
A
The Secrets of Life: A Mathematician’s Introduction to Molecular Biology
NOTE: This appendix is a reprint of Chapter 1 of the National Research Council report Calculating the Secrets of Life: Contributions of the Mathematical Sciences to Molecular Biology (National Academy Press, Washington, DC, 1995), copyright 1995 by the National Academy of Sciences.
Chapter 1 The Secrets of Life: A Mathematician’s Introduction to Molecular Biology
Eris S. Lander
Whitehead Institute for Biomedical Research and Massachusetts Institute of Technology
Michael S. Waterman
University of Southern California
|
Molecular Biology has emerged from the synthesis of two complementary approaches to the study of life—biochemistry and genetics—to become one of the most exciting and vibrant scientific fields at the end of the twentieth century. This introductory chapter provides a brief history of the intellectual foundations of modern molecular biology and defines key terms and concepts that recur throughout the subsequent chapters. |
The concepts of molecular biology have become household words. DNA, RNA, and enzymes are routinely discussed in newspaper stories, prime-time television shows, and business weeklies. The passage into popular culture is complete only 40 years after the discovery of the structure of deoxyribonucleic acid (DNA) by James Watson and Francis Crick and only 20 years after the first steps toward genetic engineering. With breathtaking speed, these basic scientific discoveries have led to astonishing scientific and practical implications: the fundamental biochemical processes of life have been laid bare. The evolutionary record of life can be read from DNA sequences. Genes for proteins such as human insulin can be inserted into bacteria, which then can inexpensively produce large and pure amounts of the protein. Farm animals and crops can be engineered to produce healthier and more
desirable products. Sensitive and reliable diagnostics can be developed for viral diseases such as AIDS, and treatments can be developed for some hereditary diseases, such as cystic fibrosis.
Molecular biology is certain to continue its exciting growth well into the next century. As its frontiers expand, the character of the field is changing. With ever growing databases of DNA and protein sequences and increasingly powerful techniques for investigating structure and function, molecular biology is becoming not just an experimental science, but a theoretical science as well. The role of theory in molecular biology is not likely to resemble the role of theory in physics, in which mathematicians can offer grand unifying theories. In biology, key insights emerge less often from first principles than from interpreting the crazy quilt of solutions that evolution has devised. Interpretation depends on having theoretical tools and frameworks. Sometimes, these constructs are nonmathematical. Increasingly, however, the mathematical sciences—mathematics, statistics, and computational science—are playing an important role.
This book emerged from the recognition of the need to cultivate the interface between molecular biology and the mathematical sciences. In the following chapters, various mathematicians working in molecular biology provide glimpses of that interface. The essays are not intended to be comprehensive up-to-date reviews, but rather vignettes that describe just enough to tempt the reader to learn more about fertile areas for research in molecular biology.
This introductory chapter briefly outlines the intellectual foundations of molecular biology, introduces some key terms and concepts that recur throughout the book, and previews the chapters to follow.
BIOCHEMISTRY
Historically, molecular biology grew out of two complementary experimental approaches to studying biological function: biochemistry and genetics (Figure 1.1). Biochemistry involves fractionating (breaking up) the molecules in a living organism, with the goal of purifying and characterizing the chemical components responsible for carrying out a particular function. To do this, a biochemist devises an assay for
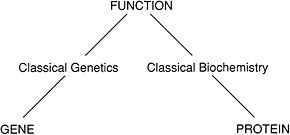
FIGURE 1.1 Genetics and biochemistry began as independent ways to study biological function.
measuring an “activity” and then tries successive fractionation procedures to isolate a pure fraction having the activity. For example, a biochemist might study an organism’s ability to metabolize sugar by purifying a component that could break down sugar in a test robe.
In vitro (literally, in glass) assays were accomplished back in the days when biologists were still grappling with the notion of vitalism. Originally, it was thought that life and biochemical reactions did not obey the known laws of chemistry and physics. Such vitalism held sway until about 1900, when it was shown that material from dead yeast cells could ferment sugar into ethanol, proving that important processes of living organisms were “just chemistry.” The catalysts promoting these transformations were called enzymes.
Living organisms are composed principally of carbon, hydrogen, oxygen, and nitrogen; they also contain small amounts of other key elements (such as sodium, potassium, magnesium, sulfur, manganese, and selenium). These elements are combined in a vast array of complex macromolecules that can be classified into a number of major types: proteins, nucleic acids, lipids (fats), and carbohydrates (starches and sugars). Of all the macromolecules, the proteins have the most diverse range of functions. The human body makes about 100,000 distinct proteins, including:
-
enzymes, which catalyze chemical reactions, such as digestion of food;
-
structural molecules, which make up hair, skin, and cell walls;
-
transporters of substances, such as hemoglobin, which carries oxygen in blood; and
-
transporters of information, such as receptors in the surface of cells and insulin and other hormones.
In short, proteins do the work of the cell. From a structural standpoint, a protein is an ordered linear chain made of building blocks known as amino acids (Figures 1.2 and 1.3). There are 20 distinct amino acids, each with its own chemical properties (including size, charge, polarity, and hydrophobicity, or the tendency to avoid packing with water). Each protein is defined by its unique sequence of amino acids; there are typically 50 to 500 amino acids in a protein.
FIGURE 1.2 Proteins are a linear polymer, assembled from 20 building blocks called amino acids that differ in their side chains. The diagram shows a highly stylized view of this linear structure.

FIGURE 1.3 Examples of different representations of protein structures focusing on (left) chemical bonds and (right) secondary structural features such as helices and sheet-like elements. Reprinted, by permission, from Richardson and Richardson (1989). Copyright © 1989 by the Plenum Publishing Corporation.
The amino acid sequence of a protein causes it to fold into the particular three-dimensional shape, having the lowest energy. This gives the protein its specific biochemical properties, that is, its function. Typically, the shape of a protein is quite robust. If the protein is heated, it will be denatured (that is, lose its three-dimensional structure), but it will often reassume that structure (refold) when cooled. Predicting the folded structure of a protein from the amino acid sequence remains an extremely challenging problem in mathematical optimization. The challenge is created by the combinatorial explosion of plausible shapes, each of which represems a local minimum of a complicated nonconvex function of which the global minimum is sought.
CLASSICAL GENETICS
The second major approach to studying biological function has been genetics. Whereas biochemists try to study one single component purified away from the organism, geneticists study mutant organisms that are intact except for a single component. Thus a biochemist might study an organism’s ability to metabolize sugar by finding mutants that have lost the ability to grow using sugar as a food source.
Genetics can be traced back to the pioneering experiments of Gregor Mendel in 1865. These key experiments elegantly illustrate the role of theory and abstraction in biology. For his experiments, Mendel started with pure breeding strains of peas—that is, ones for which all offspring, generation after generation, consistently show a trait of interest. This Choice was key to interpreting the data.
One of the traits that he studied was whether the pea made round or wrinkled seeds. Starting with pure breeding round and wrinkled strains, Mendel made a controlled cross to produce an F1 generation. (The ith generation of the cross is denoted Fi.) Mendel noted that all of the F1 generation consisted of round peas; the wrinkled trait had completely vanished. However, when Mendel crossed these F1 peas back to the pure breeding wrinkled parent, the wrinkled trait reappeared: of the second generation, approximately half were round and half were wrinkled. Moreover, when Mendel crossed the F1 peas to themselves, he found that
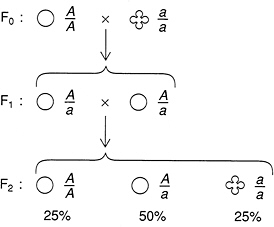
FIGURE 1.4 Mendel’s crosses between pure breeding peas with round and wrinkled seeds revealed the telltale binomial ratio 1:2:1 in the second generation that led Mendel to infer the existence of discrete particles of inheritance.
the second generation showed 75 percent round and 25 percent wrinkled (Figure 1.4).
On the basis of these and other experiments, Mendel hypothesized that traits such as roundness are affected by discrete factors—which today we call genes. In particular, Mendel suggested the following:
-
Each organism inherits two copies of a gene, one from each parent. Each parent passes on one of the two copies, chosen at random, to each offspring. (These important postulates are called Mendel’s First Law of Inheritance.)
-
Genes can occur in alternative forms, called alleles. For example, the gene affecting seed shape occurs in one form (allele A) causing roundness and one form (allele a) causing wrinkledness.
-
The pure breeding round and wrinkled plants carried two copies of the same allele, AA and aa, respectively. Individuals carrying two copies of the same gene are called homozygotes. The F1 generation consists of individuals with genotype Aa, with the round trait dominant over the wrinkled trait. Such individuals are called heterozygotes.
-
In the cross of the F1 generation (Aa) to the pure breeding wrinkled strain (aa), the offspring were a 1:1 mixture of Aa:aa according to which allele was inherited from the F1 parent. In the cross between two F1 parents (Aa), the offspring were a 1:2:1 mixture of AA:Aa:aa according to the binomial selection of alleles from the two parents.
It is striking to realize that the existence of genes was deduced in this abstract mathematical way. Probability and statistics were an intrinsic part of early genetics, and they have remained so. Of course, Mendel did not have formal statistical analysis at his disposal, but he managed to grasp the key concepts intuitively. Incidentally, the famous geneticist and statistician R.A. Fisher analyzed Mendel’s data many years later and concluded that they fit statistical expectation a bit too well. Mendel probably discarded some outliers as likely experimental errors.
It was almost 35 years before biologists had an inkling of where these hypothetical genes resided in the cell (in the chromosomes) and almost 100 years before they understood their biochemical nature.
MOLECULAR BIOLOGY
As suggested in Figure 1.1, the biochemical and the genetic approaches were virtually disjoint: the biochemist primarily studied proteins, whereas the geneticist primarily studied genes. Much like the great unifications in mathematics, molecular biology emerged from the recognition that the two apparently unrelated fields were, in fact, complementary perspectives on the same subject.
The first clues emerged from the study of mutant microorganisms in which gene defects rendered them unable to synthesize certain key macromolecules. Biochemical study of these genetic mutants showed that each lacked a specific enzyme. From these experiments the hypothesis became clear that genes somehow must “encode” enzymes. This (Nobel-Prize-winning) notion was dubbed the “one gene-one enzyme” hypothesis, although today it has been modified to “one
gene-one protein.” Of course, the mystery remained: How do genes encode proteins?
The answer depended on finding the biochemical nature of the gene itself, thereby uniting the fields. To purify the gene as a biochemical entity, one needed a test tube assay for heredity—something that might seem impossible. Fortunately, scientific serendipity provided a solution. In a famous series of bacteriological studies, Griffith showed 50 years ago that certain properties (such as pathogenicity) could be transferred from dead bacteria to live bacteria. Avery et al. (1944) were able to successively fractionate the dead bacteria so as to purify the elusive “transforming principle,” the material that could confer new heredity on bacteria. The surprising conclusion was that the gene appeared to be made of DNA.
The notion of DNA as the material of heredity came as a surprise to most biochemists. DNA was known to be a linear polymer of four building blocks called nucleotides (referred to as adenine, thymine, cytosine, and guanine, and abbreviated as A, T, C, and G) joined by a sugar-phosphate backbone. However, most knowledgeable scientists reckoned that the polymer was a boring, repetitive structural molecule that functioned as some sort of scaffold for more important components. In the days before computers, it was not apparent how a linear polymer might encode information. If DNA contained the genes, the structure of DNA became a key issue.
In their legendary work in 1953, Watson and Crick correctly inferred the structure of most DNA and, in so doing, explained the main secret of heredity. While some viruses have single-stranded DNA, the DNA of humans and of most other forms of life consists of two antiparallel chains (strands) in the form of a double helix in which the bases (nucleotides) pair up to form base pairs in a certain way (Figure 1.5) so that the sequence of one chain completely specifies the sequence of the other: an A on one chain always corresponds to a T on the other, and a G to a C. The sequences are complementary. The fact that the information is redundant explains the basis for the replication of living organisms: the two strands of the double helix unwind, and each serves as a template for the synthesis of a complete double helix that is passed on to a daughter cell. This process of replication is carded out by enzymes called DNA polymerases. Mutations are changes in the nucleotide sequence in DNA. Mutations can be induced by external
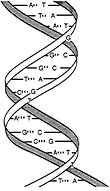
FIGURE 1.5 The DNA double helix consists of anti-parallel helical strands, with complementary bases (G–C and A–T).
forces such as sunlight and chemical agents or can occur as random copying errors during replication.
There remained the question of how the 4-letter alphabet of DNA could “encode” the instructions for the 20-letter alphabet of protein sequences. Biochemical studies over the next decade showed that genes correspond to specific stretches of DNA along a chromosome (much like individual files on a hard disk). These stretches of DNA can be expressed at particular times or under particular circumstances. Typically, gene expression begins with transcription of the DNA sequence into a messenger molecule made of ribonucleic acid (RNA) (Figure 1.6A). This transcription process is carded out by enzymes called RNA polymerases. RNA is structurally similar to DNA and consists of four building blocks, the nucleotides denoted A, U, C, and G, with U (uracil) playing the role of T. The messenger RNA (mRNA) is copied from the DNA of a gene according to the usual base pairing rules (a U in RNA corresponds to an A in DNA, an A, corresponds to a T, a G to a C, and a C to a G). The messenger RNA copied from a gene is single-stranded and is just an unstable intermediate used for transmitting information from the cell nucleus (where the DNA resides) to the cytoplasm (where protein synthesis occurs). The mRNA is then translated into a protein by a remarkable molecular machine called the ribosome.
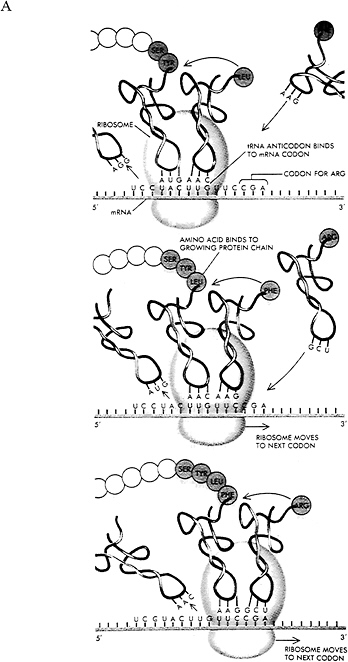
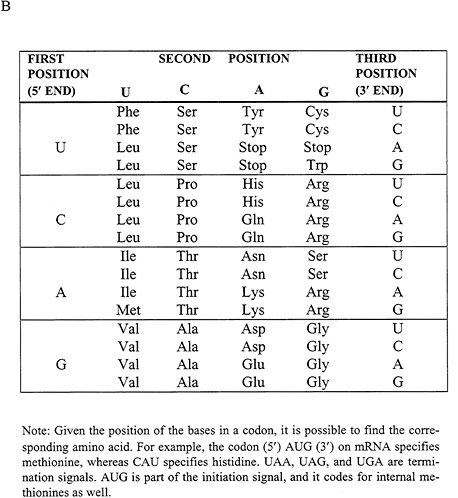
FIGURE 1.6 After messenger RNA is transcribed from the DNA sequence of a gene, it is translated into protein by a remarkable molecular device called the ribosome. (A) Ribosomes read the RNA bases and write a corresponding amino acid sequence. The correct amino acid is brought into juxtaposition with the correct nucleotide triplet through the mediation of an adapter molecule known as transfer RNA. (B) The table showing the correspondence between triplets of bases and amino acids is called the genetic code. Reprinted from Recombinant DNA: A Short Course by Watson, Tooze, and Kurtz (1994). Copyright © 1994 James D. Watson, John Tooze, and David T. Kurtz. Used with permission of W.H. Freeman and Company.
The ribosome “reads” the linear sequence of the mRNA and “writes” (i.e., creates) a corresponding linear sequence of amino acids of the encoded protein. Translation is carried out according to a three-letter code: a group of three letters is a codon that specifies a particular amino acid according to a look-up table called the genetic code (Figure 1.6B). There are 43 different codons. The codons are read in contiguous, nonoverlapping fashion from a defined starting point, called the translational start site. Finally, the newly synthesized amino acid chain spontaneously folds into its three-dimensional structure. (For a recent discussion of protein folding, see Sali et al., 1994.)
The details of' the genetic code were solved by elegant biochemical tricks, which were necessary because chemists had only the ability to synthesize random collections of RNA having defined proportions of different bases. With some combinatorial reasoning, this proved to be sufficient. For example, if the ribosome is given an mRNA with the sequence UUUUU …, then it makes a protein chain consisting of only the amino acid phenylalanine (Phe). Thus UUU must encode phenylalanine. By examining more complex mixtures, researchers soon worked out the entire genetic code.
Molecular biology provides the third leg of the triangle, relating genetics and biochemistry (Figure 1.7).
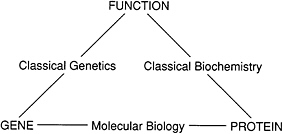
FIGURE 1.7 Molecular biology connected the disciplines of genetics and biochemistry by showing how genes encoded proteins.
THE RECOMBINANT DNA REVOLUTION
By 1965, molecular biology had laid bare the basic secrets of life. Without the ability to manipulate genes, however, the understanding was more theoretical than operational. In the 1970s, this situation was transformed by the recombinant DNA revolution.
Biochemists discovered a variety of enzymes, made by bacteria that allowed one to manipulate DNA at will. Bacteria made restriction enzymes, which cut DNA at specific sequences and served as a defense against invading viruses, and ligases, which join DNA fragments. With these and other tools (which are now all readily available from commercial suppliers), it became possible to cut and paste DNA fragments at will and to introduce them into living cells (Figure 1.8). Such cloning experiments allow scientists to reproduce unlimited quantities of specific DNA molecules and have led to detailed understanding of individual genes. Moreover, producing recombinant DNA molecules that contain bacterial DNA instructions for making a particular human protein (such as insulin) gave birth to the biotechnology industry.
A key development was the invention of DNA sequencing, the process of determining the precise nucleotide sequence of a cloned DNA molecule. With DNA sequencing, it became possible to read the sequence of any gene in stretches of 300 to 500 nucleotides at a time. DNA sequencing has revealed striking similarities among living creatures as diverse as humans and yeast, with far-reaching consequences for our understanding of molecular structure and evolution. DNA sequencing has also led to an information explosion in biology, with public databases still expanding at a rapid exponential rate. In early 1993, there were over 100 million bases of DNA in the public databases. For reference, the entire genome of the intestinal bacteria Escherichia coli (E. coli) consists of about 4.6 million bases, and the human genome sequence has roughly 3 billion bases.
In recent years a powerful new technique called the polymerase chain reaction (PCR) has been added to the molecular biologist’s tool kit (Figure 1.9). PCR allows one to directly amplify a specific DNA sequence without resort to cloning. To perform PCR, one uses short DNA molecules called primers (typically about 20 bases long) that are complementary to the sequences flanking the region of interest. Each
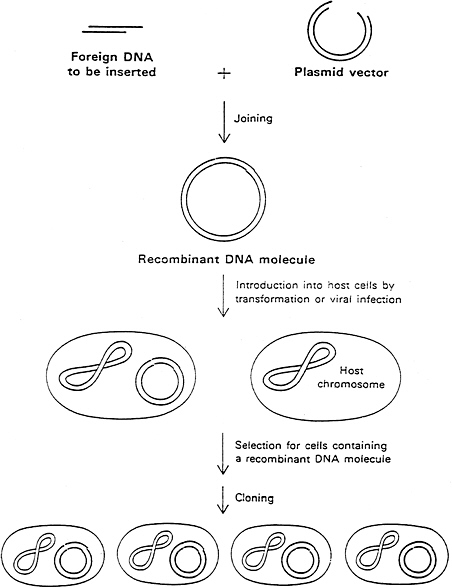
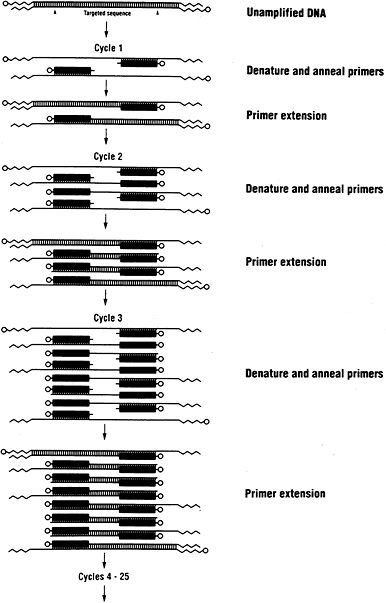
FIGURE 1.9 The polymerase chain reaction (PCR) allows exponential amplification of DNA. The method involves successive rounds of copying (using the enzyme DNA polymerase) between two synthetic primers corresponding to nearby DNA sequences. Each round doubles the number of copies. Courtesy of the Perkin-Elmer Corporation. Reprinted from the National Research Council (1992).
primer is allowed to pair with a base in the complementary region and is then extended to contain the full sequence from the region by using the enzyme DNA polymerase. In this fashion a single copy of the region gives rise to two copies. By iterating this step n times, one might make 2n copies of the region. In practice, one can start with a small drop of blood or saliva and obtain a millionfold amplification of a region. Not surprisingly, PCR has found myriad applications, especially in genetic diagnostics.
MOLECULAR GENETICS IN THE 1990s
With the tools of recombinam DNA, the triangle of knowledge (see Figure 1.7) has been transformed, to use a mathematical metaphor, into a commutative diagram (Figure 1.10). It is possible to traverse the diagram in any direction—for example, to find the genes and proteins underlying a biological function or to find the protein and function associated with a given gene.
A good illustration of the power of the techniques is provided by recent studies of the inherited disease cystic fibrosis (CF). CF is a recessive disease, the genetics of which is formally identical to wrinkledness in peas as studied by Mendel: if two non-affected carriers of the recessive CF gene a (that is, heterozygotes with genotype Aa) marry, one fourth of their offspring will be affected (that is, will have genotype aa). The frequency of the disease-causing allele is about 1/42 in the Caucasian population, and so about 1/21 of all Caucasians are careers. Since a marriage between two careers produces 1/4 affected children, the disease frequency in the population is about 1/2000 (≈ 1/4×1/21×1/21).
Although CF was recognized relatively early in the century, the molecular basis for the disease remained a mystery until 1989. The first breakthrough was the genetic mapping of CF to human chromosome 7 in 1985 (Figure 1.11). Genetic mapping involved showing that the inheritance pattern of the disease in families is closely correlated with the inheritance pattern of a particular DNA polymorphism (that is, a common spelling variation in the DNA), in this case on chromosome 7.
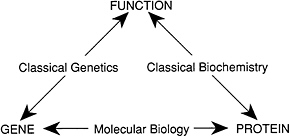
FIGURE 1.10 Recombinant DNA provided the ability to move freely in any direction among gene, protein, and function, thereby converting the triangle of Figure 1.7 into a commutative diagram.
The correlation does not imply that the polymorphism causes the disease, but rather that the polymorphism must be located near the site of the disease gene. Of course, “near” is a relative term. In this case, “near” meant that the CF gene must be within 1 million to 2 million bases of DNA along the chromosome. The next step was the physical mapping and the DNA sequencing of the CF gene itself, which took four more years to accomplish. This involved starting from the nearby polymorphism and sequentially isolating adjacent fragments in a tedious process called chromosomal walking until the diseases gene was reached. Once the disease gene was found, its complete DNA sequence was determined. (A description of how one knows that one has found the disease gene is beyond the scope of this introducation.)
From the DNA sequence, it became clear that the CF gene encoded a protein of 1,480 amino acids and that the most common misspelling in the population (accounting for about 70 percent of all CF alleles) was a three-letter deletion that removed a single codon specifying an amino acid, a phenylalanine at position 508 of the protein. On the basis of this finding, it became possible to perform DNA diagnostics on individuals to see if they carried the common CF mutation.
Even more intriguingly, the sequence gave immediate clues to the structure and function of the gene product. When the protein sequence was compared with the public databases of previous sequences, it was found to show strong similarities to a class of proteins that were membrane-bound transporters—molecules that reside in the cell membrane, bind adenosinetriphosphate (ATP), and transport substances
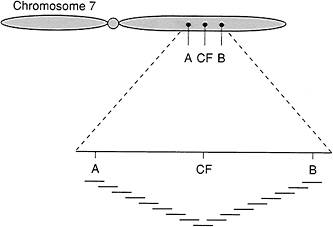
FIGURE 1.11 Chromosomal walking from flanking genetic markers to the gene responsible for cystic fibrosis. The distance covered totaled more than 1 million DNA bases.
into and out of the cell (Figure 1.12A). By analogy, it was even possible to infer a likely three-dimensional shape for the CF protein (Figure 1.12B). In this way, computer'based sequence analysis shed substantial light on the structure and function of this important disease gene.
With the recent advent of gene therapy—the ability to use a virus as a shuttle to deliver a working copy of a gene into cells carrying a defective version—clinical trials have been started to try to cure the disease in the lung cells of CF patients. The path from the initial discovery of the gene to potential therapies has been stunningly short in this case.
THE HUMAN GENOME PROJECT
With the identification of the CF gene as well as a number of other successes, it has become clear that molecular genetics has developed a powerful general paradigm that can be applied to many inherited diseases and will have a profound impact on our understanding of human health. Unfortunately, the paradigm involves many tedious laboratory steps: genetic mapping (finding a polymorphism closely linked to the
disease gene), physical mapping (isolating the consecutive fragments of DNA along the chromosome), and DNA sequencing (typically performed in pieces of only 300 to 500 letters at a time). It would be inefficient to repeat these steps for each of the more than 4,000 genetic traits and diseases already, known. To accelerate progress, molecular geneticists have seen the value of building infrastructure—a common set of maps, tools, and information—that can be applied to all genetic problems. This recognition led to the creation of the Human Genome Project (National Research Council, 1988), an international effort to analyze the structure of the human genome (as well as the genomes of certain key experimental 'model systems, such as E. coli, yeast, nematodes, fruit flies, and mice).
Because most molecular biological methods are applicable only to small fragments of DNA, it is not practical to sequence the human genome by simply starting at one end and proceeding sequentially. Moreover, because the current cost of sequencing is about $1 per base, it would be expensive to sequence the 3 × 109 bases of the human chromosomes by conventional methods. Instead, it is more sensible to construct maps of increasing, resolution and to develop more efficient sequencing technology. The current goals of the Human Genome Project include development of the following tools:
-
Genetic maps. The goal is to produce a genetic map showing the location of 5,000 polymorphisms that can be used to trace inheritance of diseases in families. As of this writing, the goal is nearly complete.
-
Physical maps. The goal is to produce a collection of overlapping pieces of DNA that cover all the human chromosomes. This goal is not completed yet but should be by 1996.
-
DNA sequence. The ultimate goal is to sequence the entire genome, but the intermediate steps include sequencing particular regions, generating more efficient and automated technology, and developing better analytical methods for handling DNA information.
With the vast quantities of information being generated, the Human Genome Project is one of the driving forces behind the expanding role

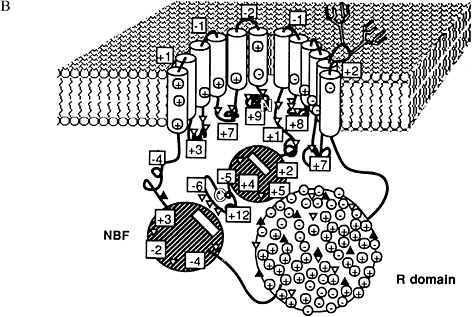
FIGURE 1.12 (A) The protein sequence of the cystic fibrosis gene showed striking similarities to a variety of proteins known to transport molecules across cell membranes. (B) Based on these similarities, it was possible to construct a basic molecular model of the architecture of the CF protein. Reprinted, by permission, from Riordan et al. (1989). Copyright © 1989 by the American Association for the Advancement of Science.
for mathematics, statistics, and computer science in modem molecular biology.
COMING ATTRACTIONS
The chapters of this book describe important applications of mathematical, statistical, and computational methods to molecular biology. These methods are developing rapidly, and, mainly because of this situation, the presentations in this book are intended to be introductory sketches rather than scholarly reviews. Without claiming to be a complete survey, this book should convey to readers some of the exciting uses of mathematics, statistics, and computing in molecular biology. Other introductions to various aspects of molecular biology can be found in Watson et al. (1994), Streyer (1988), U.S. Department of Energy (1992), Watson et al. (1987), Lewin (1990), and Alberts et al. (1989).
Chapter 2 (“Mapping Heredity”) describes how statistical models can be used to map the approximate location of genes on chromosomes. Gene mapping was mentioned above foi the case of the cystic fibrosis gene. The problem becomes especially challenging—and mathematics plays a bigger role—when the disease does not follow simple Mendelian inheritance pattems—for example, when it is caused by multiple genes or when the trait is quantitative rather than qualitative in nature. This is an important subject for the Human Genome Project and its applications in modem medical genetics.
The next three chapters focus on the analysis of DNA and protein sequences. As new genes are sequenced, they are routinely compared with public databases to look for similarities that might indicate common evolutionary origin, structure, or function. As databases expand at ever-increasing rates, the computational efficiency of such comparisons is crucial. Chapter 3 (“Seeing Conserved Signals”) describes combinatorial algorithms for this problem. Because coincidences abound in such comparisons, careful statistical analysis is needed. Chapter 4 (“Heating Distant Echoes”) discusses the application of extremal statistics to sequence similarity. For closely related sequences, sequence comparison also sheds light on the process of evolution. Chapter 5 (“Calibrating the Clock”) discusses the applications
of stochastic processes to such evolutionary analysis. The discovery and reading of genetic sequences have breathed new life into the study of the stochastic processes of evolution. The chapter focuses on one of the most exciting new tools, the use of the coalescent to estimate times to the most recent common ancestor.
Geometric methods applied to DNA structure and function are the focus of the next three chapters. Watson and Crick’s famous DNA double helix can be thought of as local geometrical structure. There is also much interesting geometry in the more global structure of DNA molecules. Chapter 6 (“Winding the Double Helix”) uses methods from geometry to describe the coiling and packing of chromosomes. The chapter describes the.supercoiling of the double helix, in terms of key geometric quantities—link, twist, and writhe—that are related by a fundamental theorem. Chapter 7 (“Unwinding the Double Helix”) employs differential mechanics to study how stresses on a DNA molecule cause it to unwind in certain areas, thereby allowing access by key enzymes needed for gene expression. Chapter 8 (“Lifting the Curtain”) uses topology to infer the mechanism of enzymes that recombine DNA strands, providing a glimpse of details that cannot be seen via experiment.
Finally, Chapter 9 (“Folding the Sheets”) discusses one of the hardest open questions in computational biology: the protein-folding problem, which concerns predicting the three-dimensional structure of a protein on the basis of the sequence of its amino acids. Probably no simple solution will ever be given for this central problem, but many useful and interesting approximate approaches have been developed. The concluding chapter surveys various computational approaches for structure prediction.
Together, these chapters provide glimpses of the roles of mathematics, statistics, and computing in some of the most exciting and dynamic areas of molecular biology. If this book tempts some mathematicians, statisticians, and computational scientists to learn more about and to contribute to molecular biology, it will have accomplished one of its goals. Its two other goals are to encourage molecular biologists to be more cognizant of the importance of the mathematical and computational sciences in molecular biology and to encourage scientifically literate people to be aware of the increasing impact of both molecular biology and mathematical and computational sciences on their
lives. If this book makes progress toward these three goals, it shall have been well worth the effort.
REFERENCES
Alberts, B., D. Bray, J. Lewis, M. Raft, K. Roberts, and J.D. Watson, 1989, Molecular Biology of the Cell, 2nd ed., New York: Garland.
Avery, O.T., C.M. McLeod, and M. McCarty, 1944, “Studies on the chemical nature of the substance inducing transformation of pneumococcal types,” J. Exp. Med. 79, 137-158.
Lewin, B., 1990, Genes IV, Oxford: Oxford University Press.
National Research Council, 1988, Mapping and Sequencing the Human Genome, Washington, D.C.: National Academy Press.
National Research Council, 1992, DNA Technology in Forensic Science, Washington, D.C.: National Academy Press.
Richardson, J. S., and D. C. Richardson, 1989, “Principles and patterns of protein conformation,” pp. 1-98 in Prediction of Protein Structure and the Principles of Protein Conformation, Gerald D. Fasman (ed.), New York: Plenum Publishing Corporation.
Riordan, J.R., J.M. Rommens, B. Kreme, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Plavsic, J-L. Chou, M.L. Drumm, M,C. Innuzzi, F.S. Collins, and L-C. Tsui, 1989, “Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA,“ Science 245 (September 8), 1066-1073.
Sali, A., E. Shakhnovich, and M. Karplus, 1994. “How does a protein fold?” Nature 369 (19 May), 248-251.
Streyer, Lubert, 1988, Biochemistry, San Francisco, Calif.: W.H. Freeman.
U.S. Department of Energy, Human Genome Program, 1992, Primer on Molecular Genetics , Office of Energy Research, Office of Health and Environmental Research, Washington, D.C.: U.S. Government Printing Office.
Watson, J.D., N. Hopkins, J. Roberts, J.A. Steitz, and A. Weiner, 1987, Molecular Biology of the Gene, Menlo Park, Calif.: Benjamin-Cummings.
Watson, J.D., J. Tooze, and D.T. Kurtz, 1994. Recombinant DNA: A Short Course, 2nd ed., New York: W.H. Freeman and Co.




























