
4
Understanding Cells
INTRODUCTION
To understand cells, one must understand the macromolecular structure of cells, the spatiotemporal patterns and mechanisms of cellular dynamics, the connections between cellular dynamics and cellular functions, and the connections between cells and higher levels of organization, such as tissues and organs. Understanding cells is intrinsically more difficult than understanding molecules because there is no cellular counterpart to the linear sequence of nucleotides and amino acids that provides much of the information necessary for predicting the structure and function of nucleic acids and proteins. Moreover, eukaryotic cells are highly compartmented and contain both a nuclear genome and one or more organellar genomes. With rapid increases in computational power and the sophistication of biophysical measurements, one can imagine constructing reasonably exact models of the dynamics of DNA and proteins, whereas all quantitative descriptions will still be approximate for cells. Thus, the main challenges for the mathematical analysis of cells are not computational but reside instead in the basic challenge of how to model features of interest. The primary challenge in this area for the next decade is the systematic formulation of reduced-order representations of cellular structure and dynamics, drawn from increasingly complex data and validated in model-driven experiments.
There is a long and successful history of mathematical modeling of cellular functions. The success stories come from systems that are rich in data and for which models can be validated or at least put in direct corre-
spondence with experiments. Models of the endocytic cycle, signal transduction cascades, and the cell division cycle have been productively used to organize data, extract “laws” for cellular processes, and even engineer cellular systems. Nevertheless, the models formulated over the past two decades build primarily on population-level measurements with poor spatial and temporal resolution. Without exception, the dynamics predicted by early models were richer than those predicted by the corresponding experimental data. For example, current models of cell cycle dynamics have more variables than can be measured experimentally. This experimental limitation is changing as a result of the rapid development of imaging techniques and high-throughput assays of cellular processes. The result should be an increased ability to evaluate models, which is the limiting step in improving them. It is now possible to collect multivariable and spatiotemporally resolved data on cellular processes ranging from molecular trafficking and signal transduction to integrated responses such as the cell division cycle and cell migration.
In spite of these improvements in experimental capabilities, the quantitative models that emerge will not be as resolved and detailed as the models used in, for example, the aerospace and semiconductor industries. This difference reflects the intrinsic variability of biology, the immense spatiotemporal complexity of cells, and our incomplete knowledge of cellular processes. In many areas of the physical sciences, a coarse model can be very simple and yet fairly accurate, and one only needs to develop heterogeneous, multivariable, spatially resolved models when dealing with second- or higher-order effects. In contrast, even the simplest cellular models must be extracted from heterogeneous, multivariable, spatially resolved experimental data. Learning how to manage these data and mine them to extract computationally manageable models of cellular functions is the key challenge to quantitative understanding of cells.
Exemplification of These Issues
In the early 1980s, Steven Wiley and his colleagues formulated kinetic models of receptor-mediated ligand internalization (Wiley and Cunningham, 1981). The models were initially developed for the epidermal growth factor (EGF) receptor, a key regulator of cell and tissue functions across species (Wiley, 2003). The models described the kinetics of ligand-receptor binding, internalization, recycling, and degradation. The mathematical models were in the form of small systems of ordinary differential equations that were integrated in time and coupled with standard optimization and parameter estimation routines to extract the model parameters (Figure 4.1).
Based on experiments with radioactively labeled EGF ligands, the
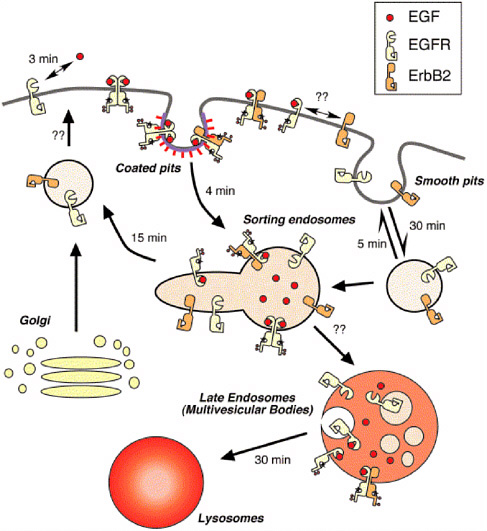
FIGURE 4.1 Trafficking of ErbB receptor family. Only the epidermal growth factor receptor (EGFR) and ErbB2 proteins are shown for clarity, but the behaviors of ErbB3 and ErbB4 are similar to that of ErbB2. Activated EGFR and EGFR:ErbB2 heterodimers are internalized through a coated pit pathway, but other members of the ErbB family are probably internalized by a smooth pit pathway. The numbers next to the arrows represent the approximate mean time of the specific process. The time constants for heterodimerization and formation of multivesicular bodies are unknown. The mean time for lysosomal degradation is a combination of the time necessary for multivesicular body formation and for lysosomal fusion. Reprinted from Experimental Cell Research, 284, H.S. Wiley. Trafficking of ErbB receptors and its influence on signaling, pp. 78-88 (2003), with permission from Elsevier.
models could extract the rate constants for different processes, such as endocytic uptake or recycling. Before this modeling effort, the only quantitative measures of ligand-receptor dynamics in cells were related to ligand-receptor interactions. The models of Wiley and colleagues produced new quantitative measures of ligand-receptor dynamics. In this way the biological effects of various ligands could be interpreted in terms of the quantitative differences of a larger number of rate constants for the multiple steps in the endocytic pathway. The model was also critical for suggesting the functional roles of the different parts of the EGF receptor. The EGF receptor is a large protein that combines multiple functions, including ligand binding, receptor phosphorylation, internalization, endocytic sorting, and recycling. By analyzing the ligand uptake data in cells that express mutant receptors and fitting these data to models, the functional roles of specific residues could be identified by changes in the rate constants of specific cellular processes (Wiley et al., 1991).
This modeling and experimental approach has been validated by a number of experiments and used to parse the dynamics of internalization and trafficking for other ligand-receptor systems (Wiley et al., 2003). In the meantime, the experimental tools used to study these processes have changed. It is now possible to visualize multiple steps of ligand-receptor interactions and the endocytic cycle at the single-cell level and in real time (Sorkin et al., 2000). Furthermore, many new molecules have been identified in each of the steps of the endocytic cycle. For example, the recruitment of the ligand-bound receptor to the coated clathrin pits and the transfer of receptors to early endocytic compartments rely on tens of proteins. Protein-protein interactions in this system can be assayed using powerful biophysical techniques, and new components can be discovered by high-throughput proteomic approaches (Blagoev et al., 2003). In connection with this increased appreciation of the underlying molecular complexity of the system, it becomes necessary to rethink the mechanistic meaning of the endocytic rate constants predicted by the original model. How should the current model be changed to incorporate new data? Should the new models necessarily have more variables and parameters? Or, alternatively, can the old models be “parameterized” by new interactions? Given the structural complexity of living cells and the significant cell-to-cell variations, it is unlikely that useful models will account for every protein discovered in the endocytic cycle. But, for this and every other cellular system, it remains an open question how to use the new and much richer data to formulate the simplest model that can be used to correlate data and formulate new experiments. The main point is that, at this time, the data are richer than the models, which was not the case in the 1980s and 1990s.
CELLULAR STRUCTURES
In the same way that knowledge of the composition and structure of biomolecules is the key to understanding their biological function, detailed knowledge of the structure of cells is a prerequisite for the quantitative understanding of cellular functions. Mathematics plays an important role in characterizing the intracellular architecture (Figure 4.2). At one
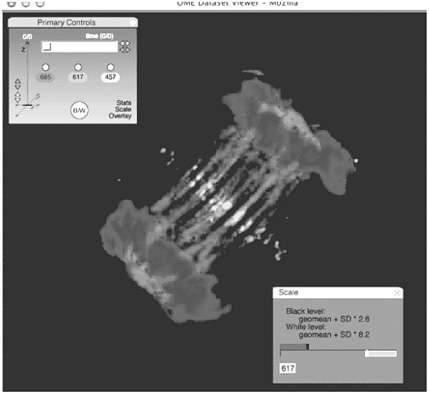
FIGURE 4.2 Applications for quantitative imaging. The image shows an XlK2 cell during the process of cytokinesis stained for DNA, microtubules, and the aurora-B protein kinase. Although the image demonstrates the relative localization of different cellular components and structures, quantitative analysis reveals specific characteristics that can be used to assay the effects of inhibitors on expressed proteins. For example, integrating the signal from a DNA-specific fluorophore might reveal defects in chromosome segregation during mitosis. Measuring the overlap of microtubules and aurora-B (using, for example, a cross-correlation analysis) within a subregion of a dividing cell might be a means of assessing effectors of cytokinesis. The image is displayed within the Open Microscopy Environment Image Viewer. The viewer includes support for displaying multidimensional image data (top left) and some of the associated metadata about each image (bottom right). SOURCE: Swedlow et al., 2003.
level, the mathematical sciences have enabled progress through contributions to the development of instrumentation and other tools. For instance, the explosion of information about the spatiotemporal dynamics in cells would have not have been possible without earlier progress in imaging and data-processing algorithms. Tools such as deconvolution microscopy rely on robust numerical algorithms. Moreover, sophisticated data-processing algorithms have become more accessible to biologists through packages such as Matlab and Metamorph. New data-related challenges are emerging. For example, the assembly of large imaging datasets requires advances in bioinformatics and data mining. The informatic aspects of intracellular imaging are therefore receiving increased attention (Swedlow et al., 2003; Young et al., 2004).
Quantitative imaging enables the formulation of data-driven models of intracellular dynamics and transport. The most notable examples include the quantitative analysis of the dynamics of Golgi to plasma membrane transport (Hirschberg et al., 1998) and nucleocytoplasmic shuttling (Smith et al., 2002). In both cases, green fluorescent protein (GFP)-based imaging provided data of unprecedented spatiotemporal resolution; nevertheless, it was possible to formulate simple compartmental models based on a small number of linear ordinary differential equations. Newer models enable the identification of the rate-limiting steps of the process and the formulation of testable hypotheses. Conclusions about the mechanisms in each case were based on the analysis of a small number of cells. It is unlikely that models of intracellular protein transport and trafficking will remain simple as knowledge of the processes grows and incorporates information on cell-to-cell variation. More sophisticated models that use nonlinear partial differential equations based on the geometry derived from imaging have been used to describe the intracellular dynamics of calcium and metabolites (Slepchenko et al., 2003). In each case, the main challenge in assessing the validity of quantitative predictions lies in careful analysis of the underlying assumptions, such as the use of Fickian diffusion to model the intracellular transport of proteins and small molecules.
Recent years have witnessed the discovery of a large number of highly organized, coherent, dissipative structure in cells, including waves of intracellular calcium and metabolites and protein concentration waves accompanying the division of bacterial cells (Kindzelskii and Petty, 2002; Schuster et al., 2002). While the general phenomenology of these structures is understood from the standpoint of nonlinear dynamics and physicochemical pattern formation, how these structures arise and how they are maintained and used by cells is a topic of intense research. Mathematical analysis of these processes requires significant extensions of the theory
and computational methods for control of spatially distributed nonlinear systems.
In addition to studying and modeling intracellular structures, it is also important to model the possible mechanisms for their emergence from macromolecules. A number of experiments suggest that simple physicochemical principles can drive the emergence of cellular life (Szostak et al., 2001; Hanczyc et al., 2003; Chen et al., 2004). Indeed, experiments with nucleation of lipid vesicles by clays and the competition of protocells containing RNA polymerase suggest that the mechanisms of the formation of cells and intracellular compartments can be systematically studied in the test tube. Molecular simulations of these processes and population balance modeling of the evolution of primitive cellular compartments may provide the link between models at the molecular and cellular scales.
DISCOVERY OF CELLULAR NETWORKS AND THEIR FUNCTIONS
Since networks of interacting proteins control all cellular functions, understanding cellular functions requires quantitative analysis of the spatiotemporal dynamics of these networks in the cellular environment (Figure 4.3). Efforts to elucidate their dynamics can be subdivided into the analysis of network topology, dynamics, spatial organization, and function. Most of the progress recently has been in the area of deducing the network topology, using the classical techniques of cellular and molecular biology, large-scale molecular profiling experiments, or bioinformatics approaches (Brent and Finley, 1997; Chen and Xu, 2003; Ideker, 2004; Irish et al., 2004; Schulze and Mann, 2004; Xia et al., 2004; Yeger-Lotem et al., 2004). While there is significant room for the validation and perfection of each of these approaches, there is an urgent need to compare the networks predicted by distinct methods (Greenbaum et al., 2003). Because of the difficulties associated with generating high-quality data on cellular dynamics, much less work has been done in the analysis of network dynamics. For instance, some of the most interesting results associated with network dynamics have required construction of special experimental systems that include fluorescent reporters for a large number of bacterial genes coupled with high-resolution analysis of bacterial responses over a broad range of experimental conditions. This approach has led to the validation of the network motifs predicted on the basis of bioinformatics analysis and has identified the dynamic and functional roles of these motifs (Shen-Orr et al., 2002; Kalir and Alon, 2004; Zaslaver et al., 2004).
The simplest use of mathematical models for intracellular networks is to integrate data and test if they fit together. For example, mechanistic
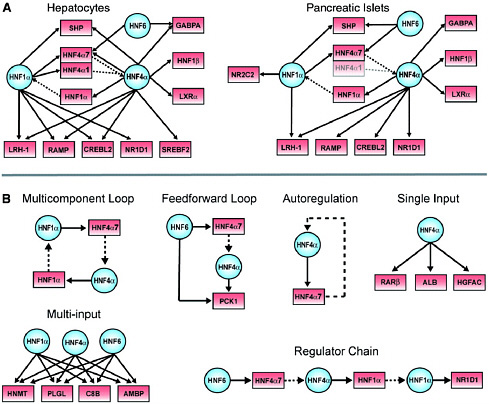
FIGURE 4.3 Transcriptional regulatory networks and motifs. (A) HNF1α, HNF6, and HNF4α are at the center of tissue-specific transcriptional regulatory networks. In these examples selected for illustration, regulatory proteins and their gene targets are represented as circles and boxes, respectively. Solid arrows indicate protein-DNA interactions, and genes encoding regulators are linked to their protein products by dashed lines. The HNF4α1 promoter is poorly expressed in pancreatic islets and is shaded to reflect this. The HNF4α7 promoter, also known as the P2 promoter, is the predominant promoter in pancreatic islets and was recently implicated as an important locus for human diabetes susceptibility. For clarity, some gene promoters have been designated by the names of their protein products (e.g., HNF1α for TCF1, SHP for NR0B2, HNF4α7 for HNF4A P2, and HNF1β for TCF2). (B) Examples of regulatory network motifs in hepatocytes. For instance, in the multicomponent loop, HNF1α protein binds to the promoter of the HNF4α gene, and the HNF4α protein binds to the promoter of the HNF1α gene. These network motifs were uncovered by searching binding data with various algorithms; details on the algorithms used and a full list of motifs found are available. SOURCE: Odom et al., 2004.
models of the cell division cycle in fission yeast summarize the dynamic behavior of multiple mutants and make testable experimental predictions (Tyson et al., 2001). Most of the “realistic” models of intracellular networks, including the widely publicized models of the lysis-lysogeny switch in bacteriophage lambda and models of growth factor signaling, were severely overparameterized (Arkin et al., 1998; Schoeberl et al., 2002). On the one hand, the models have to be large to be able to predict the effects of genetic or biochemical perturbations of network components, and on the other, the models must have the lowest required number of parameters and processes to explain the observed phenomenology.
At this time, there are no standards for assessing the complexity of a model and whether it is indeed a minimal representation of data. Furthermore, current models are characterized by high levels of uncertainty, both structural and parametric. This situation is not surprising, given that even the most comprehensive models may be missing entire parts of a network, may neglect its spatial organization and temporal evolution, and may employ approximate functional forms for various cellular processes. While some tools for dealing with these issues can be borrowed from linear control systems (Csete and Doyle, 2002), new theoretical and computational approaches are required to analyze highly uncertain and nonlinear systems. These new approaches require solving problems in simulation, system identification, parameter estimation, and experimental design.
Recently, robustness has emerged as an important principle for model screening and validation (Barkai and Leibler, 1997; Stelling et al., 2004). In a nutshell, the relative plausibility of two models for the same process can be assessed by comparing the size of the parametric perturbations that can be tolerated by the models without qualitatively changing the predicted behavior. The rationale for using robustness as a screen is that evolution seems to favor the most robust mechanisms. For example, two models of the cell division cycle can be compared on the basis of the size of the regions of the parameter space that predict the limit cycle behavior (Morohashi et al., 2002). Analysis of robustness requires tools that can be used to compare models with different numbers of parameters and even different mathematical structures. The method of mathematically controlled comparisons is a very important development in this direction (Alves and Savageau, 2000). On the experimental side, the model-driven analysis of the robustness of cellular systems requires quantitative characterization of natural and induced parameter variations in cells (Houchmandzadeh et al., 2002; Jones et al., 2004). Quantitative and multivariable analysis of cell-to-cell variations is becoming possible thanks to advances in flow cytometry and live cell imaging (Irish et al., 2004).
The number of systems that have been characterized over the entire range from the biochemical description of the network to its dynamics and function is still very small. One of the best examples is the result of efforts by Ferrell and co-workers, who have analyzed the function of the mitogen-activated protein kinase (MAPK) network, a three-stage enzymatic cascade conserved from yeasts to humans. In an elegant sequence of papers, Ferrell et al. have shown that the multistage nature of the cascade enables it to act as a switch that is insensitive to small inputs at the top layer of the cascade and is fully activated when the threshold value for the input has been crossed (Huang and Ferrell, 1996; Ferrell, 1997; Ferrell and Machleder, 1998; Bagowski and Ferrell, 2001; Ferrell and Xiong, 2001; Xiong and Ferrell, 2003). This prediction, based on extensive computational analysis of the cascade model, has been validated through in vitro experiments with purified components of the network. The biochemical and modeling work set the stage for the analysis of MAPK dynamics in the frog oocyte maturation response. In this response system, it became apparent that the MAPK cascade is embedded in a positive feedback circuit that sharpens the threshold-detection capabilities of the circuit and mediates the irreversibility of the cell’s maturation response to hormones. This insight was enabled by the large size of the frog oocyte, which made it possible to carry out single-cell biochemical assays of cellular responses, once again underscoring the importance of examining cellular responses at a single-cell level. Other examples of quantitative analysis of network dynamics at the single-cell level are now available (Irish et al., 2004; Jones et al., 2004; Lahav et al., 2004; Nelson et al., 2004; Raser and O’Shea, 2004).
The reductionist approach to cellular networks, which focuses on single modules such as the MAPK cascade or the EGF receptor pathway, has been reasonably successful. However, it must be realized that these modules do not operate in isolation and are affected by the large number of other processes occurring simultaneously. For example, genetic and biochemical evidence indicates that the MAPK cascade is coupled to essentially every other signal transduction pathway in cells. Quantitative understanding of cross talk in biochemical networks is necessary in order to probe these “cellular context” effects. While a modeling approach to the problem can start with simulations of coupled signaling or with genetic models, the eventual success of these models will depend on the availability of convenient experimental systems where pathway cross talk can be analyzed at the quantitative level.
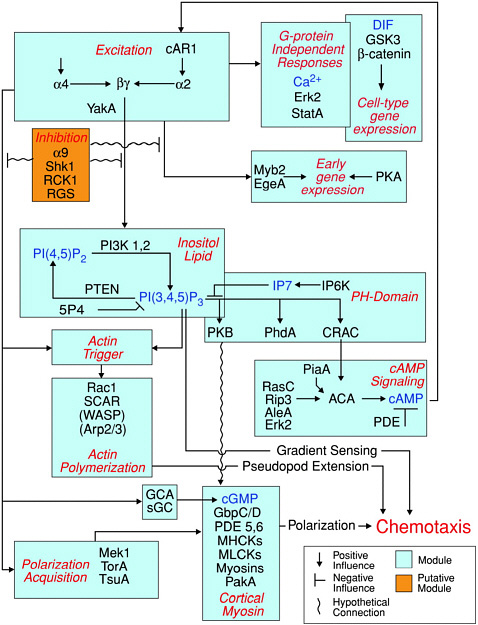
FIGURE 4.4 Modular view of the chemoattractant-induced signaling pathway in Dictyostelium. Except for those in parentheses, the proteins depicted in this pathway have been shown to be involved in chemotactic signaling through analysis of cells in which the genes have been deleted. SOURCE: Manahan et al., 2004.
FROM NETWORKS TO CELLULAR FUNCTIONS
After the analysis of gene and protein networks, the next goal in the quantitative understanding of cells is modeling integrated responses and functions, such as cell differentiation, migration, and the DNA damage response (Figure 4.4). Phenomenological models have been successful in correlating data and discovering qualitative trends in cellular responses. For example, simple biophysical and rheological models have been used to explain the biphasic dependence of cell migration speed on the adhesiveness of the substrate on which the cell is migrating (Lauffenburger and Horwitz, 1996). Simple birth-death processes have been used to model cell division and cell differentiation (Loeffler and Wichmann, 1980).
The earlier phenomenological models must be extended to integrate increasing amounts of information about each of these responses. In the case of cell migration, the intracellular rheology and motile behavior of cells is under the control of signal transduction and cytoskeletal networks that can now be monitored in real time and with increasing spatial resolution (Soll et al., 2000). Each of the biophysical and biochemical modules in cell migration, from signal transduction by integrins to the spatiotemporal dynamics of actin polymerization, is the subject of an extensive modeling effort (Grimm et al., 2003; Manahan et al., 2004). Notably, the modeling formalisms necessary to describe the integrated response have to be heterogeneous, in the sense that they differ in the amount of mechanistic detail incorporated in each specific model and in the mathematical structure of the model. For instance, a stochastic model of actin polymerization has to be coupled to deterministic models of the interactions between integrins and adhesive peptides. The main challenges for the development of quantitative models of cellular responses are related to the multiscale nature of each particular response and to the significant structural and parametric uncertainty of the current models. Integrated models of cell migration, currently being developed by the Cell Migration Consortium (http://www.cellmigration.org/), can be used as surrogates to test whether they can manifest the behavior predicted by earlier phenomenological models (Horwitz et al., 2002).
Perhaps the richest example of a cell-migration phenomenon that has been productively analyzed with the aid of quantitative models is bacterial chemotaxis (see Box 4.1). The ability of many bacteria to swim toward potential food sources or to evade noxious chemicals involves a complete cycle of signal detection and signal transduction and an elaborate response by cellular motility systems. To mount an effective chemotactic response, bacterial cells must not only detect the relevant chemical but also regulate behavior on the basis of spatial and temporal gradients in the signal. A different phenomenon, which also involves global changes in the regula-
tion of bacterial genes in response to chemical signals, is quorum sensing (for a review, see Daniels et al., 2004). In this case, bacteria in a growing population both generate the chemical signal and respond to it. No one bacterium makes enough of the chemical to trigger the response system; however, once cell densities reach a threshold level (i.e., a “quorum” is achieved), the whole population of bacteria alters its regulatory state. This system, too, has attracted increasingly sophisticated mathematical modeling (Chopp et al., 2002; Ward et al., 2001, 2003). Some of this research is explicitly directed at exploring the potential of novel antibacterial drugs that would disrupt quorum sensing (Anguige et al., 2004); this concept is appealing since the regulatory change that many bacteria undergo when cell densities are high leads to increased expression of gene products that severely damage the tissues of an infected host.
Many instances of signal transduction and information processing in cells have been based on population-averaged data derived, for example, from Western blotting for the analysis of protein modification, as exemplified by Hoffmann et al. (2002) and Schoeberl et al. (2002). Currently, multicolor flow cytometry and high-throughput protein localization assays can monitor these processes at single-cell resolution and correlate the information obtained with responses measured at a single-cell level, such as the migratory tracks of individual cells or the differentiation responses of single cells. These data are enabling the systematic analysis of the heterogeneity of cellular responses (Krooshoop et al., 2003; Abraham et al., 2004; Irish et al., 2004). Statistical analysis of the resulting large sets of heterogeneous data—for instance, cell trajectories, protein modification, and localization—will become an important area of future research. In the end, models of cellular responses that can be used in biotechnology and medicine are likely to be reasonably simple correlations, similar in form to but more realistic than the phenomenological models of cellular responses developed over the past two decades.
The ultimate challenge in modeling cellular responses to signals is to track cause-and-effect relationships throughout the pathways leading from signal detection to cellular response. For most biological systems, this goal is remote. For relatively simple phenomena such as bacterial chemotaxis (see Box 4.1), many of cause-and-effect links, all the way from detection of chemical gradients to cellular locomotion, are now known. However, in systems of more typical complexity—particularly in multicellular organisms—the complexity on the response side of signal-response pathways poses immense challenges to modeling techniques. Progress appears likely to involve relatively detailed modeling of the upstream processes and, when possible, the final steps, such as cell motion. However, modeling of the linkage between the front and back ends of signal-response processes will often require heuristic methods that sim-
|
Box 4.1 Bacterial chemotaxis in Escherichia coli (E. coli) is the best understood signal transduction system where one can go all the way from the molecular composition and subcellular organization of the biochemical network to the response of a single cell or population of cells (Berg, 2000). The current picture is a result of extensive genetic, biochemical, and biophysical analysis of this system over the past 50 years. The evolution of the quantitative understanding of bacterial chemotaxis shows unequivocally that helpful quantitative models are impossible without experimental innovations and are greatly enabled by the ease of genetic manipulation of the system. The phenomenon was originally described by Adler at the macroscopic level as a directed flux of bacteria in gradients of nutrients such as aspartate (see Berg (2000) for a review). Berg and colleagues, who designed a tracking microscope that generated information about the migratory tracks of single cells, described the microscopic nature of bacterial chemotaxis. In isotropic environments, the path of E. coli is composed of straight runs punctuated by brief tumbles that can change the direction of migration. In a gradient of chemoattractant, the runs between the tumbling events are increased. At the population level, this change in the microscopic behavior of a single cell generates a directed flux of cells toward a source of a chemoattractant. Gradient sensing is mediated by ligand-receptor interaction at the cell surface that induces a sequence of biochemical reactions in the cytoplasm and culminates in the generation of the diffusible cytoplasmic molecule that binds to the motor powering the bacterial flagella, biases the sense of its rotation, and in this way changes the frequency of the tumbling events. The output of the circuit, defined as the frequency of the tumbling events, reflects the temporal derivative of receptor occupancy, which can be “measured” by bacteria with very high sensitivity and speed. The circuit is characterized by a very wide dynamic range, which is mediated by negative feedback loop in the signal transduction cascade. Today, we have detailed information about the genetics and biochemistry of the chemotaxis network in E. coli, we know the three-dimensional structures of several key proteins, we can monitor their interactions in vivo and in real time, and we can reconstitute parts of the network in vitro and measure the relevant thermodynamic and rate constants. Bacterial chemotaxis has become a fruitful arena for modeling and computational analysis. Essentially every part of the circuit, from cell surface receptor to the flagellar motor, gave rise to mathematical models, ranging from structural descriptions of protein organization in the mem- |
|
brane (Bray and Duke, 2004), to the dynamics of signal transduction in the cytoplasm (Barkai and Leibler, 1997), to the kinetic theory of bacterial transport (Hillen and Othmer, 2000). The two most notable modeling efforts are the Berg and Purcell theory of ligand concentration measurement in gradient detection (Berg and Purcell, 1977) and the Barkai-Leibler model of robustness in the adaptation part of the circuit (Barkai and Leibler, 1997). Berg and Purcell used a model to test the hypothesis that bacteria can sense temporal gradients on the timescale of seconds. To test this hypothesis, they developed a very elegant stochastic biophysical theory of diffusion-limited ligand receptor binding. As a result of their analysis, they have concluded that bacteria can indeed measure concentrations and “take the temporal derivatives” of concentrations within a very short period of time. Their analysis was based on the assumption that receptors are distributed randomly over the surface of the cell. Subsequently their results were rederived by Szabo et al. (1982), using a much more transparent approach based on the homogenization theory and tested in direct Brownian dynamics simulations by Northrup (1988). The Berg-Purcell model of ligand-receptor binding in bacterial chemotaxis has become a classic in the theory of diffusion-limited reactions, continues to enjoy extensive citations, and has entered the textbooks in biophysics. At the same time, the electron microscopy images of E. coli produced in the early 1990s show that the main assumption of the theory, the random distribution of receptors, is not satisfied and that receptors are clustered in one region of the cell surface (Parkinson and Blair, 1993). Independent confirmations of this result and biochemical proof of receptor clustering gave rise to a new wave of models that attempt to explain their functional significance. At this time, it is established that receptor clustering is crucial for high sensitivity of the gradient sensing system. The dynamics and spatial organization of receptor clusters is now studied in models that are firmly grounded in the structural details of protein-protein interactions in bacterial chemotaxis (Bray and Duke, 2004). While we are still a long way from having an integrated model of bacterial chemotaxis that would integrate all the structural, genetic, and biochemical evidence, analysis of this system over the past decades sets an excellent example of the most productive integration of experiments, multiscale biophysical modeling, and mathematical analysis (Erban and Othmer, 2005). One of the natural questions is whether the biophysical mechanisms of gradient detection in E.coli can be useful for understanding these processes in other cell types. A first step in this direction was taken in a recent computational model that compared the control strategies in E. coli and B. subtilis (Rao et al., 2004). |
ply capture cause-and-effect correlations rather than providing quantitative models of actual pathways.
FROM CELLS TO TISSUES
Quantitative descriptions of cellular processes and functions form the basis for models at the tissue and organism level (Figure 4.5). Population-level modeling of cell migration is a good example of this approach (Maheshwari and Lauffenburger, 1998). Statistical analysis of the migratory tracks of single cells can be used to extract the probability density functions of cell velocities, turning frequencies, persistence time, and other variables. Such information about the properties of a single cell can be used to derive partial differential equations for the evolution of cell densities. The dynamics predicted by these equations can be quantitatively compared with the measurements of cellular fluxes (Hillen and Othmer, 2000). The same general framework, in which a microscopic description of particle motion is used to predict the evolution of particle ensembles, is encountered many times in the natural sciences—for example, in the derivation of the kinetic theory of gases or in the equations of fluid motion from the detailed description of molecular motion. The rules of cellular motion are much more complex than those governing the molecules in an ideal gas. However, cellular trajectories can be visualized with much greater ease than the trajectories of interacting molecules in gases or fluids (Othmer et al., 1988; Painter and Sherratt, 2003). An integrated program of this kind was implemented for bacterial and animal cells in the 1980s (Farrell et al., 1990). Today, similar analyses can be complemented with increasingly detailed information about the coupling between intracellular processes, such as signal transduction or cytoskeletal dynamics, and cellular responses, such as proliferation and migration. Multiscale models for the evolution of cell densities are being constructed to describe E. coli chemotaxis (Bren and Eisenbach, 2000; Erban and Othmer, 2005). Analysis of these models poses many challenging problems for multiscale theory and numerical analysis.
Modeling of tissue patterning is another example of analysis at the tissue level that is based on extensive studies of cellular processes. One of the mechanisms for generating cell diversity in embryogenesis is based on patterning of an epithelial layer, whereby a lattice of initially identical cells is presented with a spatial gradient of a ligand that binds to cell surface receptors and induces gene expression in target cells (Tabata and Takei, 2004). The level of cell surface receptor occupancy can be directly translated into the transcriptional response of the target cell. In this way, the spatial gradient of an extracellular ligand can be translated into a spatial pattern of gene expression in a layer of “naive” cells. Morphogen gra-
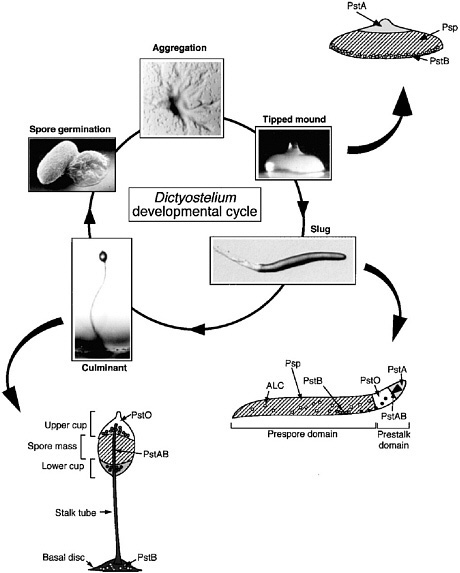
FIGURE 4.5 Dictyostelium development cycle and position of cell types within the multicellular differentiating organism. SOURCE: Kimmel and Firtel, 2004.
dients are established by the combination of localized secretion of ligands, their extracellular transport, binding to cell surface receptors, and intracellular trafficking processes. Computational models can be used to identify the relevant spatial and temporal scales in the generation of the morphogen gradients and to evaluate the relative feasibility of competing
hypotheses (Lander et al., 2002; Kruse et al., 2004). These models can be based on earlier quantitative models of ligand-receptor dynamics in cells and on the current imaging experiments in developing tissues (Lauffenburger and Linderman, 1993).
Experimental models such as those for bacterial quorum sensing and slime mold aggregation enable biochemical and genetic analyses of the emergence of multicellular behavior in populations of seemingly identical cells (Taga and Bassler, 2003; Chisholm and Firtel, 2004). Detailed understanding of cell-to-cell communication is the key to developing integrative models of these processes (Nagano, 2000; Dockery and Keener, 2001). In all current biology textbooks, cell communication proceeds in a unidirectional way, where the signal is received by the cell and interpreted to direct cellular responses. In reality, cell-cell communication protocols are not unidirectional. Cells can both receive and respond to extracellular signals; hence they actively modify their environments. At the same time, cells are generating and responding to mixtures of signals. Modeling of such cell-cell communication protocols is complicated by the experimental difficulties associated with quantifying the dynamics and spatial regulation of cell communication signals. For example, mammalian cells can secrete a large number of soluble growth factors. The molecular identity of these signals and their potential roles in spatiotemporal information processing by cells is only beginning to be understood (Werb and Yan, 1998).
In addition to studying the processes through which single cells assemble into spatial patterns and tissues, it is important to study how tissues devolve to cells, as they do, for example, in the epithelial-mesenchymal transition (EMT), one of the critical steps in tumorigenesis. Genetic studies show that relatively small networks of genes can mediate EMT (Hahn and Weinberg, 2001). Translating this information into the integrative descriptions of epithelial dynamics poses an exciting and important problem for modeling.
DATA INTEGRATION
Rapid technical advances in genomics and proteomics have led to an extraordinary proliferation of data, which offers an unprecedented opportunity to understand how organisms function but poses significant challenges as well. Experimental design, hypothesis testing, and conceptual model building all require biologists to collect, evaluate, and integrate large amounts of information of many disparate kinds. There is a need to create user-friendly tools to assist researchers in designing and testing new hypotheses against the quickly growing, distributed knowledge base and to facilitate the development of experimentally verified
models through the accumulation of validated hypotheses stored in databases designed from the ground up to support hypothesis testing.
Three kinds of conceptual and bioinformatics challenges appear in today’s data-rich environment: (1) information retrieval and integration, (2) knowledge representation, and (3) hypothesis testing and model building. The first and second are closely related: How can we retrieve and express the many qualitatively different kinds of information available in databases and the published literature in a representation that informs experimentation? Developing common ontologies for biological objects and processes is essential for supporting the intercommunication of diverse databases (Schulze-Kremer, 1998; Ashburner et al., 2000) and for enabling the automated annotation and extraction of information from the published literature (Andrade et al., 1999; Fleischmann et al., 1999; Friedman et al., 2001; Stephens et al., 2001; Yakushiji et al., 2001). An ontology also provides the foundation for constructing higher-level representations of biological systems (Rzhetsky et al., 2000; Peleg et al., 2002).
The third challenge is to create and verify testable conceptual representations of the biological system. A conceptual framework for representing biological systems must accommodate the modularity and temporal evolution of biological networks, as well as handle their nonlinearity, plasticity, redundancy, and degeneracy. Conceptual models vary from the simple Boolean networks pioneered by Kaufmann (1969, 1993), Liang et al. (1998), and Akutsu et al. (2000a, 2000b) to Bayesian networks (Friedman et al., 2000; Hartemink et al., 2001; Pe’er et al., 2001), as well as highly concrete (McAdams and Arkin, 1998; Judd et al., 2000) and quantitative (Sveiczer et al., 2000) models. Incorporating disparate kinds of information about biological systems into a common conceptual framework remains a major stumbling block for validating ideas about real biological networks, and current efforts focus largely on just one or two categories of information (Rzhetsky et al., 2000; Hartemink et al., 2001; Wessels et al., 2001). There is a need to develop a hypothesis representation language that can assist in integrating experimental information at the logical level, as well as approaches to aggregating validated hypotheses into increasingly quantitative models.
Most currently available bioinformatics tools support the analytical tasks of the biologist. These tools are very useful and effective for certain specific tasks, such as identifying patterns, categorizing information, and simultaneously probing multiple data sources for similarities. Such tasks usually comprise the early steps of the discovery process. However, synthesis and evaluation of the information remain the task of the individual. Kuchinsky and his colleagues (2002) argue that this synthesis task can be broken down into steps: (1) keeping track of all the diverse pieces of information collected during the database searches and other retrieval activi-
ties, (2) organizing and using this information by formulating hypotheses and higher level explanations, and (3) sharing the information with colleagues and working collaboratively with colleagues to refine hypotheses. There is a need to develop a system to allow biologists to construct and verify formal language hypotheses, making use of event- and process-based description language.
Complex processes that exhibit nonlinear behavior, as biological systems do, are often more readily described by event-driven dynamics (Ho, 1989) than by differential equations. Furthermore, when biologists think about biological systems, they typically do so in terms of biological agents, events, and causal relationships between events. A symbolic discrete modeling approach, in which the discretizations are defined by events—that is, by any biological change for which there is experimental evidence of changes in the state of the system—is likely to be a useful approach.
BIOLOGICAL CONSIDERATIONS
The capacity of cells to differentiate is a hallmark of eukaryotic organisms. Differentiation is the acquisition of structurally and chemically different identities by cells over time. The capacity for self-differentiation, which transforms a single diploid cell (the zygote) into a complex, multicellular plant or animal comprising many different structural and functional tissues, is rooted in asymmetries within the initial cell and has its origins in the unequal intracellular distribution of small molecules, macromolecules, and organelles. Such inhomogeneities in cell structure can be triggered by external stimuli, such as fertilization (Green, 1993; Rossant and Tam, 2004; Swann et al., 2004) and light stimuli (Robinson et al., 1999), which set in motion a complex series of structural and compositional re-organizations that in turn generate compositionally different daughter cells, whose differences are further reinforced by differential gene expression (Kanka, 2003).
The rapid advances during the second half of the 20th century in the understanding of how DNA functions in heredity and expresses the information it encodes fostered a markedly genocentric view of eukaryotic development. This was further reinforced by a virtual flood of genome sequences and gene expression data that followed technological developments in nucleic acid sequencing and monitoring of gene expression patterns during the final decades of the century. Both embryonic development and cellular differentiation were viewed as under the control of genes, whose differential expression was orchestrated by a “developmental program.” Indeed, the notion of a developmental program has dominated thinking about development for decades (Davidson et al., 1995; Marczynski and Shapiro, 1995; Chu et al., 1998; Roberts et al., 2000).
However, actual progress in understanding developmental processes has occurred by studying finer levels of detail rather than attempting to delineate an overarching developmental program. Mutations that affect development are often in genes that code for proteins that function in inter- and intracellular signaling and structure. Moreover, there are many molecular mechanisms by which cells affect the spatial patterning, including small molecules, such as the gaseous hormones nitric oxide (plants and animals) and ethylene (plants), and intermediate-sized molecules, such as the plant gibberellins and brassinosteroids and the animal endocrine and autocrine hormones.
Patterning mechanisms also include macromolecular mechanisms, such as the intracellular transport of proteins and RNA in both plants and animals. Examples include the translational gradients established in early Drosophila embryogenesis by the localization of bicoid and oskar mRNA molecules (Micklem et al., 2000) and the intercellular interactions that underlie the subsequent development of the abdominal segmentation pattern (Immergluck et al., 1990; Ingham and Arias, 1992; Courey and Huang, 1995). Analysis of processes such as these has led to articulation of the view that all of development can be explained by local interactions (Britten, 1998). Indeed, current developmental models are increasingly couched in terms of cellular fate determination by signaling between cells and by programmed cell death (Lam et al., 2001; Ribeiro et al., 2003; Lai and Orgogozo, 2004). Morphogenetic proteins can be secreted signaling proteins that alter the fates of cells through cell-surface-receptor-mediated pathways (Tabata and Takei, 2004). Receptors at the cell surface and intracellular signaling proteins, signaling cascades (such as MAPK cascades), and protein networks mediate the activation of genes in response to extracellular signals (Imler and Hoffmann, 2002; Muller and Bossinger, 2003; Schulz and Yutzey, 2004).
Epigenetic mechanisms are conceptualized as mechanisms that stably affect gene expression without altering gene structure. Initially, epigenetic mechanisms were equated with stable, even heritable, modifications in gene expression, commonly ones that suppress gene expression. Early descriptions of plant paramutation (Brink, 1960), transposable element inactivation (McClintock, 1965), and X-chromosome inactivation in mammals (Lyon, 1961) provided the foundation for what has become an active field of epigenetic research. Because differentiated plant cells can regenerate into whole plants and nuclei from differentiated animal cells can support development of enucleated eggs, DNA in most cells is not irreversibly altered during development and differentiation. However, the epigenetic modifications in gene expression that occur during development are highly regular, impose important differences between male and female gametes, and are not easily reversed experimentally
(Wrenzycki and Niemann, 2003; Tian, 2004) although they are altered regularly during gametogenesis and early embyronic development (Gehring et al., 2004; Santos and Dean, 2004).
There has been important progress in understanding the diverse mechanisms for epigenetic modifications of gene function. These mechanisms include DNA methylation, which compromises the digital purity of the Watson-Crick model of DNA by, in effect, putting “asterisks” on certain bits in the digital code, and highly dynamic chemical modifications of the histone proteins around which DNA wraps in the chromosomes of multicellular organisms. The complexity of these mechanisms and of the pathways that regulate their operation—together with the ubiquity of epigenetic effects during development—indicates that overly genecentric models of development are likely to fail.
FUTURE DIRECTIONS
The emphasis in cell biology is shifting from phenomenological descriptions to predictive models that are consistent with the largest possible amount of data. Data integration, reduction, and multiscale modeling approaches will take center stage in dealing with the diverse data sets emerging from molecular profiling and imaging experiments. Increasingly, biologists will use these approaches to identify the important species, interactions, and processes occurring within cells. Models of cellular processes must explicitly account for the significant parametric and structural uncertainties inevitable at the current level of knowledge and experimental resolution. In general, special attention should be paid to the proper selection and validation of the mathematical formalisms used to model any given process. Advances in dynamical systems theory will be required to deal with inevitable model heterogeneity, such as that required by the combination of stochastic descriptions of gene expression and deterministic descriptions of signal transduction events.
At this time, scientists are far from having an adequate quantitative description of cellular responses, even in well-studied systems such as bacterial chemotaxis. The field must make a concerted effort to improve the quantitative analysis of such model systems in order to achieve successes that can be used as templates for modeling and analysis in less well-established experimental systems. At the same time, the field must establish new experimental systems where integrative genetic, biochemical, and cell biological experiments are possible and that can support meaningful modeling efforts. It will be necessary to establish experimental systems that can serve as testing grounds for multiscale models that can be used to understand how cellular functions emerge from molecu-
lar-scale events and how cell population or tissue-level functions emerge from cellular-scale events.
Models must span as many scales as possible, from sequence-specific information on gene expression, to intracellular biochemistry, to cellular responses. Large-scale integrative approaches require the creation and funding of interacting groups of mathematicians, computer scientists, and biologists. While models should be based on detailed analysis of specific experimental systems—for example, particular cell types—model builders should strive to make the models generalizable to other systems. For example, it is important to analyze the evolution of cellular signaling systems in animals from worms to humans, as well as in plants. What mediates the increase in the number of signaling components in particular evolutionary linkages, and what are the systems-level consequences of this increase in complexity?
Quantitative experimental analyses of intracellular fluctuations and noise are critical for understanding cellular functions and the limits of applicability of conventional deterministic and continuum approaches. This type of analysis is also important for understanding the mechanisms and functional consequences of nongenetic individuality. Analysis of cell-to-cell variations is now possible owing to advances in imaging and single-cell molecular profiling experiments. On the purely theoretical and computational side, scientists must (1) classify dynamical systems according to the ways in which they can tolerate intracellular and extracellular noise and (2) understand the processes where noise can play a constructive role.
Thus, future research must emphasize the close connection between experiments, model validation, and data integration. While this level of integration is only possible by focusing on specific experimental systems, the field must make an effort to systematize methodological advances so that scientists do not have to start from scratch every time they analyze a new cellular system.
REFERENCES
Abraham, V.C., D.L. Taylor, J.R. Haskins. 2004. High content screening applied to large-scale cell biology. Trends Biotechnol. 22(1): 15-22.
Akutsu, T., S. Miyano, and S. Kuhara. 2000a. Algorithms for identifying Boolean networks and related biological networks based on matrix multiplication and fingerprint function. J. Comput. Biol. 7(3-4): 331-343.
Akutsu, T., S. Miyano, and S. Kuhara. 2000b. Inferring qualitative relations in genetic networks and metabolic pathways. Bioinformatics 16(8): 727-734.
Alves, R., and M.A. Savageau. 2000. Extending the method of mathematically controlled comparison to include numerical comparisons. Bioinformatics 16(9): 786-798.
Andrade, M.A., N.P. Brown, C. Leroy, S. Hoersch, A. de Daruvar, C. Reich, A. Franchini, J. Tamames, A. Valencia, C. Ouzounis, and C. Sander. 1999. Automated genome sequence analysis and annotation. Bioinformatics 15(5): 391-412.
Anguige, K., J.R. King, J.P. Ward, and P. Williams. 2004. Mathematical modelling of therapies targeted at bacterial quorum sensing. Math. Biosci. 192(1): 39-83.
Arkin, A., J. Ross, and H.H. McAdams. 1998. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics 149(4): 1633-1648.
Ashburner, M., C.A. Ball, J.A. Blake, D. Botstein, H. Butler, J.M. Cherry, A.P. Davis, K. Dolinski, S.S. Dwight, J.T. Eppig, M.A. Harris, D.P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J.C. Matese, J.E. Richardson, M. Ringwald, G.M. Rubin, and G. Sherlock. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25(1): 25-29.
Bagowski, C.P., and J.E. Ferrell. 2001. Bistability in the JNK cascade. Curr. Biol. 11(15): 1176-1182.
Barkai, N., and S. Leibler. 1997. Robustness in simple biochemical networks. Nature 387(6636): 913-917.
Berg, H.C. 2000. Motile behavior of bacteria. Physics Today 53(1): 24-29.
Berg, H.C., and E.M. Purcell. 1977. Physics of chemoreception. Biophys. J. 20: 193-219.
Blagoev, B., I. Kratchmarova, S.-E. Ong, M. Nielsen, L.J. Foster, and M. Mann. 2003. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat. Biotechnol. 21(3): 315-318.
Bray, D., and T. Duke. 2004. Conformational spread: The propagation of allosteric states in large multiprotein complexes. Annual Review of Biophysics and Biomolecular Structure 33: 53-73.
Bren, A., and M. Eisenbach. 2000. How signals are heard during bacterial chemotaxis: Protein-protein interactions in sensory signal propagation. J. Bacteriol. 182(24): 6865-6873.
Brent, R., and R.L.J. Finley. 1997. Understanding gene and allele function with two-hybrid methods. Annu. Rev. Genet. 31: 663-704.
Brink, R.A. 1960. Paramutation and chromosome organization. Q. Rev. Biol. 35(2): 120-137.
Britten, R.J. 1998. Underlying assumptions of developmental models. Proc. Natl. Acad. Sci. U.S.A. 95(16): 9372-9377.
Chen, I.A., R.W. Roberts, J.W. Szostak. 2004. The emergence of competition between model protocells. Science 305(5689): 1474-1476.
Chen, Y., and D. Xu. 2003. Computational analyses of high-throughput protein-protein interaction data. Curr. Protein Pept. Sci. 4(3): 159-181.
Chisholm, R.L., and R.A. Firtel. 2004. Insights into morphogenesis from a simple developmental system. Nat. Rev. Mol. Cell. Biol. 5(7): 531-541.
Chopp, D.L., M.J. Kirisits, B. Moran, and M.R. Parsek. 2002. A mathematical model of quorum sensing in a growing bacterial biofilm. J. Ind. Microbiol. Biot. 29(6): 339-346.
Chu, S., J. DeRisi, M. Eisen, J. Mulholland, D. Botstein, P.O. Brown, and I. Herskowitz. 1998. The transcriptional program of sporulation in budding yeast. Science 282(5389): 699-705.
Courey, A.J., and J.D. Huang. 1995. The establishment and interpretation of transcription factor gradients in the Drosophila embryo. BBA-Gene Struct. Expr. 1261(1): 1-18.
Csete, M.E., and J.C. Doyle. 2002. Reverse engineering of biological complexity. Science 295(5560): 1664-1669.
Daniels, R., J. Vanderleyden, and J. Michiels. 2004. Quorum sensing and swarming migration in bacteria. FEMS Microbiol. Rev. 28(3): 261-289.
Davidson, E.H., K.J. Peterson, and R.A. Cameron. 1995. Origin of bilaterian body plans: Evolution of developmental regulatory mechanisms. Science 270(5240): 1319-1325.
Dockery, J.D., and J.P. Keener. 2001. A mathematical model for quorum sensing in Pseudomonas aeruginosa. Bull. Math. Biol. 63(1): 95-116.
Erban, R., and H.G. Othmer. 2005. From signal transduction to spatial pattern formation in E. coli: A paradigm for multi-scale modeling in biology. Multiscale Model. Simul. 3(2): 362-394.
Farrell, B.E., R.P. Daniele, D.A. Lauffenburger. 1990. Quantitative relationships between single-cell and cell-population model parameters for chemosensory migration responses of alveolar macrophages to C5a. Cell Motil. Cytoskeleton 16(4): 279-293.
Ferrell, J. 1997. How responses get more switch-like as you move down a protein kinase cascade. Trends Biochem. Sci. 22(8): 288-289.
Ferrell, J., and W. Xiong. 2001. Bistability in cell signaling: How to make continuous processes discontinuous, and reversible processes irreversible. Chaos 11(1): 221-236.
Ferrell, J.J., and E. Machleder. 1998. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science 280(5385): 895-898.
Fleischmann, W., S. Moller, A. Gateau, and R. Apweiler. 1999. A novel method for automatic functional annotation of proteins. Bioinformatics 15(3): 228-233.
Friedman, C., P. Kra, H. Yu, M. Krauthammer, and A. Rzhetsky. 2001. GENIES: A natural-language processing system for the extraction of molecular pathways from journal articles. Bioinformatics 17(Suppl 1): S74-S82.
Friedman, N., M. Linial, I. Nachman, and D. Pe’er. 2000. Using Bayesian networks to analyze expression data. J. Comput. Biol. 7(3-4): 601-620.
Gehring, M., Y. Choi, and R.L. Fischer. 2004. Imprinting and seed development. Plant Cell 16: S203-S213.
Green, D.P. 1993. Mammalian fertilization as a biological machine: A working model for adhesion and fusion of sperm and oocyte. Hum. Reprod. 8(1): 91-96.
Greenbaum, D., C. Colangelo, K. Williams, and M. Gerstein. 2003. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 4(9): 117.
Grimm, H.P., A.B. Verkhovsky, A. Mogilner, and J.J. Meister. 2003. Analysis of actin dynamics at the leading edge of crawling cells: Implications for the shape of keratocyte lamellipodia. Eur. Biophys. J. 32(6): 563-577.
Hahn, W.C., and R.A. Weinberg. 2001. Rules for making human tumor cells. N. Engl. J. Med. 347(20): 1593-1603.
Hanczyc, M.M., S.M. Fujikawa, and J.W. Szostak. 2003. Experimental models of primitive cellular compartments: Encapsulation, growth, and division. Science 302(5645): 618-622.
Hartemink, A.J., D.K. Gifford, T.S. Jaakkola, and R.A. Young. 2001. Using graphical models and genomic expression data to statistically validate models of genetic regulatory networks. Pp. 422-433 in Biocomputing 2001: Proceedings of the Pacific Symposium. Singapore: World Scientific.
Hillen, T., and H.G. Othmer. 2000. Chemotaxis equations from the diffusion limit of transport equations. SIAM J. Appl. Math. 62: 1222-1250.
Hirschberg, K., C.M. Miller, J. Ellenberg, J.F. Presley, E.D. Siggia, R.D. Phair, and J. Lippincott-Schwartz. 1998. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 143(6): 1485-1503.
Ho, Y.C. 1989. Special issue on discrete event dynamical systems: Editorial. Proc. IEEE 77(1): 24-38.
Hoffmann, A., A. Levchenko, M.L. Scott, and D. Baltimore. 2002. The IkB-NF-kB signaling module: Temporal control and selective gene activation. Science 298(5596): 1241-1245.
Horwitz, A.R., N. Watson, and J.T. Parsons. 2002. Breaking barriers through collaboration: The example of the Cell Migration Consortium. Genome Biol. 3(11): comment 2011.
Houchmandzadeh, B., E. Wieschaus, and S. Leibler. 2002. Establishment of developmental precision and proportions in the early Drosophila embryo. Nature 415(6873): 798-802.
Huang, C., and J.J. Ferrell. 1996. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A.. 93(19): 10078-10083.
Ideker, T. 2004. A systems approach to discovering signaling and regulatory pathways—or, how to digest large interaction networks into relevant pieces. Adv. Exp. Med. Biol. 547: 21-30.
Imler, J.L., and J.A. Hoffmann. 2002. Toll receptors in Drosophila: A family of molecules regulating development and immunity. Curr. Top. Microbiol. 270: 63-79.
Immergluck, K., P.A. Lawrence, and M. Bienz. 1990. Induction across germ layers in Drosophila mediated by a genetic cascade. Cell 62(2): 261-268.
Ingham, P.W., and A.M. Arias. 1992. Boundaries and fields in early embryos. Cell 68(2): 221-235.
Irish, J.M., R. Hovland, P.O. Krutzik, O.D. Perez, O. Bruserud, B.T. Gjertsen, and G.P. Nolan. 2004. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 118(2): 217-228.
Jones, J.T., J.W. Myers, J.E. Ferrell, and T. Meyer. 2004. Probing the precision of the mitotic clock with a live-cell fluorescent biosensor. Nat. Biotechnol. 22(3): 306-312.
Judd, E.M., M.T. Laub, H.H. McAdams. 2000. Toggles and oscillators: New genetic circuit designs. Bioessays 22(6): 507-509.
Kalir, S., and U. Alon. 2004. Using a quantitative blueprint to reprogram the dynamics of the flagella gene network. Cell 117(6): 713-720.
Kanka, J. 2003. Gene expression and chromatin structure in the pre-implantation embryo. Theriogenology 59(1): 3-19.
Kauffman, S.A. 1993. The Origins of Order: Self-organization and Selection in Evolution. New York, N.Y.: Oxford University Press.
Kaufmann, S.A. 1969. Metabolic stability and epigenesis in randomly connected nets. J. Theor. Biol. 22: 437-467.
Kimmel, A.R., and R.A. Firtel. 2004. Breaking symmetries: Regulation of Dictyostelium development through chemoattractant and morphogen signal-response. Curr. Opin. Genet. Dev. 14(5): 540-549.
Kindzelskii, A.L., and H.R. Petty. 2002. Apparent role of traveling metabolic waves in oxidant release by living neutrophils. Proc. Natl. Acad. Sci. U.S.A. 99(14): 9207-9212.
Krooshoop, D.J., R. Torensma, G.J. van den Bosch, J.M. Nelissen, C.G. Figdor, R.A. Raymakers, and J.B. Boezeman. 2003. An automated multi well cell track system to study leukocyte migration. J. Immunol. Methods 280(1-2): 89-102.
Kruse, K., P. Pantazis, T. Bollenbach, F. Jülicher, and M. González-Gaitán. 2004. Dpp gradient formation by dynamin-dependent endocytosis: Receptor trafficking and the diffusion model. Development 131(19): 4843-4856.
Kuchinsky, A., K. Graham, D. Moh, A. Adler, K. Babaria, and M.L. Creech. 2002. Biological storytelling: A software tool for biological information organization based upon narrative structure. ACM SIGGROUP Bulletin 23(2): 4-5.
Lahav, G., N. Rosenfeld, A. Sigal, N. Geva-Zatorsky, A.J. Levine, M.B. Elowitz, and U. Alon. 2004. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36(2): 147-150.
Lai, E.C., and V. Orgogozo. 2004. A hidden program in Drosophila peripheral neurogenesis revealed: Fundamental principles underlying sensory organ diversity. Dev. Biol. 269(1): 1-17.
Lam, E., N. Kato, and M. Lawton. 2001. Programmed cell death, mitochondria and the plant hypersensitive response. Nature 411(6839): 848-853.
Lander, A.D., W. Nie, and F.Y.-M. Wan. 2002. Do morphogen gradients arise by diffusion? Dev. Cell 2(6): 785-796.
Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: A physically integrated molecular process. Cell 84(3): 359-369.
Lauffenburger, D.A., and J.J. Linderman. 1993. Receptors: Models for Binding, Trafficking, and Signaling. New York, N.Y.: Oxford University Press.
Liang, S., S. Fuhrman, and R. Somogyi. 1998. REVEAL, a general reverse engineering algorithm for inference of genetic network architectures. Pp. 18-29 in Biocomputing ’98: Proceedings of the Pacific Symposium. Singapore: World Scientific.
Loeffler, M., and H.E. Wichmann. 1980. A comprehensive mathematical model of stem cell proliferation which reproduces most of the published experimental results. Cell Tiss. Kinet. 13: 543-561.
Lyon, M. 1961. Gene action in the X-chromosome of the mouse. Nature 190: 372-373.
Maheshwari, G., and D.A. Lauffenburger. 1998. Deconstructing (and reconstructing) cell migration. Microsc. Res. Tech. 43(5): 358-368.
Manahan, C.L., P.A. Iglesias, Y. Long, and P.N. Devreotes. 2004. Chemoattractant signaling in dictyostelium discoideum. Annu. Rev. Cell Dev. Biol. 20: 223-253.
Marczynski, G.T., and L. Shapiro. 1995. The control of asymmetric gene-expression during caulobacter cell-differentiation. Arch. Microbiol. 163(5): 313-321.
McAdams, H.H., and A. Arkin. 1998. Simulation of prokaryotic genetic circuits. Annu. Rev. Biophys. Biomol. Struct. 27: 199-224.
McClintock, B. 1965. The control of gene action in maize. Pp. 162-184 in Brookhaven Symposium in Biology. Upton, N.Y.: Brookhaven National Laboratory.
Micklem, D.R., J. Adams, S. Grunert, and D. St. Johnston. 2000. Distinct roles of two conserved Staufen domains in oskar mRNA localization and translation. EMBO J. 19(6): 1366-1377.
Morohashi, M., A.E. Winn, M.T. Borisuk, H. Bolouri, J. Doyle, and H. Kitano. 2002. Robustness as a measure of plausibility in models of biochemical networks. J. Theor. Biol. 216(1): 19-30.
Muller, H.A.J., and O. Bossinger. 2003. Molecular networks controlling epithelial cell polarity in development. Mech. Dev. 120(11): 1231-1256.
Nagano, S. 2000. Modeling the model organism Dictyostelium discoideum. Dev. Growth Differ. 42(6): 541-550.
Nelson, D.E., A.E. Ihekwaba, M. Elliott, J.R. Johnson, C.A. Gibney, B.E. Foreman, G. Nelson, V. See, C.A. Horton, D.G. Spiller, S.W. Edwards, H.P. McDowell, J.F. Unitt, E. Sullivan, R. Grimley, N. Benson, D. Broomhead, D.B. Kell, and M.R.H. White. 2004. Oscillations in NF-kB signaling control the dynamics of gene expression. Science 306(5696): 704-708.
Northrup, S.H. 1988. Diffusion-controlled ligand binding to multiple competing cell-bound receptors. J. Phys. Chem. 92: 5847-5850.
Odom, D.T., N. Zizlsperger, D.B. Gordon, G.W. Bell, N.J. Rinaldi, H.L. Murray, T.L. Volkert, J. Schreiber, P.A. Rolfe, D.K. Gifford, E. Fraenkel, G.I. Bell, and R.A. Young. 2004. Control of pancreas and liver gene expression by HNF transcription factors. Science 303(5662): 1378-1381.
Othmer, H.G., S.R. Dunbar, and W. Alt. 1988. Models of dispersal in biological systems. J. Math. Biol. 26(3): 263-298.
Painter, K.J., and J.A. Sherratt. 2003. Modelling the movement of interacting cell populations. J. Theor. Biol. 225(3): 327-339.
Parkinson, J.S., and D.F. Blair. 1993. Does E. coli have a nose? Science 259: 1701-1702.
Pe’er, D., A. Regev, G. Elidan, and N. Friedman. 2001. Inferring subnetworks from perturbed expression profiles. Bioinformatics 17(Suppl. 1): 215-224.
Peleg, M., I. Yeh, and R.B. Altman. 2002. Modelling biological processes using workflow and Petri Net models. Bioinformatics 18(6): 825-837.
Rao, C.V., J.R. Kirby, and A.P. Arkin. 2004. Design in bacterial chemotaxis: A comparative study in Escherichia coli and Bacillus subtilis. PLOS Biology 2(2): 239-252.
Raser, J.M., and E.K. O’Shea. 2004. Control of stochasticity in eukaryotic gene expression. Science 304(5678): 1811-1814.
Ribeiro, C., V. Petit, and M. Affolter. 2003. Signaling systems, guided cell migration, and organogenesis: Insights from genetic studies in Drosophila. Dev. Biol. 260(1): 1-8.
Roberts, C.J., B. Nelson, M.J. Marton, R. Stoughton, M.R. Meyer, H.A. Bennett, Y.D. He, H. Dai, W.L. Walker, T.R. Hughes, M. Tyers, C. Boone, and S.H. Friend. 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287(5454): 873-880.
Rossant, J., and P.P.L. Tam. 2004. Emerging asymmetry and embryonic patterning in early mouse development. Dev. Cell 7(2): 155-164.
Rzhetsky, A., T. Koike, S. Kalachikov, S.M. Gomez, M. Krauthammer, S.H. Kaplan, P. Kra, J.J. Russo, and C. Friedman. 2000. A knowledge model for analysis and simulation of regulatory networks. Bioinformatics 16(12): 1120-1128.
Santos, F., and W. Dean. 2004. Epigenetic reprogramming during early development in mammals. Reproduction 127(6): 643-651.
Schoeberl, B., C. Eichler-Jonsson, E.D. Gilles, and G. Müller. 2002. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 20(4): 370-375.
Schulz, R.A., and K.E. Yutzey. 2004. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev. Biol. 266(1): 1-16.
Schulze, W.X., and M. Mann. 2004. A novel proteomic screen for peptide-protein interactions. J. Biol. Chem. 279(11): 10756-10764.
Schulze-Kremer, S. 1998. Ontologies for molecular biology. Pp. 693-704 in Biocomputing ’98: Proceedings of the Pacific Symposium. Singapore: World Scientific.
Schuster, S., M. Marhl, and T. Hofer. 2002. Modelling of simple and complex calcium oscillations: From single-cell responses to intercellular signalling. Eur. J. Biochem. 269(5): 1333-1355.
Shen-Orr, S.S., R. Milo, S. Mangan, and U. Alon. 2002. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 31(1): 64-68.
Slepchenko, B.M., J.C. Schaff, I. Macara, and L.M. Loew. 2003. Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 13(11): 570-576.
Smith, A.E., B.M. Slepchenko, J.C. Schaff, L.M. Loew, and I.G. Macara. 2002. Systems analysis of Ran transport. Science 295(5554): 488-491.
Soll, D.R., E. Voss, O. Johnson, and D. Wessels. 2000. Three-dimensional reconstruction and motion analysis of living, crawling cells. Scanning 22(4): 249-257.
Sorkin, A., M. McClure, F. Huang, and R. Carter. 2000. Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr. Biol. 10(21): 1395-1398.
Stelling, J., U. Sauer, Z. Szallasi, F.J. Doyle III, and J. Doyle. 2004. Robustness of cellular functions. Cell 118(6): 675-685.
Stephens, M., M. Palakal, S. Mukhopadhyay, R. Raje, and J. Mostafa. 2001. Detecting gene relations from Medline abstracts. Pp. 483-495 in Biocomputing 2001: Proceedings of the Pacific Symposium. Singapore: World Scientific.
Sveiczer, A., A. Csikasz-Nagy, B. Gyorffy, J.J. Tyson, and B. Novak. 2000. Modeling the fission yeast cell cycle: Quantized cycle times in wee1-cdc25Delta mutant cells. Proc. Natl. Acad. Sci. U.S.A. 97(14): 7865-7870.
Swann, K., M.G. Larman, C.M. Saunders, and F.A. Lai. 2004. The cytosolic sperm factor that triggers Ca2+ oscillations and egg activation in mammals is a novel phospholipase C: PLCzeta. Reproduction 127(4): 431-439.
Swedlow, J.R., I. Goldberg, E. Brauner, and P.K. Sorger. 2003. Informatics and quantitative analysis in biological imaging. Science 300(5616): 100-102.
Szabo, A., D. Shoup, S.H. Northrup, and J.A. McCammon. 1982. Stochastically gated diffusion-influenced reactions. J. Chem. Phys. 77: 4484-4493.
Szostak, J.W., D.P. Bartel, and L. Luisi. 2001. Synthesizing life. Nature 409(6818): 387-390.
Tabata, T., and Y. Takei. 2004. Morphogens, their identification and regulation. Development 131(4): 703-712.
Taga, M.E., and B.L. Bassler. 2003. Chemical communication among bacteria. Proc. Natl. Acad. Sci. U.S.A. 100(2): 14549-14554.
Tian, X.C. 2004. Reprogramming of epigenetic inheritance by somatic cell nuclear transfer. Reprod. Biomed. Online 8(5): 501-508.
Tyson, J.J., K. Chen, and B. Novak. 2001. Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2(12): 908-916.
Ward, J.P., J.R. King, A.J. Koerber, J.M. Croft, R.E. Sockett, and P. Williams. 2003. Early development and quorum sensing in bacterial biofilms. J. Math. Biol. 47(1): 23-55.
Ward, J.P., J.R. King, A.J. Koerber, P. Williams, J.M. Croft, and R.E. Sockett. 2001. Mathematical modelling of quorum sensing in bacteria. IMA J. Math. Appl. Med. Biol. 18(3): 263-292.
Werb, Z., and Y.B. Yan. 1998. Cell biology: A cellular striptease act. Science 282(5392): 1279-1280.
Wessels, L.F., E.P. van Someren, and M.J. Reinders. 2001. A comparison of genetic network models. Pp. 508-519 in Biocomputing 2001: Proceedings of the Pacific Symposium. Singapore: World Scientific.
Wiley, H.S. 2003. Trafficking of the ErbB receptors and its influence on signaling. Exp. Cell Res. 284(1): 78-88.
Wiley, H.S., and D.D. Cunningham. 1981. A steady-state model for analyzing the cellular-binding, internalization, and degradation of polypeptide ligands. Cell 25(2): 433-440.
Wiley, H.S., J.J. Herbst, B.J. Walsh, D.A. Lauffenburger, M.G. Rosenfeld, and G.N. Gill. 1991. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 266(17): 11083-11094.
Wiley, H.S., S.Y. Shvartsman, and D.A. Lauffenburger. 2003. Computational modeling of the EGF-receptor system: A paradigm for systems biology. Trends Cell Biol. 13(1): 43-50.
Wrenzycki, C., and H. Niemann. 2003. Epigenetic reprogramming in early embryonic development: Effects of in-vitro production and somatic nuclear transfer. Reprod. Biomed. Online 7(6): 649-656.
Xia, Y., H. Yu, R. Jansen, M. Seringhaus, S. Baxter, D. Greenbaum, H. Zhao, and M. Gerstein. 2004. Analyzing cellular biochemistry in terms of molecular networks. Annu. Rev. Biochem. 73: 1051-1087.
Xiong, W., and J.E.J. Ferrell. 2003. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature 426(6925): 460-465.
Yakushiji, A., Y. Tateisi, Y. Miyao, and J. Tsujii. 2001. Event extraction from biomedical papers using a full parser. Pp. 408-419 in Biocomputing 2001: Proceedings of the Pacific Symposium. Singapore: World Scientific.
Yeger-Lotem, E., S. Sattath, N. Kashtan, S. Itzkovitz, R. Milo, R.Y. Pinter, U. Alon, and H. Margalit. 2004. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc. Natl. Acad. Sci. U.S.A. 101(16): 5934-5939.
Young, D.W., S.K. Zaidi, P.S. Furcinitti, A. Javed, A.J. van Wijnen, J.L. Stein, J.B. Lian, and G.S. Stein. 2004. Quantitative signature for architectural organization of regulatory factors using intranuclear informatics. J. Cell Sci. 117(21): 4889-4896.
Zaslaver, A., A.E. Mayo, R. Rosenberg, P. Bashkin, H. Sberro, M. Tsalyuk, M.G. Surette, and U. Alon. 2004. Just-in-time transcription program in metabolic pathways. Nat. Genet. 36(5): 486-491.
Zilberman, D., X.F. Cao, L.K. Johansen, Z. Xie, J.C. Carrington, and S.E. Jacobsen. 2004. Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr. Biol. 14(13): 1214-1220.





























