
3
Potential Biological Targets of Polio Antiviral Drugs
THE PREVIOUS CHAPTER DELINEATED several qualities that a successful polio antiviral drug would need to have to be valuable in the eradication program. As part of the “ideal” drug profile (low cost, safety, preference for oral administration, and so on), a successful antipolio drug should reduce virus replication to a level that prevents transmission of the virus from host to host. The goal of this chapter is to describe the biological targets against which polio antiviral compounds are most likely to be successful.
Viruses offer a number of chemotherapeutic targets for the prevention or treatment of infection. Poliovirus is no exception and appears particularly suited for targeting antiviral drugs because it is one of the most thoroughly studied viruses.
The design of antiviral drugs against RNA viruses like poliovirus is complicated by the viruses’ extreme genetic variability. Polioviruses, like all RNA viruses, are so genetically variable that they are best described as “quasi-species” (Eigen 1993; Holland et al. 1982, 1992; Domingo et al. 1988). They exist as heterogeneous mixtures of related genomes; the individual viral particles are not all genetically identical. A consensus sequence can be defined for the poliovirus genome, but an infection will actually consist of thousands of genomes, each differing slightly from the consensus. The reason for this phenomenon, which has profound biological consequences, is a high inherent error rate (about 1 nucleotide of every 10,000 is misincorporated during genome replication) combined with a lack of proof-reading and editing mechanisms. High error frequency has also been
documented in poliovirus replication (Crotty et al. 2000; de la Torre et al. 1990). Quasi-species present a dynamic equilibrium of many viral geno-types in which the identity of the wild type is maintained only because of strong selection against a continuously appearing spectrum of mutants. As has been apparent in the development of drugs against many RNA viruses, the existence of a pool of variants makes it likely that mutants resistant to a given drug (“escape” mutants) already exist in the population before the drug is even used (Coffin 1995).
The speed with which drug resistance will emerge is difficult to predict since it depends on the target chosen for inhibition and the circumstances in which the drug would be used. However, an ideal polio antiviral drug will have features that slow the emergence of resistant mutants. One way to minimize or avoid the rapid emergence of escape mutants is to design a drug so that resistant viral variants are much less fit than nonresistant viruses. For example, oseltamivir, a drug that inhibits influenza neuraminidase, binds to a highly conserved active site, so variants resistant to the inhibitor also have greatly reduced neuraminidase activity and replicate poorly (Carr et al. 2002). An even more sophisticated strategy is to design the drug so that resistant mutants actually interfere with nonresistant viruses. Resistant mutants with this characteristic are said to have a “dominant negative” effect. For example, a mutation resulting in a misfolded protein that binds to and interferes with the function of normally folded proteins would be likely to have such a dominant negative effect (Crowder and Kirkegaard 2005).
SYNOPSIS OF POLIOVIRUS PATHOGENESIS AND REPLICATION
Poliovirus is a highly contagious virus that belongs to the genus Enterovirus of the large family of Picornaviridae (Stanway et al. 2002). Picornaviruses have been estimated to cause an astounding 6 billion human infections per year, and they give rise to a wide array of serious, even lethal, diseases (Melnick 1996). Enteroviruses alone (about 100 serotypes) are responsible for 1 billion infections per year. Enterovirus infections, including those caused by poliovirus, are largely covert, but the vast incidence of infections translates into a large number of clinical cases.
This report is not intended as an exhaustive review of all steps in cellular poliovirus replication or pathogenesis. Some details of the interaction of the virion (an individual virus particle) with the receptor CD155, of polyprotein synthesis and processing, and genome replication will be pro-
vided in sections below discussing drug targets. Reviews of all aspects of poliovirus replication and pathogenesis can be found in Molecular Biology of Picornaviruses (Semler and Wimmer 2002).
Poliovirus, the prototype of the enteroviruses, has a single-stranded genome (about 7,500 nucleotides) of plus strand polarity (that is, it serves as mRNA in the infected cell) whose nucleotide sequence has been known since 1981 (Kitamura et al. 1981; Racaniello and Baltimore 1981). The genome is protected by a rigid protein shell consisting of multiples of 4 polypeptides. There is no lipid envelope (Rossmann 2002).
On entry into a host cell, which is facilitated by the cellular receptor CD155 (also known as Pvr) (Koike et al. 1990; Mendelsohn et al. 1989), the viral genome expresses all its genetic information in the cytoplasm. Expression proceeds first by translation, which yields a polyprotein that is proteolytically cleaved by viral proteinases encoded in the polyprotein (Wimmer et al. 1993). This is followed by genome replication, encapsidation, and release from infected cells (Fig. 1) (Paul 2002; Agol 2002). Starting with genome RNA, the entire replication cycle can be reproduced in a cell-free extract (Molla et al. 1991).
Cytoplasmic replication of poliovirus is stringently dependent upon membranous structures that the virus builds from cellular membranes during the first phase of the infectious cycle. The poliovirus proteins 2BC and 2CATPase, mapping to the center of the viral genome, are predominantly responsible for the rearrangement of cellular vesicles to form numerous new vesicles (Egger et al. 2002) and autophagosomes (Jackson et al. 2005) that are the sites of genome replication.
Individual steps of the replication cycle at the cellular level have been studied in great detail, which will aid in the identification of targets for poliovirus antiviral drugs. Currently, the forerunners are small molecules that interfere with the entry of the virion into the host cell (drugs that insert themselves into the capsid) and small molecules that inhibit the action of the virus-encoded proteinase 3Cpro. Likewise, the RNA-dependent RNA polymerase 3Dpol is an attractive target for chemotherapeutic intervention.
Pathogenesis
In contrast with cellular replication, our knowledge of the mechanism of poliovirus pathogenesis is limited. Infection occurs by the oral-fecal route (Minor 1997; Bodian and Horstman 1965; Sabin 1956; Bodian 1955). As
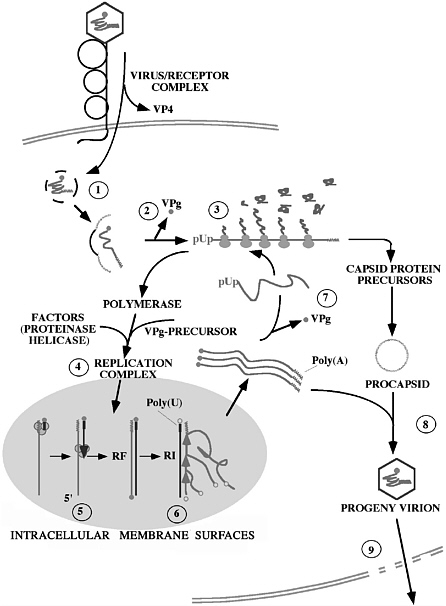
FIGURE 1 Schematic representation of the cellular replication cycle of poliovirus. (1) After binding to the receptor CD155, the virion is transported into the cell and uncoated. (2) The 5’-terminal VPg covalently-attached protein is thought to be cleaved from the incoming genome by a cellular enzyme after which (3) the RNA is translated end-to-end into a polyprotein. The latter is subsequently processed into numerous functional proteins (see Fig. 3). (4) A membrane-associated replication complex is being formed by viral and cellular proteins (possibly involving a helicase). (5) The plus stranded genome RNA is transcribed into a minus strand under formation of a double-stranded “replicative form” (RF). (6) Minus strands then function as template for the synthesis of plus strands. (7) Newly synthesized plus stranded RNA has the choice of re-entering genome replication (4), serving as mRNA in translation (7), or associating with procapsids (8) to form mature virions that are released from the decaying cell (9). The nucleus is not involved in the replicative cycle. For details, see (Semler and Wimmer 2002).
an enteric virus, poliovirus replicates in the gastrointestinal (GI) tract for 3 to 4 weeks, but occasionally for several months (Alexander et al. 1997; Horstmann et al. 1946). Rarely, at rates of 10−3 to 10−2, the virus invades the central nervous system (CNS) to target predominantly motor neurons, and this invasion leads to paralysis or death (Racaniello 2001; Mueller et al. 2005; Minor 1997; Nathanson and Martin 1979). Why motor neurons are targeted by poliovirus is not understood.
There is another significant gap in our knowledge of poliovirus pathogenesis. Although there is little doubt that the virus replicates in sites of the GI tract—most likely in secondary lymphatic tissues, such as tonsils and Peyer’s patches (Nathanson 2005; Horie et al. 1994; Wenner et al. 1959; Bodian 1955; Sabin 1956; Bodian 1952; Sabin and Ward 1941; Fairbrother and Hurst 1930)—the nature of the cells that support poliovirus proliferation in the GI-associated lymphoid tissues (GALT) or elsewhere remains obscure. The lack of detailed understanding of the sites of poliovirus replication may be an obstacle to drug development in that those sites cannot currently be specifically targeted for monitoring drug activity.
TARGETS FOR POLIO ANTIVIRALS
A. Capsid-Binding Agents
The poliovirus capsid is an icosahedron consisting of 60 copies each of capsid polypeptides VP1, VP3, VP2, and VP4 (Filman et al. 1989; Hogle et al. 1985). A unit of these four capsid polypeptides first assembles to form a “protomer” (VP1, VP3, and VP0, the latter being the precursor of VP2 and VP4); five protomers then form a pentamer, and 12 pentamers assemble into the viral procapsid that finally undergoes the maturation cleavage of VP0 to VP2 and VP4 on entry of the genome. A deep “canyon” surrounds the apex of each pentamer of the virus; at each of the 60 protomer-protomer interfaces there is a small hydrophobic pocket at the canyon floor (Fig. 2). This pocket is a promising target of antipoliovirus drugs (Rossmann 2002).
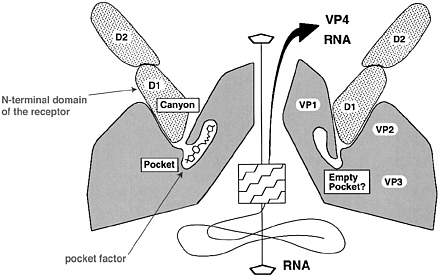
FIGURE 2 Proposed binding of an immunoglobulin-like receptor into the virion canyon followed by capsid rearrangement and genome release. Shown is the apical region of the virion with the prominent canyon at the 5-fold axis. The pocket at the canyon bottom is normally filled with a small hydrophobic compound that is thought to be released upon uncoating. In this figure, the N-terminal domain of the receptor (here ICAM-1, the receptor for the majority of rhinoviruses) enters the canyon, intruding almost down to the canyon floor. In the binding of poliovirus to CD155, the interaction between the N-terminal receptor domain and virion is largely restricted to the canyon rim. Both ICAM-1 and CD155 consist of 5 and 3 immunoglobulin-related domains, respectively, referred to as D1 (N-terminal), D2, D3, etc. The interaction between receptor and canyon destabilizes the virion, allowing release of the small capsid polypeptides VP4 and the genome (right side of the scheme). If the natural pocket factor is replaced by hydrophobic compounds that lodge more stably in the pocket (for example, binding of the WIN51711 compound into the poliovirus pocket), the rearrangement and uncoating processes are inhibited. (Modified from Rossmann et al. 2000.)
Normally, a small hydrophobic compound (such as sphingosine or other cellular components) that allows flexibility of the capsid polypeptides occupies the small canyon pocket. Drug candidates that replace the hydrophobic compound in the pocket stabilize the virion in such a way that uncoating is inhibited (Rossmann et al. 2000; Rossmann 2002; Joseph-McCarthy et al. 2001). Insertion of the drug is thought to interfere with the capsid polypeptide rearrangements necessary for genome release.
Several pharmaceutical companies have identified and studied a considerable number of hydrophobic capsid-binding compounds. The best
known is pleconaril (WIN63843), a drug that was developed by Sterling Winthrop with great promise for the treatment of enteroviral and rhinoviral infections. Pleconaril, however, was not licensed by the Food and Drug Administration (FDA) because of undesirable side effects when it was administered in combination with other medications (i.e., oral contraceptives). Nevertheless, because of the wealth of data gathered over the last 20 years, small molecules with the propensity for occupying the hydrophobic canyon pocket are strong candidates for the development of an antipoliovirus drug. A number of candidates identified during the development of pleconaril may have more activity against poliovirus than against rhinoviruses. Indeed, several potential candidates currently under investigation by ViroDefense, Inc. are further discussed in Chapter 4.
B. Drugs Directed Against Non-Structural Proteins
It is reasonable to choose proteins encoded in the non-structural region of the genome as targets for anti-poliovirus drugs. However, poliovirus can undergo genetic recombination (Wimmer et al. 1993; Romanova et al. 1980; Hirst 1962) as a second mechanism for generating drug resistance and this ability is highly relevant to the use of antiviral medications in poliovirus eradication. In a cell infected with a single serotype, recombinants are formed with frequencies as high as 10−4 (Kirkegaard and Baltimore 1986). Since homologous recombination is the mechanism of genome exchange(s) (Kirkegaard and Baltimore 1986) lower frequencies of recombination (10−5 to 10−6) are observed amongst different polioviruses serotypes (Tolskaya et al. 1983). Kew and his colleagues have recently discovered that circulating vaccine-derived polioviruses (cVDPVs), which have caused yearly outbreaks of poliomyelitis since 2001 (see Table 1), are predominantly recombinants between polioviruses and non-human enteroviruses (Kew et al. 2005, 2002). Candidates of the non-human enteroviruses are those coxsackie viruses (C-CAV) (Kew et al. 2002) that are in the same cluster as poliovirus (Stanway et al. 2002). Recombination between poliovirus, type 1 (Mahoney) [or poliovirus, type 1 (Sabin)] and C-CAV type 20 can be readily demonstrated to occur in tissue culture cells at a frequency of 10−6 (Ping et al. unpublished results). Since cross-over occurs predominantly near the middle of the genomes, the cVDPV recombinants, which have acquired coding sequences of nonstructural proteins of C-CAVs, may be resistant to drugs directed at the poliovirus encoded enzymes (e.g., proteinases or RNA polymerase). Therefore, if these targets are chosen for
antipoliovirus drug development, it may be necessary for these drugs to be broadly reactive, including having activity against polypeptides of the C-cluster coxsackie viruses.
1. Inhibitors of the Virus-Encoded Proteinases
Poliovirus expresses its entire protein-related genetic information as a single polypeptide, the polyprotein (Baltimore et al. 1969). This polyprotein is proteolytically processed by two viral proteinases, 3Cpro/3CDpro (Ypma-Wong et al. 1988; Svitkin et al. 1979) and 2Apro (Skern et al. 2002; Toyoda et al. 1986) into some 29 polypeptides, of which some are relatively stable precursor molecules that have essential functions in replication (Fig. 3) (Leong et al. 2002). As is common in viral replication, virus-encoded polypeptides often have more than one essential function and that is true also for the viral proteinases, 3Cpro/3CDpro and 2Apro.
Proteinases 3C/3CDpro
The relatively stable processing intermediate 3CDpro is not only a proteinase but also an RNA-binding protein essential for genome replication. Once cleaved, 3CDpro yields the smaller proteinase 3Cpro and the viral RNA polymerase 3Dpol. Both 3Cpro and 3CDpro can cleave at glutamine-glycine bonds, the scissile peptide bond in poliovirus polyprotein processing (Skern et al. 2002; Nicklin et al. 1988); however, only 3CDpro can efficiently cleave the capsid precursor P1 (Ypma-Wong et al. 1988). Antiviral agents that target these proteinases, however, are likely to interfere with both 3Cpro and
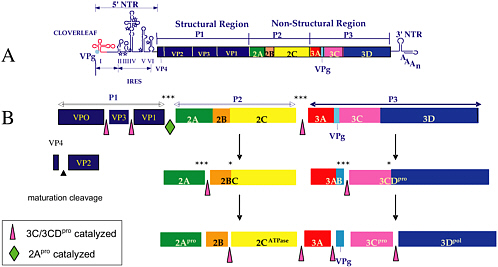
FIGURE 3 Genome organization of poliovirus and proteolytic processing of the polyprotein. (A) Schematic representation of the genome. Starting with the 5’-terminal genome-linked protein VPg, the 5’ non-translated region is divided into cloverleaf, a structure essential for genome replication, and IRES, a structure essential for internal initiation of protein synthesis. The open square indicates the polyprotein divided into polypeptides, which are end products of cleavage. The 3’ non-translated region consists of two stem-loop structures and poly(A) (Agol 2002). (B) Pathway of proteolytic cleavages by two virus-encoded proteinases 2Apro (cleavage at open diamond) and 3C/ 3CDpro (cleavages at open triangles) (Leong et al. 2002). *** Indicates rapid cleavages; * slow cleavages, the kinetics determined predominantly by the sequence at the scissile bond (Nicklin et al. 1988; Hellen et al. 1991). Precursor proteins such as 2BC and 3CDpro fulfill important functions in the replication cycle (see text). The mechanism of the maturation cleavage (filled triangle) is not understood.
3CDpro functions. The high-resolution crystal structure of poliovirus 3Cpro has been solved which is valuable for drug development targeted at the enzyme (Mosimann et al. 1997). It is a cysteine proteinase with a serine proteinase fold and structural similarities to chymotrypsin. Available evidence suggests that all 3Cpro proteinases of enteroviruses and rhinoviruses exhibit a highly conserved active site (Binford et al. 2005). 3C/3CDpro proteinase inhibitors have been shown to be active against many enteroviruses and rhinoviruses, which not only may be a prerequisite of drugs directed at these enzymes (because of the recombinant cVDPVs discussed above) but also may be an incentive for investment by the commercial sector (Patick et al. 1999). Development spearheaded by Pfizer has led to human rhinovirus type 2 (HRV2) 3Cpro proteinase inhibitors. The most advanced compounds include rupintrivir (Phase II trials), an intranasally administered compound, and compound 1 (Phase I), an orally bioavailable compound. Inhibition of poliovirus by rupintrivir has not been tested but is predicted based on strong homology between the 3C protease of poliovirus and other picornaviruses.
As an RNA-binding protein, 3CDpro, with poly(rC) binding protein (PCBP), forms a stable ribonucleoprotein complex with the cloverleaf structure at the 5’ end of the genome that is essential for genome replication (Parsley et al. 1997; Andino et al. 1990). 3CDpro is also a cofactor in the formation of a ribonucleoprotein complex between the RNA polymerase 3Dpol, a small viral protein VPg, and a cis-acting replication element (cre stem loop) mapping to the coding region of 2CATPase. This complex functions in the initiation of viral RNA synthesis (Yang et al. 2004; Paul et al. 2000). Addition of purified 3CDpro to the cell-free replication system (Molla et al. 1991) stimulates virion synthesis 100-fold, an observation suggesting the essential role of this viral protein in poliovirus proliferation (Franco et al. 2005).
Proteinase 2Apro
Whereas 3C/3CDpro catalyze the majority of proteolytic cleavages of the polyprotein, 2Apro catalyzes only one essential cleavage: that between the capsid precursor and the nonstructural proteins (Fig. 3) (Toyoda et al. 1986). 2Apro, however, is also involved in genome replication by an unknown mechanism (Molla et al. 1993). The crystal structure of poliovirus 2Apro is not known, but it is probably closely related to the known structure of HRV2 2Apro. The crystal structure of HRV2 2Apro, like that of
3Cpro, has also revealed a cysteine proteinase with a serine proteinase fold (Petersen et al. 1999). Anti-2A inhibitors are likely to display the additional advantage of suppressing genetic reversion because of the intracellular dominance of uncleaved capsid-2A precursors, inasmuch as it has recently been shown that unprocessed VP1-2A precursors have a dominant negative effect on capsid formation (Crowder and Kirkegaard 2005).
3C/3CDpro and 2Apro also catalyze proteolytic degradation of numerous cellular proteins, and this contributes to rapid cell death (Weidman et al. 2003; Kuechler et al. 2002). Inhibition of the poliovirus proteinases, therefore, probably would not only prevent viral replication but also have a positive effect on the survival of the infected cell.
Poliovirus proteinases are excellent substrates for drug development because they are essential for key stages in the replication cycle. Moreover, they exhibit minimal similarity to known mammalian enzymes. Indeed, proteases as targets for chemotherapeutic intervention—for example against HIV—have the advantage of being well explored by pharmaceutical companies.
2. Inhibitors of the Poliovirus RNA-Dependent RNA Polymerase 3Dpol
Poliovirus genome replication follows a common strategy used by all single-stranded plus-strand RNA viruses; the plus-stranded genomic RNA is transcribed into minus-strand copies, which then serve as templates for progeny genomic RNA (Wimmer et al. 1993). In poliovirus replication, all newly synthesized genomes can serve as further templates for genome replication, as mRNA in translation, or as substrate for encapsidation (Paul 2002).
The synthesis of both plus-stranded and minus-stranded RNAs is catalyzed by 3Dpol, a strictly primer dependent polymerase (Paul 2002; Flanegan and Baltimore 1977). The primer is the uridylylated form of the small viral protein VPg (VPg-pUpU), which is synthesized by the polymerase itself. Following uridylylation, VPg-pUpU is elongated to either plus or minus stranded RNAs (Paul 2002; Paul et al. 1998).
There are potentially two approaches to targeting 3Dpol. First, direct inhibition of its two enzymatic functions would block several steps in genome replication, including VPg uridylylation, initiation, or elongation. Indeed, it is possible that different compounds may be identified that selectively inhibit uridylylation of VPg or elongation (polymerization) of the polynucleotide strand.
A second approach is to target the polymerase in such a way as to induce a change in error rate. As noted above, the characteristic high error rate of RNA viruses is an important factor in their ability to adapt to new environments and resist the action of drugs. There is evidence that the optimal mutation rate for RNA viruses may have a narrow range; therefore, it may be possible to inhibit virus replication by either increasing or decreasing the mutation rate (Vignuzzi et al. 2005; Pfeiffer and Kirkegaard 2005; Crotty et al. 2000). An increase in error rate should lead to lethal mutagenesis, whereas a decrease in error rate (high fidelity replication) should reduce the ability of the virus to adapt to environmental changes (see box). Some polymerase inhibitors are likely to have the additional advantage of suppressed genetic reversion due to the intracellular dominance of inhibitor-bound polymerase molecules.
The solution of the complete three-dimensional structure of poliovirus 3Dpol provides greater opportunities for structure-based inhibitor design (Thompson and Peersen 2004). Several companies have successfully targeted the RNA-dependent RNA polymerase of hepatitis C virus—an effort resulting in compounds that are now in clinical trials (Wu et al. 2005). Some of the drugs under development against the HCV polymerase have also shown inhibitory activity against poliovirus (Olsen et al. 2004) but whether they are viable starting compounds against poliovirus infection is not known.
Nucleoside analogues with mutagenic potential, such as ribavirin, have been identified, but additional work is necessary to establish whether they are effective antipoliovirus drugs. It is recommended that screening of libraries be undertaken to identify nonnucleoside compounds that inhibit polymerase function both by interfering with replication and by altering the mutation rate of poliovirus.
C. Other Possible Targets for Antiviral Development
1. In Addition to the Capsid-Binding, Proteinase, and Polymerase Targets Described Above, the Following Poliovirus Components Could Be Promising Targets for Antiviral Drugs:
-
2C is an ATPase that is crucial for viral replication (Pfister and Wimmer 1999). High-throughput assays to screen for inhibition of its activity could be readily designed.
|
Genetic Diversity May Contribute to Poliovirus Pathogenesis Results of recent studies (Vignuzzi et al. 2005; Pfeiffer and Kirkegaard 2005) that were designed to test key postulates of the quasispecies theory suggest that population diversity may have a role in poliovirus pathogenesis, specifically in the ability of the virus to spread to the central nervous system (CNS). Although wild-type virus can access and replicate in the CNS, a high fidelity virus was restricted from systemic spread. The results show that the diversity of the quasispecies itself may be a critical determinant of pathogenesis. Notably, treatment of the high fidelity viruses with mutagenic nucleoside analogues, which restored genomic diversity, was shown also to restore the ability to spread to the CNS (Vignuzzi et al. 2005). Furthermore, the study indicated that there is positive interaction between variants in the quasispecies: one mutant may allow other mutants to enter the brain. Thus, complexity of the viral quasispecies enables the virus population to spread systemically and to access the CNS, perhaps because of complementary functions of different subpopulations that facilitate adaptation to new environments. Some variants in the population may facilitate the colonization of the gut, another set of mutants may serve as immunological decoys that trick the immune system, and yet another may facilitate crossing the blood-brain barrier. Those findings have important consequences for antiviral therapeutic strategies. Nucleoside-based mutagens, such as the anti-HIV drug zidovudine (AZT), take advantage of the RNA polymerases’ high error rate to induce the virus to incorporate faulty nucleosides as it replicates. One way that viruses acquire resistance to nucleoside-based mutagens is by having mutations that improve the fidelity of their RNA polymerases. The results of the experiments suggest that a resistant poliovirus with a high fidelity polymerase would lose the ability to spread to the CNS. |
-
RNA structures, such as the noncoding regions and the cis acting replication element (cre) are crucial for genome replication and might be excellent targets.
-
3A, a small protein required for RNA replication, is the target for enviroxime, a promising compound for further characterization.
-
2. Non-Small Molecule Based Antiviral Strategies
The antiviral strategies described above call for the development of small molecules that would bind to, block, or inhibit the various poliovirus targets. Pharmaceutical companies and regulatory institutions have experience with small-molecule development, testing, and regulation, and these approaches are most likely to be successful in the short term. However, a number of novel therapeutic approaches could be quickly adapted for the treatment of poliovirus if they are successfully developed for the treatment of other diseases.
siRNA mediated inhibition of poliovirus replication
Small interfering RNAs (siRNAs) are small strands of RNA, 21-23 nucleotides long, that are designed to bind to the complementary portion of a target messenger RNA and thereby signal the cell that the target RNA should be degraded (Yeung et al. 2005). The attractiveness of such a strategy against RNA viruses is clear: siRNAs could be used to induce infected cells to digest the viral RNA and halt replication. siRNA reagents are being developed by several companies (Anylan, Isis, and SiRNA) to treat a variety of human diseases, especially cancer. The problem of delivering the siRNAs in sufficient quantity to the appropriate target cells has not been solved. Several siRNA targets in poliovirus genomes have been identified (Gitlin et al. 2002). Such a strategy has the advantage of targeting several sequences simultaneously, thus making it difficult for the virus to acquire resistance. If the current problems with delivery in humans were resolved, siRNA might be a viable alternative for prophylaxis of and therapy for poliovirus infections.
Morpholinos
Morpholinos are similar to siRNAs in that they are short strands of nucleic acid designed to bind to specific complementary stretches on target RNA (Arora et al. 2004). Morpholinos differ from siRNAs in having backbones that are not the usual sugars and phosphates found in natural nucleic acids and therefore are immune to DNase degradation by cellular enzymes. Their binding blocks the activity of the target RNA, and this makes them ideal as therapeutic agents against RNA viruses if the capability to deliver the morpholinos to target cells in sufficient quantity can be developed.
-
Protective antibodies
The administration of hyperimmune globulin can provide immediate protective immunity against infectious agents and toxins. The development of antibodies as therapeutic agents has been under way since the 1980s. Early results of using antibodies made by infecting mice were somewhat disappointing. Recent results with humanized monoclonal antibodies (murine antibodies engineered to resemble human antibodies) are more encouraging, and more than 10 monoclonal antibodies have been licensed (Reichert 2001). Results of experiments in the early 1950s indicated that passive immunization could prevent poliomyelitis (Rinaldo 2005) and suggested that passive administration of antipoliovirus antibodies could be an attractive approach if the technology improves and costs fall.
BASIC RESEARCH NEEDS
Many of the virus-specific processes in poliovirus proliferation offer targets for chemotherapeutic intervention. Among them, the capsid and the viral proteinase 3Cpro/3CDpro have been subject to extensive studies with the specific aim of generating antiviral drugs. Those efforts, however, were directed predominantly at nonpolio enteroviruses and the related human rhinoviruses. The close genetic and biochemical kinship of human enteroviruses and rhinoviruses suggests that compounds developed previously against these picornaviruses might serve as superb immediate candidates for the development of antipoliovirus agents.
In spite of a wealth of information related to intracellular poliovirus replication, the details of individual steps (such as uptake of virus into the cell, regulation of polyprotein processing, the switch from translation to genome synthesis, regulation of plus-strand and minus-strand synthesis, rearrangement of cellular organelles, and encapsidation and virus maturation) are obscure. Similarly, the sites of replication in the GI tract after oral infection and the mechanisms by which infection progresses to poliomyelitis remain largely unknown. More detailed understanding of these processes would probably identify additional targets. The successful development of polio antiviral drugs will rely heavily on continuous strong support of poliovirus research.
















