiveness of these interventions in developing countries is needed.
Japanese Viral Encephalitis
Japanese viral encephalitis is the leading cause of viral encephalitis in Asia, where it is responsible for at least 50,000 cases of clinical disease each year,
primarily among children (Siraprapasiri et al., 1997). Case fatality is as high as 20 percent, and the frequency of neuropsychiatric sequelae among survivors is thought to be high, though specific data are lacking. Following an infectious mosquito bite, the virus replicates in the lymph nodes, spreads to the central nervous system and propagates in the brain, leading to seizures, cognitive and motor disabilities, and progressive coma (Siraprapasiri et al., 1997). Effective vaccines have been developed against the viral agent causing Japanese encephalitis. One is a mouse-brain derived vaccine that has been incorporated effectively into the national childhood vaccination program of Thailand (Siraprapasiri et al., 1997). The high cost of this vaccine and the potential for serious neurological sequelae, however, are barriers to its widespread use in endemic and epidemic situations (Siraprapasiri et al., 1997).
Measles
Measles is an acute viral disease that is still a leading cause of death worldwide, largely because of its occurrence among children under age 5 in developing countries. Rarely (about 1/1,000 cases), measles infection causes encephalitis, which can result in long-term nervous system sequelae among survivors. While Vitamin A deficiency has been shown to increase the severity of measles infection, it is thought the infection can, in turn, exacerbate Vitamin A deficiency and lead to blindness (Strebel, 1998). Vaccination using live, attenuated measles virus produces long-lasting immunity. Eradication of measles is theoretically feasible, given the effectiveness of available vaccines and the likelihood that humans are the only reservoir capable of sustaining transmission of the measles virus. Widespread vaccination has successfully prevented the spread of measles in a number of developing countries, and is considered one of the most cost-effective public health interventions ever undertaken (Strebel, 1998). However, measles continues to be a major contributor to childhood death and disease worldwide. Global eradication of this cause of developmental disability will require sustained efforts.
Poliomyelitis
Polio was eradicated from the Western Hemisphere, the Western Pacific region, and Eastern Europe following a concerted international initiative (WHO, 2000b). This enteroviral disease, however, continues to threaten children in tropical Africa and to a lesser extent in South and Southeast Asia. Once established in the intestines, poliovirus can enter the blood stream and invade the central nervous system. As it multiplies, the virus destroys motor neurons and leads to irreversible paralysis. Immunization programs have effectively eradicated poliomyelitis from much of the world, but the disease remains endemic in much of sub-Saharan Africa and parts of South and Southeast Asia. Reported immuniza-
tion coverage with the oral polio vaccine is still low in most African countries (WHO, 2001b). Although worldwide eradication of polio as a cause of childhood paralysis can be achieved by vaccination during infancy, meeting this goal will require major commitments that may be difficult to sustain in the face of the decline of the disease in much of the world (WHO, 2001b).
Trachoma and Leprosy
Trachoma is a bacterial disease of the conjunctiva caused by Chlamydia trachomatis (Cook, 1998). Repeated infections, which often begin in childhood, result in blindness in adulthood. Trachoma is endemic in many impoverished areas of the world where access to clean water is compromised. An estimated 5.9 million people worldwide have become blind or are at immediate risk for blindness as a result of trachoma infection (Cook, 1998). Improvements in hygiene, including access to clean water and education to promote frequent face washing, are highly cost-effective in the prevention of blindness due to trachoma (Helen Keller International, 2001). Leprosy is another neglected disease with neurological effects that continues to affect large numbers of children throughout the developing world (Jain et al., 2002).
Conclusions
Children in developed countries have benefited for decades from interventions such as maternal vaccination to prevent congenital rubella, pediatric vaccinations to prevent potentially brain-damaging childhood infections such as Haemophilus influenza Type b, and early detection and effective management of bacterial infections that can lead to meningitis or hearing loss. In addition, antiretroviral therapies have become available in developed countries to prevent pediatric HIV transmission. Unfortunately, cost and attitudinal and logistical barriers prevent these interventions from reaching children at greatest risk in the developing world. Extension of such interventions to low-income countries is a necessary step toward the reduction of international inequalities in child health.
To effectively respond to the impacts of infectious causes of developmental disabilities worldwide, proven methods of prevention must be implemented and expanded within primary health care systems in low-income countries. Specific interventions should be tailored to local epidemiology and resources and needs, and should include vaccination programs with high coverage to prevent conditions such as congenital rubella, bacterial meningitis and poliomyelitis, development of laboratory facilities and networks to facilitate accurate diagnoses, and commitment of resources to prevent other infectious diseases, such as pediatric AIDS, malaria, neurocysticercosis, leprosy, viral encephalitis, and trachoma. Additional recommendations articulated in the Institute of Medicine report on
Neurological, Psychiatric and Developmental Disorders: Meeting the Challenge in the Developing World (Institute of Medicine, 2001) are as follows:
-
Increase training and expertise at all levels of health care, as well as in the educational and research sectors, in the intersection between infectious disease control and child development.
-
Develop and maintain Internet cababilities to facilitate international communication among those involved in the implementation of primary prevention and rehabilitation programs for children with developmental disabilities in low-income countries.
-
In the context of the successes of current primary health care child survival initiatives in low-income countries, it is essential that increased emphasis be placed in low-income countries on prevention and early identification of developmental disabilities within the primary and maternal and child health care systems. Those systems must in turn be linked to and supported by secondary and tertiary medical services, as well as rehabilitation programs.
-
Develop increased capacity for evidence-based research by establishing regional coordinating centers in low-income countries to enable the conduct of clinical and community trials of the effectiveness of interventions to prevent infectious causes of developmental disabilities.
-
Support research on factors that are crucial to understanding how to prevent developmental disabilities in low-income countries, such as the etiology and prevention of adverse perinatal outcomes and the impact of maternal education and alleviation of poverty on the prevention of infections resulting in developmental disabilities.
-
Develop practical methods for surveillance of infections leading to childhood disabilities.
-
Document nervous system sequelae of cerebral malaria and their prevention.
-
Determine the cost-effectiveness of methods for the prevention of prevalent infections that result in developmental disabilities.
REFERENCES
Banatvala JE. 1998. Rubella—could do better. Lancet 351:849–850.
Beksinska ME, Mullick S, Kunene B, Rees H, Deperthes B. 2002. A case study of antenatal syphilis screening in South Africa: successes and challenges. Sexually Transmitted Diseases 29:32–37.
Belman AL. 1990. AIDS and pediatric neurology. Neurologic Clinics 8:571–603.
Breslau N, DelDotto JE, Brown GG, Kumar S, Ezhuthachan S, Hufnagle KG, Peterson EL. 1994. A gradient relationship between low birth weight and IQ at age 6 years. Archives of Pediatric and Adolescent Medicine 148:377–383.
Cook JA. 1998. Trachoma. Bulletin of the World Health Organization 76(Suppl 2):139–140.
Cutts FT and Vynnycky E. 1999. Modelling the incidence of congenital rubella syndrome in developing countries. International Journal of Epidemiology 28:1176–1184.
Dickson R, Awasthi S, Williamson P, Demellweek C, Garner P. 2000. Effects of treatment for intestinal helminth infection on growth and cognitive performance in children: systematic review of randomized trials. British Medical Journal 320:1697–1701.
Donders GG, Desmyter J, De Wet DH, Van Assche FA. 1993. The association of gonorrhoea and syphilis with premature birth and low birthweight. Genitourinary Medicine 69:98–101.
Dunn D, Wallon M, Peyron F, Petersen E, Peckham C, Gilbert R. 1999. Mother-to-child transmission of toxoplasmosis: risk estimates for clinical counselling. Lancet 353:1829–1833.
Durkin MS, Khan NZ, Davidson LL, Huq S, Munir S, Rasul E, Zaman SS. 2000. Prenatal and postnatal risk factors for mental retardation among children in Bangladesh. American Journal of Epidemiology 152:1024–1033.
European Mode of Delivery Collaboration. 1999. Elective caesarean-section versus vaginal delivery in prevention of vertical HIV-1 transmission: a randomized clinical trial. Lancet 353:1035–1039.
Fishkin PE, Armstrong FD, Routh DK, Harris L, Thompson W, Miloslavich K, Levy JD, Johnson A, Morrow C, Bandstra ES, Mason CA, Scott G. 2000. Brief report: relationship between HIV infection and WPPSI-R performance in preschool-age children. Journal of Pediatric Psychology 25:347–351.
Frank D and Duke T. 2000. Congenital syphilis at Goroka Base Hospital: incidence, clinical features and risk factors for mortality. Papua New Guinea Medical Journal 43:121–126.
Helen Keller International. 2001. Trachoma. Available at: http://www.hki.org/programs/trachoma.html.
Institute of Medicine. 2001. Neurological, Psychiatric, and Developmental Disorders: Meeting the Challenge in the Developing World. Washington, DC: National Academy Press.
International Perinatal HIV Group. 1999. The mode of delivery and the risk of vertical transmission of human immunodeficiency virus type 1: a meta-analysis of 15 prospective cohort studies. New England Journal of Medicine 340:977–987.
Jain S, Reddy RG, Osmani SN, Lockwood DN, Suneetha S. 2002. Childhood leprosy in an urban clinic, Hyderabad, India: clinical presentation and the role of household contacts. Leprosy Review 73:248–253.
Jara M, Hsu HW, Eaton RB, Demaria A Jr. 2001. Epidemiology of congenital toxoplasmosis identified by population-based newborn screening in Massachusetts. Pediatric Infectious Disease Journal 20:1132–1135.
Jones JL, Lopez A, Wilson M, Schulkin J, Gibbs R. 2001. Congenital toxoplasmosis: a review. Obstetrical and Gynecological Survey 56:296–305.
Koppe JG, Loewer-Sieger DH, de Roever-Bonnet H. 1986. Results of 20-year follow-up of congenital toxoplasmosis. Lancet 1:254–256.
Lawn JE, Reef S, Baffoe-Bonnie B, Adadevoh S, Caul EO, Griffin GE. 2000. Unseen blindness, unheard deafness, and unrecorded death and disability: congenital rubella in Kumasi, Ghana. American Journal of Public Health 90:1555–1561.
Levine MI, Chervenak FA, Whittle M, eds. 2001. Fetal and Neonatal Neurology and Neurosurgery. London: Churchill Livingston.
Levine OS, Schwartz B, Pierce N, Kane M. 1998. Development, evaluation and implementation of Haemophilus influenzae type b vaccines for young children in developing countries: current status and priority actions. Pediatric Infectious Disease Journal 17 (9 Suppl):S95–113.
Macmillan C, Magder LS, Brouwers P, Chase C, Hittelman J, Lasky T, Malee K, Mellins CA, VelezBorras J. 2001. Head growth and neurodevelopment of infants born to HIV-1 infected drug-using women. Neurology 57:1402–1411.
Neto EC, Anele E, Rubim R, Brites A, Schulte J, Becker D, Tuuminen T. 2000. High prevalence of congenital toxoplasmosis in Brazil estimated in a 3-year prospective neonatal screening study. International Journal of Epidemiology 29:941–947.
O’Shea TM and Dammann O. 2000. Antecedents of cerebral palsy in very low birthweight infants. Clinics in Perinatology 27:285–302.
Panagiotopoulos T, Antoniadou I, Valassi-Adam E. 1999. Increase in congenital rubella occurrence after immunization in Greece: retrospective survey and systematic review. British Medical Journal 319:1462–1467.
Peckham CS and Newell ML. 2001. Viral infections. Pp. 553–572 in Fetal and Neonatal Neurology and Neurosurgery, MI Levine, FA Chervenak, M Whittle, eds. London: Churchill Livingston.
Peckam CS, Chin KS, Coleman JC, Henderson K, Hurley R, Preece PM. 1983. Cytomegalovirus infection in pregnancy: preliminary findings from a prospective study. Lancet 1:1352–1355.
Plotkin SA, Katz M, Cordero JF. 1999. The eradication of rubella. Journal of the American Medical Association 281:561–562.
Remington JS, McLeod R, Desmonts G. 1995. Toxoplasmosis. Pp. 140–267 in Infectious Diseases of the Fetus and Newborn Infant, JS Remington and JO Klein, eds. Philadelphia: WB Saunders.
Roizen N, Swisher CN, Stein MA, Hopkins J, Boyer KM, Holfels E, Mets MB, Stein L, Patel D, Meier P, et al. 1995. Neurologic and developmental outcome in treated congenital toxoplasmosis. Pediatrics 95:11–20.
Siraprapasiri T, Sawaddiwudhipong W, Rojanasuphot S. 1997. Cost benefit analysis of Japanese encephalitis vaccination program in Thailand. Southeast Asian Journal of Tropical Medicine and Public Health 28:143–148.
Smith R, Malee K, Charurat M, Magder L, Mellins C, Macmillan C, Hittleman J, Lasky T, Llorente A, Moye J. 2000. Timing of perinatal human immunodeficiency virus type 1 infection and rate of neurodevelopment. The Women and Infant Transmission Study Group. Pediatric Infectious Disease Journal 19:862–871.
Southwick KL, Blanco S, Santander A, Estenssoro M, Torrico F, Seoane G, Brady W, Fears M, Lewis J, Pope V, Guarner J, Levine WC. 2001. Maternal and congenital syphilis in Bolivia, 1996: prevalence and risk factors. Bulletin of the World Health Organization 79:33–42.
Strebel PM. 1998. Measles. Bulletin of the World Health Organization 76 (Suppl. 2):154–155.
Walker DG and Walker GJ. 2002. Forgotten but not gone: the continuing scourge of congenital syphilis. Lancet Infectious Diseases 2:432–436.
Wicher V and Wicher K. 2001. Pathogenesis of maternal-fetal syphilis revisited. Clinical Infectious Diseases 33:354–363.
Wicher V, Baughn RE, Wicher K. 1994. Congenital and neonatal syphilis in guinea pigs show a different pattern of immune response. Immunology 82:404–409.
Wolf MJ, Wolf B, Beunen G, Casaer P. 1999. Neurodevelopmental outcome at 1 year in Zimbabwean neonates with extreme hyperbilirubinaemia. European Journal of Pediatrics 158:111–114.
World Health Organization. 1998. Malaria: Fact Sheet Number 94. Available at: http://www.who.int/inf-fs/en/fact094.html.
World Health Organization. 2000a. Control of Major Blinding Diseases and Disorders: Fact Sheet Number 214. Available at: http://www.who.int/inf-fs/en/fact214.html.
World Health Organization. 2000b. Global Polio Eradication Initiative. Available at: http://www.polioeradication.org/vaccines/polioeradication/all/partners/default.asp.
World Health Organization. 2001a. Haemophilus Influenzae Type b Disease. [Online] Available at: http://www.who.int/vaccines-diseases/diseases/hib.shtml.
World Health Organization. 2001b. Polio Eradication: Final 1% Poses Greatest Challenge. Available at: http://www.who.int/inf-pr-2001/en/pr2001-17.html.
HTLV-1: CLINICAL IMPACT OF A CHRONIC INFECTION
Eduardo Gotuzzo
Instituto de Medicina Tropical Alexander von Humboldt, Universidad Peruana
Cayetano Heredia
Lima, Peru
and
Kristien Verdonck
Prince Leopold Institute of Tropical Medicine, Antwerp, Belgium
Human T-cell lymphotropic virus type 1 (HTLV-1) was the first human retrovirus to be described. It was discovered simultaneously in the United States and in Japan in 1980 (Poiesz et al., 1980; Hinuma et al., 1981). As documented for all retroviruses, HTLV-1 produces a permanent cell infection. Therefore, all carriers are potential sources of transmission of the infection.
Epidemiology
Geographical Distribution
HTLV-1 has an ubiquitous distribution, with well-described endemic areas. An area is called endemic for HTLV-1 if 2–10 percent of the healthy adult population is infected. The islands of Kyushu and Okinawa, in southwestern Japan, are hyperendemic areas for HTLV-1, 15 percent of the healthy adult population carry the virus (Blattner, 1990). Moderate rates of infection have been reported in West Africa, Australia, and the Caribbean (Caribbean Epidemiology Center, 1990; Delaporte et al., 1989; Nerurkar et al., 1993). In South America, Brazil, Colombia, and Peru are HTLV-1 endemic areas (Zurita et al., 1997; Gabbai et al., 1993; Zaninovic et al., 1994); the virus is also present in Ecuador (Guderian et al., 1994), Paraguay (de Cabral et al., 1995), Chile (Cartier and Cartier, 1996) and Argentina (Bouzas et al., 1994). In Peru, the virus is highly prevalent in some population and ethnic groups. Sixteen percent of immigrants from Japan—particularly from Okinawa—are seropositive. However, in the first generation of these immigrants born in Peru, the virus is prevalent in 4 percent of the population, and is not present in the second generation (Gotuzzo et al., 1996). Similar trends were reported in Hawaii (Blattner et al., 1986) and Bolivia (Tsugane et al., 1988). A study of HTLV-1 infection in asymptomatic women in Peru found prevalence rates of 3.8 percent among Afro-American women in Chincha, a coastal town south of Lima, 1.3 percent among the Quechua population of the central highlands (Ayacucho) and 3.8 percent in the population of northern Lima (Sanchez-Palacios, 2003). In other regions in South America, in which there is a strong presence of African Americans, such as Tumaco (Colombia) and Bahia (Brazil), the prevalence of HTLV-1 ranges from 2–5 percent in the healthy adult population.
Transmission
HTLV-1 is transmitted through modes similar to those described for HIV, but there are also important differences that are explained by the requirement of infected lymphocytes for the transmission of HTLV-1.
Vertical Transmission
Intrauterine transmission of HTLV-1 is very rare, and prolonged breastfeeding seems to be the main risk factor associated with this type of transmission. In Peru, breastfeeding is the most common route of transmission of HTLV-1. In a study of 120 HTLV-1-infected Peruvian women and their offspring, infection was not detected in children who were not breastfed, but was documented in 14 percent of those who received maternal milk for less than 6 months and in 31 percent of those breastfed for more than 6 months (E. Gotuzzo, unpublished data). Moreover, in a hyperendemic area in southwestern Japan, screening pregnant women and abstaining from breastfeeding has been documented to dramatically decrease the prevalence of HTLV-1 (Katamine, 1999). HTLV-1-related disease in mothers may also be associated with the increased risk of transmission of the virus to their children, as suggested by a recent study which found that HTLV-1 is present in 43 percent of children born from mothers with strongyloidiasis, and 20 percent of children born from mothers with tropical spastic paraparesis (p < 0.01). Gender also seemed to be a factor, as HTLV-1 is transmitted to 17 percent of males and to 32 percent of females (p < 0.01) (E. Gotuzzo, submitted for publication).
Parenteral Transmission
HTLV-1 is transmitted less efficiently than HIV in whole blood transfusions. Fresh frozen plasma, which can transmit HIV, has not been associated with the transmission of HTLV-1. In addition, the efficacy of HTLV-1 transmission decreases when blood is stored for more than one week (Okochi et al., 1984). These observations point to the need for viable lymphocytes to establish infection with HTLV-1. Transmission through transfusion of whole blood has been estimated to infect between 50 and 60 percent of recipients (Larson and Taswell, 1988). A national survey in Peru indicated that 1.2 percent of 142,500 blood donors were HTLV-1 seropositive. Epidemiologic studies of the general population in Caribbean countries have consistently shown that the prevalence of HTLV-1 significantly increases with age, is higher in women, specifically in low socioeconomic strata, and correlates with a history of blood transfusion (Murphy et al., 1996). The efficacy of HTLV-1 transmission through needle sharing by intravenous drug users is very low (Gradilone et al., 1986).
HTLV-1 as a Sexually Transmitted Disease (STD)
There are several arguments indicating that HTLV-1 is an STD in Latin America. The virus has been found in semen and cervical secretions of infected people and sexual intercourse is an important factor for HTLV-1 transmission (Tajima et al., 1982). Male-female sexual transmission is more efficient than female-male transmission. Seropositivity is more prevalent in sexual risk groups such as female commercial sex workers (CSW) (Khabbaz et al., 1990) and promiscuous men engaged in homosexual activities (Bartholomew et al., 1987). In such cases, the seropositivity rate is associated with the number of sexual partners, time in prostitution activities, and presence of other STDs. Sexual transmission of HTLV-1 can be significantly reduced by the consistent use of condoms. Surveys among female CSW in Peru have shown rates of infection ranging between 7 and 25 percent (Wignall et al., 1992).
Diseases Associated with HTLV-1
Although HTLV-1 is a life-long retroviral infection, symptoms occur only in a minority of infected subjects. Classical complications include lymphoproliferative disorders, such as Adult T-cell Leukemia/Lymphoma (ATLL) and autoimmune disorders (Tropical Spastic Paraparesis). Both syndromes may occur in 1–5 percent of HTLV-1-infected subjects (Murphy et al., 1989). Several reports have suggested immune-suppression in HTLV-1-positive patients.
Adult T-Cell Leukemia/Lymphoma (ATLL)
In the 1970s, an epidemic of ATLL was described in Japan (Takatsuki et al., 1977)—the striking observation being that this phenomenon occurred in Okinawa and Kyushu and not in northern Japan. In 1980, two groups determined the relationship between HTLV-1 and ATLL; the HTLV-1 provirus is integrated into the neoplastic cell DNA and the virus can be isolated from malignant cells (Seiki et al., 1983). The clinical presentation of ATLL involves fever, lymphadenopathy, hepatosplenomegaly, bone lesions with hypercalcemia, skin lesions, and a fatal course with poor response to chemotherapy. Chronic and smoldering types of leukemia, with more lymphoma-like characteristics, slower courses, and more extensive skin involvement have also been described. Males aged 50–60 years are the group most frequently affected. Studies conducted in the HTLV-1 hyperendemic areas of Japan have estimated a lifetime risk for ATLL of 2–4 percent (Tajima and Hinuma, 1992). In Jamaica, Murphy reported the lifetime risk of ATLL to be 4 percent for those infected with HTLV-1 before 20 years of age (Murphy et al., 1996). In South America, the association between HTLV-1 and ATLL has been recognized in Brazil, Colombia, and in Argentina. Three hundred new cases of non-Hodgkin’s lymphoma are detected at the National Institute of
Cancer in Lima, Peru, each year, and 10 percent (30) of these cases are associated with HTLV-1.
Tropical Spastic Paraparesis
The general term “tropical spastic paraparesis” (TSP) was introduced by Mani et al. in 1969 for a chronic progressive paraparesis of unknown cause observed in tropical areas; however, it was not until 1985 that the association between this syndrome and HTLV-1 infection was recognized (Gessain et al., 1985). The designation HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) was proposed in 1989.
It has been reported that TSP occurs in 1–4 percent of people infected with HTLV-1 (Kaplan et al., 1990). TSP predominantly affects adult females, an observation that is also confirmed in WHO’s guidelines for diagnosis of TSP. The risk of TSP appears to be higher in Latin America than in Japan and associations have been reported between TSP and certain HLA alleles (Jeffrey et al., 1999). Furthermore, the association between HTLV-1 and TSP varies between geographical regions; in Colombia, 87 percent of TSP cases were HTLV-1-seropositive and in Peru, 55–65 percent of TSP patients were carriers of HTLV-1. On the other hand, in a study in Mexico, less than 1 percent of TSP cases were found to carry HTLV-1 (unpublished data, J. Sotelo, Inst Nal Neurol, Mexico City). These observations suggest that genetic background can influence susceptibility to TSP, and that HTLV-1 and other cofactors, that remain unknown, are involved in the pathogenesis of this disease.
An autoimmune mechanism has been proposed to explain the pathogenesis of TSP. According to this hypothesis, cytokines, such as tumor necrosis factor-α, are released against viral proteins in the surface of infected lymphocytes thereby causing chronic inflammation and tissue damage within the thoracic middle portion of the spinal cord.
The clinical symptoms of TSP consist of a gradually appearing symmetrical paraparesis with signs of pyramidal tract involvement that usually progresses slowly and relentlessly. However, some patients present rapid progression of the neurological symptoms; in these patients, higher antibody titers to HTLV-1 have been found (Nakagawa et al., 1995). In a recent retrospective study in Peru, 22 percent of TSP patients presented rapid progression, defined as less than 2 years between onset of symptoms and confinement to a wheelchair. Some patients presented more rapid progression, with the time between onset of symptoms and the inability to walk unaided as short as 6 months. As noted in a previous report (Nakagawa et al., 1995), there was an association between age of onset and rapid progression of TSP; 67 percent of patients with rapid progression were at least 50 years of age at disease onset, compared with 38 percent among slow progressors, p < 0.01. With the exception of unintentional tremor, no differences were found in clinical symptoms (38 percent of patients with rapid progression reported
tremor versus 9 percent among slow progressors, p < 0.001) (E. Gotuzzo, unpublished data).
Urinary symptoms are a frequent complaint of TSP patients. Initially, patients report difficulties to initiate voiding. Not uncommonly, patients mention the need to put external pressure on their lower abdomen in order to urinate. In severe cases, patients cannot maintain voiding without compressing the abdomen, sometimes leading to urinary retention. Recurrent urinary infections are common, probably reflecting disorders in bladder emptying. Dysfunction of the detrusor muscle has been implicated in the urinary tract involvement in TSP.
Manifestations of immune hyperactivity other than TSP—such as Sjögren’s syndrome, uveitis, arthritis, Behçet’s disease and thyroiditis—have been repeatedly observed among patients with TSP.
Currently, there is neither specific nor standardized treatment for HTLV-1 and TSP. Prolonged periods of systemic steroids appear to improve clinical symptoms of TSP and recently, antiretroviral drugs effective in treating HIV, such as lamivudine and zidovudine, have been used with relative success in treating patients with TSP (Sheremata et al., 1993). A combination of corticosteroids, antiretroviral drugs, and rehabilitation might considerably improve the quality of life of TSP patients—particularly if treatment is started early in the course of the disease in those cases with rapid progression (Araujo et al., 1995).
Association of HTLV-1 with Strongyloidiasis
Strongyloides stercoralis is a soil-transmitted intestinal nematode that has been estimated to infect at least 60 million people worldwide. Infection is often asymptomatic, but can cause nonspecific abdominal symptoms and mild diarrhea. While strongyloidiasis is generally a self-limited disease in immunocompetent hosts, S. stercoralis behaves as an opportunistic pathogen, producing disseminated and life-threatening infections (Neva, 1986) in immunocompromised hosts who are incapable of mounting an appropriate immune response. An association of disseminated S. stercoralis infection with malignant tumors, severe malnutrition, acquired immunodeficiency syndrome (AIDS), corticosteroid therapy, and renal transplantation has been well documented. Studies in Japan and Jamaica have shown a significant association between the presence of S. stercoralis infection and HTLV-1 (Nakada et al., 1984). In Sao Paulo, 12 percent of HTLV-1-positive blood donors carried strongyloidiasis, compared to 1.6 percent of the control group (Nakada et al., 1984). In our institute in Lima, Peru, 10 percent of strongyloidiasis patients are HTLV-1-positive. Another study conducted in Lima, Peru reveals a strong association between the phenomenon of hyperinfection with S. stercoralis and HTLV-1. Eighty-six percent (18 out of 21) of patients with S. stercoralis hyperinfection were infected with HTLV-1, but were not infected with other immunosuppressive diseases such as AIDS or cancer. The difference with the HTLV-1 prevalence in a carefully matched control
group (5 percent, 1 out of 21) and in a group with intestinal strongyloidiasis (10 percent, 6 of 62) was statistically significant (p < 0.001) (Gotuzzo et al., 1999). A report of decreased therapeutic efficacy of thiabendazole exists among patients with concomitant S. stercoralis-HTLV-1 infection in Okinawa (Sato et al., 1994). Terashima showed that the failure of the standard treatment against intestinal strongyloidiasis with thiabendazole or ivermectin was an important marker for suspecting HTLV-1 infection. Some reports suggest that there is a relation between strongyloidiasis and ATLL in HTLV-1-positive patients. It is not clear whether Strongyloides acts as a trigger, shortening the incubation time of leukemia, or a marker of high proviral load.
Association of HTLV-1 with Crusted Scabies
Crusted scabies, a severe form of scabies with generalized itching and massive numbers of mites, has been described among patients undergoing chemotherapy and with various immunosuppressive conditions, such as Down’s syndrome, cancer, and AIDS (Paterson et al., 1973). Several reports on the association between HTLV-1 and severe scabies have been published. In a study in 6 hospitals in Lima, Peru, 23 patients were diagnosed with Norwegian scabies over 19 months. Seventy percent of patients were serologically confirmed to have HTLV-1 infection; 9 percent were on long-term oral corticosteroid treatment; 1 patient had Down’s syndrome; 2 patients were chronically malnourished; and 2 patients had no known risk factors for crusted scabies. A study conducted in Bahia, Brazil, found a similar association between HTLV-1 and severe scabies but also identified dual infection (HIV/HTLV-1) as a risk factor for even more severe forms of scabies. The crusted form of the disease was highly predictive of double retroviral infection and seemed to be associated with severe immunodeficiency, because HIV/HTLV-1 coinfected patients were more likely to die during the study period. These data suggest that coinfection by HIV-HTLV-1 is associated with a deeper degree of immunodeficiency, which increases the risk of developing severe forms of scabies (Brites et al., 2002).
Coinfection of HTLV-1 with HIV
In spite of the similarities between HTLV-1 and HIV with respect to transmission, both epidemics are still largely separated in Peru. Nevertheless, dual infections do occur. In 1989, 19 percent of HIV-infected Peruvian men and 5 percent of HIV-infected women were found to be HTLV-1 coinfected; and in men, dual infection was associated with a higher number of sexual partners compared with HIV-only infected patients (Phillips et al., 1991). Dual infection has also been reported in Trinidad (Bartholomew et al., 1987), Brazil (Cortes et al., 1989), and the United States (Pierik and Murphy, 1991). Several studies have suggested that patients with dual infection are at higher risk of developing AIDS
(Bartholomew et al., 1987). In a prospective study of HIV-positive intravenous drug users, patients infected with both viruses were three times more likely to die from AIDS during follow-up than those infected with HIV-I alone (Page et al., 1990). In a Peruvian study, the mortality rate was 63 percent in HIV-infected patients and 80 percent in dually-infected patients. Of 50 patients who died without receiving any antiretroviral treatment, survival time was 5.02 ± 3.27 months in patients with dual infection, shorter than that of patients with HIV alone (10.07 ± 4.42 months) (Gotuzzo et al., 1992).
Association of HTLV-1 with Tuberculosis
It is well known that the incidence and clinical picture of tuberculosis (TB) is adversely affected by HIV infection. Similarly, in a Brazilian study, patients with TB and HTLV-1 exhibited a worse clinical course and a poorer prognosis than those without HTLV-1. In a study among 131 inpatients with TB in Lima, Peru, those patients infected with HIV-1 or with HTLV-1 were more likely to die during hospitalization than seronegative TB patients (RR = 2.6 for HIV [95 percent confidence interval = 1.05–6.30] and RR = 5.8 for HTLV-1 [95 percent confidence interval = 2.3–14.3]). The association was particularly strong among patients infected with both HIV-1 and HTLV-1 (RR = 6.61, 95 percent confidence interval = 2.5–17.2) (Henriquez et al., 2002).
Association of HTLV-1 with Chronic Infective Dermatitis
The term infective dermatitis was proposed by Sweet in 1966 for a relapsing eczematous condition in Jamaican children, usually associated with cutaneous infections by Staphylococcus aureus or β-hemolytic Streptococcus (Sweet, 1966). In 1990, LaGrenade described an association between chronic infective dermatitis and infection with HTLV-1 (LaGrenade et al., 1990). This syndrome has also been described in Colombia (Blank et al., 1995). Chronic infective dermatitis is commonly seen in children and is rare in adults. This disease presents with symmetric lesions on the scalp, face, armpits, and groin. These lesions improve markedly with antibiotics, but usually relapse when antibiotics are stopped.
Conclusion
HTLV-1 produces three different clinical patterns: lymphoproliferative disease (ATLL), autoimmune syndromes (TSP), and infections associated with immunosuppression (strongyloidiasis, crusted scabies and others). The infection is endemic in several countries in Latin America. HTLV-1 is transmitted mainly through breastfeeding, transfusion of whole blood, and as a STD. These modes of transmission are vulnerable to simple and effective methods of control, such as
screening of pregnant women and avoiding lactation in those infected, universal screening of blood donors, and promotion of condom use in sexual risk groups.
REFERENCES
Araujo AQ, Leite AC, Dultra SV, Andrada-Serpa MJ. 1995. Progression of neurological disability in HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Journal of the Neurological Sciences 129:147–151.
Bartholomew C, Blattner W, Cleghorn F. 1987a. Progression to AIDS in homosexual men co-infected with HIV and HTLV-I in Trinidad. Lancet 2:1469.
Bartholomew C, Saxinger C, Clark JW, Gail M, Dudgeon A, Mahabir B, Hull-Drysdale B, Cleghorn F, Gallo RC, Blattner WA. 1987b. Transmission of HTLV-1 and HIV among homosexual men in Trinidad. Journal of the American Medical Association 257:2604–2608.
Blank A, Herrera M, Lourido MA, Rueda R, Blank M. 1995. Infective dermatitis in Colombia. Lancet 346:710.
Blattner WA. 1990. Epidemiology of HTLV-1 and associated diseases. In Human Retrovirology: HTLV-1, WA Blattner, ed. New York: Raven Press.
Blattner WA, Nomura A, Clark JW, Ho GY, Nakao Y, Gallo R, Robert-Guroff M. 1986. Modes of transmission and evidence for viral latency from studies of human T-cell lymphotropic virus type I in Japanese migrant populations in Hawaii. Proceedings of the National Academies of Sciences USA 83:4895–4898.
Bouzas MB, Zapiola I, Quiruelas S, Gorvein D, Panzita A, Rey J, Carnese FP, Corral R, Perez C, Zala C, et al. 1994. HTLV Type I and HTLV Type II infection among Indians and Natives from Argentina. AIDS Research and Human Retroviruses 10:1567–1571.
Brites C, Weyll M, Pedroso C, Badaro R. 2002. Severe and Norwegian scabies are strongly associated with retroviral (HIV/HTLV-1) infection in Bahia, Brazil [letter]. AIDS 16:1292–1293.
Caribbean Epidemiology Center. 1990. Public Health Implications of HTLV-1 in the Caribbean. Weekly Epidemiological Record 65:63–65.
Cartier L and Cartier E. 1996. HTLV-I/II in Chile. Pp. 150–158 in HTLV, Truths and Questions, V Zaninovic, ed. Cali, Colombia: Fundación MAR.
Cortes E, Detels R, Aboulafia D, Li XL, Moudgil T, Alam M, Bonecker C, Gonzaga A, Oyafuso L, Tondo M. 1989. HIV-1, HIV-2 and HTLV-I infection in high risk groups in Brazil. New England Journal of Medicine 320:953–958.
de Cabral MB, Vera ME, Samudio M, Arias AR, Cabello A, Moreno R, Zapiola I, Bouzas MB, Muchinik G. 1995. HTLV-I/II antibodies among three different Indian groups from Paraguay. Journal of Acquired Immune Deficiency Syndromes and Human Retrovirology 19:548–549.
Delaporte E, Peeters M, Durand JP, Dupont A, Schrijvers D, Bedjabaga L, Honore C, Ossari S, Trebucq A, Josse R, et al. 1989. Seroepidemiological survey of HTLV-1 infection among randomized population of western central African countries. Journal of AIDS 2:410–413.
Gabbai AA, Bordin JO, Vieira-Filho JP, Kuroda A, Oliveira AS, Cruz MV, Ribeiro AA, Delaney SR, Henrard DR, Rosario J, et al. 1993. Selectivity of human T lymphotropic virus type I (HTLV-I) and HTLV-II infection among different populations in Brazil. American Journal of Tropical Medicine and Hygiene 49:664–671.
Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, de The G. 1985. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 2:407–410.
Gotuzzo E, Escamilla J, Phillips IA, Sanchez J, Wignall FS, Antigoni J. 1992. The impact of human T lymphotropic virus type I/II infection on the prognosis of sexually acquired cases of acquired immunodeficiency syndrome. Archives of Internal Medicine 152:1429–1432.
Gotuzzo E, Yamamoto V, Kanna M, et al. 1996. Human T lymphotropic virus type I infection among Japanese immigrants in Peru. International Journal of Infectious Diseases 1:75–77.
Gotuzzo E, Terashima A, Alvarez H, Tello R, Infante R, Watts DM, Freedman DO. 1999. Strongyloides stercoralis hyperinfection associated with human T cell lymphotropic virus type-I infection in Peru. American Journal of Tropical Medicine and Hygiene 60:146–149.
Gradilone A, Zani M, Barillari G, Modesti M, Agliano AM, Maiorano G, Ortona L, Frati L, Manzari V. 1986. HTLV-1 and HIV infection in drug addicts in Italy. Lancet 2:753–754.
Guderian R, Guevara A, Cooper P, Rugeles MT, Arango C. 1994. HTLV-1 infection and tropical spastic paraparesis in Esmeraldas Province of Ecuador. Transactions of the Royal Society of Tropical Medicine and Hygiene 88:399–400.
Henriquez C, Gotuzzo E, Cairampoma, et al. Impact of infection with HIV and/or HTLV-1 on the outcome of hospitalization for tuberculosis in a public hospital in Peru. Abstract at the 4th World Congress on Tuberculosis, Washington, DC, June 3–5, 2002.
Hinuma Y, Nagata K, Hanaoka M, Nakai M, Matsumoto T, Kinoshita KI, Shirakawa S, Miyoshi I. 1981. Adult T cell leukemia antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proceedings of the National Academy of Sciences USA 78:6476–6480.
Jeffrey KJ, Usuku K, Hall SE, Matsumoto W, Taylor GP, Procter J, Bunce M, Ogg GS, Welsh KI, Weber JN, Lloyd AL, Nowak MA, Nagai M, Kodama D, Izumo S, Osame M, Bangham CR. 1999. HLA alleles determine human T-lymphotropic virus-I (HTLV-1) proviral load and the risk of HTLV-1-associated myelopathy. Proceedings of the National Academy of Sciences USA 96:3848–3853.
Kaplan JE, Osame M, Kubota H, Igata A, Nishitani H, Maeda Y, Khabbaz RF, Janssen RS. 1990. The risk of development of HTLV-1 associated myelopathy/tropical spastic paraparesis among persons infected with HTLV-1. AIDS 3:1096–1101.
Katamine S. 1999. Milk-borne transmission of human T-cell lymphotropic virus Type 1 (HTLV-1) and its intervention in Nagasaki. Acta Medica Nagasakiensia 44:1–6.
Khabbaz R, Darrow WW, Hartley TM, Witte J, Cohen JB, French J, Gill PS, Potterat J, Sikes RK, Reich R, et al.. 1990. Seroprevalence and risk factors for HTLV-1 infection among female prostitutes in the United States. Journal of the American Medical Association 263:60–64.
LaGrenade L, Hanchard B, Fletcher V, Cranston B, Blattner W. 1990. Infective dermatitis of Jamaican children: a marker for HTLV-1 infection. Lancet 336:1345–1347.
Larson C and Taswell H. 1988. Human T-cell leukemia virus (HTLV-1) and blood transfusion. Mayo Clinic Proceedings 63: 869–875.
Mani KS, Mani AJ, Montgomery, RD. 1969. A spastic paraplegic syndrome in South India. Journal of the Neurological Sciences 9:179–199.
Murphy EL, Hanchard B, Figueroa JP, Gibbs WN, Lofters WS, Campbell M, Goedert JJ, Blattner WA. 1989. Modelling the risk of adult T cell leukemia/lymphoma in persons infected with human T lymphotropic virus type I. International Journal of Cancer 43:250–253.
Murphy EL, Wilks K, Hanchard B, Cranston B, Figueroa JP, Gibbs WN, Murphy J, Blattner WA. 1996. A case-control study of risk factors for seropositivity to HTLV-1 in Jamaica. International Journal of Epidemiology 25:1083–1089.
Nakada K, Kohakura M, Komoda H, Hinuma Y. 1984. High incidence of HTLV antibody in carriers of Strongyloides stercoralis [letter]. Lancet 1:633.
Nakagawa M, Izumo S, Ijichi S, Kubota H, Arimura K, Kawabata M, Osame M. 1995. HTLV-1-associated myelopathy: analysis of 213 patients based on clinical features and laboratory findings. Journal of Neurovirology 1:50–61.
Nerurkar VR, Song KJ, Saitou N, Melland RR, Yanagihara R. 1993. Interfamilial genomic diversity and molecular phylogeny of human T-cell lymphotrophic virus type I from Papua New Guinea and the Solomon Islands. Virology 196:506–513.
Neva FA. 1986. Biology and immunology of human strongyloidiasis. Journal of Infectious Diseases 153:397–406.
Okochi K, Sata H, Hinuma Y. 1984. A retrospective study on transmission of adult T-cell leukemia virus via blood transfusion: seroconversion in recipients. Vox Sanguinis 46:245–253.
Page JB, Lai SH, Chitwood DD, Klimas NG, Smith PC, Fletcher MA. 1990. HTLV-I/II seropositivity and death from AIDS among HIV-1 seropositive intravenous drug users. Lancet 335:1439–1441.
Paterson WD, Allen BR, Beveridge GW. 1973. Norwegian scabies during immunosuppressive therapy. British Medical Journal 4:211–212.
Phillips I, Hyams KC, Wignall FS, Moran AY, Gotuzzo E, Sanchez J, Roberts CR. 1991. HTLV-1 coinfection in a HIV-1 infected Peruvian population. Journal of Acquired Immune Deficiency Syndromes 4:301–302.
Pierik LT and Murphy EL. 1991. The clinical significance of HTLV-I and HTLV-II infection in the AIDS epidemic. Pp. 41–57 in AIDS Clinical Review, P Volberding and MA Jacobson, eds. New York: Marcel Dekker.
Poiesz BJ, Ruscetti FW, Gadzar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T cell lymphoma. Proceedings of the National Academy of Sciences USA 77:7415–7419.
Sanchez-Palacios C, Gotuzzo E, Vandamme AM, Maldonado Y. 2003. Seroprevalence and risk factors for human T-cell lymphotropic virus (HTLV-1) infection among ethnically and geographi-cally diverse Peruvian women. International Journal of Infectious Diseases 7:132–137.
Sato Y, Shiroma Y, Kiyuna S, Toma H, Kobayashi J. 1994. Reduced efficacy of chemotherapy might accumulate concurrent HTLV-1 infection among strongyloidiasis patients in Okinawa, Japan. Transactions of the Royal Society of Tropical Medicine and Hygiene 88:59.
Seiki M, Hattori S, Hirayama Y, Yoshida M. 1983. Human adult T-cell leukemia virus: Completed nucleotide sequence of the provirus genoma integrated in leukemia cell DNA. Proceedings of the National Academy of Sciences USA 80:3618–3622.
Sheremata WA, Benedict D, Squilacote DC, Sazant A, DeFreitas E. 1993. High-dose zidovudine induction in HTLV-1-associated myelopathy: safety and possible efficacy. Neurology 43:2125–2129.
Sweet RD. 1966. A pattern of eczema in Jamaica. British Journal of Dermatology 78:93–100.
Tajima K and Hinuma Y. 1992. Epidemiology of HTLV-I/II in Japan and the world. Gann Monograph on Cancer Research 39:129–149.
Tajima K, Tominaga S, Suchi, Kawagoe T, Komoda H, Hinuma Y, Oda T, Fujita K. 1982. Epidemiological analysis of the distribution of antibody to adult T-cell leukemia-virus-associated antigen: possible horizontal transmission of adult T-cell leukemia virus. Gann 73:893–901.
Takatsuki K, Uchiyama J, Sagawa K, Yodoi J. 1977. Adult T-cell leukemia in Japan. Pp. 73–77 in Topics in Hematology, S Sano, F Takaku, S Irino, eds. Amsterdam: Excerpta Medica.
Tsugane S, Watanabe S, Sugimura H, Otsu T, Tobinai K, Shimoyama M, Nanri S, Ishii H. 1988. Infectious status of human T lymphotropic virus type I and hepatitis B virus among Japanese immigrants in the Republic of Bolivia. American Journal of Epidemiology 128:1153–1161.
Wignall FS, Hyams KC, Phillips IA, Escamilla J, Tejada A, Li O, Lopez F, Chauca G, Sanchez S, Roberts CR. 1992. Sexual transmission of human T-cell lymphotropic virus type I in Peruvian prostitutes. Journal of Medical Virology 38:44–48.
Zaninovic V, Sanzón F, López F, Velandia G, Blank A, Blank M, Fujiyama C, Yashiki S, Matsumoto D, Katahira Y, et al. 1994. Geographic independence of HTLV-I and HTLV-II foci in the Andes Highland, the Atlantic Coast, and the Orinoco of Colombia. AIDS Research and Human Retroviruses 10:97–101.
Zurita S, Costa C, Watts D, Indacochea S, Campos P, Sanchez J, Gotuzzo E. 1997. Prevalence of human retroviral infection in Quillabamba and Cuzco, Peru: a new endemic area for human T-cell lymphotropic virus type 1. American Journal of Tropical Medicine and Hygiene 56:561–565.
PROGRESSION OF HEPATITIS C VIRUS INFECTION WITH AND WITHOUT SCHISTOSOMIASIS
Sanaa Kamal, M.D.
Ain Shams University, Cairo, Egypt
Hepatitis C virus (HCV) infects an estimated 170 million persons worldwide, and is a major cause of morbidity in developed and developing countries. In the United States, nearly 2 percent of the population is infected with this positive strand RNA virus from the flavivirus family (Seeff, 1997; Alter, 1997). In other regions, the prevalence of HCV infection is even higher reaching up to 10 percent to 31 percent of the population in some countries (Abdel Aziz et al., 2000; Habib et al., 2001). Approximately 70 percent of infected persons will go on to develop chronic hepatitis, and 15 to 20 percent will eventually develop cirrhosis. HCV-related cirrhosis is also one of the major risk factors for the development of hepatocellular carcinoma (Seeff, 1997). Despite recent improvements in treating HCV infection using interferon-alpha and ribavirin, about half of infected individuals will fail treatment or cannot be treated due to contraindications (Poynard et al., 1996; Thevenot et al., 2001). Also, for many persons worldwide, antiviral therapy is out of reach due to its prohibitive costs. Therefore, the development of an effective vaccine to prevent the continued spread of HCV infection remains an urgent goal. Likewise, a better understanding of the immunopathogenesis of this infection may facilitate the development of immunotherapeutic strategies to treat infected persons.
CD4 Response to HCV Infection
The host immune response probably plays a critical role in both control of HCV replication and liver injury. HCV infection evokes CD4+, HLA class II-restricted (Cerny and Chisari, 1999; Diepolder et al., 1996) and CD8+ (CTL) HLA class I-restricted T-cell response (Lechner et al., 2000; He et al., 1999). In the absence of an appropriate small animal model, studying the immunological events that occur in the earliest stages of infection in patients with acute hepatitis C infection thus offers the unique opportunity to identify efficient immune mechanisms of virus control and to characterize the factors determining the eventual outcome of disease. Several studies in individuals who experienced complete virologic recovery have found a significant association between a strong and maintained HCV-specific CD4+ T-cell response and viral clearance in acute hepatitis C (Gerlach et al., 1999; Kamal et al., 2001). As HCV-specific cellular immune responses are present in chronic infection, the other consideration is that the cytokine response may be qualitatively different in individuals with chronic infection as compared with acute resolved HCV. Analysis of the cytokine profile of bulk cultures as well as CD4+ T-cell clones from patients with hepatitis C revealed that viral clearance is more likely in cases displaying a T-helper 1 pro-
file (Koziel, 1999). However, most studies are based on few individuals who recovered infection either spontaneously or after interferon therapy (Gerlach et al., 1999; Lohr et al., 1998). Thus longitudinal prospective studies analyzing the early antiviral immune responses in acute hepatitis C infection are crucial for understanding the pathogenesis of the disease and potentially in vaccine design. Once chronic infection is established HCV-specific CD4+ T-cells compartmentalize in the liver and differ functionally and clontypically from those in the peripheral blood (Schirren et al., 2000; Bertoletti et al., 1997). The significance of the intrahepatic CD4+ responses and their relation to liver injury have not been comprehensively investigated since most studies focus on the peripheral compartment due to the difficulty in obtaining liver biopsies.
CD8 Response to HCV Infection
The CD8+ T-cell response is thought to play a crucial role in the course of HCV infection. In humans and in chimpanzees, a strong and broadly directed HCV-specific CTL response has been associated with viral clearance during acute HCV infection. In contrast, individuals with chronic infection are often found to have a relatively weak and narrowly directed CD8+ T-cell response against HCV (Lechner et al., 2000; He et al., 1999). Whether these responses in chronic disease are still beneficial in containing viral replication or whether they are mediators of hepatic injury and disease is unclear. In several studies, the magnitude of the HCV-specific CD8+ T-cell response has been correlated to HCV viral load and to liver histology, but the results of these studies have been controversial. Overall, the levels of responses detected by most investigators have been fairly low, using a variety of methods, compared to those found in many other viral infections.
CTL Response to HCV Infection
Despite an occasionally broadly directed CTL response, virus persists within the liver, demonstrating the typical lack of effective clearance of infected hepatocytes by this response (Cerny and Chisari, 1999; Lechner et al., 2000; He et al., 1999). In fact, CTL have been hypothesized to contribute to liver pathology, perhaps by chronic secretion of cytokines that may enhance the development of cirrhosis. The reasons that the CTL response is unable to clear infection remain unclear. Possible mechanisms include escape of the virus from CTL epitopes, dysfunctional CTL, or direct inhibition of CTL through the virus.
Schistosomiasis and HCV Infection
Schistosomiasis is a chronic helminthic disease infecting more than 200 million people worldwide (Chitsulo et al., 2000). Infection with Schistosoma mansoni is endemic in Egypt with a prevalence range of 17.5 percent to 42.9 percent (El-
Khoby et al., 2000; Hammam et al., 2000). Morbidity in humans infected with S. mansoni results primarily from deposition of parasite ova in the portal areas inducing a T-cell-dependent granulomatous response which progresses to irreversible fibrosis and severe portal hypertension in more than 60 percent of cases. S. mansoni infection in mice is characterized by a strong Th2-associated immune response coupled with a defect in Th1-cell effector function (Sabin and Pearce, 1995). Although a predominant Th2 profile was shown to be beneficial in polyparasitism, where mice infected with S. mansoni are capable of eliminating Trichuris muris infection more efficiently than non-infected mice (Curry et al., 1995), it is assumed to be harmful in most viral infections.
Concomitant schistosomiasis and HCV infection is common in Egypt and other developing countries (Kamal et al., 2000a; Angelico et al., 1997; Pereira et al., 1995). Patients with concomitant HCV and schistosomiasis exhibit a unique clinical, virological, and histological pattern manifested by virus persistence with high HCV RNA titers, higher necroinflammatory and fibrosis scores in their liver biopsies and poor response to interferon therapy (Kamal et al., 2000a; Angelico et al., 1997; Pereira et al., 1995; Kamal et al., 2000b). This results in a markedly accelerated disease course once chronic HCV infection has been established. Our understanding of the pathomechanisms leading to this accelerated disease progression in HCV/S. mansoni coinfection is still extremely limited. This coinfection should be a valuable model to study the effect of one pathogen on the pathogenesis of the other agent, especially the influence of an altered T helper cell response on the other arms of the immune response as well as the clinical outcome. The model of HCV/S. mansoni coinfection also offers a unique opportunity to define the role of HCV-specific T-cells in viral control as well as the pathogenesis of HCV-related liver disease.
Comprehensive study of the different aspects of HCV/S. mansoni coinfection has been conducted and the data were presented in several publications. These studies, described below, provide insight into the mechanisms through which infection with one pathogen can influence the immunopathogenesis and the clinical course of another.
-
Chronic HCV and S. mansoni coinfection is associated with more severe disease (Kamal et al., 2000a). One hundred and twenty-six patients with either chronic hepatitis C (group A), chronic schistosomiasis (group B), and chronic hepatitis C and schistosomiasis (group C) were enrolled and prospectively followed for 67.2 ± 22 months. HCV RNA titers were significantly higher in the coinfected group. Patients with coinfection showed higher fibrosis scores in their liver biopsies. Hepatocellular carcinoma was detected only in patients with coinfection. During follow-up, the mortality due to liver-related causes was 2 percent, 3 percent, and 48 percent in groups A, B, and C, respectively. In conclusion, patients with concomitant HCV and schistosomiasis are characterized through more advanced liver disease, higher HCV RNA titers, higher incidence
-
of cirrhosis and hepatocellular carcinoma as well as higher incidence of liver-related morbidity and mortality.
-
HCV and S. mansoni coinfection is associated with poor response to interferon therapy (Kamal et al., 2000b). Sixty-two patients (28 with chronic HCV and 34 patients with HCV and S. mansoni) were treated with interferon α-2b. The end of treatment response was 20 percent in coinfected patients versus 36 percent in monoinfected patients. The sustained response rate was 3 percent in coinfected patients versus 20 percent in monoinfected patients. In conclusion, patients with chronic hepatitis C coinfected with schistosomiasis respond poorly to interferon therapy and have higher relapse rates compared to patients with chronic HCV monoinfection.
-
Patients with HCV and S. mansoni coinfection fail to mount significant HCV-specific CD4+ T-cell responses and show alteration in the cytokine milieu along with more severe liver disease (Kamal et al., 2001a). To define if immunological mechanisms are responsible for this alteration in the natural history of HCV, the HCV-specific peripheral CD4+ T-cell responses and cytokines were analyzed in patients with chronic hepatitis C monoinfection, patients with S. mansoni monoinfection and patients with hepatitis C virus and S. mansoni coinfection. HCV-specific CD4+ proliferative responses to at least one HCV antigen were detected in 73.3 percent patients with HCV monoinfection compared to 8.6 percent coinfected with S. mansoni. Stimulation with HCV antigens produced a type 1 cytokine profile in patients with HCV monoinfection compared to type 2 predominance in patients with coinfection. In contrast, there was no difference in response to schistosomal antigens in patients with S. mansoni infection compared to those with coinfection. These findings suggest that the inability to generate HCV-specific CD4+/Th1 T-cell response plays a role in the persistence and severity of HCV infection in patients with coinfection.
-
Patients with acute hepatitis C and schistosomiasis coinfection cannot clear viremia and show rapid progression once chronic infection is established (Kamal et al., 2001b). Immune responses during the first few months of acute HCV infection seem crucial for viral control, but the relationship of these responses to natural history is poorly characterized. We prospectively investigated the HCV-specific CD4+ and cytokine responses in patients with acute HCV hepatitis with or without S. mansoni coinfection, a parasitic infection with T helper (Th) 2 immune bias. HCV-specific CD4+ proliferative responses and cytokine production in peripheral blood mononuclear cells (PBMCs) were correlated with liver biopsy results at six months and end of follow-up. Whereas 5 of 15 patients with HCV alone recovered from acute HCV, all (17/17) patients with S. mansoni coinfection progressed to histologically proven chronic hepatitis. Coinfected patients had either absent or transient weak HCV-specific CD4+ responses with Th0/Th2 cytokine production. The magnitude of the HCV-specific CD4+ response at week 12 was inversely correlated with the HCV RNA titers and the fibrosis progression rate in chronically infected patients.
We are currently conducting comprehensive and sensitive analysis of HCV-specific CTL and CD4+ responses in PBMCs and liver-infiltrating lymphocytes of patients coinfected with HCV and S. mansoni versus HCV-monoinfected patients.
Conclusion
In summary, HCV infection is a worldwide problem for which there has been insufficient success with treatment options presently available. The lack of a clear understanding of the immunological events during acute and chronic infection has hampered vaccine development and immunotherapeutic approaches to treatment. From an immunological point of view the interplay between T helper cell responses and CTL has been difficult to assess in humans, and infection with S. mansoni offers the unique situation of studying the impact of an altered response on the outcome and progression of HCV-related liver disease.
REFERENCES
Abdel Aziz F, Habib M, Mohamed M, Abdel Hamid M, Gamil F, Madkour S, Mikhail N, Thomas D, Fix A, Strickland T, Anwar W, Ismail S. 2000. Hepatitis C virus infection in a community in the Nile Delta: population description and HCV prevalence. Hepatology 32:111–115.
Alter MJ. 1997. Epidemiology of hepatitis C. Hepatology 26:62S–65S.
Angelico M, Renganathan E, Gandin C, Fathy M, Profili MC, Refai W, De Santis A, Nagi A, Amin G, Capocaccia L, Callea F, Rapicetta M, Badr G, Rocchi G. 1997. Chronic liver disease in Alexandria governorate, Egypt: contribution of schistosomiasis and hepatitis virus infections. Journal of Hepatology 26:236–243.
Bertoletti A, D’Elios MM, Boni C, De Carli M, Zignego AL, Durazzo M, Missale G, Penna A, Fiaccadori F, Del Prete G, Ferrari C. 1997. Different cytokine profiles of intrahepatic T cells in chronic hepatitis B and hepatitis C virus infections. Gastroenterology 112:193–199.
Cerny A and Chisari FV. 1999. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology 30:595–601.
Chitsulo L, Engels D, Montresor A, Savioli L. 2000. The global status of schistosomiasis and its control. Acta Tropica 77:41–51.
Curry AJ, Else KJ, Jones F, Bancroft A, Grencis RK, Dunn DW. 1995. Evidence that cytokine-mediated immune interactions induced by Schistosoma mansoni alter disease outcome in mice concurrently infected with Trichuris muris. The Journal of Experimental Medicine 181:769–774.
Diepolder HM, Zachoval R, Hoffmann RM, Jung MC, Gerlach T, Pape GR. 1996. The role of hepatitis C virus specific CD4+ T lymphocytes in acute and chronic hepatitis C. Journal of Molecular Medicine 74:583–588.
El-Khoby T, Galal N, Fenwick A, Barakat R, El-Hawey A, Nooman Z, Habib M, Abdel Wahab F, Gabr NS, Hammam HM, Hussein MH, Mikhail NN, Cline BL, Strickland GT. 2000. The epidemiology of schistosomiasis in Egypt: summary of findings in nine governorates. The American Journal of Tropical Medicine and Hygiene 62:88–99.
Gerlach T, Diepolder H, Jung M, Gruner N, Schraut W, Zachoval R, Hoffman R, Schirren A, Santantonio T, Pape G. 1999. Recurrence of hepatitis C virus after loss of virus specific CD4+ T-cell response in acute hepatitis C. Gastroenterology 117:993–941.
Habib M, Mohamed MK, Abdel-Aziz F, Magder LS, Abdel-Hamid M, Gamil F, Madkour S, Mikhail NN, Anwar W, Strickland GT, Fix AD, Sallam I. 2001. Hepatitis C virus infection in a community in the Nile Delta: risk factors for seropositivity. Hepatology 33:248–253.
Hammam HM, Allam FA, Moftah FM, Abdel-Aty MA, Hany AH, Abd-El-Motagaly KF, Nafeh MA, Khalifa R, Mikhail NN, Talaat M, Hussein MH, Strickland GT. 2000. The epidemiology of schistosomiasis in Egypt: Assiut governorate. The American Journal of Tropical Medicine and Hygiene 62:73–79.
He XS, Rehermann B, Lopez-Labrador FX, Boisvert J, Cheung R, Mumm J, Wedemeyer H, Berenguer M, Wright TL, Davis MM, Greenberg HB. 1999. Quantitative analysis of hepatitis C virus-specific CD8(+) T cells in peripheral blood and liver using peptide-MHC tetramers. Proceedings of the National Academy of Sciences USA 96:5692–5697.
Kamal SM, Madwar MA, Bianchi L, EL Tawil A, Fawzy R, Peters T, Rasenack JW. 2000a. Clinical, virological and histopathological features: long-term follow-up in patients with chronic hepatitis C co-infected with Schistosoma mansoni. Liver 20:281–289.
Kamal SM, Madwar MA, Peters T, Fawzy R, Rasenack J. 2000b. Interferon therapy in patients with hepatitis C and schistosomiasis. Journal of Hepatology 32:172–174.
Kamal SM, Bianchi L, Al Tawil A, Koziel M, El Sayed Khalifa K, Peter T, Rasenack JW. 2001a. Specific cellular immune response and cytokine patterns in patients coinfected with hepatitis C virus and Schistosoma mansoni. The Journal of Infectious Diseases 184:972–982.
Kamal SM, Rasenack JW, Bianchi L, Al Tawil A, El Sayed Khalifa K, Peter T, Mansour H, Ezzat W, Koziel M. 2001b. Acute hepatitis C with and without schistosomiasis: correlation with hepatitis C-specific CD4+ T-cell and cytokine response. Gastroenterology 121:646–656.
Koziel MJ. 1999. Cytokines in viral hepatitis. Seminars in Liver Disease 19:157–169.
Lechner F, Wong D, Dunbar R, Chapman R, Chung R, Dohrenwend P, Robins G, Phillips R, Klenerman P, Walker B. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. Journal of Experimental Medicine 1499–1512.
Lohr HF, Gerken G, Roth M, Weyer S, Schlaak JF, Meyer zum Buschenfelde KH. 1998. The cellular immune responses induced in the follow-up of interferon-alpha treated patients with chronic hepatitis C may determine the therapy outcome. Journal of Hepatology 29:524–532.
Pereira LM, Melo MC, Saleh MG, Massarolo P, Koskinas J, Domingues AL, Spinelli, Mies S, Williams R, McFarlane IG. 1995. Hepatitis C virus infection in Schistosomiasis mansoni in Brazil. Journal of Medical Virology 45:423–428.
Poynard T, Leroy V, Conhard M. 1996. Meta-analysis of interferon randomized trials in the treatment of viral hepatitis C: effects of dose and duration. Hepatology 24:278–289.
Sabin EA and Pearce EJ. 1995. Early IL-4 production by non-CD4+ cells at the site of antigen deposition predicts the development of a T helper 2 cell response to Schistosoma mansoni eggs. Journal of Immunology 155:4844–4855.
Schirren CA, Jung MC, Gerlach JT, Worzfeld T, Baretton G, Mamin M, Hubert Gruener N, Houghton M, Pape GR. 2000. Liver-derived hepatitis C virus (HCV)-specific CD4+ T cells recognize multiple HCV epitopes and produce interferon gamma. Hepatology 32:597–603.
Seeff LB. 1997. Natural history of hepatitis C. Hepatology 26:21S–28S.
Thevenot T, Rigimbeau C, Ratziu V, Leroy V, Opolon P, Poynard T. 2001. Meta-analysis of interferon randomized trials in the treatment of viral hepatitis C in naive patients: 1999 update. Journal of Viral Hepatitis 8:48–62.
INTERACTIONS OF MULTIPLE INFECTIOUS AGENTS IN MALARIA-ENDEMIC AREAS: CONCURRENT HIV/AIDS AND MALARIA
Altaf A. Lal, Ph.D.
Division of Parasitic Diseases, National Center for Infectious Diseases Centers for Disease Control and Prevention, Atlanta, GA
Establishment of microorganisms in human host populations requires structural, biologic, and molecular compatibilities between the host and pathogen. In situations where multiple infectious organisms coexist in an individual, the resulting polyparasitism could lead to increased infectivity, altered pathogen load, and modulation in pathogenesis. Burkitt’s lymphoma, which is commonly found in areas with malaria transmission, is a good example of coinfections. It has been proposed that malaria-induced immune activation may be associated with the development of these lymphomas (Whittle et al., 1984).
The introduction of HIV-1 in the human population has altered the epidemiology of several infectious diseases. A number of these organisms, termed together as opportunistic infectious agents, cause significant morbidity and mortality. HIV-1 is now a firmly established infectious agent and the potential to interact with parasitic, viral, fungal, and bacterial infectious agents is very high.
The progression from HIV-1 infection to AIDS is associated with a decline in the CD4 T-cell count and an increase in HIV-1 viral load. Although several factors may be responsible for the variability in HIV-1 disease progression, immune activation appears to be an important determinant. Immune activation leads to up-regulation of viral co-receptors, decreased β chemokine secretion, enhanced viral entry and integration, viral assembly and/or release of the viral particles, changes in the cytokine environment and various degrees of immune dysfunction, hyporesponsiveness, and apoptosis. Because all systemic and/or local concurrent infections cause various degrees of immune activation, it is very likely that they may enhance HIV infection, increase HIV replication and viral load, and even promote progression of the disease.
Several studies have focused on the interaction between HIV/AIDS and three major infectious diseases, namely malaria, sexually transmitted diseases (STDs), and tuberculosis (TB) (Bentwich et al., 2000; Chandramohan and Greenwood, 1998). The main impact of STDs has been to facilitate HIV-1 transmission, and the interaction of TB and HIV-1 has been an increase in the burden of an already major cause of morbidity and mortality. As far as malaria is concerned, although early studies did not reveal a definite interaction between malaria and HIV, there is increasing evidence now that suggests these two pathogens interact, thus modifying the pathogenesis of each disease (Bentwich et al., 2000; Chandramohan and Greenwood, 1998; Corbett et al., 2002). This presentation will focus on the interactions between HIV/AIDS and malaria.
Malaria, TB, and HIV/AIDS are important public health problems in sub-Saharan Africa and some parts of Asia. Both HIV and malaria exert their heaviest toll in sub-Saharan Africa, where the progression of HIV-related disease is considered to be most rapid. The interaction between HIV/AIDS and malaria can be viewed in the mechanistic context, where immunomodulation by one organism can impact the natural course of infection of the co-existing pathogen, and in programmatic context, where the treatment for one disease may have beneficial impact on the other disease and/or the treatment for one disease may not be effective in the presence of the co-infecting pathogen.
Initial studies of the interactions between HIV and malaria focused on the ability of malaria parasites to act as opportunistic organisms in immunosuppressed HIV-positive persons. As recent reviews demonstrate, most of the earlier studies, conducted primarily in adults, did not show an effect of HIV infection on the prevalence or severity of malaria (Chandramohan and Greenwood, 1998; Corbett et al., 2002).
Earlier studies conducted in Zaire, Uganda, Rwanda, and Zambia, showed no or marginal effect of HIV infection on malaria parasitemia (Simooya et al., 1988; Chattopadhya et al., 1991; Greenberg, 1992). However, recent studies conducted in Malawi reported increased prevalence rates of malaria parasitemia and parasite density in HIV-infected pregnant women (Chandramohan and Greenwood, 1998). The higher prevalence of malaria parasitemia was seen in HIV-infected women of all gravidities, indicating that the parity-specific immunity to malaria, which is normally associated with multigravidae, was impaired in HIV-infected women (Chandramohan and Greenwood, 1998). More importantly, these studies revealed that infants born to HIV- and malaria-positive mothers were at a significantly higher risk for low birth weight. Increased prevalence of peripheral parasitemia and placental malaria has also been seen in HIV-positive pregnant women in western Kenya, which has higher rates of malaria transmission than Malawi (Chandramohan and Greenwood, 1998). The increased prevalence of parasitemia in HIV-positive women seemed to be pregnancy associated, because parasitemia in HIV-positive women reduced to the level seen in HIV-negative women 2–6 months postpartum.
HIV infection has been shown to induce poor responses to antimalarial treatment with sulfadoxine-pyrimethamine (S/P) in pregnant women. A recent study conducted in western Kenya indicated that although a standard two-dose S/P regimen worked well in controlling peripheral parasitemia and placental malaria during pregnancy in HIV-negative women, it failed to prevent peripheral and placental parasitemia in HIV-infected women. Poor response to S/P antimalarial treatment was also reported in pregnant Malawian women with HIV-1 infection. Because parasitemia was reduced drastically after each treatment and monthly S/ P dosing worked well in both HIV-positive and HIV-negative women, it is possible that the poor treatment response was due to rapid re-infection rather than delayed parasite clearance. No difference in quinine treatment failure was seen
between HIV-positive and HIV-negative children with malaria in Kinshasa, Zaire (Chandramohan and Greenwood, 1998; Corbett et al., 2002).
Conflicting results have been reported about the effect of HIV infection on malaria antibody responses in Plasmodium falciparum-endemic areas. Earlier studies conducted in Zambia and India showed no differences between HIV-positive and HIV-negative persons in terms of the prevalence of antimalarial antibodies (Simooya et al., 1988; Chattopadhya et al., 1991). Very few HIV-positive persons, however, were involved in these studies, and no attempts were made to compare the titers of antibody response, although one did compare the optical density (OD) (Chattopadhya et al., 1991).
In contrast, a study conducted in Uganda demonstrated consistent reduction in mean OD of antibodies to synthetic peptides from the ring-infected erythrocyte surface antigen (RESA) and circumsporozoite protein (CSP) of P. falciparum and CSP of P. malariae. In addition, HIV-positive persons with AIDS had significantly lower antibody levels (mean OD) of RESA antibodies than asymptomatic HIV-positive persons (Wabwire-Mangen et al., 1989). Cellular immune responses to malaria are seemingly also affected by HIV-1 infection. Compared with HIV-negative persons, AIDS patients in Burkina Faso had lower proliferation of PBMCs to stimulation with merozoite surface protein-1 (MSP-1) and parasite culture supernatant. They also had reduced in vitro production of IFN-γ and IL-2. The immune suppression induced by HIV was probably general, because PBMCs of AIDS patients also respond poorly to phytohaemagglutinin, tuberculin purified protein derivative, and lipopolysaccharide (Migot et al., 1996).
We have recently evaluated the influence of HIV-1 on malaria antigen specific antibody responses during pregnancy. These studies have revealed that maternal and neonatal antibody levels against blood stage and sporozoite stage antigenic determinants are significantly lower among HIV-infected women compared with HIV-uninfected women. We also observed reduced maternal-fetal transplacental antibody transfer in dually infected women. In another recent study conducted in Kenya, we found elevated production of IFN-γ by maternal placental (intervillous blood) mononuclear cells (IVBMC) from multigravidae to be associated with protection against placental malaria (Moore et al., 2000). A protective role for IFN-γ in controlling infection has been demonstrated both in human studies and with animal models. Mechanistically, this cytokine has been proposed to be important in mediating asexual blood-stage parasite clearance, perhaps via its regulatory influence on phagocytic cells. The importance of this cytokine in protection against placental malaria is further supported by our recent finding that IVBMC from HIV-positive women have impaired antigen-specific IFN-γ and IL-4 responses (Moore et al., 2000). Since these cytokines are produced primarily by T-cells, we conclude that this loss of cytokine responsiveness may play a role in the increased susceptibility of HIV-positive women to placental malaria.
It has been suggested that the progression from HIV-1 infection to AIDS is more rapid in sub-Saharan African patients than in persons living in developed countries (Gilks, 1993; Mulder et al., 1994; Grant et al., 1997). In addition to the lack of access to health care and treatment, chronic immune stimulation from increased exposure to other infectious agents are probable co-factors of immune activation. Earlier investigations of the relationship between HIV-1 and malaria focused mainly on the effect of HIV-1 infection on malaria. Only one study examined the effect of malaria on HIV-1 infection, and failed to detect any effect of malaria infection on HIV-1 progression. No measurements of changes in CD4+ T-cell counts and viral load, which are two current predictors of HIV disease progression, were done in this study.
Recent in vitro and in vivo studies, nevertheless, indicate that malaria can potentially affect the course of HIV infection in several aspects. The initial evidence of a possible effect of malaria on HIV-1 infection came from a retrospective analysis of data from a cohort study of mothers and infants in rural Malawi. It was demonstrated that infants born to mothers with both placental malaria and HIV-1 infection had post-neonatal mortality 4.5 times higher than infants born to mothers with only placental malaria, and 2.7–7.7 times higher than infants born to mothers with only HIV-1 infection (Chandramohan and Greenwood, 1998; Corbett et al., 2002). This increased mortality in infants born to mothers with dual HIV and malaria was attributed to the increased transmission of HIV from mothers to infants, although no HIV testing was conducted in these infants.
Because immune activation is an important prerequisite for efficient HIV infection and viral replication, we evaluated the effect of malarial antigen stimulation on HIV-1 infection. Stimulation with soluble malarial antigens or malarial pigment from P. falciparum enhanced HIV-1 replication in PBMC from naive donors by 10- to 100-fold. The malarial antigen-upregulated HIV-1 replication was mediated through induction of TNF-α via the activation of long terminal repeat (LTR)-directed viral transcription (Xiao et al., 1998). Preliminary studies conducted with PBMC from HIV-positive individuals residing in western Kenya indicated that recall immune responses induced by soluble malarial antigens can increase HIV-1 replication (Xiao et al., unpublished observation). PBMC from 3 of 10 HIV-1 infected individuals showed active in vitro viral production after the antigen stimulation.
These in vitro observations have been confirmed by the result of a recent prospective, cohort study of 47 HIV-positive adults with active falciparum malaria and 42 HIV-positive adults without malaria in Malawi. It was shown that HIV-positive individuals with active malaria had a mean plasma HIV-1 viral load 7-fold higher than HIV-positive individuals without malaria (Hoffman et al., 1999). Plasma HIV-1 RNA concentrations did not correlate significantly with P. falciparum parasite density or the duration of fever. However, antimalarial chemotherapy with S/P resulted in a small (37 percent) but significant reduction in
HIV-1 RNA load by week 4 post-treatment in individuals with HIV and malaria coinfection.
Another potential interaction between HIV and malaria is at the invasion stage of both pathogens. HIV-1 has been recently shown to bind erythrocytes from Caucasian persons through the Duffy antigen receptor for chemokines (DARC), a receptor that is also used by the invasion of P. vivax merozoites into reticulocytes (Lachgar et al., 1998). It has been proposed that erythrocytes may function as a reservoir for HIV-1, and this binding to CD4 (–) cells via DARC by HIV may be used as a mechanism for the entry of HIV-1 into endothelial cells and neurons (Lachgar et al., 1998). Because P. falciparum-infected erythrocytes adhere to brain endothelial cells and cause brain hemorrhage, it is conceivable that the sequestration of parasitized erythrocytes in the brain with HIV viral particles attached may facilitate the entry of HIV into neurons in individuals that are DARC-positive. This may promote the occurrence of neurologic disorders, which are frequently seen in AIDS patients.
Programmatic concerns for interactions between malaria and HIV/AIDS are mainly at the level of diagnosis and treatment. Earlier diagnostic studies showed false positivity of blood samples from malaria-affected individuals during HIV testing (Biggar et al., 1996). This was probably due to nonspecificity of the early HIV diagnostic kits, because antigen cross-reactivity between retroviruses and malaria parasites has been reported (Lal et al., 1994). As far as treatment of uncomplicated and complicated malaria is concerned, blood transfusion for the treatment of severe malarial anemia and presumptive treatment of febrile illness have emerged as two important problems. Recent studies have shown that many of the presumed malarial febrile illnesses were actually the result of primary HIV-1 infection (Nwanyanwu et al., 1997). This problem may be more severe in areas with high prevalence of HIV and malaria, leading to unnecessary use of antimalarials for the treatment of fever. This overuse of antimalarials may contribute to the rapid emergence of drug resistance. As far as the transfusion-related transmission of HIV is concerned, earlier studies clearly revealed that use of unscreened blood for the treatment of severe malarial anemia was a factor in the transmission of HIV.
While this paper provides an account of published work on the interaction between malaria and HIV/AIDS, there is compelling evidence of interaction between other microorganisms and HIV/AIDS. It is likely that the interactions between several microorganisms present together in an individual may modulate the pathogenesis and transmission of major infectious agents.
From a mechanistic point of view, however, a common thread that seems to tie this interaction together is immune activation induced by infectious agents prevalent in malaria-endemic areas. Therefore, removing risk factors of immune activation (i.e., co-infectious agents) by effective use of drugs, physical interventions to interrupt transmission, such as bednets for malaria, and other prevention methods should have the dual effect of reduced risk of rapid progression of HIV-
related disease (by elimination or suppression of viral activating factors) and reduced morbidity and mortality by a co-infecting pathogenic organism.
The schematics of our current knowledge and the likely outcomes of the interaction between HIV and malaria are shown in Figure 2-1. It is very likely that multiple enteric, respiratory, bloodborne, vectorborne, and waterborne and foodborne agents may induce immunologic changes (even in asymptomatic infections) that promote infection, transmission, and clinical manifestation of illnesses of the co-infectious pathogens. It is therefore important to capture all morbidity data and conduct extensive diagnostic work in future studies so that the analysis can be controlled for the effect of different co-infectious agents.
In the context of HIV/AIDS and malaria, the available data should be considered in:
-
Promoting the development of and implementation of intervention guidelines and policies for prompt treatment of malaria with effective antimalarials; treatment would reduce the frequency of malaria-related illness and reduce the risk of rapid progression and transmission of HIV.
-
Incorporating prevention methods, such as the use of insecticide-impregnated bednets and environmental modifications in controlling malaria transmission.
-
Implementing blood screening guidelines in anemia-related blood transfusions.
Because of the increasing prevalence of major infectious diseases in many countries, even a small impact of coinfection-mediated increase in pathogenesis and transmission could have unparalleled human health consequences. Therefore, from a global health perspective, there is a need to raise awareness at the national level to the consequences of interactions of multiple infections in malaria-endemic regions of the world. These efforts need to be complemented by political commitment and funding at the national and international level for research and disease control and prevention programs for infectious diseases in malaria-endemic settings.
REFERENCES
Bentwich Z, Maartens G, Torten D, Lal AA, Lal RB. 2000. Concurrent infections and HIV pathogenesis. AIDS 14:2071–2081.
Biggar RJ, Miotti PG, Taha TE, Mtimavalye L, Broadhead R, Justesen A, Yellin F, Liomba G, Miley W, Waters D, Chiphangwi JD, Goedert JJ. 1996. Perinatal intervention trial in Africa: effect of a birth canal cleansing intervention to prevent HIV transmission. Lancet 347:1647–1650.
Chandramohan D and Greenwood BM. 1998. Is there an interaction between human immunodeficiency virus and Plasmodium falciparum? International Journal of Epidemiology 27:296–301.
Chattopadhya D, Kumari S, Chatterjee R, Verghese T. 1991. Antimalarial antibody in relation to seroreactivity for HIV infection in sera from blood donors. Journal of Communicable Diseases 23:195–198.
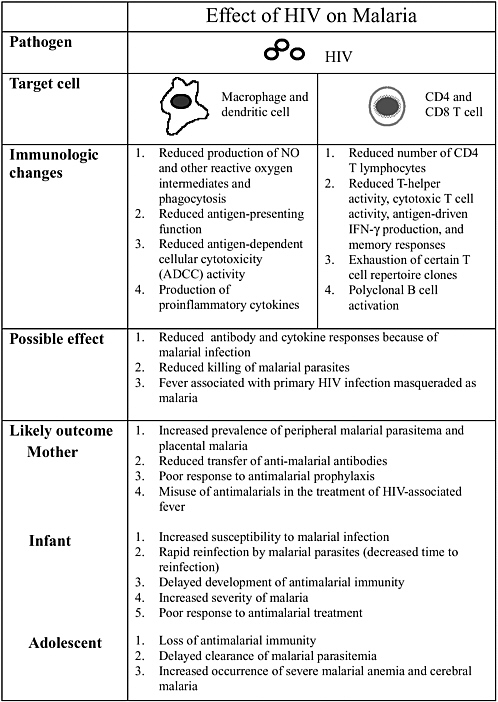
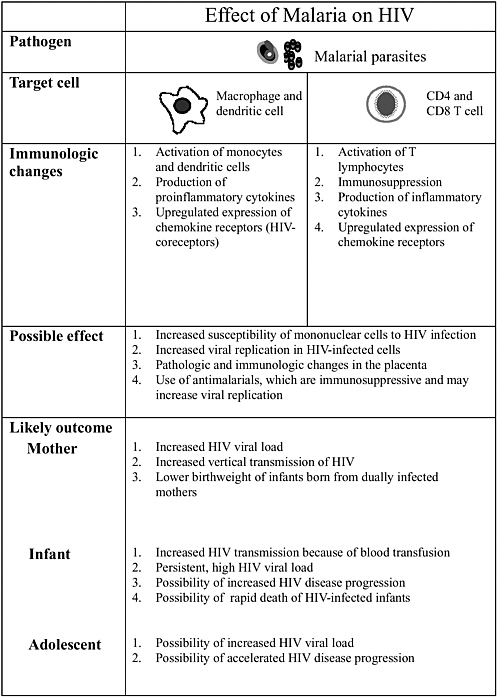
Corbett EL, Steketee RW, ter Kuile FO, Latif AS, Kamali A, Hayes RJ. 2002. HIV-1/AIDS and the control of other infectious diseases in Africa. Lancet 359:2177–2187.
Gilks CF. 1993. The clinical challenge of the HIV epidemic in the developing world. Lancet 342:1037–1039.
Grant AD, Djomand G, De Cock KM. 1997. Natural history and spectrum of disease in adults with HIV/AIDS in Africa. AIDS 11:S43–S54.
Greenberg AE. 1992. Pp. 143–148 in AIDS in the World, TW Netter, J Mann, DJM Tarantola, eds. Boston: Harvard University Press.
Hoffman IF, Jere CS, Taylor TE, Munthali P, Dyer JR, Wirima JJ, Rogerson SJ, Kumwenda N, Eron JJ, Fiscus SA, Chakraborty H, Taha TE, Cohen MS, Molyneux ME. 1999. The effect of Plasmodium falciparum malaria on HIV-1 RNA blood plasma concentration. AIDS 13:487–494.
Lachgar A, Jaureguiberry G, Le Buenac H, Bizzini B, Zagury JF, Rappaport J, Zagury D. 1998. Binding of HIV-1 to RBCs involves the Duffy antigen receptors for chemokines (DARC). Biomedicine and Pharmacotherapy 52:436–439.
Lal RB, Rudolph D, Alpers MP, Sulzer AJ, Shi YP, Lal AA. 1994. Immunologic cross-reactivity between structural proteins of human T-cell lymphotropic virus type I and the blood stage of Plasmodium falciparum. Clinical and Diagnostic Laboratory Immunology 1:5–10.
Migot F, Ouedraogo JB, Diallo J, Zampan H, Dubois B, Scott-Finnigan T, Sanou PT, Deloron P. 1996. Selected P. falciparum specific immune responses are maintained in AIDS adults in Burkina Faso. Parasite Immunology 18:333–339.
Moore JM, Ayisi J, Nahlen BL, Misore A, Lal AA, Udhayakumar V. 2000. Immunity to placental malaria. II. Placental antigen-specific cytokine responses are impaired in human immunodeficiency virus-infected women. Journal of Infectious Diseases 182:960–964.
Mulder DW, Nunn AJ, Wagner HU, Kamali A, Kengeya-Kayondo JF. 1994. HIV-1 incidence and HIV-1-associated mortality in a rural Ugandan population cohort. AIDS 8:87–92.
Nwanyanwu OC, Kumwenda N, Kazembe PN, Jemu S, Ziba C, Nkhoma WC, Redd SC. 1997. Malaria and human immunodeficiency virus infection among male employees of a sugar estate in Malawi. Transactions of the Royal Society of Tropical Medicine and Hygiene 91:567–569.
Simooya OO, Mwendapole RM, Siziya S, Fleming AF. 1988. Relation between falciparum malaria and HIV seropositivity in Ndola, Zambia. British Medical Journal 297:30–31.
Wabwire-Mangen F, Shiff CJ, Vlahov D, Kline R, Serwadda D, Sewankambo NK, Mugerwa RD, Quinn TC. 1989. Immunological effects of HIV-1 infection on the humoral response to malaria in an African population. The American Journal of Tropical Medicine and Hygiene 41:504–511.
Whittle HC, Brown J, Marsh K, Greenwood BM, Seidelin P, Tighe H, Wedderburn L. 1984. T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature 312:449–450.
Xiao L, Owen SM, Rudolph DL, Lal RB, Lal AA. 1998. Plasmodium falciparum antigen-induced human immunodeficiency virus type 1 replication is mediated through induction of tumor necrosis factor-alpha. Journal of Infectious Diseases 177:437–445.






















































