
6
Biological Pathways Leading to Preterm Birth
ABSTRACT
Preterm birth has usually been treated as a single entity, for epidemiological and statistical purposes. This traditional empiric approach, however, presupposes a single pathologic process for which treatment could be uniform. This approach has met with only limited success in the treatment and prevention of preterm labor. It is now clear that the causes of preterm labor are multifactorial and vary according to gestational age. Important common pathways leading to preterm birth include stress, systemic or maternal genital tract infections, placental ischemia or vascular lesions, and uterine overdistension. These pathways differ in their initiating factors and mediators, but ultimately, they share many common features that result in preterm uterine contractions and birth. Appropriate animal models have been very useful in describing the temporal events leading to preterm birth and the neonatal sequelae of prematurity, particularly in the setting of intrauterine infection. The use of animal models to answer specific questions related to prematurity and to describe the pathophysiological events associated with preterm birth will contribute to the development of rational and efficacious treatment and prevention strategies for preterm birth.
MECHANISMS OF PARTURITION
Parturition
The process of normal spontaneous parturition can be divided into four stages (see the reviews of Challis [2000] and Challis et al. [2000]). During most of pregnancy, the uterus remains relatively quiescent, and this corresponds to Phase 0 (quiescence) of parturition. Phase 1 (activation) involves uterine stretch and fetal hypothalamic-pituitary-adrenal (HPA) activation. Phase 2 (stimulation) refers to stimulation of the activated uterus by various substances, including corticotropin-releasing hormone (CRH), oxytocin, and prostaglandins. These different processes lead to a common pathway to parturition involving increased uterine contractility, cervical ripening, and decidual and fetal membrane activation (Romero et al., 2004a). Phase 3 (involution) corresponds to postpartum involution of the uterus. These unique phases are described below and are summarized in Figure 6-1.
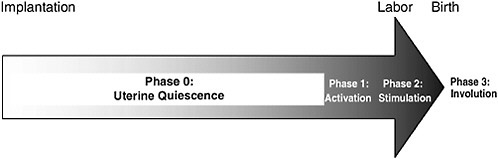
FIGURE 6-1 The stages of parturition. Following implantation, more than 95 percent of gestation is spent in Phase 0, uterine quiescence. During quiescence, myometrial contractility is inhibited by a variety of biological substances, including progesterone. Phase 1, myometrial activation, is characterized by increased expression of contraction-associated proteins, receptors for oxytocin, and prostaglandins, and increased placental estrogen biosynthesis. The signal for myometrial activation is controlled by the fetal HPA axis, which, in turn, is up-regulated by endogenous placental CRH production. Phase 2, myometrial stimulation, involves a progressive cascade of events, beginning with myometrial activation, which results in myometrial contractility, cervical ripening, and decidua and membrane activation. It is likely initiated by the same events of fetal HPA activation that initiate Phase 1. Phase 3, involution, involves placental separation and contraction of the uterus. It is primarily effected by maternal oxytocin. Implantation
Phase 0:
Quiescence
Throughout most of pregnancy the uterus remains relatively quiescent. Myometrial activity is inhibited during pregnancy by various substances, including progesterone, prostacyclin (PGI2), nitric oxide, relaxin, and parathyroid hormone-related peptide. These substances function by different mechanisms, but in general they increase intracellular levels of cyclic nucleotides (cyclic adenosine monophosphate [cAMP] or cyclic guanosine monophosphate), which in turn inhibit the release of calcium from intracellular stores or reduce the activity of the enzyme myosin light-chain kinase (MLCK). Calcium and MLCK are central to uterine contractility. Calcium is required to activate calmodulin, which in turn induces a conformational change in MLCK, allowing the enzyme to phosphorylate myosin and initiate the coupling of actin and myosin, which leads to myometrial contraction.
Rare uterine contractions that occur during the quiescent phase are of low frequency and amplitude and are poorly coordinated; these are commonly referred to as contractures in animals and Braxton-Hicks contractions in women. The poor coordination of these contractions is primarily due to an absence of gap junctions in the pregnant myometrium (Garfield, 1988). Gap junctions (and their associated proteins, called connexins) allow cell-to-cell coupling. With the onset of labor, there is a massive increase in the numbers of gap junctions, resulting in significantly enhanced electrical coupling and synchronized high-amplitude contractions throughout the myometrium.
Phase 1:
Activation
Phase 1 myometrial activation is characterized by increased levels of expression of contraction-associated proteins (CAPs), including connexin43 (CX-43; the major protein of myometrial gap junctions), and receptors for oxytocin and stimulatory prostaglandins (Lye et al., 1998). Normally, the signals for myometrial activation can come from uterine stretch as a result of fetal growth or from activation of the fetal HPA axis as a result of fetal maturation, or both.
Uterine stretch has been shown in animal models to increase CAP and oxytocin receptor gene expression in the myometrium, but the ability to do so is highly dependent on the endocrine environment. Progesterone blocks stretch-induced increases in the levels of CX-43 expression. However, with progesterone withdrawal at term (see below), uterine stretch is associated with significant increases in the levels of CX-43 expression.
Signals for myometrial activation also come from the fetal HPA axis (Liggins and Thorburn, 1994). It is currently thought that once fetal matu-
rity has been reached (as determined by as yet unknown mechanisms), the fetal hypothalamus and/or the placenta (see below) increase the level of CRH secretion, which in turn stimulates adrenocorticotropic hormone (ACTH) expression by the fetal pituitary and cortisol and androgen production by the fetal adrenals. Fetal androgens are then aromatized into estrogens by the placenta. Ultimately, this initiates a biological cascade that leads to a common pathway of parturition characterized by uterine contractility, cervical ripening and decidual/fetal membrane activation seen in Phase 2 of parturition.
Phase 2:
Stimulation
Phase 2 involves a progressive cascade of events that lead to a common pathway of parturition involving uterine contractility, cervical ripening, and decidual and fetal membrane activation. These events are characterized by fetal HPA activation, functional progesterone withdrawal, increasing maternal and fetal estrogens, and rising prostaglandins. The cascade may begin with the placental production of CRH and eventually leads to a functional progesterone withdrawal in the myometrium. The progesterone withdrawal causes increased levels of expression of estrogen receptors and promotes estrogen activity. The increased action of estrogen leads to the formation of many estrogen-dependent CAPs, such as CX-43, oxytocin receptors, and prostaglandins, that promote uterine contractility.
CRH and the “placental clock.” Corticotropin-releasing hormone (CRH) is thought to play a central role in fetal maturation and human parturition (McLean et al., 1999; reviewed by Smith R et al. [2002]). CRH, a neuropeptide of predominantly hypothalamic origin, is also expressed in the human placenta and membranes and is released into maternal and fetal compartments in exponentially increasing amounts over the course of gestation. The trajectory of the rise in CRH levels has been associated with the length of gestation (Hobel et al., 1999; Leung et al., 1999; McLean and Smith, 1999). Specifically, women destined to preterm delivery have higher concentrations of maternal CRH in plasma as early as 16 weeks of gestation and a more rapid rise in CRH levels than women who deliver at term. These findings have led some researchers to suggest that placental CRH may act as a “placental clock” that regulates the length of gestation (McLean and Smith, 1999).
Placental CRH synthesis is stimulated by glucocorticoids, in contrast to the inhibitory effect of glucocorticoids on hypothalamic CRH synthesis. Placental CRH, in turn, promotes fetal cortisol and DHEA-S production, and this positive-feedback loop is progressively amplified, thereby driving the process forward from fetal HPA activation to parturition. Placental
CRH, in turn, enhances prostaglandin production by increasing the levels of expression of prostaglandin H2 synthase (PGHS) chorion and amnion cells, creating yet another positive-feedback loop that drives the process of parturition. Paradoxically, during uterine quiescence CRH may act as a myometrial relaxant rather than as a promoter of parturition. Throughout most of pregnancy, the myometrium expresses CRH type 1 receptors that are linked by Gsα regulatory proteins to adenylate cyclase and cAMP, which would promote myometrial relaxation when they are stimulated. At the end of pregnancy, however, an alternative splice variant of the CRH receptor is expressed and the level of expression of Gsα subunits declines, which may promote a contractile phenotype (reviewed by Challis et al. [2000]).
Functional progesterone withdrawal. For most of pregnancy, uterine quiescence is maintained by the action of progesterone. It does so by blocking CAP gene expression and gap junction formation within the myometrium; inhibiting placental CRH secretion; opposing the activity of estrogen (see below); up-regulating systems (e.g., nitric oxide) that promote myometrial relaxation; and suppressing the expression of cytokines and prostaglandins. At the end of pregnancy in most mammals, maternal progesterone levels fall and estrogen levels rise. In women, however, progesterone and estrogen concentrations continue to rise throughout pregnancy until delivery of the placenta. Recent data suggest that functional progesterone withdrawal may occur in women and nonhuman primates by alterations in the levels of progesterone receptor (PR) isoforms (Smith R et al., 2002). In women, the PR-B receptor isoform functions predominantly as an activator of progesterone-responsive genes, whereas the PR-A receptor isoform acts as a repressor of PR-B function and other nuclear receptors. In the term myometrium, the onset of labor is associated with increased levels of PR-A expression relative to the levels of PR-B expression. Because PR-A suppresses the action of progesterone, the increased level of PR-A expression relative to that of PR-B decreases the responsiveness of the myometrium to progesterone, resulting in a functional progesterone withdrawal that enables parturition to proceed.
Estrogens. Unlike the placentas of most other species, the human placenta cannot convert progesterone to estrogen because it is deficient in 17-hydroxylase, which is required for this conversion. Estrogen production in the placenta depends largely on precursor androgens synthesized in the fetal zone of the fetal adrenal; approximately 50 percent of circulating maternal estrone and estradiol are derived from placental aromatization of the fetal androgen, DHEA-S. Placental CRH directly and indirectly (via fetal pituitary secretion of ACTH) stimulates the fetal zone of the fetal adrenals to
produce DHEA-S, thereby supplying the precursors needed for estrogen synthesis in the placenta.
Estrogens, in turn, enhance the expression of many estrogen-dependent CAPs, including CX-43 (gap junctions), oxytocin receptor, prostaglandin receptors, cyclooxygenase-2 (COX-2; which results in prostaglandin production), and MLCK (which stimulates myometrial contractility and labor) (Challis, 2000).
Prostaglandins. Extensive evidence supports a central role for prostaglandins in promoting uterine contractiity (Challis et al., 2000). The actions of prostaglandin are effected through specific receptors. PGE2 induces myometrial contractions by binding to EP-1 and EP-3 receptors, which mediate contractions through mechanisms that lead to increased calcium mobilization and reduced levels of production of inhibition of intracellular cAMP. Prostaglandins also enhance the production of matrix metalloproteinases (MMP) in the cervix and decidua to promote cervical ripening and decidual and fetal membrane activation. PGF2α binds to FP receptors to induce myometrial contractions. In contrast, in the lower uterine segment PGE2 induces myometrial relaxation by binding to EP-2 and EP-4 receptors that increase the level of cAMP formation.
Prostaglandins are formed from arachidonic acid by PGHS. In turn, prostaglandins are metabolized to inactive forms by the actions of PGDH. Cortisol, CRH, and estrogens stimulate PGHS activity and cortisone and CRH also inhibit PGDH expression. Thus, increases in fetal steroid hormone production following fetal HPA activation leads to a net increase in prostaglandin levels. Similarly, proinflammatory cytokines such as IL-1 and tumor necrosis factor alpha (TNF-α) up-regulate PGHS expression and down-regulate PGDH expression leading to prostaglandin synthesis associated with preterm delivery in the setting of infection.
In summary, these events initiated by fetal HPA activation and resulting in increased fetal and placental steroid biosynthesis result in a progressive cascade of biological processes lead to a common pathway of parturition involving cervical ripening, uterine contractility and decidual and fetal membrane activation.
Cervical ripening. Cervical changes precede the onset of labor, are gradual, and develop over several weeks. Cervical ripening is characterized by a decrease in the total collagen content, an increase in collagen solubility, and an increase in collagenolytic activity that results in the remodeling of the extracellular matrix of the cervix (Romero et al., 2004a). Prostaglandins, estrogens, progesterones, and inflammatory cytokines (e.g., IL-8) affect the metabolism of the extracellular matrix. PGE2 stimulates collagenolytic activity and the synthesis of subtypes of proteoaminoglycans
that are less stabilizing. Estrogen stimulates collagen degradation in vitro, and intravenous administration of 17β-estradiol induces cervical ripening. Progesterone blocks estrogen-induced collagenolysis in vitro and down-regulates IL-8 production by the uterine cervix. In addition to these hormones, nitric oxide may play a role in cervical ripening in some circumstances. Nitric oxide accumulates at sites of inflammation and can act as an inflammatory mediator at high concentrations. Nitric oxide donors (e.g., sodium nitroprusside) have been shown to induce cervical ripening, whereas nitric oxide inhibitors (e.g., L-nitro-arginine methylester) block cervical ripening.
Uterine contractility. Uterine contraction results from the coupling of actin and myosin, which depends on the phosphorylation of myosin by MLCK. MLCK is activated by calcium-calmodulin after an increase in intracellular calcium levels. This increase in generated by the actions of various uterotonins, including oxytocin and prostaglandins. Cell-to-cell coupling, which allows the myometrium to develop synchronous high-amplitude contractions during labor, is facilitated by the formation of gap junctions and their associated proteins (e.g., connexins) (Lye et al., 1998). Their formation is highly dependent on estrogen; estrogen activation, in turn, is induced by a functional progesterone withdrawal at term.
Decidual and fetal membrane activation. Decidual and fetal membrane activation refers to a complex set of anatomical and biochemical events that result in the separation of the lower pole of the membranes from the deciduas of the lower uterine segment and, eventually, in the spontaneous rupture of membranes. The precise mechanism of membrane and decidual membrane activation remains to be elucidated, but extracellular matrix-degrading enzymes such as MMP type 1 (MMP-1), interstitial collagenase, MMP-8 (neutrophil collagenase), MMP-9 (gelatinase B), neutrophil elastase, and plasmin have been implicated. These enzymes degrade extracellular matrix proteins (e.g., collagens and fibronectins), thereby weakening the membranes, which eventually leads to the rupture of membranes. Some MMPs, such as MMP-9, may induce apoptosis in the amnion.
Phase 3:
Involution
Phase 3 begins with the third stage of labor and involves placental separation and uterine contraction. Placental separation occurs by cleavage along the plane of the decidua basalis. Uterine contraction is essential to prevent bleeding from the large venous sinuses that are exposed after delivery of the placenta and is primarily affected by oxytocin.
Summary of Human Parturition
Parturition in women involves a progressive cascade of events initiated by HPA activation and increased placental CRH expression, leading to a functional progesterone withdrawal and estrogen activation, which in turn results in the expression and activation of CAPs (including oxytocin receptors), oxytocin, and prostaglandins. This biological cascade eventually leads to a common pathway involving cervical ripening, uterine contractility, decidual and fetal membrane activation, and, in the second stage, increases in maternal oxytocin. It has been hypothesized that both preterm and term labor share this common pathway and that the pathologic stimuli of parturition, as described in the following sections, may act in concert with the normal physiological preparation for labor, especially after 32 weeks of gestation. Before 32 weeks of gestation, a greater degree of pathologic stimulus may be required to initiate labor. One fundamental difference between spontaneous parturition at term and preterm labor is that whereas term labor results from physiological activation of the components of the common pathway, preterm labor arises from pathologic processes that activate one or more of the components of the common pathway of parturition. However, further research is necessary to answer fundamental questions, including the following:
-
What role do implantation errors have in the pathogenesis of preterm delivery?
-
What are the cellular, endocrine, and paracrine mechanisms that maintain uterine quiescence?
-
What are the mechanisms involved in the switch from the quiescent uterus to uterine activation and stimulation?
-
What is the basis for disparities in gestational length between ethnic groups? Does it have a biological basis, or can it be accounted for by environmental and social factors?
PATHWAYS TO SPONTANEOUS PRETERM PARTURITION
Until recently, obstetricians and epidemiologists have had a tendency to combine, for statistical purposes, all preterm births occurring between 22 and 37 weeks of gestation. The traditional empirical approach to preterm labor presupposed a single pathologic process for which treatment could be uniform.
It is now clear that the causes of preterm labor are multifactorial and vary according to gestational age. They include systemic and intrauterine infections (which are responsible for a majority of extremely preterm births), stress, uteroplacental thrombosis and intrauterine vascular lesions
TABLE 6-1 Commonly Recognized Etiologies and Pathways Leading to Spontaneous Preterm Birth
|
Maternal-fetal HPA activation |
Stress |
Maternal-fetal HPA activation |
|
Infection and inflammation |
Intrauterine Lower genital tract |
Proinflammatory cytokine and prostaglandin cascade |
|
Systemic |
Matrix metalloproteinases |
|
|
Decidual hemorrhage |
Thrombophilias, Abruptio placentae |
Thrombin |
|
Autoantibody syndromes |
Matrix metalloproteinases |
|
|
Pathologic uterine overdistension |
Multifetal gestation |
Expression of gap junctions proteins |
|
Polyhydramnios |
Prostaglandins Oxytocin receptors |
associated with fetal stress or decidual hemorrhage, uterine overdistension, and cervical insufficiency. Each pathway may be influenced by geneenvironment interactions, as discussed in Chapter 7 (Table 6-1 and Figure 6-2). It is also noted that these pathways or their relative impact may differ for ART patients. The causes for preterm birth among ART patients are multifactorial and poorly understood, except for uterine overdistension resulting from multiple gestations. The reader is referred to Chapter 5 for discussion of the impact of infertility and infertility treatment on preterm birth. Nevertheless, there is strong evidence that despite different etiologies and initiators, preterm and term labor share many common pathways in the activation of common downstream cellular and molecular effectors. This may include stimulation of the fetal HPA axis (by maturation, infection, or ischemia), in addition to endocrine, paracrine, and immune system interactions, which were summarized in the preceding section. Commonly occurring pathways of preterm parturition are described below.
Stress and the Placental Clock
Stress is increasingly being recognized as an important risk factor for preterm delivery. Stress may be simply defined as any challenge—psychological or physical—that threatens or that is perceived to threaten homeostasis (i.e., the stability of the internal milieu of the organism). The epidemiological evidence linking maternal psychological stress to prematurity is reviewed in Chapter 3. Several pathways linking maternal psychological stress and prematurity have been proposed, including neuroendocrine, immune-inflammatory, vascular, and behavioral processes.
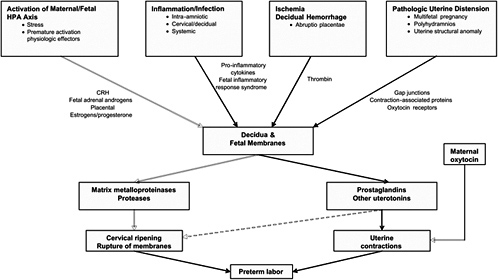
FIGURE 6-2 Overview of commonly occurring pathways to preterm birth. Although the causes of preterm birth are multiple and there are many unique upstream regulatory initiators of preterm birth, there are few downstream effectors that lead to preterm birth. These include prostaglandins or other uterotonins, MMPs, and oxytocin. This suggests that interventions for the prevention of preterm birth can be directed either to the inhibition of specific upstream initiators of a given pathway or to the blocking of downstream effectors in general.
Neuroendocrine Processes
The neuroendocrine processes linking stress to prematurity are mediated by placental CRH (reviewed by Wadhwa et al. [2001]). Placental CRH is responsive to stress. In vitro studies of human placental cells have shown that CRH is released from cultured human placental cells in a dose-response manner in response to all the major biological effectors of stress, including cortisol, catecholamines, oxytocin, angiotension II, and IL-1. In vivo studies have also found significant correlations between maternal psychosocial stress and the levels of CRH, ACTH, and cortisol in maternal plasma. Several studies have related early increases in maternal plasma CRH levels to the timing of birth. Hobel and colleagues (1999) conducted serial assessments of CRH levels over the course of gestation and found that women delivering preterm had significantly elevated CRH levels compared with those in women delivering at term, as well as a significantly accelerated rate of increase in CRH levels over the course of their gestations. In addition, they found that maternal psychosocial stress levels at midgestation significantly predicted the magnitude of increase in maternal CRH levels between midgestation and later times of gestation.
These data suggest that the relationship between maternal psychological stress and prematurity may be mediated by prematurely increased levels of expression of placental CRH. As discussed earlier in this chapter, in term parturition placental CRH activation is largely driven by the fetal HPA axis in a forward-feedback loop upon fetal maturation. In preterm parturition it may be the maternal HPA axis (as well as the sympathoadrenal-medullary [SAM] system) that drives placental CRH expression (Wadhwa et al., 2001). Maternal stress results in increased levels of biological effectors of stress, including cortisol and epinephrine, which could activate placental CRH gene expression. Placental CRH, in turn, can stimulate fetal secretion of cortisol and DHEA-S (by activation of the fetal HPA axis) and placental release of estriol and prostaglandins, thereby precipitating preterm delivery (reviewed by Hobel et al., 1998).
Immune and Inflammatory Processes
Stress can also alter neuroendocrine modulation of immune function, leading to increased susceptibility to intra-amniotic infection or inflammation. Extensive interconnections exist among the SAM system, the HPA axis, and the immune system. Under physiological conditions, the SAM system and the HPA axis suppress the body’s immunoinflammatory responses. A negative-feedback loop exists between the HPA axis and the immune system; proinflammatory cytokines (e.g., IL-1β, TNF-α, and IL-6) stimulate the HPA axis, resulting in the secretion of glucocorticoids. These
glucocorticoids, in turn, down-regulate proinflammatory cytokines and also suppress other aspects of the inflammatory response (reviewed by McEwen et al. [1997]).
During acute stress, glucocorticoids primarily suppress inflammation; but with repeated or chronic stress, glucocorticoids can enhance inflammation, including the expression of proinflammatory cytokine expression. Excessive secretion of proinflammatory and T-helper cell type 1 (Th1)-type cytokines is found in subjects undergoing life stress, and this immune activation is associated with a loss of nonspecific and specific cellular immune responses (Irwin, 1999). These cytokines, in turn, can down-regulate glucocorticoid receptors (Norbiato et al., 1997) and decrease glucocorticoid receptor affinity (Pariante et al., 1999). Evidence suggests that this cycle of immune activation, together with insufficient HPA axis restraint of cytokine secretion, is a major component of inflammatory disease progression. Thus, repeated or chronic stress could lead to a dysregulation of proinflammatory and Th1 cytokine expression, thereby predisposing an overreactive inflammatory response to stress or infection. The role of these mechanisms in contributing to preterm birth, however, has not been well studied, and may represent a fertile area for further investigation.
Behavioral Processes
Stress may induce maternal risk behaviors as a means of coping with stress. These behavioral processes may be mediated by altered neurochemistry. In animal studies, CRH and related neuropeptides play a central role in mediating motor and psychic activation, stimulus avoidance, and threat recognition responses to aversive stimulus exposure (Heinrichs and Koob, 2004). CRH may also mediate some of the neuroendocrine and behavioral effects of cocaine addiction (Sarnyai, 1998). In rhesus monkeys, strong correlations were found between behavioral hyperactivity and the CRH-dependent elements of pulsatile activity of the HPA axis. Acute cocaine administration induced dose- and time-dependent alterations in hypothalamic and extrahypothalamic-limbic CRH concentrations in rats. Cocaine withdrawal elicited anxiety-like behavior and alterations of CRH concentration in the hypothalamus, amygdala, and basal forebrain (Sarnyai, 1998).
Despite the accumulating data from animal studies, the question of whether stress increases maternal risk behaviors has not been clearly addressed in human studies. Most epidemiological studies of maternal stress and birth outcomes have treated risk behaviors as confounders, although a few clinic-based studies have found that behaviors may play a mediating role between maternal psychosocial stress and lower birth weight (see Chapter 3). A recent population-based study (Whitehead et al., 2003) found
maternal stressful life events were significantly associated with cigarette smoking during pregnancy and late entry into prenatal care, although stressful life events were not significantly associated with preterm delivery.
Uterine Overdistension
Uterine overdistension plays a key role in the onset of preterm labor associated with multiple gestations, polyhydramnios, and macrosomia. The mechanisms by which uterine overdistension might lead to preterm labor are not well understood. Uterine stretch induces the expression of gap junction proteins, such as CX-43 and CX-26 (Ou et al., 1997), as well as other contraction-associated proteins, such as oxytocin receptors (Terzidoo et al., 2005). In vitro stretch of myometrial strips also increases PGHS-2 and PGE (Sooranna et al., 2004). Stretching the muscle of the lower uterine segment has been shown to increase the levels of IL-8 and collagenase production, which in turn facilitates cervical ripening (Loudon et al., 2004; Maradny et al., 1996). The increased myometrial expression of PGHS-2 and IL-8 from uterine stretch appears to be mediated by activation of the MAPK system (Sooranna et al., 2005). An interaction between mechanical and endocrine signals during myometrial activation may exist; in vivo studies have shown increased levels of expression of CX-43 in response to mechanical stretch in an ovariectomized rat uterus, an effect that could be blocked by progesterone administration (Ou et al., 1998). Similarly, in rats uterine stretch has no effect on myometrial CX-43 expression in midpregnancy, probably as a result of the high levels of progesterone that are present at that time; progesterone withdrawal before the onset of labor allows stretch-induced CX-43 expression (Wathes and Porter, 1982).
The effect of uterine stretch on myometrial expression of G proteins (e.g., Gsα), which mediate myometrial relaxation, is not known. Human studies of uterine overdistension are lacking. One recent study found no difference between singleton and multiple gestations in the levels of expression of Gsα, PGE2 receptors, CX-43, and CX-26 in myometrium taken from nonlaboring women undergoing elective cesarean delivery at term (Lyall et al., 2002). Moreover, mechanical stretch did not alter the levels of Gsα expression in vitro, and Gsα expression was unaffected by steroid hormones. These findings suggest that the mechanisms by which uterine stretch can promote myometrial contractions in humans are complex and may involve additional factors or that multiple gestations that do not result in preterm labor may have compensatory mechanisms for the increased uterine stretch by preventing aberrant CAP expression.
Uteroplacental Thrombosis and Decidual Hemorrhage
Vascular lesions of the placenta are commonly associated with preterm birth and preterm premature rupture of membranes (PPROM) (PROM refers to premature rupture of membranes, meaning before the onset of labor at any gestational age; hence, although “preterm PROM” is apparently redundant, it is not, given the definition of PROM, and is abbreviated here PPROM). Vascular lesions of the placenta have been reported in 34 percent of women with preterm delivery, 35 percent of women with PPROM, and 12 percent of uncomplicated deliveries at term (Arias et al., 1993). These lesions may be characterized as failure of the physiological transformation of the spiral arteries, atherosis, and maternal or fetal arterial thrombosis. The proposed mechanism linking vascular lesions to preterm birth is related to uteroplacental ischemia. Although the pathophysiology remains unclear, thrombin is thought to play a central role.
Independent of its critical role in coagulation, thrombin is a multifunctional protease that elicits the contractile activity of vascular, intestinal, and myometrial smooth muscle. Thrombin activates a unique set of receptors, including protease-activated receptor 1, protease-activated receptor 3, and protease-activated receptor 4 (Bohm et al., 1998; Grand et al., 1996). These transmembrane receptors are members of the heptahelical G-proteincoupled superfamily. Interaction with thrombin results in conformational changes that produce G-protein coupling and phospholipase C activation (Bohm et al., 1998; Grand et al., 1996). Phospholipase C activation initiates the biochemical reactions that lead to the release of intracellular calcium from the endoplasmic reticulum. The combination of intracellular calcium release and the influx of extracellular calcium cause cytosolic calcium oscillations that activate calmodulin, MLCK, actin, and myosin, resulting in phasic uterine contraction (Phillippe and Chien, 1998). Through these intracellular signaling events, thrombin acts as a classic uterotonic agonist.
Thrombin stimulates increases in basal tone and phasic contractions in longitudinal myometrial smooth muscle in vitro in a dose-dependent manner (Elovitz et al., 2000). Recently, these in vitro observations have been confirmed with in vivo models by using thrombin, whole blood, and thrombin inhibitors (Elovitz et al., 2000). Both thrombin and whole blood increased myometrial contractions in a dose-dependent fashion. However, myometrial contractility was significantly reduced by the addition of heparin, a known thrombin inhibitor. These in vitro and in vivo experiments provide a possible mechanistic explanation for the increased uterine activity clinically observed in abruptio placentae and preterm birth following first-or second-trimester bleeding.
A relationship between thrombin and PPROM may also exist. MMPs
break down the extracellular matrix of the fetal membranes and the choriodecidua and contribute to PPROM, as discussed below. In vitro, thrombin significantly increases the levels of MMP-1, MMP-3, and MMP-9 protein expression in decidual cells and fetal membranes collected from uncomplicated term pregnancies (MacKenzie et al., 2004; Rosen et al., 2002; Stephenson et al., 2005). Thrombin also elicits a dose-dependent increase in decidual interleukin-8, a chemoattractant cytokine responsible for neutrophil recruitment (Lockwood et al., 2005). Overt abruptio placentae, an example of decidual hemorrhage, is also associated with a marked decidual infiltration of neutrophils, a rich source of proteases and matrix metalloproteinases (Lockwood et al., 2005). This may provide a mechanism for premature rupture of the membranes in the setting of decidual hemorrhage. Taken together, these investigations provide a mechanism for the relationship between increased intrauterine thrombin levels and PPROM.
However, confirmation of these in vitro observations in women has been difficult, largely because the direct measurement of thrombin levels is very difficult. Instead, the levels of thrombin-antithrombin III (TAT) complexes are usually measured as an indirect measurement of thrombin activation. In pregnant women TAT levels increase throughout gestation. TAT levels continue to increase with labor and reach a peak with the delivery of the placenta. A prospective cohort study found that pregnant women with preterm labor who delivered within 3 weeks of admission had significantly elevated TAT levels compared with those in control subjects (Elovitz et al., 2001). A receiver-operator-curve analysis generated in that study demonstrated that a TAT level of greater than 8.0 nanograms per milliliter had a sensitivity of 50 percent, a specificity of 91 percent, a positive predictive value of 80 percent, and a negative predictive value of 71 percent for the prediction of preterm delivery. These results add credence to the hypothesis that a significant portion of the cases of idiopathic preterm labor or PPROM may be caused by subclinical decidual bleeding. Subclinical decidual bleeding would provide the small quantities of thrombin that would significantly stimulate uterine activity. The recognition that thrombin plays an important role in contractility and in membrane degradation may help explain the association between vaginal bleeding, retroplacental hematomas, and preterm birth.
The relationship between elevated TAT levels and PPROM has also been explored in a nested case-control study in which plasma samples from women with eventual PPROM were examined in the second and third trimesters (Rosen et al., 2001). In both the second and the third trimesters, plasma TAT levels were significantly increased among women who experienced PPROM compared with the levels among women at term without PPROM.
Infection and Inflammation
Genital tract infections are strongly associated with preterm birth (Andrews et al., 2000; Goldenberg et al., 2000). These generally represent bacterial infections that ascend from the lower genital tract; viral infections have not been implicated as a significant cause of preterm birth. The sources of infection that have been linked to preterm birth include intrauterine infections, lower genital tract infections, systemic maternal infections, asymptomatic bacteruria, and maternal periodontitis.
Intrauterine infections are recognized as one of the most important and potentially preventable causes of preterm birth. These infections are thought to be responsible for up to 50 percent of extreme preterm births of less than 28 weeks of gestation, in which the rates of both neonatal mortality and neonatal morbidity are high. The prevalence of microbial invasion of the chorioamnion is 73 percent in women with a spontaneous preterm birth before 30 weeks of gestation and only 16 percent among women with indicated preterm delivery without labor (Hauth et al., 1998). The prevalence of histologic chorioamnionitis is inversely related to gestational age and occurs in 60 to 90 percent of gestations ending at between 20 and 24 weeks; microbial infection of the chorioamnion occurs in 60 percent of patients with preterm delivery (Hillier et al., 1988). Furthermore, the infections in a high proportion of women in preterm labor with evidence of microbial invasion of the amniotic fluid are refractory to standard tocolytic therapy and result in rapid preterm delivery (in 62 percent of women with evidence of microbial invasion but only 13 percent of women with sterile amniotic fluid) (Romero et al., 1991). This suggests that the pathophysiology of infection-associated preterm labor differs from that of idiopathic preterm labor.
Considerable evidence now suggests that the proinflammatory cytokineprostaglandin cascade plays a central role in the pathogenesis of infectionassociated preterm birth (Romero et al., 2005). These inflammatory mediators are produced by macrophages, decidual cells, and fetal membranes in response to bacteria or bacterial products. A role for selected cytokines in preterm labor is based on the following observations (see Gravett and Novy (1997) for a concise review of this literature): elevated concentrations of cytokines and prostaglandins in amniotic fluid are found in patients with intra-amniotic infection and preterm labor; in vitro, bacterial products stimulate the production of proinflammatory cytokines by human decidua; these cytokines, in turn, stimulate the production of prostaglandins by the amnion and the decidua; the administration of IL-1 to pregnant mice or nonhuman primates induces preterm labor, which can be prevented by the administration of IL-1 receptor antagonist protein.
Another complementary mechanism by which intrauterine infection leads to preterm birth is by activation of the fetal HPA axis. Increases in fetal cortisol and fetal adrenal androgen levels have been reported among the fetuses of women with intrauterine infections (Gravett et al., 2000; Yoon et al., 1998) and in nonhuman primates with experimental intra-amniotic infections (Gravett et al., 1996).
Gravett and colleagues (1994) have demonstrated in nonhuman primates that after experimental intra-amniotic infection with group B streptococci there are sequential increases in the levels of proinflammatory cytokines (IL-1β, TNF-α, IL-6, and IL-8), prostaglandins, and MMPs in amniotic fluid that precede increases in uterine contractility by 24 to 48 hours and that result in preterm delivery. This model provides a characterization of the temporal relationships among infection, inflammation, and labor. In complementary work with knockout mice, Hirsch and Wang (2005) have demonstrated that IL-6 is neither a sufficient nor a necessary component of this cascade to stimulate preterm labor but that IL-1β is sufficient. Thus, animal models, as discussed below, have contributed greatly to providing an understanding of the pathophysiology of infection-induced preterm birth.
The observations presented above suggest that infection-associated preterm birth is an acute event that occurs proximal to delivery. Recent evidence, however, suggests that midtrimester amniotic fluid infection with Ureaplasma urealyticum may result in preterm birth many weeks later (Greber et al., 2003; Gray et al., 1992). Furthermore, elevated midtrimester concentrations of IL-6, a proinflammatory cytokine, in amniotic fluid have been associated with preterm birth at 32 to 34 weeks of gestation (Wenstrom et al., 1996).
Although the strongest evidence associating preterm birth with infection is derived from intrauterine infections, considerable evidence also suggests that lower genital tract infections, especially bacterial vaginosis, contribute to prematurity. Bacterial vaginosis has been associated with preterm labor or delivery, amniotic fluid infection, chorioamnionitis, and postpartum endometritis. These associations have been reviewed extensively elsewhere (Kimberlin and Andrews, 1998) and are based on the findings of case-control and cohort studies that consistently demonstrate an approximate twofold increase in the rates of preterm labor or delivery among women with bacterial vaginosis (Kimberlin and Andrews, 1998); the recovery of bacterial vaginosis-associated microorganisms from the amniotic fluid of 30 percent of women with intact fetal membranes in preterm labor and subclinical amniotic fluid infection (Martius and Eschenbach, 1990); and the frequent recovery of bacterial vaginosis-associated microorganisms from the amniotic fluid of women with overt clinical amniotic fluid infection or
from the chorioamnions of women with histological chorioamnionitis or preterm delivery (Hillier et al., 1988).
Although the magnitude of the increased risk for prematurity noted in these studies is modest (an approximately twofold increased risk compared with that for women without bacterial vaginosis), the total impact upon prematurity may be much greater given the high prevalence (20 percent) of bacterial vaginosis during pregnancy. It has been estimated that up to 6 percent of the cases of preterm delivery of infants with low birth weights may be attributable to bacterial vaginosis (Hillier et al., 1995).
Thus, bacterial vaginosis represents an important and potentially preventable cause of prematurity. However, trials of antibiotic treatment for bacterial vaginosis during pregnancy have met with mixed results (see Chapter 9 for a full review). Several studies have demonstrated reductions in the rates of preterm delivery or pregnancy loss among women at increased risk of prematurity by antibiotic treatment and eradication of bacterial vaginosis, whereas others have not (Hauth et al., 1995; McDonald et al., 1997b; Morales et al., 1994; Ugwumadu et al., 2003). However, a recent meta-analysis of all trials of antibiotic treatment for bacterial vaginosis during pregnancy did not reveal a consistent reduction in the rates of preterm birth or pregnancy morbidities, in part because of heterogeneity of the data (McDonald et al., 2005; Varma and Gupta, 2006). (An analysis of these treatment trials is discussed in detail in Chapter 9.)
Preterm birth has also been associated with maternal systemic infection (and is largely attributable to the severity of maternal illness) and, more recently, with maternal periodontal disease. Periodontal disease is an anaerobic bacterial infection of the mouth that affects up to 50 percent of the population, including pregnant women. Maternal periodontal disease has been associated with several adverse pregnancy outcomes, including preterm birth, preeclampsia, and fetal loss (Boggess et al., 2003; Jeffcoat et al., 2001a,b; Offenbacher et al., 1996). In a recent review of 25 studies, 18 sound an association between periodontal disease and adverse outcomes of pregnancy, with odds ratios for preterm birth or low birth weight of 1.1 to 20 (Xiong et al., 2005). Further, three clinical trials of periodontal treatment suggested a 50 percent reduction in the risk of preterm birth.
The mechanisms responsible for preterm birth in association with periodontal disease are not completely understood. Experimental evidence from studies with rabbits suggests that the oral pathogens associated with periodontitis can gain access to the systemic circulation and can be recovered from amniotic fluid or the organism’s DNA can be recovered from the placenta (Boggess et al., 2005a). Additionally, the gram-negative anaerobes associated with periodontitis may serve as a source of the lipopolysaccharide endotoxin that increases the levels of proinflammatory mediators, including cytokines and prostaglandins.
One conundrum for infection-associated preterm birth is that the majority of women who have lower genital tract infections, systemic infections, or periodontal disease do not deliver prematurely. Hence, the host inflammatory response to a potential pathogen must play a critical role in preterm birth. Cytokines (and some Toll-like receptors) are genetically very pleomorphic. It is likely that genetic differences in inflammatory responsiveness play a major role in determining whether or not a preterm birth occurs. (This concept of the gene-environment interaction is discussed in detail in Chapter 7.)
Implantation Errors
Traditionally, preterm delivery was thought to result from events that occurred at about the time of labor onset. The finding of elevated CRH levels before midgestation in association with preterm delivery suggests that some events triggering preterm delivery may be set in motion earlier in pregnancy than was previously thought (Hobel et al., 1999; Leung et al., 1999; McLean and Smith, 1999; McLean et al., 1999). A growing body of evidence now suggests that complications that become apparent relatively late in pregnancy, including preterm delivery, may actually reflect errors that occurred much earlier in placental development, beginning with implantation of the blastocyst in the uterus.
Implantation occurs approximately 6 or 7 days after conception and consists of three stages: apposition, adhesion, and invasion (reviewed by Norwitz et al. [2001]). Successful implantation is the end result of complex synchronized interactions between a receptive uterus and an activated blastocyst. Ovarian estrogen and progesterone transform the prereceptive uterus to a receptive state via a number of locally expressed growth factors, cytokines, transcription factors, vasoactive mediators in the uterus, whereas uterine-derived catecholestrogen and embryonic chorionic gonadotrophin are involved in implantation (Cameo et al., 2004). However, many of these data are derived from animal studies, and it is difficult to determine precisely what factors are critical in human implantation. Once implantation begins, a brief interval of stable adhesion involving adhesion molecules and other proteins is followed by a much longer period during which trophoblasts invade the uterus. The molecular mechanisms that regulate trophoblastic invasion are not well understood but probably involve multiple growth factors and cytokines, including leukemia-inhibiting factor, a heparin-binding epidermal growth factor, IL-1 and its receptors, and vascular endothelial growth factor (Dey et al., 2004; Kayisli et al., 2004; Norwitz et al., 2001). IL-1 appears to play a key role in implantation. For example, IL-1 induces the expression of COX-2 and MMP-9, both of which are central to implantation and decidualization (Fazleabas et al., 2004). Addition-
ally, IL-1α also stimulates the production of uterine IL-10, a Th2-type cytokine that plays a crucial role in the maintenance of pregnancy, in part by inhibiting the synthesis of Th1-type cytokines and suppressing the activities of natural killer-like cells and other inflammatory cells at the uteroplacental interface (Kelly et al., 2001; Vigano et al., 2003). However, data derived from humans remains incomplete, and there is a need for more research in this area.
A direct causal link between implantation problems and spontaneous preterm delivery has not been established in animal or human studies; nonetheless, indirect evidence suggests that this may be an important area for further research. The nonpregnant endometrial cavity is frequently colonized by microorganisms (Arechavaleta-Velasco et al., 2002; Romero et al., 2004b), and subclinical endometrial infection or inflammation may impair implantation or placentation, possibly by eliciting an antitrophoblast immune response that results in apoptosis, reduced trophoblast invasion and remodeling of the deciduas and uterine arterial vessels, and the arrest of early embryonic development (Romero et al., 2004b). This raises the possibility that inflammation in the endometrium around the time of implantation may contribute to subsequent preterm delivery. Furthermore, a normal pregnancy is characterized by a shift from a proinflammatory Th1-type response toward an anti-inflammatory Th2-type response (Marzi et al., 1996). One untested hypothesis is that women with microbial invasion of the endometrium may develop a persistent proinflammatory response in the endometrium (a Th1-type bias). This, in turn, could predispose the woman toward damage of the conceptus, implantation failure, spontaneous abortion, and preterm delivery. Of note, pregnancies complicated by preeclampsia, an important cause of indicated preterm delivery, also show reduced trophoblastic invasion and spiral artery remodeling as well as increased trophoblast apoptosis. No study, however, has directly linked preeclampsia to peri-implantation endometrial infection or inflammation. A recent study showed that modest undernutrition (defined as caloric restriction) in sheep commencing before conception and continuing for only 30 days thereafter induces premature delivery (Bloomfield et al., 2003). The undernourished ewes had higher ACTH levels throughout gestation, with a precocious rise in cortisol levels in half of them. This raises the possibility that the placental clock can be set by undernutrition at about the time of conception, resulting in accelerated maturation of the fetal HPA axis, leading to preterm birth.
Summary of the Pathways to Preterm Birth
Preterm birth has many potential pathways (Figure 6-2). In the past, obstetricians and epidemiologists have had a tendency to combine, for statistical purposes, all preterm births occurring between 22 and 37 weeks of
gestation. This has obscured the opportunity to study preterm birth as a final common end point and has led to uniform, largely empirical, and unsuccessful treatment strategies. It is now clear that the causes of preterm labor and multifactorial and vary according to gestational age. Each pathway to preterm labor can be characterized by its own unique upstream initiators of preterm parturition. Nonetheless, all share common downstream effectors of preterm contractions. For example, whether it is related to stress or infection, fetal HPA activation plays a role. Similarly, whether they are related to infection, uterine overdistension, or PPROM, MMPs play a role. Finally, regardless of the unique initiating circumstances myometrial contractility is mediated by prostaglandins. The recognition that preterm delivery is the common end result of a myriad of unique initiators provides an opportunity for research into unique upstream and common downstream interventions that can be used to reduce the risks of preterm birth.
Important areas of research on the pathways to preterm birth remain
-
the need for better understanding of human implantation and placentation,
-
the need for improved early diagnostic tests to better discriminate between the many pathways to preterm birth,
-
recognition that preterm birth is multifactorial and represents only a common end point for a myriad of unique, independent etiologies, and
-
the development of rational intervention strategies that target unique upstream initiators and common downstream effectors of preterm birth.
Finding 6-1: Preterm parturition has heterogeneous origins that result in common biological pathways and that lead to relatively few clinical presentations (e.g., preterm labor, preterm rupture of membranes, and cervical insufficiency).
PREMATURE RUPTURE OF MEMBRANES
Regardless of the etiology or mechanistic pathway to spontaneous preterm labor, preterm birth is usually preceded by PPROM. PPROM accounts for approximately 40 percent of preterm births (Shubert et al., 1992) and represents a final common pathway to preterm birth. Thus, an understanding of the mechanistic pathways leading to PPROM is important in understanding the biological basis of prematurity. Preterm prelabor rupture of membranes has been associated with intrauterine infection, tobacco use, abruption, multiple gestations, previous PPROM, previous cervical surgery or laceration, a short cervix as detected by ultrasound, genetic connective tissue disorders, and vitamin C deficiency (Asrat, 2001; Asrat et al., 1991;
Barbaras, 1966; Major et al., 1995; Odibo et al., 2002; Sadler et al., 2004; Spinello et al., 1994; Wideman et al., 1964).
Potential Mechanisms of PPROM
The fetal membranes (amnion and chorion) abut the maternal decidua and rest upon a collagenous basement membrane of type II and IV collagen. Beneath this layer is a fibrous layer that contains collagen types I, III, V, and VI. Thus, collagen provides major structural strength for the membranes. Membrane rupture is a process similar to wound healing, a process in which collagen is degraded (Malak and Bell, 1994). MMPs are the only family of enzymes that act to degrade collagen and play a major role in tissue remodeling. MMP-1 and MMP-8 are collagenases that act to degrade collagen types I, II, and III. MMP-2 and MMP-9 are gelatinases that degrade collagen types IV and V.
The activities of MMPs are regulated at several levels but, most importantly, are regulated by tissue inhibitors of MMPs (TIMPs). A balance between activators and tissue inhibitors of metalloproteinases controls metalloprotease activity. An increased ratio of MMP-9 to TIMP type 1 (TIMP-1) is associated with a decrease in the tensile strength of fetal membranes. Menon and Fortunato (2004) reviewed the role of MMPs in PPROM, with the overall hypothesis that a host inflammatory response inappropriately activates MMPs in the (ECM). Specifically, MMP type 1 to 3, 8, 9, and 14 messenger RNAs are detected in the amnion and chorion; and their levels increase in the amniotic fluid in PPROM (Menon and Fortunato, 2004). MMP-9 concentrations are increased in the amniotic fluid of patients with PPROM and to a lesser extent in patients with preterm birth (Fortunato et al., 2000a). Increased levels of pro-MMP-9 are found overlying the cervix in term gestation. MMP types 2 and 9 degrade type IV collagen, which is found in the basement membranes of the extracellular matrix (ECM).
Epidemiological, histological, and microbiological studies indicate that changes in fetal membrane production of MMP may be caused by infection or inflammation (reviewed by Menon and Fortunato [2004]). In vitro studies demonstrate an increase in MMP levels and a decrease in TIMP levels when the amniochorion is exposed to bacterial products (Fortunato et al., 1999). Several species of bacteria produce collagenases and decrease the bursting load and elasticity and work to rupture the membranes in vitro (MacGregor et al., 1987). Infection has been associated with an increase in MMP levels and a decrease in TIMP levels in the amniotic cavity. MMP-9 levels are increased in the amniotic fluid of women with intrauterine infections (Fortunato et al., 1999). MMP-2 levels are increased in membranes exposed to lipopolysaccharide in vitro (Fortunato et al., 2000b).
The mechanisms by which infection causes PPROM are likely multifactorial. Bacteria may directly secrete proteases that degrade collagen (MacGregor et al., 1987). Some bacterial species produce phospholipase A2, which acts to increase the levels of arachidonic acid, a prostaglandin precursor (Bejar et al., 1981). PGE2 decreases collagen synthesis in fetal membranes. Prostaglandin increases MMP-1 and MMP-3 levels in fibroblasts. Proinflammatory cytokines such as IL-1 and TNF-α also increase MMP levels and decrease TIMP levels in cultured amniocytes (So, 1993). In nonhuman primates, intrauterine infection with group B streptococci stimulates both proinflammatory cytokine and MMP-9 production (Vadillo-Ortega et al., 2002). Reactive oxygen species (ROS) resulting from immune cell signaling may also increase MMP levels and contribute to PPROM (Woods, 2001). Many clinical risk factors for PPROM, such as smoking, vaginal bleeding, cocaine use, and intra-amniotic infection, may also increase ROS levels by a variety of mechanisms. Exposure to superoxide increases MMP-9 activity and stimulates the release of arachidonic acid, a precursor to PGE2.
Amniotic fluid concentrations of MMP may also be increased in PPROM in the absence of infection. In vitro data indicate that both thrombin and thrombin receptor agonist peptide type 14 also increase MMP-9 concentrations (Stephenson et al., 2005). Thrombin increases MMP-9 levels in cultures with amniochorion. Thrombin also increases MMP-3 levels, and progesterone decreases this effect in decidual cell cultures (MacKenzie et al., 2004). Stretching of the membranes in multifetal gestations or hydramnios may result in PPROM by increasing PGE2, IL-8, and MMP-1 activities (Maradny et al., 1996).
The mechanisms by which decreased collagen content causes rupture of the fetal membranes are unknown. Rupture of the membranes may be mediated by apoptosis or programmed cell death following degradation of the extracellular matrix. Human fetal membranes have more apoptotic cells near the rupture site in women with premature rupture of membranes (Leppert et al., 1996). MMP-2 gene activation coincides with increased apoptosis in fetal membranes in PPROM (Fortunato et al., 2000a). The MMP-2 gene promoter has a transcription factor binding site that can be bound by protein p53, an intracellular regulator of apoptosis (Bian and Sun, 1997). Fetal membranes have high levels of expression of several components of the FAS-caspase apoptosis pathway. Women with PPROM had higher levels of proapoptotic proteins such as p53, Bax, and caspase, whereas the membranes of women with preterm labor had higher levels of Bcl2, an antiapoptotic protein. Investigators have also found that FAS and TNF-α-mediated apoptotic pathways are up-regulated in PPROM but not in preterm labor without premature rupture of membranes (reviewed by Menon and Fortunato, 2004).
ANIMAL MODELS FOR PRETERM BIRTH AND NEONATAL SEQUELAE
Although most animal species do not have significant rates of spontaneous preterm birth, there is much interest in the use of relevant animal models to elucidate the mechanisms of preterm birth and the neonatal sequelae of prematurity and to develop rationale and efficacious treatment and prevention strategies. However, in choosing an appropriate animal model, a necessary caveat is that many species differ from humans in the length of gestation and the number of fetuses, the type of placentation, the hormonal control of parturition, and the timing of fetal organ maturation (Figure 6-3 and Table 6-2). For example, lower mammalian species such as rats, rabbits, and mice have short gestational periods with multiple fetuses and delayed maturation of the fetal brain. Sheep, another commonly used experimental model, have a longer gestation and a singleton pregnancy, similar to humans, but have different placentation; and parturition is initiated by abrupt increases in cortisol levels, which is not seen in humans. In all of these species, parturition is preceded by systemic progesterone withdrawal, which is not seen in humans (refer to the works of Challis et al. [2000] and Elovitz and Mrinalini [2004] for reviews). Finally, there may be species differences in implantation and placentation. Further research in this area is critical. Recent research with nonhuman primates suggests that they have a reproductive biology that is the most similar to that of humans and represent the most appropriate model with which to study preterm birth; however, the cost and restricted availability of nonhuman primates limit their use (Elovitz and Mrinalini, 2004).
Despite their differences in reproductive biology, mice continue to play a major role in prematurity-related research. Their ready availability, low cost, and ability to be genetically manipulated (i.e., gene-knockout models are available) make the mouse model appealing. However, their short length of gestation (19 to 20 days) and delayed fetal maturation, especially of the central nervous system, limit the ability to generalize observations for mice to humans.
Much can be learned about the mechanisms of preterm birth by use of appropriate animal models. However, each species has distinct advantages and disadvantages that must be carefully considered in asking and answering important research questions relevant to preterm birth and neonatal sequelae.
Animal Models of Preterm Birth
Spontaneous preterm birth occurs infrequently in most species. This has limited research in this area to specific pharmacological or environmen-
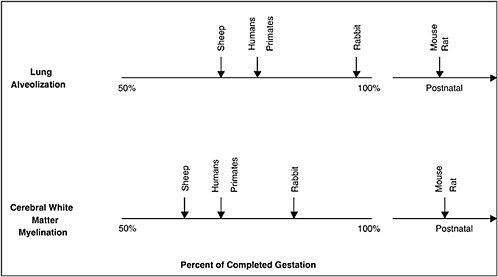
FIGURE 6-3 Comparative gestational ages at critical maturational steps in fetal development among different species relevant to neonatal morbidity. Vertical arrows indicate when lung alveolar development or preoligodendrocyte development (oligodendrocytes are responsible for myelination) begins. This schema indicates that maturation of the lungs and cerebrum, two organs with important contributions to neonatal morbidity and mortality, in sheep and nonhuman primates most closely parallel development in humans. In contrast, rabbits and rodents experience equivalent maturational sequences only near term or postnatally, limiting their role in ascertaining the relationships among the causes of prematurity and its consequences.
TABLE 6-2 Reproductive Characteristics of Animal Models Used to Study Parturition
tal interventions that result in preterm birth. Pharmacological administration of RU-486, a progesterone antagonist, leads to preterm delivery in rodents but not in nonhuman primates (Dudley et al., 1996; Garfield et al., 1987; Haluska et al., 1994). Administration of cortisol (Grigsby et al., 2000) or maternal starvation (protein and caloric restriction) (Kumarasamy et al., 2005) leads to preterm delivery in sheep.
The most compelling data from animal models are derived from studies of the role of infection and inflammation in preterm birth. Research with nonhuman primates has demonstrated that following experimental intraamniotic fluid infection, sequential increases in the levels of proinflammatory cytokines, prostaglandins, and MMPs precede the onset of preterm labor by 24 to 48 hours (Gravett et al., 1994; Vadillo-Ortega et al., 2002).
This research has also shown that pregnancy can be significantly prolonged by treatment with antibiotics and immunomodulators but not with antibiotics alone (Gravett et al., 2003).
Mice, rats, rabbits, sheep, and nonhuman primates have all been used as models for infection-induced preterm delivery. Regardless of the inciting stimuli (e.g., lipopolysaccharide, live microorganisms, or cytokines) or the route of administration, all of these models have confirmed the central role of the inflammatory response and proinflammatory cytokines in infection-induced preterm birth (see the review by Elovitz and Mrinalini [2004]).
The inflammatory system is redundant; that is, many proinflammatory cytokines act to up-regulate other proinflammatory cytokines. Hence, the role of individual cytokines in preterm labor has been difficult to ascertain. Recent work by Hirsch and Wang (2005), however, with genetically altered gene-knockout mice has demonstrated a central role for IL-1α but not IL-6, in infection-induced preterm labor. That important work demonstrates the utility of animal models in elucidating the mechanisms of preterm labor and points the way to effective intervention strategies.
Animal Models of Neonatal Sequelae
Two of the most important neonatal sequelae of preterm birth, especially in the setting of infection and inflammation, are periventricular white matter disease (PWMD) and neonatal lung disease (Dammann et al., 2005). Animal models have been particularly useful in elucidating the role of prematurity in these adverse neonatal outcomes.
Periventricular Leukomalacia and Cerebral White Matter Lesions
PWMD is detected in a significant proportion of premature infants and is strongly associated with adverse outcomes, including motor, perceptual, visual, behavioral, and cognitive disorders (see Chapter 10 for a further discussion of PWMD). The incidence of PWMD in preterm infants ranges from 3 to 20 percent, depending on the method of diagnosis and the extent of prematurity (Blumenthal, 2004). Approximately 10 percent of very low birth weight infants develop cerebral palsy, and 90 percent of these cases are thought to be due to PWMD (Blumenthal, 2004; Hack and Taylor, 2000; Wood et al., 2000).
PWMD includes a spectrum of cerebral injuries, ranging from focal cystic necrotic lesions (periventricular leukomalacia) to extensive, diffuse white matter lesions. Focal lesions occur in the deep white matter and are characterized by the necrosis of all cellular elements (axons, oligodendrocytes [OLs], and astrocytes), with subsequent cyst formation. Diffuse le-
sions, on the other hand, are characterized by more widespread, cell-specific injury to OL precursors (pre-OLs), with the subsequent impairment of myelinogenesis.
The pathogenesis of PWMD in the premature brain has been studied extensively in both in vitro and in vivo models (Back and Rivkees, 2004; Hagberg et al., 2002; Inder et al., 2004). A complex interplay of factors related to cerebrovascular immaturity appears to predispose the preterm periventricular white matter to injury. The major interacting factors include an underdeveloped vascular system, impaired cerebrovascular regulation, and pre-OL populations that are more vulnerable to oxidative stress and injury (Back and Rivkees, 2004).
Underdeveloped Vascular System
The vascular supply to the brain principally consists of the long and short penetrating arteries, neither of which is fully developed in the premature brain. Any decrease in cerebral blood flow can therefore lead to ischemia in the “watershed” areas of the white matter. Decreased blood flow to the long penetrating arteries results in severe ischemia and subsequent focal damage to the deep white matter, whereas decreased blood flow to the short penetrating arteries leads to moderate ischemia and subsequent diffuse pre-OL-specific damage in the border zones between the long penetrating arteries and at the end zones of the short penetrating arteries (subcortical areas).
A number of different animal models have been developed to evaluate the role of hypoperfusion in PWMD. These models have used transient or permanent unilateral or bilateral carotid artery ligation, combined hypoxiaischemia, umbilical cord occlusion, or hemorrhagic hypotension. In the vast majority of these models, however, both the white matter and the gray matter are affected. Two models in which the distribution and the morphology of PWMD more closely resemble those in the brains of human infants born preterm include a neonatal dog model in which bilateral ligation of the common carotids is used (Yoshioka et al., 1994) and a fetal sheep model in which hemorrhagic hypotension is used (Matsuda et al., 1999).
A physiological correlate of these anatomic factors is the observation of extremely low level of blood flow to the cerebral white matter in premature infants compared with that in term infants and adults (Altman et al., 1988; Greisen, 1986). This suggests that there is a minimal margin for safety for blood flow to cerebral white matter in such infants. Direct experimental evidence that human periventricular white matter is selectively susceptible to hypotension and ischemic injury, however, is lacking.
Impairment of Cerebrovascular Regulation
Studies have indicated that a significant proportion of ventilated premature infants (up to 53 percent) have impairment of cerebrovascular regulation (Tsuji et al., 2000). In these infants the cerebral circulation is pressure passive, and therefore, as the blood pressure falls, so, too, does cerebral blood flow. Again, a minimal margin of safety may exist, leading to ischemia in the watershed areas of the white matter. In a child or an adult, an intact cerebrovascular regulation system is in place that keeps the cerebral blood flow constant over a wide range of blood pressures through appropriate compensatory vessel dilation and vasoconstriction (Volpe, 2001).
Studies with preterm lambs indicate that at an early stage during maturation of the cerebrovascular autoregulatory system, the range of blood pressures over which cerebral blood flow is maintained constant, although present, is particularly narrow (Papile et al., 1985; Szymonowicz et al., 1990). This indicates that even premature infants with newly intact cerebrovascular autoregulation would be vulnerable to modest declines in blood pressure.
The relationship between cerebral blood flow and PWMD is further supported by clinical studies that demonstrate a correlation between PWMD and neonatal events expected to cause cerebral ischemia (severe hypotension, marked hypocarbia, hypoplastic left heart syndrome, patent ductus arteriosus with retrograde cerebral diastolic flow, and severe illness requiring extracorporeal membrane oxygenation) (Volpe, 2001).
Pre-OL Vulnerability
Before 32 weeks of gestation in humans, 90 percent of OLs are in an early stage of development and are known as pre-OLs (Back et al., 2001). Several lines of evidence from both in vitro and in vivo experimental models, outlined below, support the role of targeted pre-OL death in the pathogenesis of PWMD. Pre-OLs are exquisitely sensitive to injury and death by a number of different mechanisms.
Studies with neonatal and fetal rats, rabbits, and sheep have provided both direct and indirect evidence of increases in the levels of oxygen free radicals in the developing brain following hypoxia-ischemia (Bagenholm et al., 1997, 1998; Hasegawa et al., 1993; Rosenberg et al., 1989). The generation of free radicals is most pronounced during the initial period of reperfusion. The type of free radical involved varies somewhat by experimental model but principally involves superoxide anion and hydrogen peroxide. In vitro and in vivo neonatal rodent models of hypoxia-ischemia have demonstrated that pre-OLs are highly susceptible to free radical attack, whereas mature OLs are resistant (Back et al., 2002). Studies with
neonatal piglets subjected to hypoxia-ischemia have shown that the mechanism of pre-OL death from free radical attack is apoptosis (Yue et al., 1997). This has been confirmed in in vitro studies and has been suggested by autopsy studies of the brains of human infants born preterm (Back et al., 1998; Gilles et al., 1983).
In addition to being more vulnerable to free radicals, pre-OLs tend to accumulate free radicals, whereas mature OLs do not. Information derived from animal models (mouse, rat, and lamb models) and limited analyses of autopsied brains of human infants born preterm suggest that there is a delay in the development and the reactivity of antioxidant defenses, especially those involving glutathione peroxidase and catalase (Juurlink, 1997; Volpe, 2001). These enzymes are involved in the detoxification of hydrogen peroxide. If hydrogen peroxide accumulates and iron ions (Fe2+) are present, the Fenton reaction will take place, producing the deadly hydroxyl radical.
Glutamate has also been implicated in rodent models of PWMD (Deng et al., 2004; Follett et al., 2000; Liu et al., 2002). Hypoxia-ischemia in the preterm infant brain leads to coagulation necrosis and the disruption of axons. Axon disruption leads to the leakage of glutamate into the extracellular space. In addition, with an altered brain energy supply, the level of glutamate uptake by astrocytes and neurons is reduced. Additional sources of glutamate include the reversal of glutamate transporter function in astrocytes and OLs and cytokine effects on astrocytes, among other factors.
Glutamate can cause the destruction of pre-OLs by either receptor-mediated or non-receptor-mediated mechanisms (Volpe, 2001). Activation of the α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) and kainite type of glutamate receptor has been shown to lead to pre-OL death in culture and in rodent models of PWMD (Follett et al., 2000; Gan et al., 1997). This occurs only in developing OLs and not in mature OLs. In an immature rat model, Follet and colleagues (2000a,b) showed that such injuries could be prevented by the administration of a receptor antagonist such as 6-nitro-7-sulfamoylbenzo(f)quinoxaline-2,3-dione or topiramate, an anticonvulsant drug.
In a non-receptor-mediated mechanism, glutamate causes glutathione depletion in pre-OLs, leading to free radical generation and subsequent cell death. This is mediated by activation of a glutamate-cystine exchange transporter, whereby glutamate uptake results in the efflux of cystine, intracellular cystine depletion, and impaired glutathione synthesis (Oka et al., 1993).
Maternal or Fetal Infection or Inflammation and Cerebral White Matter Lesions
Recently, the role of intrauterine infections in the pathogenesis of periventricular leukomalacia and cerebral palsy has become a major focus
of research (Dammann and Leviton, 1998; Grether and Nelson, 1997; Leviton, 1993). A growing body of evidence suggests that maternal genital tract infections, particularly intrauterine and intra-amniotic infections, may be important and potentially preventable causes of periventricular white matter damage and cerebral palsy. The association between intrauterine infection and cerebral palsy is supported by human observational studies and animal experimental studies. Bejar and colleagues (1988) observed that the risk of cerebral white matter damage was 9.4-fold greater among preterm neonates with purulent amniotic fluid than among those with nonpurulent fluid. Similarly, Grether and Nelson (1997) observed cerebral white matter damage in association with maternal intrapartum fever or chorioamnionitis. A recent meta-analysis of 30 human observational studies by use of a random-effects model reported that clinical chorioamnionitis was significantly associated with both cerebral palsy (relative risk [RR] 1.9; 95% confidence interval [CI] 1.4–2.5) and periventricular leukomalacia (RR 3.0; 95% CI 2.2–4.0) in preterm infants (Wu and Colford, 2000). Among term infants, a significant association was also found between clinical chorioamnionitis and cerebral palsy (RR 4.7; 95% CI 1.3–16.2) Clinical studies have reported elevated concentrations of proinflammatory cytokines, including IL-6, IL-1, and TNF-α, in amniotic fluid and elevated concentrations of IL-6 in umbilical cord plasma among neonates with periventricular leukomalacia or cerebral palsy (Nelson et al., 1998; Yoon et al., 1996, 1997a); increased expression of IL-6 and TNF-α within the brain lesions of infants who died with periventricular leukomalacia has also been reported (Yoon et al., 1997b).
Animal models consisting of pregnant rabbits with experimental intrauterine infection have demonstrated brain white matter lesions characterized by increased karyorrhexis, rarefaction, disorganization of the white matter, and increased apoptosis in the cerebral cortex (Yoon et al., 1997c). Finally, in a feline model of Escherichia coli endotoxin-induced cerebral white matter injury, daily intraperitoneal injections of endotoxin resulted in injury of the telencephalic white matter of newborn kittens (Gilles et al., 1976). One shortcoming of these animal studies is a lack of detailed monitoring of adverse systemic events associated with endotoxin, including hypoglycemia, acidosis, and hypotension. Subsequent studies with rabbit pups (Ando et al., 1988) and neonatal dogs (Young et al., 1983) demonstrated that cerebral white matter lesions occurred within 1 to 3 days of endotoxin exposure in the setting of transient acute arterial hypotension. Hence, the diverse forms of white matter pathology induced by endotoxin or infection might be due, at least in part, to systemic vascular effects.
Neonatal Lung Disease
The premature infant faces primarily two lung-related injuries: acute injury (respiratory distress syndrome [RDS]) and chronic or progressive lung injury (bronchopulmonary dysplasia [BPD]) (also see Chapter 10 for a discussion). A complex interplay of factors plays into the risk of these injuries, including incomplete development (immaturity), mechanical ventilation, oxidative stress, and inflammation (Zoban and Cerny, 2003).
Neonatal RDS is an acute lung process due to a deficiency in surfactant. It ranks as the sixth most common cause of death among newborns in the United States and occurs almost exclusively in premature infants, occurring in excess of 80 percent of infants born before 27 weeks of gestation (Bancalari, 2002; Lemons et al., 2001). The incidence and severity are dependent on the infant’s gestational age at birth and birth weight. Although the outcome of RDS has improved in recent years, the incidence and severity of complications continue to present significant morbidities. Complications may include pneumothorax; intraventricular hemorrhage; chronic lung disease (BPD); or even respiratory failure, leading to death.
BPD, also known as neonatal chronic lung disease, is another important cause of respiratory illness in preterm newborns. BPD is usually defined by the requirement of oxygen at 36 weeks since the mother’s last menstrual period and is characterized by small airway injury, dilated alveolar ducts, and decreased alveolarization. The incidence of BPD is directly related to birth weight, with rates of 7 percent in infants weighing 1,241 to 1,500 grams, 15 percent in infants weighing 1,001 to 1,240 grams, 34 percent in infants weighing 751 to 1,000 grams, and 52 percent in infants weighing 501 to 750 grams (Lemons et al., 2001).
Normal Lung Development
Fetal lung development is divided into the pseudoglandular, canalicular, and saccular-alveolar stages on the basis of histological appearance (Cardoso and Williams, 2000; Zeltner and Burri, 1987). The pseudoglandular stage takes place at between 7 and 17 weeks of gestation, the canalicular stage takes place at between 16 and 24 weeks of gestation, and the saccular-alveolar stage takes place at between 24 and 40 weeks of gestation. The saccular and alveolar stages overlap beginning at approximately 32 weeks of gestation, when secondary septation begins the process of alveolarization. Microvascular development occurs in parallel with alveolarization and continues for several months after term birth. Alveolarization and the associated vascularization are the critical terminal stages of lung development. The increase in alveoli, lung volume, and lung
surface area establishes the anatomic potential for gas exchange and thus the potential for fetal viability.
Surfactant production by type II alveolar cells begins at about 34 weeks of gestation. Normal production is achieved by the time that the fetal lungs are mature, at about 37 weeks of gestation. Pulmonary surfactant is a complex lipoprotein made up of six phospholipids and four apoproteins (Jobe and Ikegami, 2001). Functionally, dipalmitoylphosphatidylcholine, or lecithin, is the principal phospholipid. The components of surfactant are synthesized in the Golgi apparatus of the endoplasmic reticulum of type II alveolar cells. The surfactant is then secreted by exocytosis, ultimately forming monolayers in the air-liquid interface inside the alveoli. Surfactant is necessary for reducing the surface tension of the liquid lining and therefore decreasing the pressure needed to keep the alveoli inflated. A deficiency in surfactant leads to high surface tension, making it difficult to inflate the alveoli. Alveolar collapse subsequently occurs, leading to diffuse atelectasis and decreased compliance. Any disruption in the process of lung development can lead to both short-term and long-term effects on lung function in newborns, as described below.
Surfactant Deficiency
Surfactant deficiency is the primary cause of RDS and has been demonstrated in both animals (sheep) and humans. Studies with preterm rabbits have found that surfactant-deficient lungs do not accumulate much gas on inflation until pressures exceed 25 centimeters of water (cm H2O) (Jobe, 1993). Surfactant treatment results in a striking decrease in the opening pressure to about 15 cm H2O. This decrease in opening pressure allows more units to open and therefore leads to more uniform inflation and a decreased risk of overdistension.
Neonatal surfactant therapy reduces the rate of mortality from RDS and overall infant mortality rates (Soll and Morley, 2001). In addition, surfactant treatment decreases the incidence of pneumothorax, oxygen requirements, and ventilatory requirements during the first several days of life. Although neonatal surfactant therapy has had an impressive effect on the incidence of RDS, it has not been seen to have an effect on BPD. Presumably, infants who survive RDS with surfactant treatments are those who are more likely to go on to develop BPD. Further studies are necessary to test this hypothesis.
Disruption of Alveolarization
Recent pathologic analyses of very low birth weight human infants who have died of BPD demonstrate an arrest of alveolar development, with the
lungs containing fewer and larger alveoli (Thibeault et al., 2000). Numerous exposures common to the care of the premature infant have been shown to disrupt normal alveolarization. These exposures include hypoxia or hyperoxia, mechanical ventilation, nutritional deficits, glucocorticoids, and inflammatory mediators.
Both hypoxia and hyperoxia disrupt septation and therefore reduce the surface area for gas exchange. Rats exposed to increased oxygen concentrations are found to have severely disrupted alveolarization (Randell et al., 1989; Thibeault et al., 1990). Abnormalities, including decreased pulmonary septation, increased terminal space diameter, and decreased surface area, persist even after recovery.
Mechanical ventilation can also interfere with alveolar and vascular development, as has been demonstrated in preterm baboons and sheep (Jobe and Bancalari, 2001). Coalson and colleagues (1999) studied preterm baboons and found that mechanical ventilation with 100 percent oxygen severely reduced the number of alveoli. The same interference with septation was noted after surfactant treatment and ventilation but without exposure to large amounts of supplemental oxygen.
Glucocorticoid treatment can cause a profound arrest in alveolarization, as seen in many animal models, including rodents, monkeys, and sheep. In preterm mice and rats, glucocorticoid treatment has been found to cause permanent abnormalities in alveolar and vascular development (Massaro and Massaro, 2000). In preterm monkeys, glucocorticoid administration during the saccular stage of lung development leads to a decrease in the mesenchyme, a lower gas volume, and fewer alveoli (Johnson et al., 1978). In preterm sheep, glucocorticoid treatment is associated with a decrease in the numbers of alveoli; however, the alveoli increase in size (Ikegami et al., 1996, 1997; Willet et al., 2001).
Maternal glucocorticoid treatment has both an acute and a chronic effect on the fetal monkey lung (Johnson et al., 1978, 1981). Glucocorticoid treatment in preterm monkeys causes mesenchynal thinning and a large increase in maximal lung gas volumes. In term monkeys, however, the alveolar number, lung surface area, and lung gas volumes are decreased with glucocorticoid treatment. These results suggest that glucocorticoids are associated with an acute increase in lung gas volume (early lung maturation) but have an adverse effect on subsequent alveolarization and lung growth (Johnson et al., 1981).
Mechanical Ventilation and Lung Injury
Diffuse atelectasis and decreased compliance lead to alveolar hypoventilation and an imbalance in ventilation-perfusion. The ultimate outcome is worsening hypoxemia, requiring aggressive mechanical ventila-
tion. Studies done with preterm animal models (predominantly baboons and lambs) have demonstrated that the strategy of ventilation plays an important role in the development of the progression of lung injury (Albertine et al., 1999; Coalson et al., 1999; Yoder et al., 2000). Ventilatory factors that have been found to increase lung injury include high tidal volumes, a lack of surfactant before the institution of ventilation, and inadequate endexpiratory pressure.
High tidal volumes lead to regional overinflation of alveoli and airways (volutrauma). In neonates with RDS, only a small portion of the lung may be recruited and available for ventilation. If only a small portion of the lung is being ventilated, a normal tidal volume for weight will be too large and will result in the disruption of structural elements, such as the pulmonary capillary endothelium, the alveolar and airway epithelia, and the basement membrane.
This concept has been tested in studies with preterm lambs, in which as few as six manual inflations of 35 to 40 milliliters per kilogram of body weight before surfactant treatment and mechanical ventilation led to increased lung injury and a decrease in the response to surfactant therapy (Bjorklund et al., 1997). In contrast, Wada and colleagues (1997) found, again in studies with preterm lambs, that surfactant treatment before the institution of assisted ventilation decreased the degree of lung injury, presumably by promoting more uniform lung inflation.
The premature lung, which is deficient in surfactant, is prone to collapse (atelectasis). Ventilation below a normal functional residual capacity leads to cyclic opening and closing of lung units, which adds to lung injury. This type of injury can be minimized by recruiting lung volumes with positive end-expiratory pressure (PEEP) and then maintaining a higher than normal functional residual capacity with high-frequency oscillatory ventilation. The combination of PEEP and oscillatory ventilation has been shown to improve surfactant function, decrease lung injury, and improve survival in rats (Chiumello et al., 1999).
Oxidative Stress and Lung Injury
The premature lung is particularly susceptible to free radical-induced injury because of the delay in the development and the reactivities of antioxidant defenses (Bracci, 1997; Saugstad, 1990). Oxygen-induced injury is caused by the overproduction of free radicals, such as superoxide, hydrogen peroxide, and perhydroxyl. The presence of free radicals overwhelms the immature antioxidant system, leading to the oxidation of enzymes, the inhibition of proteins and DNA synthesis, decreased surfactant production, and lipid peroxidation, all of which play a role in lung injury.
Multiple pathways of ROS generation have been found both in humans and in animal models. These include ischemia-reperfusion and hypoxanthine oxidase reactions, the metabolism of catecholamines, the arachidonic acid cascade, and mitochondrial metabolism. An additional source of free radicals in the lungs is thought to be phagocyte activation. The increases in phagocyte numbers and interleukin concentrations and the increased levels of free radical release seen in the bronchoalveolar fluid of premature infants with both acute and chronic lung disease indicate that oxygen toxicity and inflammation are involved in the development of lung injury (Zoban and Cerny, 2003).
Infection-Inflammation and Lung Injury
Increasing evidence indicates that infection and inflammation may play a role in the development of lung injury. The role of inflammation in RDS is suggested by several studies with preterm animal models. Infectioninflammation is also associated with a disruption in alveolus formation. This has been demonstrated in studies with both mice and sheep. In an experimental model of chorioamnionitis in sheep, a single dose of endotoxin given 7 days before preterm delivery or a continuous 28-day intraamniotic infusion of endotoxin significantly decreased the numbers of alveoli (Willet et al., 2000). In transgenic mouse models, the overexpression of proinflammatory cytokines during the period of postnatal alveolarization has been found to disrupt alveolus formation (Jobe, 1999).
Studies done with preterm lambs have found that atelectasis (which is due to surfactant deficiency) initiates a cytokine-mediated inflammatory cascade that promotes the recruitment of neutrophils into the lung and that leads to endothelial cell damage and subsequent pulmonary edema. The depletion of neutrophils, however, prevents the pulmonary edema (Carlton et al., 1997; Naik et al., 2001). Proteins in the edema fluid are thought to worsen the lung injury by inactivating surfactant and exacerbating the surfactant deficiency.
Mechanical ventilation plays an important role in the inflammatory response of the preterm lung. Ventilation affects the numbers of inflammatory cells and the expression of soluble mediators within the lungs. This has been demonstrated with several animal models.
In rabbits subjected to saline lavage, injurious mechanical ventilation increased the levels of lung neutrophil accumulation and chemiluminescence (an indicator of neutrophil priming), increased the levels of inflammatory mediators (platelet-activating factor and thromboxane B2) in bronchoalveolar lavage fluid, and increased the levels of TNF-α expression by alveolar macrophages (Clark et al., 2001).
Summary of Pathophysiological Mechanisms
An understanding of the relevant pathophysiological mechanisms of preterm birth is necessary to develop rationale and efficacious intervention strategies in preventing and treating preterm birth and its sequelae in neonates. Animal models provide a unique opportunity to study longitudinally the causes and consequences of preterm birth in a controlled experimental environment that cannot be created in women. However, many animal models have important limitations based on the length of gestation, the endocrine events of parturition, the presence of multiple fetuses, and fetal developmental milestones. Experimental animal models must be developed that address specific questions on the basis of their similarities or relevance to the research questions important in human preterm birth. These include the use of
-
nonhuman primates to describe the temporal or longitudinal relationships among the mediators of preterm labor;
-
genetically altered mice to ascertain the roles that putative mediators of preterm birth has in leading to prematurity; and
-
sheep or nonhuman primates to ascertain the relationships among the various pathways to preterm birth and their sequelae in neonates, such as cerebral white matter injury and bronchopulmonary dysplasia.






































