
2
Genome Databases Today
GENOME DATABASES
Genome Sequencing
An organism’s genome is the sum of its entire genetic potential, stored as an encoded sequence of the nucleotides adenine, thymine, guanine, and cytosine (A, T, G, and C) that make up its nucleic acids. Bacteria are prokaryotic: they have no organized nucleus, and most of their genes—units of heredity—are in a single large circular chromosome floating free in the cell, although some do have multiple circular chromosomes, and a few have linear chromosomes. Smaller loops of extrachromosomal DNA called plasmids can also be present. Plasmids can be passed between cells, and the instructions they contain can allow bacteria to quickly acquire properties they would not otherwise have, such as resistance to various antibiotics. The cells of more complex organisms, the eukaryotes, store most of their genomic DNA on tightly organized paired chromosomes in a membrane-bound nucleus. A few genes in eukaryotes are outside the chromosomes, in energy-processing cellular organelles called mitochondria or in chloroplasts. Viruses, which contain relatively few genes in their genomes, are parasites that pirate prokaryotic or eukaryotic cells’ replication and protein-synthesis machinery to reproduce.
Some genes encode proteins. The cells use the DNA sequences in these genes to make the corresponding sequences of amino acids. The amino acid sequences in turn determine the proteins’ structures and functions, which can be structural or functional components of cells or used to cata-
lyze the production of virtually every other building block of life. Some genes are regulatory and are involved in controlling the activity of other genes. The environmental and chemical sensing mechanisms that regulate gene activity are extraordinarily complex and are the target of a great deal of research.
The fundamental principles used today to sequence DNA were developed in the middle 1970s. The speed with which sequencing can be carried out has increased exponentially and has been largely driven by the development of automated sequencing machines and new technologies. The per-nucleotide cost of sequencing has similarly decreased; it fell by about 2 orders of magnitude between 1998 and 2003 and reached about 2 cents per nucleotide by early 2004. The increase in speed and decrease in cost are expected to continue in much the same way that the power and cost of computer processing chips have changed over the years (Carlson, 2003) (see Figure 1). Indeed, the power of sequencing technology is now
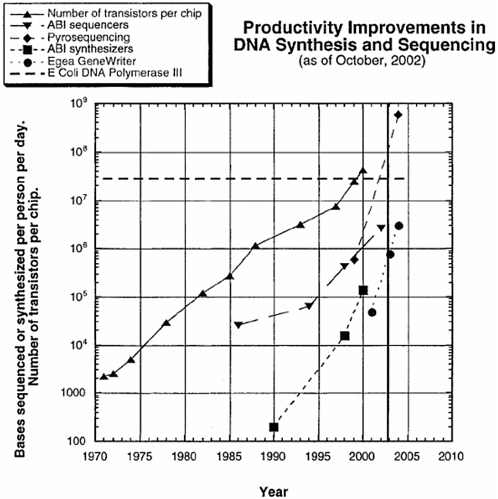
FIGURE 1 Productivity improvements in DNA synthesis and sequencing. SOURCE: Reprinted, with permission, from Carlson, 2003.
such that obtaining sequence data is no longer considered to be research, but merely a routine technical procedure carried out in the course of research, largely with fully automated and easy-to-use equipment operated by technicians. Many laboratories own automated sequencers or use central sequencing facilities in their research institutions for routine sequencing tasks. Others contract the work out to private companies or sequencing centers around the world.
The first complete genome sequence of a virus was determined in 1975 when the 3,569-nucleotide genome of MS2, an RNA virus that infects bacteria, was sequenced (Fiers et al., 1976). By the end of 2003, the complete genome sequences of more than 1,100 viral species were available in public databases. The genomes of bacteria and eukaryotic species are far larger, but in recent years determination of these sequences has also become routine. The Institute for Genomic Research (TIGR), a nonprofit institution in Rockville, Maryland, that has been a major participant in many whole-genome sequencing projects, has built a powerful infrastructure for determining DNA sequences accurately and quickly. In 1995, TIGR scientists published the first complete genome sequence of a free-living organism, the pathogenic bacterium Haemophilus influenzae, which contains 1.8 million nucleotides (Fleischmann et al., 1995). By November 2003, complete sequences of 140 bacteria had been deposited in genome databases worldwide, and at least 181 more were being determined. The genomes of dozens of other eukaryotic organisms had also been completed by then, including plants, animals, insects, fungi, and the human.
Genome Data and Analysis
The primary data that DNA sequencing generates consist of a long list of the letters A, T, C, and G in what looks like no order. For whole genomes, the list can be very long. The human genome is more than 3 billion nucleotides long, for example, and the genome of Yersinia pestis, the bacterium that causes plague—and that devastated Europe in the Middle Ages—has about 4 million nucleotides.
To keep track of sequence data, the Los Alamos National Laboratory in 1982 opened a data repository called GenBank. The purpose was to create a single repository that would allow easy access to all sequence data as they became available. GenBank moved to the National Center for Biotechnology Information (NCBI) on the National Institutes of Health (NIH) campus in Bethesda, Maryland, in 1988 and has grown to an extraordinary degree in recent years. It now contains more than 30 million gene sequences from more than 130,000 species, comprising more than 36 billion nucleotides (GenBank, http://www.ncbi.nlm.nih.gov/Genbank/genbankstats.html).
Since the middle 1980s, GenBank has coordinated its activities, policies, and data with two other large genome-sequence repositories overseas: the European Bioinformatics Institute (EBI) and the DNA Data Bank of Japan (DDBJ). Under the terms of the International Nucleotide Sequence Database Collaboration (INSDC), these three repositories exchange sequence data daily, and thereby each maintain essentially the same set of sequence data as workers around the world submit new data daily.
In addition to those three repositories, however, many other sites provide access to genome data. Some sites are comprehensive, such as those run by TIGR; the Whitehead Institute in Cambridge, Massachusetts; and the Wellcome Trust Sanger Institute in Cambridge, England. Others specialize in particular organisms or topics. WormBase.org, for example, is devoted entirely to the genome of Caenorhabditis elegans, a simple nematode often used in basic-science experiments, and the Jackson Laboratory in Bar Harbor, Maine, maintains an extensive on-line library of information and tools relevant to the mouse genome. In addition, numerous private facilities periodically download all new genome data submissions.
Raw gene sequences, however, would be of little use without computers and analytic tools to decipher them. Bioinformatics specialists have been working to create and improve such tools since gene sequences were first obtained. In those early years, sequences consisted of relatively short single genes. Researchers compared the amino acid sequences of proteins from different species by printing them out, cutting them into strips, and examining them by eye to find similarities and differences.
Today, however, entire genomes consisting of thousands of genes have been sequenced, and powerful programs running on large, networked computer systems are needed to analyze, compare, and interpret the data and store the results in an accessible form. The first level in genome analysis is called annotation. After assembling the entire sequence, computers scan the data for landmarks, such as start and stop signals for genes that encode proteins. The protein sequences of putative structural genes are predicted, and, if possible, a potential physiologic or regulatory function of each encoded protein is assigned on the basis of similarity to known proteins. Three-dimensional structural models of proteins encoded by genes can also be constructed. And the full-length genome is analyzed for the presence of entire biochemical and regulatory pathways.
Just as computer-based translations of human language can yield peculiar results, the results of computerized genome annotation are often flawed. NCBI, EBI, TIGR, and other organizations employ teams of experts to constantly check and edit the results of computer-generated annotation. Borrowing a word from museum-exhibit management, genome scientists refer to this editing of computer-generated genome sequence annotations as curation. The resulting curated annotations are stored in standard
formats in databases for easy access. The entire set of annotations for one genome can then be compared with those derived from other whole genomes. As new analytic tools become available, existing sequence data are constantly reanalyzed by both machines and people to keep them as up to date and accurate as possible.
Although annotation and curation are the first steps in making sequence data comprehensible, scientists need sophisticated tools to make efficient use of them. A remarkable array of such tools has been placed in the public domain, and new ones are under development constantly as genomics expands. NCBI, EBI, DDBJ, TIGR, the Sanger Institute, and other institutions provide many Internet-accessible analytic tools that scientists can use to query genome and other databases to solve questions relevant to their work (see, for example, the list of tools available at NCBI at http://www.ncbi.nlm.nih.gov/About/tools/index.html). The tools can also be downloaded free with annotated data for use on users’ personal computers and shared over local computer systems. Thus, the vast majority of known genome sequences and the tools to analyze them are freely available to anyone in the world who has a computer and Internet access.
In addition to gene sequences, biologists have constructed databases that contain many other kinds of data, including protein amino acid sequences, three-dimensional structures, protein functions, organism taxonomy, and protein-protein interactions. The scientific literature is indexed, and abstracts made available, through freely accessible databases maintained by the National Library of Medicine. Most newly published scientific articles are available in electronic form, although many journals limit full access to paid subscribers. In addition, databases have recently been established to catalog and make available experimental data on changing gene-expression patterns obtained in microarray experiments. Those experiments often generate far more data than can be easily interpreted by a single laboratory; the databases are intended to let other scientists access and interpret the data for their own work. EBI, for example, has a Web site called ArrayExpress for this kind of information (http://www.ebi.ac.uk/arrayexpress/), and the Microarray Gene Expression Data Society was founded in 1999 to facilitate the exchange of such data (http://www.mged.org/index.html).
Database-Access Policy
Science thrives on free and open exchange of ideas, results, data, and materials. It is a long-standing principle of scientific practice that all experimental protocols and data be completely described at the time of publication of a scientific finding. That allows others to evaluate thoroughly whether the analysis was done correctly, to repeat the experiment
if they so desire, and to use the published work to further their own research.
GenBank and other international gene sequence databases were set up to allow the free and open exchange of genome data. Consequently, the policy of the INSDC has always been to offer worldwide users open, unfettered access to all data, including genome annotations (http://www.ebi.ac.uk/embl/Documentation/INSD_policies.html). Moreover, privacy policies for all three of the member institutions state that no personally identifiable information is collected about what data a user might access or how these data might be analyzed. The largest nonprofit organizations that provide genome information and analytic tools, such as the Sanger Institute and TIGR, have similar policies mandating free, unfettered, and anonymous access.
The open-access policies are guided by broader U.S. government policy statements concerning the release of results of scientific research funded by the federal government. National Security Decision Directive 189 (NSDD-189), promulgated by the Reagan administration in 1985, states that access to fundamental research results should be unrestricted to the greatest possible extent. If such access is deemed a threat to national security, the research results should be formally classified as secret. Classified documents are available only to people who have undergone an approval process controlled by the government. Individual classified documents are then made available on a need-to-know basis and are subject to regulations on the locations and situations in which they may be viewed and stored. Recently, however, there has been considerable discussion of “sensitive but unclassified” information. How a sensitive-but-unclassified label might be used to categorize the products of life-science research remains to be seen. The issue is discussed further in the recent National Research Council report Biotechnology Research in an Age of Terrorism (NRC, 2003a). For the present, policies at the major U.S. funding agencies for life-science research still adhere to the principles set forth in NSDD-189 and long-standing scientific practice, and they require that grant recipients make research results and data publicly available. The NIH Grants Policy Statement, for example, says that “it is NIH policy to make available to the public the results and accomplishments of the activities that it funds” (NIH grants Web site, http://grants1.nih.gov/grants/policy/nihgps_2001/part_iia_6.htm).
Scientific journals also strongly support openness as a scientific norm and require the deposition of primary gene sequence data into a free and open database as a condition of publication of research results based on them. Science, for example, states that authors must “agree to honor any reasonable request for materials and methods necessary to verify the conclusions of experiments reported, and must also agree to make the data
upon which the study rests available to the scientific community. For large data sets such as DNA sequences, Science advises authors that this means deposition in GenBank or some other open database prior to publication of the paper…” (Science web site, http://www.sciencemag.org/feature/contribinfo/home.shtml). Most life-science journals have similar policies.
USE OF GENOMICS IN MODERN LIFE-SCIENCE RESEARCH
Genome data have become indispensable to the conduct of much life-science research—to the point where not many life scientists would consider starting a project without thinking about how existing genome data could be used in their experimental design. The growing importance of genomics cuts across all divisions of the life sciences to include biomedical, agricultural and environmental-biology topics; basic and applied research; and science in academic, government and industrial laboratories. Just as no entrepreneur would start a business without thinking about how to use computer technology, most biological scientists today do not go into a laboratory without incorporating available genome data into their plans.
Although access to whole-organism genome sequences has become vital to life-science research, the data do not immediately provide understanding of any organism’s natural properties, nor do they furnish a road map for manipulating the organism to give it new properties. A credible attempt to do that requires substantial experience, knowledge, training, and a great deal of patient thoughtful experimentation. And because biological systems are so intricate and finely tuned, attempts to manipulate genome structure rarely work out as the experimenter expects. Nonetheless, the growing library of genome data is an extraordinarily potent research tool. Malefactors might make use of genome data on organisms engineered by others (and selected for particular traits of interest); this would almost certainly be easier than trying to create new phenotypes themselves, but the full implications of the modification may not be clear from the publicly available data.
Genome data allow investigators to use and develop experimental tools that are far more potent than those available just a few years ago. One of the most powerful is the whole-genome microarray (Duggan et al., 1999). DNA consists of two strands that bind to one another when their sequences are complementary (see Figure 2). In the 1970s, molecular biologists developed techniques that allowed them to isolate a specific DNA sequence, make multiple copies of it, and add it to an experimental solution to “probe” for the presence of the complementary sequence. If present, the complement would bind to the probe, and the resulting double-stranded molecule could be readily detected. Finding the complementary sequence might, for example, indicate that the gene associated
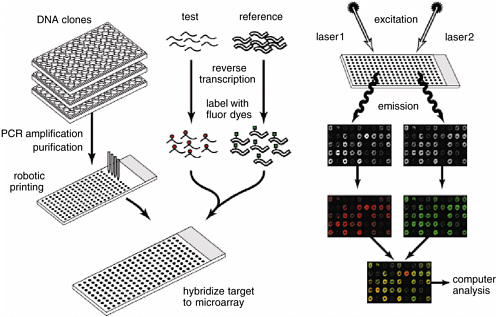
with it was being transcribed and the protein it encoded was being synthesized.
Microarray technology developed in the 1990s allows an investigator to carry out thousands of DNA probe experiments simultaneously. A microarray is generally a set of single-stranded DNA sequences, each representing a single genetic feature bound at known locations onto a suitable surface, often an ordinary microscope slide. Each bound sequence acts as a specific target for the presence of its complementary sequence found in an experimental solution, for example, material extracted from cells or from animal tissue. The presence of combined target and complement is commonly detected by using sensitive fluorescent markers (to mark the DNA in the experimental solution) and a laser scanning device. Microarrays can be constructed by using the same principle as an inkjet printer; an area scarcely the size of a coin can contain individual samples of DNA sequences corresponding to every one of the thousands of genes in an organism’s genome. Scientists now routinely use microarrays to determine how gene-expression patterns change in cells in response to experimental variables or to study the DNA sequence of a microbial agent. Studies in differential gene expression are performed in organisms as diverse as bacteria and human cells and, indeed, can be used to monitor the changes that occur when cells are infected by parasites, become cancerous, or are just growing normally. They have made it possible to detect unique molecular “signatures” of biological events. This powerful technology depends entirely on the availability of genome data, and today a single scientist performing a single experiment can obtain data that would have taken years to obtain with older techniques—if they would have been collectible at all.
The impact of genomics goes well beyond microarrays and other technical advances that improve the efficiency of data collection. As the entire genomes of many organisms have been sequenced and made widely available, research scientists have begun to analyze genomes as an individual complex biological system and compare them with each other. This kind of comparative analysis, called systems biology, will greatly accelerate understanding of how entire organisms work and how organisms interact with one another. In particular, it will initially facilitate understanding of bacterial genomes (Rappuoli and Covacci, 2003). As more genomes are sequenced, the power of comparative genomics will increase our ability to understand larger biological systems (Kanehisa and Bork, 2003). Two examples—development of meningococcus B vaccine candidates and the recent experience with the SARS coronavirus—illustrate how genome data and contemporary experimental techniques have permitted the rapid development of new products and tools to fight infectious diseases. These examples focus on human health but the techniques used are equally
applicable to the development of countermeasures to plant and animal pathogens.
Meningococcus B Vaccine
Neisseria meningitidis (the meningococcus) is a bacterial species that can cause meningitis (an infectious disease of the fluid and membranes surrounding the brain and spinal cord) and septicemia (infection of the blood). Those infections are fatal if untreated. The bacterium is spread through intimate contact and airborne droplets.
Several distinct strains of the meningococcus have been identified, and vaccines for many of them have been developed with standard methods (Moxon and Rappuoli, 2002). All vaccines rely on the principle that after the immune system has been primed to mount a protective response to an agent, it can mobilize quickly to defeat infection when the immunized person is exposed to the agent again (Grifantini et al., 2002). Classical vaccines are based on the administration of killed or attenuated versions of pathogenic agents and increasingly on the use of purified molecules from cultured bacteria that can elicit a protective immune response when injected into a susceptible person.
In the case of N. meningitidis, it was known that the injection of a polysaccharide capsule from the meningococcal cell wall worked well as a vaccine for preventing meningococcal disease caused by different bacterial types—A, C, Y, and W135 (Pizza et al., 2000; Adu-Bobie et al., 2003). However, the corresponding capsular polysaccharide from type B meningococci was ineffective for protection because it is very similar to a molecule that humans also produce; the immune system usually fails to produce antibodies against such “self” antigens and thus avoids harming the host. Despite years of effort, biomedical scientists failed to find a protective molecule that would induce immunity to type B meningococcal disease (Moxon and Rappuoli, 2002).
The complete genome of N. meningitidis type B was sequenced in 2000 (Tettelin et al., 2000). Scientists at Chiron Corporation then used the sequence in “reverse vaccinology.” That is, they worked from gene to protein to vaccine candidate rather than purifying various bacterial constituents for testing as protective antigens. Reverse vaccinology uses the analysis of every gene in the type B meningococcal genome, looking for gene products that might encode proteins likely to be “seen” by the immune system during infection; these are molecules predicted to be either on the bacterial surface or secreted by the microorganism into its surroundings. Of the 600 such type B genes identified in their computer analysis, they inserted 350 into E. coli that then manufactured the corresponding encoded type B proteins. The recombinant proteins were puri-
fied and injected into mice. Blood serum from the immunized mice was then analyzed to see which might contain antibodies that would bind to the N. meningitides type B cell surface and kill the bacteria. They discovered 28 candidate antigens that could induce this killing (bactericidal) activity. Five of them are now in initial clinical testing—less than 3 years after the genome data first became available and after more than 2 decades of failure with standard pregenomic vaccine-development methods. It is possible that products could enter the marketplace within 5 years.
That work is the first example of how a genomic approach can lead to novel vaccine candidates. It will certainly not be the last. Similar reverse-vaccinology efforts are currently under way to apply the strategy to several other microbial pathogens, including those of malaria, plague, and anthrax (Mora et al., 2003).
Those efforts and a second generation of vaccine-discovery strategies aimed at pathogenic microorganisms make use of proteomics and wholegenome microarray analysis of gene transcription to find potential protective antigens. Proteomics refers to the rapidly advancing ability to isolate and analyze large numbers of proteins from a cell efficiently. It provides additional structure and function data about various candidate protein antigens that will help to identify the most promising among them. In addition, microarray analysis of bacterial gene activation when the bacteria first encounter host cells can complement the kind of genome analysis described above. Specifically, these experimental approaches allow identification of genes that are not active in cell culture but produce their encoded protein only when the bacteria are actively interacting with an infected host. For example, when this type of analysis was applied to N. meningitides type B, several additional protective antigens were discovered that are made only in the presence of human cells. Those antigens could not have been characterized with genome analysis alone or by simply isolating various proteins from cultured N. meningitidis cells grown on ordinary laboratory growth media (Grifantini et al., 2002). It requires the use of genome information and carefully executed laboratory experiments.
SARS Coronavirus
The disease that became known as severe acute respiratory syndrome (SARS) first came to world attention on March 12, 2003, when the World Health Organization (WHO) issued a global health alert about an atypical pneumonia in Viet Nam, Hong Kong, and Guangdong Province, China. The global research community responded vigorously with all the tools of modern science. Within 6 weeks, the virus that causes the disease, dubbed SARS coronavirus (SARS-CoV), had been isolated and cultured
and its 29,727-nucleotide genome completely sequenced and posted on the Internet. In the months that followed, dozens more SARS-CoV isolates were sequenced and published.
The availability of the sequence data quickly put to rest fears that SARS was the result of a laboratory-fabricated agent. The sequence data also allowed research scientists throughout the world to begin immediately to analyze viral structure, function, and the molecular basis of how it might cause illness. The sequence quickly revealed that the new virus was related to other coronaviruses and provided key insights into its potential pathogenic mechanisms. The sequence data were also crucial to global efforts to develop candidate vaccines, antiviral drugs, and especially accurate, sensitive diagnostic tests.
Vaccines. Within 3 months of the initial WHO alert, workers in academic, government, and industrial laboratories had created several SARS-vaccine candidates and were moving to test them in animal models. Many more vaccine candidates have since been created. In most cases, vaccine development relied entirely on knowledge of the viral sequence. Anti-SARS DNA vaccines, for example, are based on DNA sequences that encode portions of a viral protein. Those DNA sequences are injected directly into a vaccine recipient, whose cells take up the injected DNA and express the viral protein in a way that stimulates the immune system. Scientists have also created several live attenuated vaccine candidates by inactivating specific genes in the viral genome. Without sequence information, those vaccine strategies could not have been pursued so quickly. Several of the candidate vaccines have shown initial promise, and some are in preclinical testing in nonhuman primates.
Drug Therapies. In a search for antiviral drugs, the goal is to find a compound that can disrupt the viral life cycle without harming the infected host. Screening efforts were begun immediately after SARS emerged, looking for compounds that could prevent viral replication in cell culture. Many compounds screened were obvious choices; they included every known drug that might have antiviral activity. The choice of other potential antiviral-drug candidates, however, relied on insights provided by the SARS sequence. For example, analysis of the sequence made it clear that SARS-CoV enters cells by fusing with their outer membrane. That immediately suggested that drugs that inhibit membrane fusion might be active against SARS. Furthermore, knowledge of the amino acid sequence and the three-dimensional structures of the SARS proteins involved in fusion provided clues for targeted development of more-efficient fusion inhibitors.
The sequence data also provided clues about how to design drugs that could interfere with other viral proteins. For example, the SARS virus
contains a protease enzyme that cleaves and thereby activates many other viral proteins. Possession of the sequence of the protease allowed protein chemists to quickly construct three-dimensional molecular models, which revealed it to be structurally similar to a protease from rhinoviruses, a separate family of viruses that cause the common cold. Research workers at the pharmaceutical company Pfizer took particular note of that similarity because they had recently designed and synthesized a series of peptides intended to inhibit the rhinovirus protease. Some of those compounds were found to partially block SARS-CoV replication in cell culture. Pfizer scientists then began to refine the SARS protease structural model and to design new drug candidates that might bind the SARS protease more tightly and therefore more effectively inhibit its activity and serve as a potent anti-SARS therapeutic agent.
Diagnostics. The initial symptoms of SARS, like those of many other viral infections, are fever, malaise, and other nonspecific “flu-like” symptoms. Thus, fast and accurate diagnostic tests are needed to separate SARS patients from those with less serious illnesses. The availability of the SARS sequence has greatly accelerated diagnostic development. One standard method for detecting virus in a clinical sample involves the use of antibodies that can bind to viral proteins. Creating SARS-specific antibodies requires purification of viral proteins from cultured virus, which are injected into animals to produce antibodies. The availability of the SARS sequence allows the use of more-efficient genetic-engineering techniques to make recombinant versions of the SARS proteins in bacteria to produce antibodies that can be used in diagnostic tests.
Two of the most promising new diagnostic strategies, polymerase chain reaction (PCR) and diagnostic DNA microarrays, rely entirely on sequence data. In PCR, DNA sequences that are complementary to specific viral sequences are synthesized in the test tube and added to a clinical sample with appropriate transcription enzymes. If SARS viral sequences are present in clinical material, such as sputum, they are easily identified with specific enzyme-detection methods. Thanks to the availability of the SARS sequence, many PCR kits and procedures for SARS detection have been developed around the world.
GENOMICS AND BIOTERRORISM
Modern Technology
As noted above, access to sequence data and the associated tools needed to analyze them are indispensable tools for life-science research. But genome databases are also of interest to anyone who might want to
enhance pathogens for destructive purposes. Just as any scientist planning research aimed at finding new cures for infectious diseases would tap the power of genomics, so would any malefactor setting out to create engineered pathogens for use as biological weapons.
In the future, newly engineered agents will be a growing concern, but even natural pathogens can be used to carry out devastating attacks. The naturally occurring forms of the “category A” infectious agents that the Centers for Disease Control and Prevention considers to be the worst potential bioterror threats—those of anthrax, smallpox, botulism, plague, tularemia, and viral hemorrhagic fevers—already have inherent properties that give them terrible destructive potential. The most difficult step in carrying out an attack with such an infectious agent is neither obtaining a starter culture of the organism nor expanding it to produce a quantity needed to conduct an effective bioterror attack. Instead, the highest hurdle is preparing and disseminating the agent so that it can be delivered effectively to a dispersed target population; for contagious agents, this problem is less difficult because infection of a relatively small number of people can spread widely. (That is not the case for smallpox, however, which is known to exist only in two high-security laboratories and would therefore be very difficult for a terrorist to obtain.) For an attack intended to affect many people simultaneously, the delivery vehicle of choice would probably be an aerosol or the food chain. If effectively executed under even less than optimal conditions, such an attack could be catastrophic. Even a small-scale attack can have serious consequences, as shown by the 1984 Salmonella typhimurium salad bar attacks in The Dalles, Oregon, and the better known 2001 anthrax attacks (Torok et al., 1997). Those consequences can include illness, death, and social and economic disruption. Although the dissemination of an infectious agent for either a large-scale or a small-scale bioterrorism attack may be difficult, it is important to acknowledge that relatively unsophisticated dissemination methods are effective.
Regulations on access to genome data would not affect the ability of a terrorist to carry out an attack with naturally occurring pathogens. The techniques used to prepare agents for this kind of attack were mastered by workers in biological-warfare programs in the 1950s, and a large, unclassified technical literature relevant to the methods required to aerosolize pathogens already exists. Although the technical hurdles that would confront a bioterrorist intending to deploy a naturally occurring agent to cause large numbers of casualties are substantial, they are much lower than those associated with enhancing the virulence of a known pathogenic species with genetic manipulation. Thus, an attack with a natural pathogen is still the most likely; however, given the developments in biotechnology described in this report, a more sophisticated attack with an engineered pathogen is a serious concern.
Potential Malefactors
Those who might attempt to make an engineered pathogen that has enhanced properties are in several categories. Nations conducting dedicated biological-warfare research and development programs are likely to have access to substantial funding and the requisite expertise. However, attempting to create an enhanced pathogen would not be beyond the capabilities of a lone person with the appropriate background, a relatively modest budget, and a destructive agenda (Carlson, 2003). A nationstate or a group might be able to recruit a scientist who already has access to facilities and could use them for biological-weapons research. Subnational terrorist groups, such as Al Qaeda, or apocalyptic religious groups, such as Aum Shinrikyo, which released the nerve gas Sarin in the Tokyo subway in 1995 and experimented with biological weapons, might also attempt such a project (Lifton, 1999). Press reports assert that Al Qaeda is attempting to achieve biological-weapons capability, although of a conventional variety involving naturally occurring pathogens and toxins (Petro and Relman, 2003). It is possible that fanatical religious groups with apocalyptic fantasies, such as Aum Shinrikyo, would be interested in developing enhanced pathogens intended to cause an indiscriminate global catastrophe (Kaplan, 2000).
As the technology for manipulating DNA becomes more widespread and easier to use, some observers have suggested that large numbers of amateur experimenters might begin to dabble in the molecular engineering of organisms (Carlson, 2003). One technology columnist, for example, went so far as to suggest that perhaps “bathtub biotech” will do for biology what garage hackers did for information technology (Schrage, 2003).
Maybe bioinformatics and the diffusion of genetic engineering technologies and techniques will inspire a new generation of bio-hackers. Certainly the technologies are there for those inclined to genetically edit their plants or pets. Maybe a mouse or E. coli genome becomes the next operating system for hobbyists to profitably twiddle.
Whether many “bio-hackers” will actually emerge remains to be seen. Most amateurs today would be unlikely to achieve much through manipulation of microbial genomes, which is far more difficult than many people outside the scientific community recognize. In addition to relevant biological training, a potential terrorist interested in engineering new pathogens would need access to appropriately equipped experimental facilities. Unexpected difficulties often arise in this type of work. For some organisms, genetic systems are not well understood, so many details must be determined from scratch. And random and targeted gene insertions or deletions can have unintended consequences that change the phenotype of an organism in ways not anticipated.
Apart from ability, the proportion of amateurs who would be interested in deliberate manipulation of pathogens is not known. Given the record of destructive computer viruses created by computer hackers, however, the possibility that the tools needed to carry out 21st-century genetic engineering will be available to hobbyists the world over is unsettling and underscores the fact that individuals or groups that might want to enhance an organism’s potential to cause harm theoretically have powerful tools within their reach. Large-scale bioterrorism is unlikely, but the possibility of such a rare devastating event dictates that we not dismiss it and that we be vigilant.
How Genome Data Might Be Misused
One way that governments, groups, or individuals might misuse genome data would be to conduct primary research on pathogen enhancement, starting with hypotheses that they generate themselves from genome analysis and pursuing them experimentally in the laboratory. Creating a genetic construct that an experimenter believes might be more virulent than its naturally occurring form is not very demanding technically. However, experimental evidence has shown that enhancing pathogenicity is quite difficult. Manipulations of biological systems rarely turn out as planned; attempts to change one property, even if successful, usually have consequences that the experimenter does not want. Pathogens have been highly refined by nature over many millennia, and even minor manipulation can lead to unexpected consequences. The scientific community does not understand virulence and pathogenesis well enough to predict the results of genetic engineering reliably. If the genome is changed to overproduce a known virulence factor, for example, the organism could be unchanged phenotypically or not be able to infect its host efficiently in a real-world setting. For example, Isberg and Falkow in the 1980s showed that the gene for a protein they called invasin, when transferred from Yersinia pestis into E. coli, permitted entry of E. coli into cultured mammalian cells (Isberg and Falkow, 1985); this was discussed in the book Germs: Biological Weapons and America’s Secret War (Miller et al., 2002). However, not everyone who read the Isberg and Falkow paper understood that the inheritance of invasin did not turn E. coli into a pathogen. When investigators put the invasin gene into Salmonella and Shigella to determine its effects on virulence, it had no effect (Voorhis et al., 1991). In addition, although invasin is active in the closest known relative of Yersinia pestis, it is inoperative in Yersinia pestis itself. (Rosqvist et al., 1988)
Instead of pursuing enhancement strategies of their own, however, malefactors might limit themselves to replicating or adapting published results that have revealed how pathogens can be enhanced. As the
example above illustrates, replicating the same type of genetic modification in a new organism may or may not have a similar result. Such experiments generally are in the category of “functional genomics” because they go beyond obtaining gene-sequence data to tie specific genome information to the implied specific functions, capabilities, or vulnerabilities of an organism. Successful manipulation of microorganisms to create more-efficient biological weapons probably requires methodical investigation of the types of changes that individual kinds of microorganisms can handle, a long time spent on trial-and-error experiments, or much good luck. The cases presented here are only examples. They illustrate that it is no easy feat, but certainly is possible, to use genome data to design an enhanced biological weapon.
As stated above, results that have immediate implications for pathogen enhancement or weapons development have been called contentious research (Epstein, 2001) or are said to be in a gray zone where the benefits of publication might not outweigh the dangers. At the workshop one speaker suggested that the Nature Biotechnology paper “Engineering hypervirulence in a mycoherbicidal fungus for efficient weed control” (Amsellem, 2002) might fall into this zone because it uncovered unanticipated lethality for tomato and tobacco plants. This presenter also discussed his own views that the gray zone has an evolving nature, as well as differing shades of gray, and called for continuing review and discussion of the gray zone and of whether any monitoring or control efforts would be beneficial. He called for absolutely transparent discussions that involve multiple communities (scientific, intelligence, public, and policy).
Another example of such gray zone research was publicized in late October 2003 when scientists at St. Louis University extended previously published work in which the mousepox virus was made hypervirulent and capable of overcoming an effective vaccine (Washington Post, 10/31/03, pg A1). In 2001, an Australian research group had shown that insertion of a gene for the mouse version of an immune regulator called interleukin-4 (IL-4) into the mousepox virus increases the virus’s virulence; cells infected with the modified virus produce excess IL-4, which “jams” the IL-4 signal and thereby disrupts the normal immune response to infection. As part of a broader effort to explore possible countermeasures to address engineered pox viruses, the St. Louis scientists extended the Australians’ work, inserting the IL-4 gene into a different part of the mousepox genome so that it came under the control of different regulatory sequences that increased the amount of IL-4 generated in infected cells. The result was an extraordinarily potent virus that killed every one of the mice it infected, including those previously vaccinated. It is interesting to note, however, that, unlike the wild-type virus, the altered virus reportedly was not transmitted from animal to animal.
Those results are significant in two ways. First, they provide an obvious starting point for anyone who might want to create and release an enhanced pox virus. Mousepox is closely related to several viruses that can cause disease in humans, including viruses that cause smallpox, cowpox, and monkeypox. It would not be difficult for a skilled scientist or technician to use the published results to carry out an analogous genetic manipulation of one of those viruses; the effect of the manipulation of the other pox viruses on virulence and on the ability to overcome vaccine in these is not known. Second, they clearly demonstrate the important principle that gene sequences in a host can be as important to people who intend to enhance a pathogen as gene sequences in the pathogen itself. In this case, replicating the work requires the human IL-4 gene sequence. It should be noted that the effect of the modification in the mousepox virus was to make the host more susceptible. That requires an understanding of the host in addition to an understanding of the virus. A number of microbial pathogens cause disease by manipulating host immune function. One important implication is that a potential bioterrorist could use human, animal, or plant genome sequences to create a more dangerous pathogen.


















