
3
Federal Regulatory Landscape
The current regulatory scheme for research with human subjects is a patchwork of regulations and enforcement mechanisms that do not lend themselves to broad or easy application, particularly with regard to research involving prisoners. The environment when the existing regulations were adopted resulted in a set of regulations promulgated by the U.S. Department of Health and Human Services (DHHS) that were intended to be restrictive with respect to research involving prisoners: The default position is that no such research should occur, and the four or five categories of research allowed under the regulations are essentially exceptions to that general rule. The Office for Human Research Protections (OHRP) applies these regulations with the assumption that if the research described does not appear to fit into any given category, it cannot be approved, even if it otherwise seems beneficial and appropriate.
The restrictiveness of the DHHS regulations regarding prisoners may have had the unintended effect of creating widely varying regulatory schemes applicable to research involving prisoners, because of the unwillingness of other federal agencies to adopt the same set of regulations. Under the current framework, although they can voluntarily agree to more, research institutions are only required to abide by DHHS-promulgated regulations when they conduct research funded by the DHHS (including DHHS agencies such as the National Institutes of Health [NIH], the Food and Drug Administration [FDA], the Centers for Disease Control and Prevention [CDC], and the Substance Abuse and Mental Health Services Administration, which is actually a very small portion of all research involving
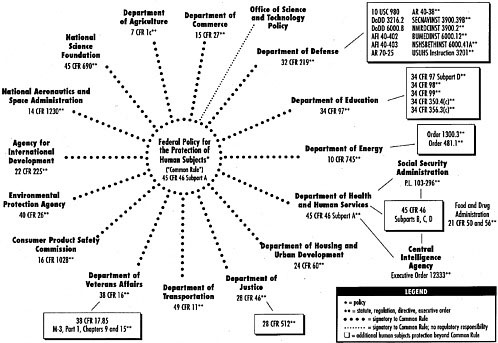
prisoners conducted in the United States). For the regulations to apply to other federally funded research, absent the consent of research institutions, it was necessary for every other department or agency funding such research to incorporate the DHHS-promulgated regulations into that department or agency’s own regulations. Sixteen other departments and agencies of the federal government adopted the generally applicable Common Rule regarding research with human subjects, thus partially accomplishing a goal of uniformity in the ethical regulations applicable to federally funded research.
However, perhaps because of the restrictiveness of the regulations, nearly all of the same departments or agencies did not adopt the additional protections for prisoners. At least one of the departments adopting the Common Rule, the Bureau of Prisons (BOP) at the Department of Justice (DOJ), instead adopted its own regulations that apply to all research with prisoners in BOP custody (not merely research that is funded by the DOJ). Additionally, the FDA promulgated its own rules, consistent to the extent practical with the Common Rule, governing clinical research associated with the products it regulates. However, the FDA’s attempt to adopt parallel regulations that were essentially the same as DHHS’s prisoner protections1 was the subject of a lawsuit brought by prisoners wishing to participate in such research. Therefore, the FDA does not have provisions comparable to DHHS Subpart C for prisoner populations.
Outside of DHHS and its agencies, only the Central Intelligence Agency (CIA) and the Social Security Administration (SSA) have adopted the DHHS’s prisoner protections (Figure 3-1).
In sum, regarding research involving prisoners as human research subjects, the applicable regulations are far from uniform and range from no protection at all (for research that is not funded by one of the 17 agencies that have adopted the Common Rule), to basic Common Rule protection, to heightened, overlapping, and possibly inconsistent regulations (e.g., for persons in BOP custody participating in therapeutic clinical trials). This chapter describes the components of the patchwork of regulations: the Common Rule, Subpart C (the DHHS’s protections for prisoners as research subjects), and the alternative regulations applied by other departments.
ADOPTION OF DHHS HUMAN SUBJECTS PROTECTION REGULATIONS
The first federal protections for human subjects were issued in 1966 by the NIH. This document, Clinical Investigations Using Human Subjects, served as the Public Health Service’s policy and was an initial attempt to
protect human subjects. It required prospective review of human subjects research, focusing on the rights of potential participants by balancing risks and benefits while ensuring appropriate informed consent procedures (Public Health Service [PHS], 1966).
These NIH policies, which initially applied only to extramural research, were later raised to regulatory standards for the entire Department of Health, Education, and Welfare (DHEW) in 1974. These regulations were later modified in 1981 and codified as Title 45 Part 46 of the Code of Federal Regulations. Revisions have occurred several times since then; the most recent changes took effect in 1991 with the development of the Federal Policy for the Protection of Human Subjects, also known as the Common Rule.
The Common Rule
The Common Rule is incorporated as Subpart A of 45 C.F.R. Part 46, the basic DHHS regulations for the protection of human research subjects. The regulatory framework outlined in the Common Rule applies to 17 federal agencies that are involved in conducting or funding human subjects research.2 The Common Rule provides guidelines on conducting certain types of research with human subjects. Specifically, it discusses issues such as review by institutional review boards (IRBs), informed consent, balancing risks and benefits, protecting privacy, and additional requirements for approval. Failure to adhere to these regulations can result in sanctions. Agency or department support of the research can be suspended or terminated, or additional conditions can be imposed on the individual project or on the research organization or institution.
Human subjects are defined as persons about whom a research investigator obtains either (1) identifiable private information or (2) data as a result of an intervention or interaction with the person.3 The Common Rule defines research as any “systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.”4
The Common Rule also identifies certain categories of research that are exempt. Notably, these exemption categories cannot be applied to any research involving prisoners; therefore, both the general protections (Subpart A) and the heightened protections (Subpart C) provided under 45 C.F.R. Part 46 still apply.5
|
2 |
See Figure 3-1. The FDA adopted a modified version of the Common Rule applicable to research involving all products it regulates. |
|
3 |
45 C.F.R. § 46.102(f). |
|
4 |
45 C.F.R. § 46.102(d). |
|
5 |
45 C.F.R. § 46.101(i), n.1. |
IRBs
A number of protections for human subjects involved in research are specified in the Common Rule. IRBs are required to review and approve any nonexempt research that involves human subjects, with its membership specified, functions defined, and review processes outlined.6 The Common Rule also includes criteria for IRB approval of research and identifies certain categories of research that can be approved on an expedited basis.7 It also specifies that, when “some or all of the subjects are likely to be vulnerable to coercion or undue influence,” the IRB must find that “additional safeguards have been included to protect the rights and welfare of those subjects.”8 IRBs are empowered to suspend or terminate research that has been approved and are required to maintain records that document all IRB activities.9
In some cases, IRB review of individual research projects can be expedited if the project involves no more than minimal risk and fits certain categories (e.g., collection of small amounts of blood, analysis of existing materials) or involves minor changes to a previously approved research project. Minimal risk for the Common Rule is defined as:
The probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.10
Under expedited procedures that bypass the full IRB process, the IRB chair or one or more experienced IRB members who is designated by the chair can review and approve the research. Although regulations do not preclude the use of expedited review for research involving prisoners, the OHRP recommends that such research be reviewed by the full committee.
Informed Consent
Informed consent processes are also defined in the Common Rule. The basic disclosure requirements for obtaining informed consent are (1) a description of the study and its purposes; (2) identification of any foreseeable risks or discomforts to the participant; (3) a description of any benefits that could be expected; (4) disclosure of alternative treatments that may
also be beneficial; (5) a description of how confidentiality of records will be maintained; (6) for treatment involving more than minimal risk, an explanation of the potential consequences resulting from participation in the research; (7) contact information for answering questions; and (8) a statement that the individual’s participation in the research must be voluntary, that refusal to participate will not result in a penalty or loss of benefits to which the individual is otherwise entitled, and that the individual may withdraw at any time.11 Additional information may also be required, depending on the specific nature of the research.12 The investigator must document that the person agreed to participate in the research project by obtaining the individual’s signature or the signature of an authorized representative.13
Institutional Assurances
Generally, the Common Rule requires that the departments or agencies applying the Common Rule obtain some form of written assurance from all research institutions engaging in covered research that the research complies with the regulations.14 If a research organization frequently conducts research supported by one of the signatory federal agencies, it may apply for a federal-wide assurance (FWA), a special kind of assurance process administered by the DHHS. The research organization seeking an FWA certifies that (1) all research will be performed in accordance with the ethical principles in the Belmont Report and (2) for any research that the organization conducts for which it receives any federal funds,15 certain procedures will be followed that ensure compliance with the Common Rule and any other applicable subparts the organization chooses to sign on to.
In lieu of requiring its own form of written assurance, a department or agency other than DHHS may accept the FWA.16 In that case, once the FWA is approved by the DHHS, it allows individual research proposals to be approved by the organization and its local IRB rather than by the individual federal department or agency that is funding the research, and any reports required are made both to the department or agency and the OHRP.17 Note that most organizations that conduct federally funded re search hold an FWA.
Subpart C: Prisoners as Research Subjects
Beyond the Common Rule contained in Subpart A, additional subparts of 45 C.F.R. Part 46 provide further and more specific protection for certain particularly vulnerable populations: pregnant women, fetuses, and neonates (Subpart B); prisoners (Subpart C); and children (Subpart D). Subpart C, “Additional Protections Pertaining to Biomedical and Behavioral Research Involving Prisoners as Subjects,” the focus of this project, was first finalized in 1978. These additional protections were developed in response to the National Commission’s Report and Recommendations: Research Involving Prisoners (NCPHSBBR, 1976). They represent further safeguards that must be met when conducting research with this vulnerable population group. To date, Subpart C has only been adopted by the DHHS, the CIA, and the SSA.
Key Definitions Within Subpart C
Subpart C defines a prisoner as any person who is “involuntarily confined or detained in a penal institution” as a result of violating a criminal or civil statute, a person who is committed to other facilities as an alternative to criminal prosecution or incarceration, or someone who is detained pending arraignment, trial, or sentencing.18 Research with this population must present no more than minimal risk. Here, that is defined as:
The probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical, dental, or psychological examination of healthy persons.19
Subpart C identifies four categories of research that are permitted with prisoners.
-
Study of the possible causes, effects, and processes of incarceration and of criminal behavior, provided that the study presents no more than minimal risk and no more than inconvenience to the subjects;
-
Study of prisons as institutional structures or of prisoners as incarcerated persons, provided that the study presents no more than minimal risk and no more than inconvenience to the subjects;
-
Research on conditions particularly affecting prisoners as a class (for example, vaccine trials and other research on hepatitis which is much more prevalent in prisons than elsewhere; and research on social and psychologi-
-
cal problems such as alcoholism, drug addiction, and sexual assaults) provided that the study may proceed only after the Secretary has consulted with appropriate experts, including experts in penology, medicine, and ethics, and published notice in the Federal Register, of the intent to approve such research; or
-
Research on practices, both innovative and accepted, which have the intent and reasonable probability of improving the health or well-being of the subjects. In cases in which those studies require the assignment of prisoners in a manner consistent with protocols approved by the IRB to control groups which may not benefit from the research, the study may proceed only after the Secretary has consulted with appropriate experts, including experts in penology, medicine, and ethics, and published notice in the Federal Register of the intent to approve such research.20
There has been confusion among researchers and correctional departments regarding the exact meaning of the categories stated previously and the specific circumstances in which they should be applied (See later discussion on the Subpart C Subcommittee of Secretary’s Advisory Committee on Human Research Protections [SACHRP] on page 81). In October 2002, the secretary of the DHHS published a notification in the Federal Register proposing to waive certain provisions of Subpart C to epidemiological research involving prisoners that involved no more than minimal risk and no more than inconvenience to potential subjects.21 The regulations, which became final in June 2003, allow for epidemiological research on specific diseases that describes the prevalence or incidence of the disease by identifying all cases, including prisoner cases, or studies of potential risk factor associations for these diseases in which prisoners were included in the population of interest but were not the sole study group.22
Implementation of an epidemiological research project involving prisoners requires that an IRB must approve the research, document that one of the two conditions described previously has been met, and determine that the research involves minimal risk and no more than inconvenience to the prisoner-subjects.23
Additional Requirements for IRBs
When research is proposed that involves prisoners, IRBs must approve each individual project. IRBs for prisoner research are not only required to
meet the conditions of the Common Rule (outlined previously) but also must have among its membership a prisoner or prisoner representative.
Further, the IRB must find that the research proposal meets both the Common Rule requirements as well as additional requirements specific to the prisoner setting. These additional requirements are as follows:
-
the research is within one of the four permissible categories of research for prisoners;
-
benefits that accrue as a result of the prisoner’s participation should not be so great in comparison to what is available in the correctional environment that the ability to provide informed consent is impaired;
-
risks are commensurate with those that would be accepted by nonprisoner volunteers;
-
selection procedures are fair and not subject to arbitrary intervention by either prison authorities or other prisoners;
-
the consent form is written in language that is easily understood by the prisoner;
-
the person’s participation in the research project will not be a consideration in parole or probation decisions; and
-
adequate provisions are made for follow-up care, should it be needed, once the research study ends.24
OHRP Certification
Once the IRB has found that the research meets the criteria described previously and approves the study, OHRP certification must be obtained for research in Categories i through iv and for epidemiological waivers as well (Table 3-1). If the OHRP certifies that the category is appropriate and that the criteria have been met, the research is approved. This certification step adds an average of 3–4 weeks to the review process.
Report of the SACHRP Subcommittee
In 2003, the SACHRP asked its Subpart C Subcommittee to review the text and application of Subpart C, primarily to determine whether the current DHHS interpretation and application of Subpart C’s requirements should be modified. Among the topics the subcommittee addressed were:
-
the definition of the term prisoner under Subpart C;
TABLE 3-1 Approximate OHRP Prisoner Certifications January 2000– October 12, 2005
-
the application of research protections to those who become incarcerated after agreeing to participate in a nonprisoner study;
-
issues with identifying a prisoner representative for prisoner-research IRBs and particularly in multisite studies;
-
conduct of expedited review in prisoner research;
-
the definition of minimal risk under Subpart C (which is different from the Subpart A definition); and
-
the requirement of secretarial review when prisoners in the control group are merely provided the standard of care (SACHRP, 2005).
Definition of Prisoner
The subcommittee recommended that a modified Subpart A analysis apply when a subject who is enrolled in a study may not be fully within the definition of prisoner for the duration of the study. First, the subcommittee affirmed that the interpretation of the term prisoner should remain defined by the words of the regulation and not be expanded to include other individuals whose liberty is restricted, such as those in community correctional facilities or on probation or parole. Although these individuals deserve heightened protection, the subcommittee recommended that the DHHS rely on Subpart A’s protections for individuals “vulnerable to coercion or undue influence” without including them as prisoners under Subpart C. Likewise, when an individual is incarcerated after enrolling in a study, the concerns about coercion and undue influence are not as great, and it may be difficult to modify the research protocol to comply with Subpart C. Therefore, the subcommittee suggested that Subpart A’s general requirement of heightened protection instead apply. The subcommittee recommended that an IRB should review a
researcher’s request to continue the research when an individual subsequently becomes incarcerated, taking into account the new conditions of incarceration but without fully engaging in a new Subpart C approval process.
Prisoner IRB Representative
The subcommittee discussed a variety of problems with identifying a representative who would be skilled and knowledgeable enough to be effective but not so unlike the rest of the IRB as to be marginalized. The subcommittee recommended that the OHRP should assist IRBs in searching for an appropriate prisoner representative, which might include family members of prisoners, former prisoners (especially people in recovery from substance addiction who have also had experience as prisoners), and service providers who assist in the correctional process. The OHRP should provide functional criteria that might help IRBs (and investigators, who are also responsible for the composition of an IRB that will properly evaluate ethical issues) identify persons who can be an effective voice for prisoners within the IRB. With respect to multisite studies, the subcommittee recommended that, although Subpart C only requires one prisoner representative on a central IRB for multisite research, the IRB must nevertheless consider the individual circumstances of each prison site, which can vary widely. With respect to expedited review, the subcommittee recommended that, if expedited review of a protocol is required, a prisoner representative be one of the reviewers.
Defining Minimal Risk and Benefit to Participant
The subcommittee considered two issues regarding the distinction between using other healthy prisoners as the ethical baseline as opposed to other healthy persons generally. First, the subcommittee affirmed that the different definition of minimal risk in the Subpart C regulations compared with Subpart A regulations was appropriate. The Subpart C regulations specify that the determination of minimal risk must be in comparison to the ordinary experience of a healthy person, interpreted as meaning a healthy person outside the prison environment. The subcommittee cautioned that the greater situational risk in the prison setting should not influence the baseline for the IRB’s decision; rather, the minimal risk should be compared with the risk to a healthy person in a safe environment. The OHRP should provide guidance, using examples, of how the minimal risk might be viewed in different protocols.
At the same time, the subcommittee viewed the current OHRP interpretation of when a protocol does not provide a benefit to the participant as overly restrictive. The OHRP’s position is that using standard of care as a control arm does not provide any benefit to the participant and thus re-
quires secretarial review and expert panel consideration. The subcommittee’s view is that, because the participant receives the standard of care and does ultimately benefit from the results of the research, even if not immediately, such a control arm should not require heightened review. The subcommittee recommended that only when the control group is placebo only (and thus deviating from the standard of care) should the protocol be considered to include an arm not benefiting from the research.
The subcommittee also pointed out the problems with the jurisdiction of Subpart C. Because it has been adopted by so few agencies, it has limited application to federally funded research. In addition, it does not automatically apply to institutions that have signed an FWA unless they specifically request that it be part of their obligation. Because of these two enormous gaps in coverage, most research involving prisoners does not fall under the special protections of Subpart C.
Recommendations for Further Consideration by the IOM
In addition to its recommendations on these issues, the subcommittee noted with approval that the IOM had been charged with studying the human research protections for prisoners. The subcommittee recommended the IOM committee’s consideration of:
-
the need for a requirement that research only be conducted in prisons providing standard of care to the general population (and how best to get such services in place);
-
the interpretation of the requirement that follow-up care be provided when the prisoner has been released from confinement; and
-
the limited jurisdiction of Subpart C (i.e., to DHHS-supported research only).
OTHER FEDERAL HUMAN SUBJECTS PROTECTIONS
The full panoply of DHHS protections for prisoners in Subpart C presently apply only to research funded by the DHHS, the CIA, and the SSA. Some of the other 14 departments and agencies that have adopted the Common Rule accept the OHRP-approved FWA as assurance of compliance with ethical regulations regarding human research subjects. However, those departments and agencies have not adopted Subpart C, so the assurance will only require certification of compliance with the Common Rule (Subpart A).25 Although institutions holding an FWA and engaging in re-
search funded by one of those other departments or agencies may voluntarily extend their protections to include those under the other subparts (including Subpart C), the OHRP estimates that only about 60 percent of institutions holding an FWA have done so.
Moreover, prisoner research that is funded by another department or agency (other than DHHS) falls outside of the protections of OHRP oversight even if the institution has requested in its FWA that Subpart C apply, because the OHRP does not monitor the institution’s compliance with a voluntary assurance regarding Subpart C. Additionally, an organization that does not receive its funding from any of these sources generally will not hold an FWA and would not be required to comply with the Common Rule or any of the subparts.26
In evaluating the effectiveness of Subpart C, it is useful to compare other human research subjects protections to these regulations. In particular, Subpart D contains DHHS’s regulations regarding children, and provides a different framework for assessing the risks and benefits (and according appropriate protections). Within the DHHS, the FDA has promulgated additional human subjects protections regarding research conducted on drugs and medical devices (but has not succeeded in attempting to regulate such research in the prison context). In contrast, the BOP has established a set of regulations regarding all research conducted with the prisoners in its custody.
Subpart D
Recall that there are four categories of permissible research established in Subpart C: (1) study of the possible causes, effects, and processes of incarceration (presenting no more than minimal risk); (2) study of prisons as institutional structures or of prisoners as incarcerated persons (presenting no more than minimal risk); (3) research on conditions particularly affecting prisoners as a class (after consultation with experts and notice in the Federal Register); and (4) research on practices, both innovative and accepted, that have the intent and reasonable probability of improving the health or well-being of the subjects, when there is a control group that is nontherapeutic, after consultation with experts and notice in the Federal Register.27 If biomedical or behavioral research does not fall into one of these categories as described, it is not permitted.28
Subpart D, although similar in some ways to Subpart C, takes a different approach to the definition of categories of permissible research involv-
|
26 |
See Figure 3-1. However, various human subjects protections may still apply, independently of the funding source, as discussed in more detail below. See Table 3-3. |
|
27 |
45 C.F.R. § 46.306(a)(2). |
|
28 |
45 C.F.R. § 46.306(b). |
ing children (IOM, 2004). Specifically, Subpart D gradually increases the protections as the risk-benefit scale tilts more toward risk and, at the top end of the scale, has a case-by-case escape clause for research that is not otherwise approvable but that presents an exceptional opportunity to learn about a problem particularly affecting children.29
As to the risk-benefit analysis, the protection is tailored as shown in Table 3-2.
Thus, the scheme gradually steps up the requirements for approval as the risk increases and the prospect of direct benefit to the individual decreases.
Moreover, this scheme allows for appropriate research that might fall through the cracks under the Subpart C framework. First, IRBs may find Subpart D’s descriptions of categories easier to understand than those of Subpart C because they specify how the risks and benefits are to be analyzed and how the protections should be increased to match. Second, the framework is more flexible in that 45 C.F.R. § 46.407 allows for research that does not neatly fit into one of the previous three categories, if, after expert consultation and public review and comment, the secretary finds it is both sufficiently important and well designed and is conducted in accordance with sound ethical principles.
Overall, the Subpart D framework is a more natural fit with the overarching ethical framework. Rather than determining in advance that certain kinds of research appropriately balance risks and benefits and forbidding all others, as in Subpart C, Subpart D allows the IRB discretion to determine the balance of risk to the individual with the prospect of benefit (direct or indirect) to the individual, requires appropriate assurance of informed and voluntary participation and draws a line when the risk outweighs the benefit to such an extent that it can be presumed that individuals would not consent if their consent was completely voluntary.
Other DHHS Agencies: FDA Regulations
As noted previously, the FDA has adopted a modified form of the Common Rule in 21 C.F.R. Part 50, Subpart A, as well as regulations regarding research with children as subjects in Subpart D. Apart from the FDA’s definition of the scope of its regulations, the differences between these and the Common Rule are, for present purposes, minimal.30 Rather,
TABLE 3-2 Subpart D Framework
|
Risk-Benefit |
IRB Finding/Protection |
|
No greater than minimal risk (§ 46.404)a |
|
|
More than minimal risk but either
|
|
|
More than minimal risk and no prospect of direct benefit but likely to yield generalizable knowledge about the subject’s disorder or condition (§46.406) |
|
|
Research not otherwise approvable but presents an “opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children” (§ 46.407) |
IRB finds:
If the Secretary, after consultation with experts and opportunity for public review and comment, finds:
|
|
aNote that minimal risk is defined in the same manner in Subpart D as in Subpart A (the Common Rule). The definition of minimal risk in Subpart C is different, as noted previously. SOURCE: 45 C.F.R. Part 46, Subpart D. |
|
the FDA regulations on protection for human research subjects are interesting primarily because of their scope and secondarily because of the reason why they do not contain a Subpart C.
Unlike DHHS as a whole, which only enforces its requirements in DHHS-funded research, the FDA has the authority to regulate a broad category of research governing medical treatments and devices regardless of
the source of funding or the FDA’s ability to control the subjects or direct the research. Specifically, the FDA’s regulations apply to “all clinical investigations regulated by the [FDA]31 … as well as clinical investigations that support applications for research or marketing permits for products regulated by the [FDA].”32 Thus, the FDA’s regulations reach all research regarding the application, safety, and effectiveness of any drug, medical device, biological product, nutritional supplement, food or color additive, or other product subject to FDA approval. Moreover, because compliance with the FDA standards is required of all research that will be used to support an effort to gain FDA approval, it is in the interest of the research sponsor who intends to use the research to support an FDA application to use care in developing research protocols that comply with these regulations; supportive research can be rendered worthless because of noncompliance.
Nevertheless, although FDA regulations contain a Subpart D governing children as research subjects that is similar to Subpart D of the DHHS regulations, the FDA regulations do not contain a Subpart C governing research with prisoners as subjects. The FDA developed such regulations and posted them in the Federal Register in 1978, and it adopted a final rule on the regulations applicable to prisoner research in 1980.33 Before the regulations became effective, however, a group of prisoners brought suit in federal court to have these regulations declared invalid as violating the prisoners’ rights to participate in medical research. The FDA decided to delay the effective date of the regulations until five months after the court’s decision in the case, and it ultimately settled the case by declaring the regulations indefinitely suspended. According to the notices posted in the Federal Register, research was being conducted on a small number of persons in a small number of prisons, so it was not worth the FDA’s time and expense to litigate the suit to uphold the validity of the regulations.34 Since then, the indefinitely suspended regulations have been removed from the Code of Federal Regulations, and Subpart C of 21 C.F.R. Part 50 is simply “reserved.”
DOJ Regulations
As a general matter, the DOJ (including its research and development arm, the National Institute of Justice) has adopted the Common Rule at 28 C.F.R. Part 46 (NIJ).35 With respect to research involving prisoners in the custody of the BOP, however, the DOJ was concerned that Subpart C did not adequately address the level of risk in the third category of Subpart C (research on conditions particularly affecting prisoners as a class) and did not fully consider the difficulty in ensuring confidentiality of prisoners’ personal information in the prison environment. For those reasons, as well as to conform the review process to other BOP and local prison procedures, the department developed its own regulations that apply to prisoners in BOP custody. The resulting BOP policy is found in two separate program statements: PS 1070.07 regarding research (BOP, 1999)36 and PS 6031.01 regarding patient care (BOP, 2005).
Addressing the DOJ’s concern about the kinds of research allowed, the BOP regulations forbid nontherapeutic medical research or pharmaceutical trials and cosmetic research. However, they do allow the following:
-
therapeutic medical research, including clinical trials, “that may be warranted for a specific inmate’s diagnosis or treatment” if the research is (1) approved by the prison’s medical director, (2) conducted with prior written consent, and (3) “conducted under conditions approved by the Department of Health and Human Services,” which presumably means that it is conducted in accordance with Subpart C;
-
research regarding disease prevalence, response to accepted therapeutic interventions, behavioral, and other nonmedical research pursuant to the program statement on research. (See Table 3-3.)
Thus, research of medical treatments, including clinical trials of pharmaceuticals and medical devices, is either not permitted or, at least in theory, is conducted in compliance with Subpart C requirements. Research involving federal prisoners is conducted pursuant to regulations other than Subpart C only when it involves behavioral, epidemiological, or other nonmedical research.37 Of course, prisoners in BOP custody account for less than 10 percent of the total incarcerated population, so the remainder of
TABLE 3-3 Summary of Regulations Applicable to Research Even If It Is Not Federally Funded by Any Agency
incarcerated persons (of whom nearly two-thirds are housed in state prisons and over one-third in local jails) are not covered by these regulations.38
The BOP program statement on research contains many protections similar to those in Subparts A and C but with more detail on the subject of confidentiality, some differences in informed consent, and more concern that the research protocol be approved by all levels of prison administration.
Confidentiality
The BOP research program statement specifically provides that personal identifiable information may not be released without the subject’s prior written consent and, in particular, may not be admitted as evidence or used for any other purpose in any judicial, administrative, or legislative proceeding. At the same time, as part of the process of informed consent, the subject must be told that confidentiality may not be guaranteed as to information that the subject intends to commit a crime, harm him- or herself or someone else, or leave a facility in which he or she is incarcerated.
The concern for confidentiality of records also places specific limits on the researcher. Records that contain information that may be traced to an individual generally must not be placed on any electronic retrieval system. Additionally, nonemployee researchers can only have access to BOP records if the information does not identify the individual and will only be used “as a statistical research or reporting record,” unless the information is available under the Freedom of Information Act.
Informed Consent
BOP policies relating to informed and voluntary consent appear to have been tailored to their assessment of the requirements of the prison setting. First, the disclosures that are required for informed consent are modified slightly from the Common Rule requirements. In some cases, the disclosure is required to be more specific. For instance, where the Common Rule requires disclosure of “which procedures are experimental,” the BOP regulations require a somewhat more specific disclosure of “the purpose of each procedure” and further require “identification of the principal investigator(s),” which is not specifically required by the Common Rule. As noted previously, the program statement regarding confidentiality contains an exception for when an inmate threatens to harm him- or herself or others, or to leave the facility. Certain other information that is required
under the Common Rule’s informed consent disclosure requirement, such as alternative treatments and information regarding treatment for research-related injury, is not expressly required to be disclosed under the BOP program statement on research,39 perhaps because such disclosures are less relevant in a setting in which medical treatment is controlled by the institution. Nevertheless, the BOP program statement allows the possibility that other information might be required “as needed to describe adequately the nature and risks of the research.”
More striking on the issue of informed (and voluntary) consent is the BOP program statement’s treatment of incentives for participating in research. Where Subpart C provides a standard against which to measure incentives—that incentives should not be “of such a magnitude that his or her ability to weigh the risks of the research against the value of such advantages in the limited choice environment of the prison is impaired”40— BOP policy simply forbids any incentives for research subjects in BOP custody. However, “soft drinks and snacks to be consumed at the test setting may be offered,” and steps may be taken to avoid prisoners being put at a disadvantage (e.g., because of work schedule) by participating in the research.
Review of Research Protocols
Review under the BOP research program statement reflects further BOP control over the research protocol and review process. The review is conducted at three levels. First, an application meeting the detailed informational requirements set forth in the policy is submitted to the warden, who convenes a local research review board (LRRB).41 The LRRB not only reviews the proposal for compliance with the research policy but also consults with prison operational staff and evaluates the research protocol’s compliance with prison policies. The warden takes the LRRB review, formulates recommendations, and forwards the application to the regional director, who then sends the application to a central IRB, called the Bureau Research Review Board (BRRB). After BRRB review and recommendations, the chief of the Office of Research and Evaluation (ORE), who chairs
the BRRB, sends the proposal to the director of the BOP, who has the final authority to approve or disapprove all research proposals. Finally, the warden has the opportunity to review the project in its final form, consult with the LRRB, and request reconsideration if necessary. Furthermore, each research project is reviewed on an annual basis, and the director has the authority to terminate a project at any time if it violates the policy or “may prove detrimental to the inmate population.”
Perhaps in recognition of the difficulty of convening a fully qualified local IRB, and because the LRRB’s decision is always reviewed by the BRRB, membership requirements for LRRBs are not as strict as in Subpart C and, in fact, need not necessarily meet the requirements of an IRB as set out in Subpart A. As a general rule, the LRRB “is encouraged, but not required” to meet the membership requirements set forth in the Common Rule. However, the program statement specifies that the LRRB must have the chief psychologist at the prison as the chairperson, and representatives of departments that will be involved with the project must serve as consultants to the LRRB. When the facility allows more than one research project per year, it is “specially encouraged” to require membership, including a prisoner’s representative, that would comply with DHHS’s Subpart C.
The BRRB is necessarily an IRB and, as described by the research policy requirements, meets Subpart C requirements. The BRRB is composed of the chief of the ORE, who serves as the chair; at least four other members; and one alternate, who serves in the event of a conflict of interest of a member. A majority cannot be BOP employees, and the membership must include an individual with legal expertise (usually someone from the BOP general counsel’s office) and “a representative for inmates whom the Director determines is able to identify with inmate concerns and evaluate objectively a research proposal’s impact on, and relevance to, inmates and to the correctional process” (who is generally a prison chaplain). The implementation guidelines further specify that the members shall have varying backgrounds, genders, and racial/cultural makeup, shall not be associated with the conduct of the research, and shall include at least one scientist and one nonscientist. Thus, the research program statement’s description of the BRRB meets Subpart C requirements and also sets out a few additional requirements not contained in Subpart C.
Overall, the BOP guidelines are a useful tool for comparison with Subpart C because they govern all research involving prisoners in federal custody, whether or not it is funded by the DOJ (or DHHS). Moreover, they reflect the BOP’s decisions regarding which aspects of Subpart C are not feasible or are unnecessary and which aspects are inadequate, because they either do not provide enough protection for subjects in the prison environment or are not specific enough about what is required.
ANALYSIS
The SACHRP Subpart C Subcommittee requested that the IOM consider the limited reach of the DHHS regulations under the current regime. The committee agrees that the limited reach of the regulations, combined with the patchwork of different regulatory schemes, inhibits the impact of the regulations to the detriment of prisoners involved in research.
Recommendation 3.1 Establish uniform guidelines for all human subjects research involving prisoners. Congress should mandate a uniform set of guidelines for human research participant protection programs42 for research involving prisoners.
All human subjects research involving prisoners should be regulated by the same ethical standards irrespective of the source of funding, the supporting agency, or the type of correctional facility (federal, state, local, or private) or program that houses the prisoner. Under the current system of research regulation, this would mean that all 17 federal agencies that are signatories to the Common Rule, any additional federal agencies, and any nonfederal sponsors of research would be required to comply with a newly drafted subpart C.43 All research involving prisoners, therefore, would be registered with the OHRP. There is no justification for variability across agencies and facilities regarding their approaches to protecting the rights, health, and dignity of prisoners participating in human subjects research, individuals who are among the most vulnerable human subjects of research.
The primary policy forming the basis of the DHHS regulations regarding the protection of human subjects is at 42 U.S.C. § 3515b (Prohibition on Funding Certain Experiments Involving Human Participants):
None of the funds appropriated by this Act or subsequent Departments of Labor, Health and Human Services, and Education, and Related Agencies Appropriations Acts shall be used to pay for any research program or project or any program, project, or course which is of an experimental nature, or any other activity involving human participants, which is determined by the Secretary or a court of competent jurisdiction to present a
danger to the physical, mental, or emotional well-being of a participant or subject of such program, project, or course, without the written, informed consent of each participant or subject, or a participant’s parents or legal guardian, if such participant or subject is under eighteen years of age. The Secretary shall adopt appropriate regulations respecting this section.44
In addition, the Public Health Service Act contains more specific statutes requiring IRB review as a precondition to funding.45 Because the primary statute is explicit about being limited to research funded by these departments, the comprehensive regulations promulgated by the DHHS regarding human participants are, in part, limited to research funded by the DHHS, which the secretary has full and unquestionable authority to regulate.
However, most of the Common Rule was also drafted to apply to research not funded by the DHHS but regulated by the department.46 The catch is that the regulation defines research subject to regulation quite narrowly as “those research activities for which a federal department or agency has specific responsibility for regulating as a research activity (for example, investigational new drug requirements administered by the FDA).”47 Regulation in such a narrow area is likewise on quite solid ground as a natural extension of its authority to regulate the research activity.
The jurisdictional limits of these regulations demonstrate a conservative approach to regulation, limiting the scope of the regulations to those areas where the DHHS’s authority is unquestionable. Two issues remain: whether the DHHS presently has some implicit authority to regulate beyond these two narrow areas (if it were willing to go beyond the specific authorization) and, if not, whether Congress can grant it such authority.
Existing Authority for Broader Regulation
The authority for any regulation promulgated by an executive department such as the DHHS must be traced to a statute authorizing the DHHS to create regulations in that area. In turn, the statutory authority for the executive department to create rules and regulations in a certain subject area must be traced to a specific constitutional authority for the federal government to oversee that area, because the federal government has no general power to regulate. The DHHS authorizing statute is not entirely clear (although it does not foreclose the possibility of regulation), but it is
clear that Congress could, if it so chose, expressly expand DHHS’s authority to regulate human research subjects protection.
The DHHS’s authorizing statute is actually an enactment of the 1953 reorganization plan transmitted by President Eisenhower, transferring the responsibilities of the Federal Security Agency to the newly created DHEW.48 The DHEW was created “to improve the administration of the vital health, education, and social-security functions now being carried on in the Federal Security Agency.”49 The SSA and the Department of Education were originally agencies within the DHEW; the Department of Education was created in 1979 (at which point the department was renamed DHHS), and the SSA was separated from the DHHS in 1994. Thus, the remaining function of the DHHS is dedicated to various health- and safety-related activities.
Can the DHHS Be Granted Broader Authority?
Nevertheless, for reasons similar to why the FDA is constitutionally possible, it is possible for the DHHS to be given authority to regulate research involving human subjects.
The Constitution of the United States
The congressional spending power derives from the federal Constitution, Article I, Section 8, clause 1, which provides in pertinent part:
Section 8. The Congress shall have power to lay and collect taxes, duties, imposts and excises, to pay the debts and provide for the common defense and general welfare of the United States; but all duties, imposts and excises shall be uniform throughout the United States….
The Scope of the Congressional Spending Power
For generations, the “spending power” has provided the legal basis for legislation touching upon myriad subjects. The court has long recognized the broad authority conferred under the spending clause. “[W]hen money is spent to promote the general welfare, the concept of welfare or the opposite is shaped by Congress….” Helvering v. Davis, 301 U.S. 619 (1937).
South Dakota v. Dole, 483 U.S. 203 (1987), remains the primary precedent for the authority of Congress to pass spending clause legislation. In
that case, the Supreme Court upheld a federal statute that reduced federal highway funding to states with a minimum drinking age below 21. The court found that the legislation was sufficiently related to the federal interest in promoting safe highway travel and concluded that the statute did not exceed Congress’s spending power under the U.S. Constitution, Article I, Section 8, clause 1, Id. at 208–209. The Supreme Court affirmed the broad scope of the spending power, though it acknowledged that congressional authority is limited in the following ways:50
The first of these limitations is derived from the language of the Constitution itself: the exercise of the spending power must be in pursuit of “the general welfare.” In considering whether a particular expenditure is intended to serve general public purposes, courts should defer substantially to the judgment of Congress. Second, we have required that if Congress desires to condition the States’ receipt of federal funds, it “must do so unambiguously…., enabl[ing] the States to exercise their choice knowingly, cognizant of the consequences of their participation.” Third, our cases have suggested (without significant elaboration) that conditions on federal grants might be illegitimate if they are unrelated “to the federal interest in particular national projects or programs.” Finally, we have noted that other constitutional provisions may provide an independent bar to the conditional grant of federal funds.
South Dakota v. Dole, 483 U.S. 203, 207–208 (citations and footnote omitted). Considering the legislation at issue—limiting the drinking age—was considered sufficiently related not only to the general welfare and to the states’ receipt of federal highway funds, these “limitations” are evidently not exceedingly rigorous. In sum, it is clear that Congress has well established and broad authority to condition federal funding upon acceptance of specified conditions that, in a general sense, pertain to the purpose of the legislation.51
The spending power is a familiar basis for congressional action. There are numerous examples of legislation—for example, Title VI of the Civil Rights Act, Title IX of the Education Amendments, § 504 of the Rehabilitation Act, and The Religious Land Use and Institutionalized Persons Act of 2000, 42 U.S.C. § 2000cc, et seq. (2000)—all of which contain conditions based on the spending clause power. One particularly pertinent example of spending power legislation is the Prison Rape Elimination Act of 2003 [PREA] (42 U.S.C. § 15601, et. seq.). Section 15605 (d) of PREA provides in pertinent part:
(d) Applications.
(1) In general. To request a grant under this section, the chief executive of a State shall submit an application to the Attorney General at such time, in such manner, and accompanied by such information as the Attorney General may require.
(2) Contents. Each application required by paragraph (1) shall—
(A) include the certification of the chief executive that the State receiving such grant—
(i) has adopted all national prison rape standards that, as of the date on which the application was submitted, have been promulgated under this Act [42 U.S.C. § 15601, et seq.] …
This legislation conditions the release of federal funds upon fairly demanding and relatively intrusive terms, but because of the conditional nature of spending power legislation, it likely would withstand a legal challenge under principles articulated by the U.S. Supreme Court.52
Spending power legislation could easily be used to require compliance with the DHHS regulations in state as well as federal institutions. Every state receives federal assistance for corrections (U.S. Census Bureau, 2006), and that assistance is itself based, at least in part, on Spending Clause legislation.53 Hence, if Congress were so inclined, it could enact legislation (or amend the existing legislation) to require the state agency’s adoption of a regulatory scheme governing the involvement of prisoners in research as a
condition for receiving the federal funds. There are many rationales that would likely be sufficient justification for such legislation. For example, the federal government’s interest in “set[ting] uniform national standards in regulating health and safety,” as articulated in Gonzales v. Oregon, 126 S. Ct. 904, 923 (2006), and in particular, in ensuring reliable results from health sciences research, would be sufficient. This assisted suicide case addressed Congress’s more limited authority to directly impose requirements, but the interest articulated by the Court would be sufficient to impose conditional requirements. Additionally, the PREA was based in part on the federal government’s interest both in ensuring states do not violate prisoners’ federal civil and constitutional rights by their deliberate indifference to the problem of prison rape, and its interest in ensuring the “effectiveness and efficiency” of federally funded research and grant programs relating to health care and other prisoner-related research, which (Congress stated) were compromised (directly and indirectly) by state officials’ failure to monitor and address the problem of prison rape.54 Clearly these rationales would also support legislation requiring states to impose (at a minimum) the ethical limitations on research required by the DHHS as a condition of receiving the same federal funding.
The regulations could only be imposed in this manner if the states did in fact accept the federal funding. However, because the federal government only seeks to require the states to establish the same ethical regulations as those promulgated by the DHHS, and not to spend money on any new programs, the committee is confident that the states would choose simply to enact the DHHS regulations rather than foregoing substantial federal monies to avoid compliance.
Alternatives to Comprehensive Regulation
If a change in the statutory authority for these regulations is not possible (either constitutionally or practically), the DHHS might, at a minimum, work with the FDA (and perhaps the BOP) to develop regulations for research involving prisoners, which the FDA might then consider adopting (either in whole or in a modified form) for all research within its jurisdiction. The result would be uniform regulation by the FDA of all research relating to pharmaceuticals, medical devices, and other products within the FDA’s jurisdiction, both in state and federal prisons, supplemented by the DHHS’s regulation of federally supported research. Although not ideal, this framework may still reduce the amount of patchwork regulation applicable to research involving prisoners and be a step toward uniformity of ethical
standards used in biomedical research in the prison setting. However, the more desirable approach, which the committee recommends (i.e., to establish a uniform set of guidelines for all research involving prisoners), will require congressional action.
REFERENCES
BJS (Bureau of Justice Statistics). 2001. Justice Variable Passthrough Data, 1997. NCJ 190359. Washington, DC: U.S. Department of Justice.
BJS. 2005. Prison and Jail Inmates at Midyear 2004. NCJ 208801. Washington, DC: U.S. Department of Justice.
BOP (Bureau of Prisons). 1999. Program Statement 1070.07. [Online]. Available: http://www.bop.gov/DataSource/execute/dsPolicyLoc [accessed March 27, 2006].
BOP. 2005. Program Statement 6031.01. [Online]. Available: http://www.bop.gov/DataSource/execute/dsPolicyLoc [accessed March 27, 2006].
Gorey J. 2005. Statement of Task: Committee on Ethical Considerations to DHHS Regulations for Protection of Prisoners Involved in Research. Presented at the Institute of Medicine Meeting 4 (October 19, 2005) of the Committee on Ethical Considerations to DHHS Regulations for Protection of Prisoners Involved in Research, Washington, DC.
IOM (Institute of Medicine). 2003. Responsible Research: A Systems Approach to Protecting Research Participants. Washington, DC: The National Academies Press.
IOM. 2004. The Ethical Conduct of Clinical Research Involving Children. Washington, DC: The National Academies Press.
NBAC (National Bioethics Advisory Commission). 2001. Ethical and Policy Issues in Research Involving Human Participants. [Online]. Available: http://www.georgetown.edu/research/nrcbl/nbac/human/overvol1.pdf [access-ed March 27, 2006].
NCPHSBBR (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research). 1976. Report and Recommendations: Research Involving Prisoners. Washington, D.C. U.S. Department of Health, Education, and Welfare, Pub. No. (OS) 76-131.
NIJ (National Institute of Justice). Introduction to Human Subject and Privacy Protections at the National Institute of Justice [Online]. Available: http://www.ojp.usdoj.gov/nij/funding/humansubjects/hs_01.html [accessed April 11, 2006].
PHS (Public Health Service). 1966. Clinical investigations using human subjects. In: Final Report (Supplemental Vol. I). Washington, DC: U.S. Government Printing Office. Pp. 475–476.
SACHRP (Secretary’s Advisory Committee on Human Research Protections). 2005. Report of the Subpart C Subcommittee to SACHRP, April 18, 2005 [Online]. Available: http://www.hhs.gov/ohrp/sachrp/mtgings/present/SubpartC.htm [accessed March 27, 2006].
U.S. Census Bureau. 2006. Federal Aid to States for Fiscal Year 2004. U.S. Government Printing Office, Washington, D.C.




























