
Part IV
GENOMIC APPROACHES AND NEW INSIGHTS ON DIVERSITY
Because Mayr was not a geneticist, we do not count among his direct legacies our current era of genomics. But, in some respects, genomic studies of biological diversity are just the next step on a ladder that Mayr helped to hoist. Furthermore, it is fair to ask whether genomic tools are changing our view of biological diversity. One example of the way our view has changed is provided in the article by Ochman et al. (Chapter 12) that is mentioned above. Another example lies in the paper by James Lake and colleagues, “Decoding the Genomic Tree of Life” (Chapter 14), who use genomic data to reconstruct the process by which eukaryotes arose from prokaryotes. Unlike typical phylogenetic events, such as the splitting of lineages, eukaryotes appear to have arisen by the fusion of genomes. The authors describe the development and application of a new phylogenetic method, called “conditioned reconstruction,” which is designed to detect fusion events.
As the number of sequenced genomes grows, so will the number and availability of tools for identifying the genes responsible for important variation. This is a major point of the paper by Scott Edwards et al. (Chapter 6) that was discussed above. Two other papers in this volume demonstrate some of the latest techniques for finding genes responsible for phenotypes of interest. Stuart Macdonald and Anthony Long, in “Prospects for Identifying Functional Variation across the Genome” (Chapter 15), describe a new method for reducing the number of single nucleotide polymorphisms that are required in association mapping studies for genes that contribute to traits that have recently been under natural selection. The idea follows
from the expectation that recent selection will have shaped divergence, and especially polymorphism, in and around the relevant sites. Using population genetic predictions of the response to selection of linked sites, it should be possible to conduct genomic scans of variation and divergence to identify the subset of polymorphic sites upon which to base an association mapping study. They demonstrate the method by looking at polymorphism within and divergence between Drosophila species.
Trudy Mackay et al., in “Genetics and Genomics of Drosophila Mating Behavior” (Chapter 16), also used a Drosophila model to identify genomic sites with interesting functions—mating behavior, in this case. Traditionally, genes that are directly involved in reproduction are not the easiest to study genetically, simply because mutants often have low reproductive success. These authors took the artificial selection approach and generated, over the course of 20 generations, two lines of Drosophila melanogaster that had high and low mean values for mating speed. They then conducted a microarray study to see which genes differed in expression level between the two divergently selected lines of flies.
The final paper in the volume takes an explicitly forward look and describes the ongoing and future changes that are happening to the biological sciences. With genomic sequences for many organisms having been available for several years, many biologists are turning to the highly integrated study of cellular processes and networks, a field that is called Systems Biology (Hood, 2003). Mónica Medina, “Genomes, Phylogeny, and Evolutionary Systems Biology” (Chapter 17), writes about how this nascent field is being shaped by the availability of genome sequences throughout the tree of life and of the kinds of questions about the evolution of networks that we can anticipate. Surely, just as Systems Biology emerges and qualitatively new kinds of insights emerge about how cells function, so too will emerge the field of Evolutionary Systems Biology with concomitant insights on the evolution of cell function.
REFERENCE
Hood, L. (2003) Systems biology: Integrating technology, biology, and computation. Mech. Ageing Dev. 124, 9–16.
14
Decoding the Genomic Tree of Life
ANNE B. SIMONSON,†§ JACQUELINE A. SERVIN,* RYAN G. SKOPHAMMER,† CRAIG W. HERBOLD,* MARIA C. RIVERA,†§ AND JAMES A. LAKE*†‡§
Genomes hold within them the record of the evolution of life on Earth. But genome fusions and horizontal gene transfer (HGT) seem to have obscured sufficiently the gene sequence record such that it is difficult to reconstruct the phylogenetic tree of life. HGT among prokaryotes is not random, however. Some genes (informational genes) are more difficult to transfer than others (operational genes). Furthermore, environmental, metabolic, and genetic differences among organisms restrict HGT, so that prokaryotes preferentially share genes with other prokaryotes having properties in common, including genome size, genome G + C composition, carbon utilization, oxygen utilization/sensitivity, and temperature optima, further complicating attempts to reconstruct the tree of life. A new method of phylogenetic reconstruction based on gene presence and absence, called conditioned reconstruction, has improved our prospects for reconstructing prokaryotic evolution. It is also able to detect past genome fusions, such as the fusion that appears to have created the first eukaryote. This genome fusion between a deep branching eubacterium, possibly an ancestor of the cyanobacterium and a proteobacterium, with an archaeal eocyte (crenarchaea), appears to be the result of an early symbiosis. Given new tools and new
genes from relevant organisms, it should soon be possible to test current and future fusion theories for the origin of eukaryotes and to discover the general outlines of the prokaryotic tree of life.
Today there is enormous interest in discovering the tree of life. But as we get closer to reconstructing it, new experimental and theoretical challenges appear that cause us to reexamine our goals. New obstacles may initially seem insurmountable, but in reality they enrich our understanding of the evolution of life on Earth.
One of the most recent evolutionary mechanisms to challenge our view of genome evolution is the massive horizontal gene transfer (HGT) that has recently become so apparent (Campbell, 2000; Doolittle, 1999a; Gogarten et al., 2002; Karlin et al., 1997; Koonin et al., 2001; Lawrence and Ochman, 1998, 2001; Rivera et al., 1998). This genetic crosstalk theoretically has the potential to erase much of the history of life that has been recorded in DNA. Indeed, some scientists think that HGT has already effectively erased the phylogenetic history contained within prokaryotic genomes (reviewed in Doolittle, 1999b).
Although sympathetic to many of these points, we think the best way to decide whether the tree of life is knowable is to try one’s hardest to determine it. This article reviews the progress made using whole-genome analyses but does so primarily from the unique perspective of our laboratory. When Darwin uttered his famous quote, “The time will come I believe, … when we shall have fairly true genealogical trees of each great kingdom of nature,” (1887) he was not describing prokaryotic life. Rather, he probably envisioned understanding the trees of animal and plant life. In that sense, part of his dream is already a reality. We currently understand the major radiations of the bilateral animals (Aguinaldo et al., 1997; Halanych et al., 1995), and the relationships linking the major plant groups are starting to be understood (Karol et al., 2001; Pryer et al., 2001, 2002; Nickrent et al., 2000; Soltis et al., 1999). This review, however, focuses on understanding the radiations that occurred even before those of the plants and animals, namely the enigmatic evolution of prokaryotes and the emergence of eukaryotes.
The origin of the eukaryotes was a milestone in the evolution of life, because eukaryotes are utterly different from prokaryotes in their spatial organization. Eukaryotes, for example, possess an extensive system of internal membranes that traverse the cytoplasm and enclose organelles, including the mitochondrion, chloroplast, and nucleus. This compartmentalization has required a number of unique eukaryotic innovations. The most dramatic innovation is the nucleus, a specific compartment for storing and transcribing DNA, for processing DNA and RNA, and possibly even for translating mRNAs (Hentze, 2001). The nucleus is unique to
eukaryotes, hence it and the nuclear genome are the defining characters for which eukaryotes are named (eu, good or true; karyote, kernel, as in nucleus).
The prokaryotes, with their simple cellular organization, are generally thought to have preceded the eukaryotes (although see Poole et al., 1999). Which prokaryotic groups branched first, however, is not clear, because the root of the tree of life is uncertain and in flux due to a concern that artifacts of phylogenetic reconstruction may have unduly influenced the location of even the root that has the most experimental support (Penny and Poole, 1999; Philippe and Forterre, 1999).
THE HGT REVOLUTION
The possibility of analyzing complete genomes awakened interest in prokaryotic genome evolution and profoundly changed our understanding of genome evolution. Before the first genomes were sequenced, there was nearly unanimous scientific agreement that prokaryotic genomes were evolving clonally, or approximately so. In other words, as generation after generation of bacteria divided, each bacterium would contain the DNA it inherited from its parent, except that occasionally a single DNA nucleotide might have mutated, causing a minor change in the daughter genome. Thus it was thought that the family tree derived from any one gene would look like the family tree from any other gene. Diploid eukaryotic cells with two copies of each gene per cell slightly complicated this picture, but they, too, were thought to be evolving clonally. Most researchers felt comfortable with the premise that reliable organismal trees could be calculated from sequences of individual genes. In particular, rRNA genes were favored, because rRNA was easy to sequence, and it was assumed trees calculated from rRNA would probably be the same as those calculated from any other genes. However, it was not acknowledged that HGT had the potential to significantly alter gene trees. For example, if a gene were horizontally transferred from a prokaryote to a human, then the tree reconstructed from that gene would place humans in the midst of prokaryotes. Furthermore, each gene tree would show a different set of relationships. (Sometimes one keeps track of whether the transferred genes are new to the genome or whether they replace existing genes. Although this distinction can be important, in this paper, we will refer to both types of exchange as HGT.) Because so much attention was focused on the approximately clonal evolution of rRNA in the pregenomic era, only a few genes other than rRNA were sequenced from multiple organisms, and HGT was largely overlooked.
Once complete genomes were available, the pace of discovery accelerated, as highlighted in early analyses of complete, or nearly complete,
genome studies from the laboratories of R. Doolittle (Doolittle and Handy, 1998), W. F. Doolittle (Brown and Doolittle, 1999), Gogarten (Gogarten et al., 1999), Golding (Ribeiro and Golding, 1998), Ochman (Lawrence and Ochman, 1998), and ourselves (Rivera et al., 1998). These and even more recent studies of the evolution of life, based on analyses of complete genomes, described below, revealed the flaws in the old view of clonal evolution. Scientific opinion has now shifted and favors a significant role for HGT in prokaryotic genome evolution.
HGT HAS PROFOUNDLY AFFECTED OUR UNDERSTANDING OF PROKARYOTIC GENOME EVOLUTION
Three remarkable new findings, based on analyses of whole genomes, have engendered appreciation for the important role of HGT in prokaryotic evolution. First, HGT is now generally recognized to be rampant among genomes (rampant at least on a geological timescale). Second, not all genes are equally likely to be horizontally transferred. Informational genes (involved in transcription, translation, and related processes) are rarely transferred, whereas operational genes (involved in amino acid biosynthesis, and numerous other operational activities) are readily transferred. Third, biological and physical factors appear to have altered HGT. These include intracellular structural constraints among proteins (the complexity hypothesis), interactions among organisms, and interactions with the physical environment. These three findings are described below.
EVIDENCE FOR EXTENSIVE HGT
As early as 1996, the complete sequence of the methanogen Methanococcus janaschii (Bult et al., 1996) revealed that its genome consisted of certain groups of genes that were much more similar to eukaryotic genes than those from bacteria, whereas other groups of genes were much more closely related to their bacterial homologs. Koonin et al. (1997) substantiated that the M. jannaschii genes for translation, transcription, replication, and protein secretion were more similar to eukaryotes than to bacteria. They interpreted this finding to mean that archaea were a chimera of eukaryotic and eubacterial genes (Koonin et al., 1997). Using whole-genome phylogenetic methods, our laboratory discovered the presence of two superclasses of genes in prokaryotes that had different relationships to eukaryotic genes. In that study (Rivera et al., 1998) of the Escherichia coli, Synechocystis PCC6803 (a cyanobacterium), M. jannaschii, and Saccharomyces cerevisiae genomes (Blattner et al., 1997; Bult et al., 1996; Goffeau et al., 1996; Kaneko et al., 1996), the M. jannaschii informational genes, consisting of gene products responsible for such processes as trans-
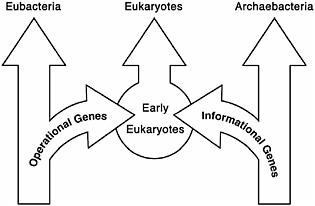
FIGURE 14.1 Early genome studies indicated that eukaryotes were a mixture of eubacterial and archaebacterial genes with an unusual distribution. The operational genes were primarily from the eubacteria, and the informational genes were from the archaebacteria.
lation and transcription, were found to be most closely related to those found in eukaryotes. The operational genes of the eukaryote, responsible for the day-to-day operation of the cell (operational genes), on the other hand, were most closely related to their counterparts found in E. coli and Synechocystis (Rivera et al., 1998). Of the yeast genes analyzed, approximately one-third were informational genes, and two-thirds were operational genes. This provided good evidence that the 16S rRNA tree does not reflect the evolution of all of the genes in a genome and also supplied evidence that early eukaryotes were a chimera of eubacteria and archaebacterial genes. A stylized illustration of these results is shown in Fig. 14.1. Recently, a thorough comprehensive analysis involving large numbers of genomes and genes has documented the strength of this correlation (Esser et al., 2004).
Further evidence for extensive HGT came from the observation that another methanogen, Methanobacterium thermoautotrophicum, contains several regions that have an ≈10% lower G + C content than the G + C content of the whole genome on average (Smith et al., 1997). ORFs in these regions exhibit a codon usage pattern atypical of M. thermoautotrophicum, suggesting that the DNA sequences may have been acquired by HGT (Smith et al., 1997).
Additional evidence for HGT came from a thermophilic relative of the methanogens, Archaeoglobus fulgidus. ORFs in the functional categories of translation, transcription, replication, and some essential biosynthetic pathways in this prokaryote are very similar to those in M. jannaschii. However, these two genomes differ in many of their opera-
tional genes, such as those for environmental sensing, transport, and energy metabolism (Klenk et al., 1997). The tryptophan biosynthesis pathway in A. fulgidus seems very closely related to the eubacterium Bacillus subtilis, even though these two are separated by large distances on the 16S tree (Klenk et al., 1997). These observations suggested that the extent of gene exchange that has occurred in the methanogens and their relatives is tremendous.
Among the extreme thermophiles, some of which live in temperatures in excess of the boiling temperature of water, HGT is equally prevalent (Makarova et al., 1999). Lecompte et al. (2001) compared the three closely related proteomes from the high-temperature methanogen relatives Pyrococcus abyssi, Pyrococcus furiosus, and Pyrococcus horikoshii. In their gene analysis, the ORFs encoding translation proteins and transcription proteins (informational genes) fairly consistently indicated that the distances among the three species were uniform, as would happen if these genes were evolving approximately clonally. However, most other ORFs (mainly operational genes) gave a wide distribution of distances. The existence of a distribution was interpreted as evidence of HGT (Lecompte et al., 2001), because the horizontal transfer of genes from closely and distantly related organisms would be expected to correspond to heterogeneous distances. In addition, P. furiosus is capable of transporting and metabolizing maltose/maltodextrin, properties that are absent in P. horikoshii. Of two maltose/maltodextrin import systems in P. furiosus, one has the greatest similarity to the transport system in E. coli, a finding most parsimoniously explained as a lateral transfer of the entire system from E. coli to P. furiosus (DiRuggiero et al., 2000; Maeder et al., 1999). Comparison between P. furiosus and P. abyssi has revealed linkage between restriction-modification genes. Because codon usage is different in various organisms, the codon biases of some restriction-modification systems in the Pyrococcus genomes suggest that these systems have been acquired by horizontal transfer (Chinen et al., 2000).
HGT is also widely prevalent in the eubacteria [see the article by Ochman et al., “Examining Bacterial Species Under the Specter of Gene Transfer and Exchange” (Chapter 12)]; this has been demonstrated in Aquifex aeolicus, where little consistency was seen among trees reconstructed from a number of operational genes (Deckert et al., 1998). Comparative analyses of E. coli ORFs showed that 675 E. coli ORFs have greatest similarity to Synechocystis, 231 to M. jannaschii, and 254 to the eukaryote S. cerevisiae (Blattner et al., 1997). Using skewed base composition and codon usage as a measure of an alien gene, Ochman and coworker (Lawrence and Ochman, 1998) argued that 755 of 4,288 E. coli ORFs have been horizontally acquired in 234 lateral transfer events, because E. coli diverged from Salmonella ≈100 million years ago (Lawrence and Ochman, 1998).
Classically, the three principal molecular mechanisms known to produce horizontal transfer are transformation, conjugation, and transduction. Numerous authors have found evidence of transduction. For example, the B. subtilis genome harbors a number of foreign genes, as evidenced by many prophage-like regions encompassing ≈15% of the genome (Kunst et al., 1997). Like its close relative B. subtilis, Bacillus halodurans, an alkaliphilic prokaryote, also possesses regions with a G + C content similar to that of some viruses (Takami et al., 2000). As a consequence of this similarity, those DNA sequences were proposed to have been obtained by lateral transfer (Takami et al., 2000). The genome of Clostridium acetobutylicum contains genes missing in B. subtilis. These genes have a number of different phylogenetic relationships. For example, 49 genes reveal an immediate relationship between C. acetobutylicum and eukaryotes, and another 195 are most closely related to archaeal extremophiles (Nolling et al., 2001).
The cyanobacterium Synechocystis PCC6803 is another bacterium whose genome supports extensive HGT among prokaryotes. The genome of Synechocystis contains a number of insertion sequence (IS) elements. The DNA in the vicinity of the IS elements displays features of E. coli DNA, indicative of horizontal genetic acquisitions (Cassier-Chauvat et al., 1997).
ALTHOUGH HGT IS RAMPANT, IT IS NOT RANDOM: THE COMPLEXITY HYPOTHESIS
In a subsequent phylogenetic analysis (Jain et al., 1999), our laboratory examined the frequency of horizontal/lateral transfer of operational genes among six prokaryotic proteomes, E. coli, Synechocystis PCC6803, B. subtilis, A. aeolicus, M. jannaschii, and A. fulgidus, using three different topology-based tests of gene ortholog relationships to measure the extent of HGT in informational and operational genes. All three tests showed that operational genes have been continually transferred much more frequently among prokaryotes since the last common ancestor of life or cenancestor (Fitch and Upper, 1987). To explain at least partially why operational genes undergo HGT more frequently than informational genes, we proposed the complexity hypothesis (Jain et al., 1999), which posits that informational genes are less likely to undergo horizontal transfer, because their products are members of large complexes with many intricate interactions. Operational genes, on the other hand, are generally not parts of large complexes, and thus are more readily transferred. Obviously the complexity hypothesis is not the sole factor relating differential horizontal transfer rates between informational and operational genes, because many other factors, including environmental ones, can also
modify horizontal transfer. At the same time, the data are forcing us to recognize that gene exchange is not simply occurring within species, but extensive exchanges also occur within larger groups of prokaryotes consisting of multiple species as well.
HGT ACCELERATES GENOME INNOVATION AND EVOLUTION
It is becoming clear that HGT has had great impact on the evolution of life on Earth. It is a key agent, perhaps the major agent, responsible for spreading genetic diversity among prokaryotes by moving genes across species boundaries (Jain et al., 2003). By rapidly introducing newly evolved genes into existing genomes, HGT circumvents the slow step of ab initio gene creation and thereby accelerates genome innovation (the acquisition of novel genes by organisms), although not necessarily gene evolution. We refer to a collection of organisms that can share genes by HGT but need not be in physical proximity as an exchange community. In effect, when organisms are exchanging genes, genome innovation is increased in proportion to the effective population sizes of their exchange groups.
We were interested in the structure of exchange communities and in the environmental and other factors that help define them. In an analysis of ≈20,000 genes contained in eight free-living prokaryotic genomes, we assessed which geographic, environmental, and internal parameters have influenced genetic exchange by HGT and found that HGT is not random but depends critically upon these internal and environmental factors. The statistically significant parameters were similar genome sizes, genome G + C compositions, carbon utilization methods, oxygen tolerance, and maximum, optimal, and minimum temperatures (Jain et al., 2003). By identifying and quantifying those parameters, we were able to delineate exchange community boundaries, estimate the effective population size of exchange groups, and thereby estimate the extent to which HGT has accelerated genome innovation. By correlating the extent of HGT among specific organisms with the degree of phylogenetic clustering of those organisms observed on all possible gene trees, one can determine the effect of various environmental or other parameters on HGT. We found that HGT preferentially occurs among organisms that have environmental and genomic factors in common, a phenomenon we termed positive associativity (Jain et al., 2003). In short, like prokaryotes preferentially exchanged genes by HGT with like prokaryotes. It is difficult to ascertain precisely how much HGT has accelerated prokaryotic genome innovation, but the acceleration is significant. It has been estimated there are 109 prokaryotic species on Earth containing 1030 prokaryotes (Whitman et al., 1998). The sizes of exchange communities are unknown, but some of the
parameters characterizing them are not too different from those of some terrestrial ecosystems. The median prokaryotic population of 12 diverse soil ecosystem types, as reviewed by Whitman, Coleman, and Wiebe (1998), is ≈1028 prokaryotes, suggesting an average exchange group could contain 107 species. Allowing 3 orders of magnitude for the inexactness of our estimate, the increase in innovation afforded by HGT could be as small as 104, but even this would constitute a huge HGTdependent increase in innovation. This means that a species exchanging genes only with other members of its species would take 10,000 years to obtain the amount of genome innovation that would occur for an average exchange group in just 1 year. Indeed, HGT may be responsible for a remarkable increase in genome innovation that greatly exceeds anything that could have been accomplished by clonal evolution.
HGT GREATLY COMPLICATES RECONSTRUCTING THE UNIVERSAL TREE OF LIFE
W. Ford Doolittle recently reviewed the state of “Phylogenetic Classification and the Universal Tree” in a thoughtful analysis (Doolittle, 1999b). He points out the specific challenges to classification that HGT presents as follows, “If, however, different genes give different trees, and there is no fair way to suppress this disagreement, then a species (or phylum) can ‘belong’ to many genera (or kingdoms) at the same time: There really can be no universal phylogenetic tree of organisms based on such a reduction to genes.” In other words, Doolittle (1999b) suggests that the gene mixing resulting from HGT is so extensive that it might preclude one from ever reconstructing the tree of life. Although it would be disingenuous to pretend that the difficulties are not sizable, our laboratory is pursuing an alternative strategy. We agree that HGT is extensive and imposes limits to phylogenetic reconstruction. However, we also think the only way to discover whether HGT could destroy Darwin’s dream of understanding the great kingdoms of nature is to assume that it cannot, and then make every effort to try to determine the tree of life. Some of the barriers to reconstructing the tree of life and the progress being made to surmount them are discussed below.
PITFALLS IN RECONSTRUCTING THE TREE OF LIFE
Consider what has happened to the once-ebullient field of rRNA phylogenies. For years, phylogenies based on rRNAs were the holy grail of microbial phylogenetics. To be sure, rRNA-based phylogenies have been responsible for many successes, including the new animal phylogeny and demonstrations that the mitochondrion and chloroplast are endosym-
bionts (Adoutte et al., 1999, 2000; Aguinaldo et al., 1997; Gray, 1999; Halanych et al., 1995; Schwarz and Kossel, 1980). However, prokaryotic phylogenies are another story. One has only to read the latest Bergey’s Manual (Boone and Castenholz, 2001) to realize that the tree of prokaryotic life is fuzzy and unresolved, so much so that rRNA-based trees, although capable of identifying to which phylum a prokaryote belongs, in most cases cannot determine how the phyla are related to each other. Furthermore, our ability to determine phylogenies accurately depends upon how extensive HGT has been. If very little or no HGT has occurred, then current methods of analysis will allow one to reconstruct the clonal tree of life. At the other extreme, if all genes undergoHGTonce per year, then coherent gene trees will be unobtainable. Between these extremes lies a continuum of results, so that perhaps the question we should be asking is, how much phylogenetic information can one obtain, and how can it best be analyzed?
HOW CAN ONE RECONSTRUCT THE TREE OF LIFE IN THE PRESENCE OF HGT?
Presences and absences of genes and gene products have been used for more than two decades to support parsimonious conclusions about the tree of life (Charlebois et al., 2000; Dickerson, 1980; Lake et al., 1982; Woese et al., 1986). In these analyses, the absences and presences of genes were used as character states, much in the way that nucleotides A, C, G, and T are used as character states in sequence analyses. With the availability of complete genomes, useful methods have been developed for whole-genome analyses (Fitz-Gibbon and House, 1999; Montague and Hutchison, 2000; Snel et al., 1999; Tekaia et al., 1999). However, when analyzed using parsimony and simple distance-based methods, these analyses can be significantly influenced by HGT (Eisen, 2000; House and Fitz-Gibbon, 2002).
Recently the prospects of recovering the tree of life in the presence of HGT have improved with the development of a new mathematical algorithm, conditioned reconstruction (CR), for whole-genome-based phylogenetic reconstructions (Lake and Rivera, 2004). Like some other whole-genome methods, CR analyses also use the absences and presences of genes as character states but, through the use of a reference genome, they can obtain additional information that is not available in other types of analyses. For example, by restricting the analyses to only the genes present in a reference genome R, one can also estimate the number of gene pairs that are missing in both genomes A and B. This is critical information that is not available without the reference genome, and it allows one to use a very general class of mathematical (Markov) models to reconstruct the tree of life.
In CR, the dynamic deletions and insertions of genes that occur during genome evolution, including the insertions introduced by HGT, actually help provide the information needed to reconstruct phylogenetic trees. CR appears to have the potential to reconstruct deeper branchings in the tree of life than is possible with sequence analyses, because whole gene characters evolve more slowly than nucleotides, amino acids, and even gene inserts.
At the same time, it is important to recognize that CRs are not a panacea. It is difficult to assign the gene ortholog sets used by CR analyses accurately, because the process is greatly complicated by the need to distinguish orthologs from paralogs and to simultaneously recognize recently duplicated genes (Lake and Rivera, 2004). Currently available methods to identify gene ortholog sets are still rudimentary, and new methods are just beginning to be developed. Because CR can be no better than the ortholog sets that it is based on, much improvement is needed in this area.
Although CR analysis provides a new tool for investigating the tree of life, other methods are also likely to provide important information about deep divergences in the tree of life. These include such important emerging techniques as phylogenetic analyses of concatenated gene sequences (Baldauf et al., 2000; Brown et al., 2001) or of sets of gene sequences (Esser et al., 2004; Raymond et al., 2002), particularly of informational genes, and the analyses of more slowly evolving sequence-related characters such as gene inserts, gene fusions, and even structural domains (Gupta and Singh, 1994; Stechmann and Cavalier-Smith, 2002; Yang et al., 2005). Like CRs, these methods also have their limitations, and much work remains to be done to improve these promising techniques as well.
One of the most remarkable properties of CR is that it can rigorously identify the merger of genomes, a process that until now could not be analyzed using gene sequence. A recently published application of this method has provided evidence that the eukaryotic genome was actually formed by a fusion of the genomes from two disparate prokaryotes.
EVIDENCE THAT AN ANCIENT GENOME FUSION FORMED THE FIRST EUKARYOTE
Various theories have been proposed for the origin of the nuclear genes of eukaryotes. These include the autogenous-, chimeric-, and genome-fusion theories. To obtain a better understanding of eukaryotic origins, we analyzed 10 complete genomes using the CR method (Rivera and Lake, 2004). The sample was comprised of two eukaryotic genomes and eight prokaryotes representing the diversity of prokaryotic life. An additional 24 prokaryotic genomes were studied in supplementary studies. The results from one analysis are shown in Fig. 14.2. In this analysis, the

FIGURE 14.2 CRs provide evidence for the ring of life. The genomes are from two yeasts, Y1 (Schizosaccharomyces pombe) and Y2 (S. cerevisiae); a gammaproteobacterium, P (Xylella fastidiosa); a bacillus, B (Staphylococcus aureus MW2); a halobacterium, H (Halobacterium sp. NRC-1); a methanococcus, M (Methanosarcina mazei Goe1); an eocyte, T (Sulfolobus tokodaii); and an archaeoglobium not shown, the conditioning genome (A. fulgidus DSM4304). Cumulative probabilities are shown at the right of each tree. Fully and partially resolved rings are Lower Left and Lower Right, respectively. [Reproduced with permission from Rivera and Lake (2004) (Copyright 2004, Nature Publishing Group).]
five most probable trees are from a set of three Bacteria, three Archaea, and two eukaryotes. The cumulative probabilities of these five trees are shown at the right of each tree. We initially thought that the resolution of the tree was disappointingly poor, because the most probable tree was supported by a low bootstrap value (70% approximately corresponds to the 95% confidence level), and the other trees were supported by even lower values.
However, when the five most probable unrooted trees are aligned by
shifting each to the left or the right until their leaves match, they form a repeating pattern indicating that the five trees are simply permutations of an underlying cyclic pattern. (The five most probable unrooted trees are shown with leaves pointing upward to emphasize that each is part of a repeating pattern.) This suggested that they are derived from the single cycle graph (Lake and Rivera, 2004), or ring, shown in Fig. 14.2 Lower Left. When that ring is cut at any of the five central arcs and then unfolded, the resulting unrooted tree will correspond to one of the five most probable trees. In other words, the data are not tree-like; they are ring-like.
Previously, a combinatorial analysis of the genomic fusion of two organisms had shown that the CR algorithm recovers all permutations of the cycle graph (Lake and Rivera, 2004). Hence these results can be interpreted in a manner analogous to the interpretation of restriction digests of a circular plasmid or the mapping of a circular chromosome, as implying a ring of life. The fully resolved ring shown in Fig. 14.2 Lower Left is fully consistent with all five of the resolved trees shown in Fig. 14.2 Upper. That ring explains 96.3% of the bootstrap replicates, and the partially resolved ring in Fig. 14.2 Lower Right explains almost all (99.2%) of the bootstrap replicates. These and other control experiments provide robust evidence for the completely resolved ring (Fig. 14.2 Lower Left) and even stronger evidence for the less-resolved ring (Fig. 14.2 Lower Right).
Analyses of this type supported the ring, but other experiments were still necessary to identify the fusion organism. In particular, it was necessary to show that it was the eukaryotes, rather than a prokaryote, that resulted from the genome fusion that closed the ring of life. Hence the identity of the fusion organism was explicitly tested by systematically eliminating the eukaryotes and the individual prokaryotes for the ring of life. The ring opened into a tree only when both eukaryotes were simultaneously deleted from the analysis, indicating the eukaryotic genome had inherited genes from its prokaryotic fusion partners. This then demonstrated that eukaryotes are indeed the products of genome fusions. Furthermore, statistical support for the ring remained high for all possible choices of conditioning genome. From these results and other studies not discussed here, we inferred that the eukaryotic nuclear genome was formed from the genome fusion of either a proteobacterium or a member of a large photosynthetic clade that includes the Cyanobacteria and the Proteobacteria, with an archaeal eocyte as shown schematically in Fig. 14.3.
IMPLICATIONS OF THE RING OF LIFE
Various theories have been proposed for the origin of eukaryotes. These include autogenous, chimeric, and genome fusion theories. The
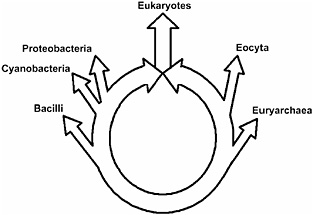
FIGURE 14.3 A schematic diagram of the ring of life. The eukaryotes include all eukaryotes plus the two eukaryotic root organisms, the operational and informational ancestors. Ancestors defining major prokaryotic groups are represented by branching points from the ring. Archaea (Woese et al., 1990), shown on the bottom right, includes the Euryarchaea, the Eocyta, and the informational eukaryotic ancestor. Karyota (Lake, 1988), shown on the upper right of the ring, includes the Eocyta and the informational eukaryotic ancestor. The upper left circle includes the Proteobacteria (Woese et al., 1990) and the operational eukaryotic ancestor. The most basal node on the left represents the photosynthetic prokaryotes and the operational eukaryotic ancestor.
results derived in the CR analyses argue against autogenous theories, i.e., tree of life theories, in which eukaryotes evolved clonally from a single, possibly very ancient, prokaryote. Chimeric theories refer to the acquisition of genes by eukaryotes from multiple sources through unspecified mechanisms. The data presented here argue against them, except of course chimeric theories that specifically propose genome fusions.
At least half a dozen genome fusion theories have been proposed in which the eukaryotic genome originated from two diverse genomes (Gupta et al., 1994; Horiike et al., 2001; Lake and Rivera, 1994; Lake et al., 1982; Martin and Muller, 1998; Moreira and Lopez-Garcia, 1998). These are strongly supported by CR analyses. By default, an endosymbiosis (Margulis, 1970) between two prokaryotes is probably the mechanism responsible for the genome fusion observed here, although the fusion signal may have been augmented by gene contributions from eukaryotic organelles. Symbiotic relationships are fairly common among organisms living together and, in rare cases, this leads to endosymbiosis, the intracellular capture of former symbionts (Margulis, 1970). Given a genome fusion, and in the absence of other mechanisms that could produce fusions, one concludes that an endosymbiosis was the probable cause.
Although the data reviewed here solidly support the ring of life, it is important to recognize that CR analysis is a new technique, and its usefulness is still being explored. Currently, the resolution in CR trees is still relatively low. At the same time, it seems unlikely that the ring could be caused by low phylogenetic resolution, because the ring signal monitored in CR analyses is fundamentally different from the parsimony signals that are generated by poorly resolved trees (Lake and Rivera, 2004).
The ring of life is consistent with and confirms and extends a number of previously reported results. It implies that prokaryotes predate eukaryotes, because two preexisting prokaryotes contributed their genomes to create the first eukaryotic genome. This likely places the root of the ring below the eubacterial– and eocytic–eukaryotic last common ancestors, as shown in Fig. 14.3. This partial rooting of the ring of life is consistent with the eukaryotic rooting implied by the EF-1α insert that is present in all known eukaryotic and eocytic EF-1α sequences and lacking in all paralogous EF-G sequences (Gupta, 1998; Rivera and Lake, 1992).
The ring of life also explains some previously confusing observations and raises new ones. Because the eukaryotic genome resulted from a fusion, it is expected that in some gene trees, eukaryotes will be related to Bacteria, whereas in other gene trees, eukaryotes will be related to Archaea, in accord with the results of others (Brown and Doolittle, 1997; Feng et al., 1997; Gupta, 1998; Martin et al., 1996). The observations of ourselves and others (Esser et al., 2004; Rivera et al., 1998), that the informational genes of eukaryotes are primarily derived from Archaea and the operational genes are primarily derived from Bacteria, are also consistent with the ring. Those observations suggest that the operational genes have come from the eubacterial fusion partner and the informational genes, from the archaeal fusion partner. The ring of life does not explain why the fusion happened, but it provides a broad phylogenetic framework for testing theories for the origin and evolution of the eukaryotic genome. The genome fusion that created the ring of life may in some ways be the ultimate HGT.
ACKNOWLEDGMENTS
We thank M. Kowalczyk for illustrations. This work was supported by grants from the National Science Foundation, the National Aeronautics and Space Administration Astrobiology Institute, the Department of Energy, and the National Institutes of Health (to J.A.L.).
REFERENCES
Adoutte, A., Balavoine, G., Lartillot, N. & de Rosa, R. (1999) Animal evolution—the end of the intermediate taxa? Trends Genet. 15, 104–108.
Adoutte, A., Balavoine, G., Lartillot, N., Lespinet, O., Prudhomme, B. & de Rosa, R. (2000) The new animal phylogeny: Reliability and implications. Proc. Natl. Acad. Sci. USA 97, 4453–4456.
Aguinaldo, A. M. A., Turbeville, J. M., Linford, L. S., Rivera, M. C., Garey, J. R., Raff, R. A. & Lake, J. A. (1997) Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387, 489–493.
Baldauf, S. L., Roger, A. J., Wenk-Siefert, I. & Doolittle, W. F. (2000) A kingdom-level phylogeny of eukaryotes based on combined protein data. Science 290, 972–977.
Blattner, F. R., Plunkett, G., Bloch, C. A., Perna, N. T., Burland, V., Riley, M., ColladoVides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F., et al. (1997) The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462.
Boone, D. R. & Castenholz, R. W. (2001) The Archaea and the Deeply Branching and Phototrophic Bacteria. Bergey’s Manual of Systematic Bacteriology, ed. Garrity, G. M. (Springer, New York), Vol. 1.
Brown, J. R. & Doolittle, W. F. (1997) Archaea and the prokaryote-to-eukaryote transition. Microbiol. Mol. Biol. Rev. 61, 456–502.
Brown, J. R. & Doolittle, W. F. (1999) Gene descent, duplication, and horizontal transfer in the evolution of glutamyl- and glutaminyl-tRNA synthetases. J. Mol. Evol. 49, 485–495.
Brown, J. R., Douady, C. J., Italia, M. J., Marshall, W. E. & Stanhope, M. J. (2001) Universal trees based on large combined protein sequence data sets. Nat. Genet. 28, 281–285.
Bult, C. J., White, O., Olsen, G. J., Zhou, L. X., Fleischmann, R. D., Sutton, G. G., Blake, J. A., FitzGerald, L. M., Clayton, R. A., Gocayne, J. D., et al. (1996) Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273, 1058–1073.
Campbell, A. M. (2000) Lateral gene transfer in prokaryotes. Theor. Popul. Biol. 57, 71–77.
Cassier-Chauvat, C., Poncelet, M. & Chauvat, F. (1997) Three insertion sequences from the cyanobacterium Synechocystis PCC6803 support the occurrence of horizontal DNA transfer among bacteria. Gene 195, 257–266.
Charlebois, R. L., Singh, R. K., Chan-Weiher, C. C.-Y., Allard, G. C. C., Confaloniere, F., Curtis, B., Duget, M., Erauso, G., Faguy, D., Gaasterland, T., et al. (2000) Gene content and organization of a 281-kbp contig from the genome of the extremely thermophilic archaeon, Sulfolobus solfataricus P2. Genome 43, 116–136.
Chinen, A., Uchiyama, I. & Kobayashi, I. (2000) Comparison between Pyrococcus horikoshii and Pyrococcus abyssi genome sequences reveals linkage of restriction-modification genes with large genome polymorphisms. Gene 259, 109–121.
Darwin, F. (1887) The Life and Letters of Charles Darwin (John Murray, London).
Deckert, G., Warren, P. V., Gaasterland, T., Young, W. G., Lenox, A. L., Graham, D. E., Overbeek, R., Snead, M. A., Keller, M., Aujay, M., et al. (1998) The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 392, 353–358.
Dickerson, R. E. (1980) Structural conservatism in proteins over three billion years: Cytochrome with a touch of collagen. In Diffraction and Related Studies, ed. Srinivasan, R. (Pergamon, Oxford), Vol. 1, pp. 227–249.
DiRuggiero, J., Dunn, D., Maeder, D. L., Holley-Shanks, R., Chatard, J., Horlacher, R., Robb, F. T., Boos, W. & Weiss, R. B. (2000) Evidence of recent lateral gene transfer among hyperthermophilic Archaea. Mol. Microbiol. 38, 684–693.
Doolittle, R. F. & Handy, J. (1998) Evolutionary anomalies among the aminoacyl-tRNA synthetases. Curr. Opin. Genet. Dev. 8, 630–636.
Doolittle, W. F. (1999a) Lateral genomics. Trends Genet. 15, M5–M8.
Doolittle, W. F. (1999b) Phylogenetic classification and the universal tree. Science 284, 2124–2128.
Eisen, J. A. (2000) Assessing evolutionary relationships among microbes from whole-genome analysis. Curr. Opin. Microbiol. 3, 475–480.
Esser, C., Ahmadinejad, N., Wiegand, C., Rotte, C., Sebastiani, F., Gelius-Dietrich, G., Henze, K., Kretschmann, E., Richly, E., Leister, D., et al. (2004) Genome comparisons speak to the origin of mitochondria and eukaryotes. Mol. Biol. Evol. 21, 1643–1660.
Feng, D. F., Cho, G. & Doolittle, R. F. (1997) Determining divergence times with a protein clock: Update and reevaluation. Proc. Natl. Acad. Sci. USA 94, 13028–13033.
Fitch, W. M. & Upper, K. (1987) The phylogeny of tRNA sequences provides evidence for ambiguity reduction in the origin of the genetic code. Cold Spring Harbor Symp. Quant. Biol. 52, 759–767.
Fitz-Gibbon, S. T. & House, C. H. (1999) Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 27, 4218–4222.
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J. D., Jacq, C., Johnston, M., et al. (1996) Life with 6000 genes. Science 274, 546, 563–567.
Gogarten, J. P., Murphey, R. D. & Olendzenski, L. (1999) Horizontal gene transfer: Pitfalls and promises. Biol. Bull. 196, 359–361.
Gogarten, J. P., Doolittle, W. F. & Lawrence, J. G. (2002) Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19, 2226–2238.
Gray, M. W. (1999) Evolution of organellar genomes. Curr. Opin. Genet. Dev. 9, 678–687.
Gupta, R. S. (1998) Protein phylogenies and signature sequences: A reappraisal of evolutionary relationships among archaebacteria, eubacteria, and eukaryotes. Microbiol. Mol. Biol. Rev. 62, 1435–1491.
Gupta, R. S. & Singh, B. (1994) Phylogenetic analysis of 70-Kd heat-shock protein sequences suggests a chimeric origin for the eukaryotic cell-nucleus. Curr. Biol. 4, 1104–1114.
Gupta, R. S., Aitken, K., Falah, M. & Singh, B. (1994) Cloning of giardia-lamblia heat-shock protein Hsp70 homologs—implications regarding origin of eukaryotic cells and of endoplasmic-reticulum. Proc. Natl. Acad. Sci. USA 91, 2895–2899.
Halanych, K. M., Bacheller, J. D., Aguinaldo, A. M. A., Liva, S. M., Hillis, D. M. & Lake, J. A. (1995) Evidence from 18s ribosomal DNA that the lophophorates are protostome animals inarticulate. Science 267, 1641–1643.
Hentze, M. W. (2001) Protein synthesis—believe it or not—translation in the nucleus. Science 293, 1058–1059.
Horiike, T., Hamada, K., Kanaya, S. & Shinozawa, T. (2001) Origin of eukaryotic cell nuclei by symbiosis of Archaea in baceria is revealed by homology-hit analysis. Nat. Cell Biol. 3, 210–214.
House, C. H. & Fitz-Gibbon, S. T. (2002) Using homolog groups to create a whole-genomic tree of free-living organisms: An update. J. Mol. Evol. 54, 539–547.
Jain, R., Rivera, M. C. & Lake, J. A. (1999) Horizontal gene transfer among genomes: The complexity hypothesis. Proc. Natl. Acad. Sci. USA 96, 3801–3806.
Jain, R., Rivera, M. C., Moore, J. E. & Lake, J. A. (2003) Horizontal gene transfer accelerates genome innovation and evolution. Mol. Biol. Evol. 20, 1598–1602.
Kaneko, T., Sato, S., Kotani, H., Tanaka, A., Asamizu, E., Nakamura, Y., Miyajima, N., Hirosawa, M., Sugiura, M., Sasamoto, S., et al. (1996) Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 3, 109–136.
Karlin, S., Mrazek, J. & Campbell, A. M. (1997) Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 179, 3899–3913.
Karol, K. G., McCourt, R. M., Cimino, M. T. & Delwiche, C. F. (2001) The closest living relatives of land plants. Science 294, 2351–2353.
Klenk, H. P., Clayton, R. A., Tomb, J. F., White, O., Nelson, K. E., Ketchum, K. A., Dodson, R. J., Gwinn, M., Hickey, E. K., Peterson, J. D., et al. (1997) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature 390, 364–370.
Koonin, E. V., Mushegian, A. R., Galperin, M. Y. & Walker, D. R. (1997) Comparison of archaeal and bacterial genomes: Computer analysis of protein sequences predicts novel functions and suggests a chimeric origin for the archaea. Mol. Microbiol. 25, 619–637.
Koonin, E. V., Makarova, K. S. & Aravind, L. (2001) Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 55, 709–742.
Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., Bertero, M. G. , Bessieres, P., Bolotin, A., Borchert, S., et al. (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390, 249–256.
Lake, J. A. (1988) Origin of the eukaryotic nucleus determined by rate-invariant analysis of ribosomal RNA sequences. Nature 331, 184–186.
Lake, J. A. & Rivera, M. C. (1994) Was the nucleus the 1st endosymbiont. Proc. Natl. Acad. Sci. USA 91, 2880–2881.
Lake, J. A. & Rivera, M. C. (2004) Deriving the genomic tree of life in the presence of horizontal gene transfer: Conditioned Reconstruction. Mol. Biol. Evol. 21, 681–690.
Lake, J. A., Henderson, E., Clark, M. W. & Matheson, A. T. (1982) Mapping evolution with ribosome structure: Intralineage constancy and interlineage variation. Proc. Natl. Acad. Sci. USA 79, 5948–4952.
Lawrence, J. G. & Ochman, H. (1998) Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. USA 95, 9413–9417.
Lecompte, O., Ripp, R., Puzos-Barbe, V., Duprat, S., Heilig, R., Dietrich, J., Thierry, J. C. & Poch, O. (2001) Genome evolution at the genus level: Comparison of three complete genomes of hyperthermophilic Archaea. Genome Res. 11, 981–993.
Maeder, D. L., Weiss, R. B., Dunn, D. M., Cherry, J. L., Gonzalez, J. M., DiRuggiero, J. & Robb, F. T. (1999) Divergence of the hyperthermophilic archaea Pyrococcus furiosus and P. horikoshii inferred from complete genomic sequences. Genetics 152, 1299–1305.
Makarova, K. S., Aravind, L., Galperin, M. Y., Grishin, N. V., Tatusov, R. L., Wolf, Y. I. & Koonin, E. V. (1999) Comparative genomics of the archaea (Euryarchaeota): Evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 9, 608–628.
Margulis, L. (1970) Origin of the Eukaryotic Cells (Yale Univ. Press, New Haven, CT).
Martin, W. & Muller, M. (1998) The hydrogen hypothesis for the first eukaryote. Nature 392, 37–41.
Martin, W., Mustafa, A. Z., Henze, K. & Schnarrenberger, C. (1996) Higher-plant chloroplast and cytosolic fructose-1,6-bisphosphatase isoenzymes: Origins via duplication rather than prokaryote-eukaryote divergence. Plant Mol. Biol. 32, 485–491.
Montague, M. G. & Hutchison, C. A. (2000) Gene content phylogeny of herpesviruses. Proc. Natl. Acad. Sci. USA 97, 5334–5339.
Moreira, D. & Lopez-Garcia, P. (1998) Symbiosis between methanogenic archaea and delta-proteobacteria as the origin of eukaryotes: The syntrophic hypothesis. J. Mol. Evol. 47, 517–530.
Nickrent, D. L., Parkinson, C. L., Palmer, J. D. & Duff, R. J. (2000) Multigene phylogeny of land plants with special reference to bryophytes and the earliest land plants. Mol. Biol. Evol. 17, 1885–1895.
Nolling, J., Breton, G., Omelchenko, M. V., Makarova, K. S., Zeng, Q. D., Gibson, R., Lee, H. M., Dubois, J., Qiu, D. Y., Hitti, J., et al. (2001) Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 183, 4823–4838.
Ochman, H. (2001) Lateral and oblique gene transfer. Curr. Opin. Genet. Dev. 11, 616–619.
Penny, D. & Poole, A. (1999) The nature of the last universal common ancestor. Curr. Opin. Genet. Dev. 9, 672–677.
Philippe, H. & Forterre, P. (1999) The rooting of the universal tree of life is not reliable. J. Mol. Evol. 49, 509–523.
Poole, A., Jeffares, D. & Penny, D. (1999) Early evolution: prokaryotes, the new kids on the block. BioEssays 21, 880–889.
Pryer, K. M., Schneider, H., Smith, A. R., Cranfill, R., Wolf, P. G., Hunt, J. S. & Sipes, S. D. (2001) Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature 409, 618–622.
Pryer, K. M., Schneider, H., Zimmer, E. A. & Banks, J. A. (2002) Deciding among green plants for whole genome studies. Trends Plant Sci. 7, 550–554.
Raymond, J., Zhaxybayeva, O., Gogarten, J. P., Gerdes, S. Y. & Blankenship, R. E. (2002) Whole-genome analysis of photosynthetic prokaryotes. Science 298, 1616–1620.
Ribeiro, S. & Golding, G. B. (1998) The mosaic nature of the eukaryotic nucleus. Mol. Biol. Evol. 15, 779–788.
Rivera, M. C. & Lake, J. A. (1992) Evidence that eukaryotes and eocyte prokaryotes are immediate relatives. Science 257, 74–76.
Rivera, M. C. & Lake, J. A. (2004) The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 431, 152–155.
Rivera, M. C., Jain, R., Moore, J. E. & Lake, J. A. (1998) Genomic evidence for two functionally distinct gene classes. Proc. Natl. Acad. Sci. USA 95, 6239–6244.
Schwarz, Z. & Kossel, H. (1980) Primary structure of 16s rDNA from Zea-mays chloroplast is homologous to Escherichia-coli 16s ribosomal-RNA. Nature 283, 739–742.
Smith, D. R., Doucette-Stamm, L. A., Deloughery, C., Lee, H. M., Dubois, J., Aldredge, T., Bashirzadeh, R., Blakely, D., Cook, R., Gilbert, K., et al. (1997) Complete genome sequence of Methanobacterium thermoautotrophicum Delta H: Functional analysis and comparative genomics. J. Bacteriol. 179, 7135–7155.
Snel, B., Bork, P. & Huynen, M. A. (1999) Genome phylogeny based on gene content. Nat. Genet. 21, 108–110.
Soltis, P. S., Soltis, D. E., Wolf, P. G., Nickrent, D. L., Chaw, S. & Chapman, R. L. (1999) The phylogeny of land plants inferred from 18S rDNA sequences: Pushing the limits of rDNA signal? Mol. Biol. Evol. 16, 1774–1784.
Stechmann, A. & Cavalier-Smith, T. (2002) Rooting the eukaryote tree by using a derived gene fusion. Science 297, 89–91.
Takami, H., Nakasone, K., Takaki, Y., Maeno, G., Sasaki, R., Masui, N., Fuji, F., Hirama, C., Nakamura, Y., Ogasawara, N., et al. (2000) Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res. 28, 4317–4331.
Tekaia, F., Lazcano, A. & Dujon, B. (1999) The genomic tree as revealed from whole proteome comparisons. Genome. Res. 9, 550–557.
Whitman, W. B., Coleman, D. C. & Wiebe, W. J. (1998) Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 95, 6578–6583.
Woese, C. R., Pace, N. R. & Olsen, G. J. (1986) Are arguments against archaebacteria valid. Nature 320, 401–402.
Woese, C. R., Kandler, O. & Wheelis, M. L. (1990) Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria and Eucarya. Proc. Natl. Acad. Sci. USA 87, 4576–4579.
Yang, S., Doolittle, R. F. & Bourne, P. E. (2005) Phylogeny determined by protein domain content. Proc. Natl. Acad. Sci. USA 102, 373–378.





















