
5
Exploring and Controlling the Inner Workings of a Molecule
Can we control the inner workings of a molecule? New 21 st-century tools place us on the verge of the new science of coherent control. We will soon not just observe but also control physical phenomena on all of the timescales relevant to atomic and molecular physics, chemistry, biology, and materials science. This new era of control is enabled by key advances in laser technology, which let us generate light pulses whose shape, intensity, and color can be programmed with unprecedented flexibility. Our ability to control the positions, velocities, and relative spatial orientations of individual atoms and molecules has led to a stunning array of precision measurement technologies and devices based on AMO science, leading to an enormous range of experiments that reveal qualitatively new phenomena. In this section, the committee focuses on the emerging ability to observe the inner workings of atoms and molecules on their natural timescales, and to manipulate them to achieve desired effects. Such new capabilities will allow us to visualize the complex motion of electrons and atomic nuclei through the course of chemical reactions, providing new insight into the mechanisms that determine the reaction rates and products. Accompanying our ability to observe is the ability to control: Lasers can now be used to control the outcome of selected chemical reactions. This capability may ultimately develop into powerful tools for creating new molecules and materials tailored for applications in health care, nanoscience and technology, environmental science, energy, and national security.
WHICH TIMESCALES ARE IMPORTANT?
Key events in our lives—graduations, births, or anniversaries—make us acutely aware of the passage of time. Most of our personal time markers are measured in years. At the same time, cell phones, personal digital assistants, and e-mail place ever-increasing demands on our time—seeming to accelerate the pace of our already fast-paced world, where we struggle to preserve even a free nanosecond. In nature, the important time markers span an even broader range—from the dizzy-
ing attosecond (10−18 s) timescale, the time it takes an electron to orbit an atom, to the timescale of ≈14 billion years, the age of our universe. Fascination with the passage of time is a fundamental aspect of human endeavor, driving us to attempt to understand our world and indeed our universe.
The relevant timescales for the atomic and molecular processes of interest (see Figure 5–1) cover 19 orders of magnitude—a factor of 10 billion billion. The complex folding of a protein molecule can take milliseconds (ms) (10−3 s) or longer. On the other hand, a millisecond is a very long time for an atomic collision, unless the atoms are cooled to billionths of a degree above absolute zero, as discussed in Chapter 2. The small molecules in the air we breathe undergo collisions with each other approximately every 100 picoseconds (ps) (10–10 s), and they tumble or rotate in space approximately every 1 to 10 ps (10−11 to 10−12 s). The atoms within
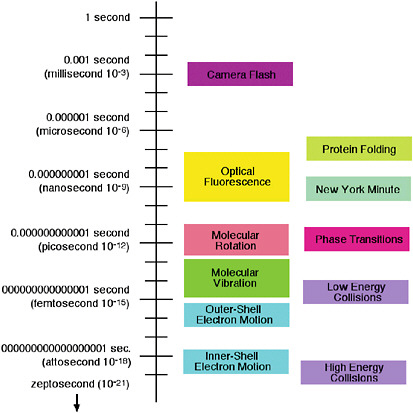
FIGURE 5–1 Characteristic timescales of atomic and molecular motions.
molecules act as if they were balls bound to each other with springs and vibrate with a period of 10 to 1,000 femtoseconds (fs) (10–12 to 10−14 s) depending on the strength of the spring (i.e., the nature of the bond between the atoms). This period also corresponds to the duration of a typical room-temperature collision between two molecules. Still faster are the orbital motions of the slowest-moving electrons of atoms and molecules, which occur on a timescale of approximately 10 attoseconds (as) (10–17 s), while the orbital motions of the fastest electrons of heavy atoms occur in hundreds of zeptoseconds (zs) (10–19 s). Much of what we know about atoms and molecules comes from observing motion, starting with the pioneering direct observation of the Brownian motion of dust colliding with air molecules, explained by Einstein in a famous paper published just a century ago. The frontiers in this field as we approach 2010 are to capture motion not just between molecules, but within them, to observe the basic processes of chemistry and biology on the scale of a single molecule.
MOLECULAR MOVIES
Motion pictures help us dissect and understand fast phenomena. About the time physicists were puzzling over Brownian motion in the late 19th century, Eadweard Muybridge used a sequence of photographic exposures of 1 millisecond to prove that a galloping horse sometimes had all four hooves off the ground (Box 5–1). The basic technique established by Muybridge is still used to capture and slow down motion today and helps to illustrate the challenge of capturing the motion within a molecule. A key ingredient in making any motion picture is the ability to freeze the action by recording images with a shutter speed much faster than the motion of the object of interest. In atomic and molecular motion, as elsewhere in high-speed photography, the mechanical shutter has been replaced by a short pulse of light, which acts as a stroboscope. The picosecond or faster processes within molecules require very short pulses which can only be produced by a laser. The speed of the motion we can freeze is limited by the duration of the laser pulse. Thus, the rotational motion of the molecules in a gas cell can be captured by illuminating the gas with laser pulses that are a fraction of a picosecond in duration, while freezing the vibrational motion requires pulses of a few femtoseconds and freezing the motion of electrons as they move about the molecule requires subfemtosecond, or attosecond, laser pulses. Such motion can be captured with laser technology developed during the last decade.
The earliest direct observations of molecular vibrations were performed using a sequence of two pulses, in which the first pulse excited the molecule and the second pulse was used to probe the resulting molecular response as a function of the time interval between the pulses. This pump-probe approach has made it possible to
|
BOX 5–1 Stopping Time In the experiments that produced the data at the right of Figure 5–1–1, an x-ray pulse of a few at-toseconds in duration is used to knock an electron out of a tightly bound inner orbital of krypton atoms to produce krypton ions. The “hole” in the inner orbital is not stable and is quickly filled by an electron from an outer orbital, with the concurrent ejection of a second electron. How fast does this hole-filling/ electron ejection, or Auger process, take place? To find the answer, a second laser pulse of femtosecond duration is introduced at a variable time delay after the first pulse. This pulse can modify the energy of the Auger electron, but only if the electron is still close to the ion, i.e., in the process of escaping. The figure shows a series of electron spectra recorded as a function of the delay of the second laser. If the probe pulse comes before the first pulse, or very long after it (here, “very long” is only 40 fs), the spectra show no signs of the second pulse: The Auger electron has either not yet been produced (before) or is long gone (after). However, at very short times after the first pulse (~0 to 15 fs), a new feature (highlighted in red) is observed in the spectra. This corresponds to electrons with energies modified by the probe laser and marks the appearance of the Auger electron. The Auger process is found to occur with a half-life of 7.9 fs, perhaps the fastest process ever directly measured.  FIGURE 5–1–1 Left: In the mid-20th century, Harold Edgerton used a strobed flash with a duration of –0.000001 s to film a golf swing by Bobby Jones, allowing countless duffers to reassess their games. SOURCE: Palm Press, © Harold & Ester Edgerton Foundation. Right: At the dawn of the 21st century, attosecond scientists recorded the evolution of the electronic structure of a krypton ion with a time resolution of ~0.000000000000001 s following excitation by a sub-fs x-ray pulse. Such studies may one day unravel the complex correlated electron motion that drives many chemical reactions and determines the properties of novel new materials. SOURCE: F.Krausz, Ludwig Maximilian University, Munich, Germany, and Max Planck Institute of Quantum Optics. |
observe the change in a molecule in solution or in a gas as its atoms stretch apart and contract or break apart to produce fragments. These developments marked the beginnings of the new field of ultrafast chemical dynamics, also known as femtochemistry, and were recognized by the 1999 Nobel prize in chemistry in 1993 (see Figure 5–2). Since this early work, the time resolution of such observations has
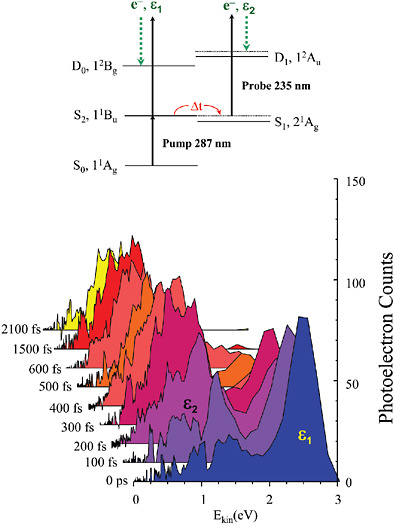
FIGURE 5–2 Time evolution of molecular excited states. Excited states of polyatomic molecules often have a mixed character that results in interesting effects when the molecule absorbs ultraviolet light. Some of these processes can be extremely fast (subattosecond), but others are comparatively slow. This experiment shows the evolution with time of a process that occurs on the picosecond timescale in the excited states of the 24-atom molecule all-trans-2, 4, 6, 8-decatetraene. A femtosecond pulse excites the S2 state, which is mixed with excited vibrational levels of the S1 state. After a variable time delay, a second femtosecond pulse ejects an electron from the excited state, and the kinetic energy of the electron is measured. This energy spectrum provides a fingerprint of the electronic and vibrational character of the intermediate level. As can be seen above, the spectrum recorded for a time delay of 0.6 ps is very different from the spectrum recorded with no delay. This series of spectra indicates that although the electron is initially in the S2 state, it rapidly switches over into the S1 state. SOURCE: A.Stolow, Steacie Institute for Molecular Sciences, Ottawa, Canada.
rapidly advanced from hundreds of femtoseconds to less than 5 femtoseconds, and the increase in the speed of the strobe continues today. At the time of this writing, strobes as short as 100 attoseconds had been reported. On the attosecond timescale, not only the motion of the nuclei of molecules but also the motion of the electrons themselves can be detected. Motion pictures of the electrons will be especially valuable, because electrons form the glue that holds molecules together. Their motion is the fundamental physical basis for chemistry. Why do some atoms bind and others do not? Why do reactions take the time they do, and why do molecules bend one way but not another? Watching the steps in the dance of electrons will provide an enormous wealth of new insight into the mechanisms of chemistry.
THEORETICAL COMPUTATION OF ULTRAFAST MOLECULAR PHYSICS
In this first decade of the 21st century, why is there still a need to do experiments to understand molecular physics? Aren’t the forces of nature responsible for molecular binding and atomic motion well understood? Why can’t the behavior of molecules simply be calculated based on a first-principles understanding of the underlying physics? If engineers can design and build a commercial aircraft using only computers, and without any test flights, then why can’t scientists dispense with experiments? The fact is that the calculation of the chemical dynamics in any but the simplest systems remains an extremely difficult problem. The difficulty is in quantum mechanics itself, which is a far more challenging theory than classical physics. Progress will require both increases in computer power and new theoretical approaches to reduce the size and difficulty of the calculations. Chapter 7 explores the prospects for quantum computers, which may someday permit the simulation of far more complex quantum problems. For now and into the next decade, ultrafast laser experiments and x-ray lasers will lead the way to forge a partnership with theory. One of the most enticing new ways to advance theory through experiments is the emerging field of quantum control.
QUANTUM CONTROL
It is one thing to observe the inner workings of a molecule as it expresses its native behavior; it is quite another thing to exploit what we have learned from these movies to direct or control specific molecular behavior of our own choosing. Such control has been a dream of scientists and engineers since atoms and molecules were identified as the building blocks of matter. It is important not only because it allows us to enhance desired reaction products, minimize by-products, and create new kinds of molecules, but also because of what it can reveal about the fundamental character and behavior of atoms and molecules.
Controlling Chemical Reactions: A Short History
The invention of the laser in the early 1960s raised great hope that this was the tool needed to control molecules. This hope was based on the knowledge that visible and near-ultraviolet laser light can be used to excite the outermost electrons in the molecule into different configurations, which could lead to control of chemical reactivity because these so-called valence electrons determine many of the properties of the molecule. For example, if a vibration could be set up in a specific bond, it was reasoned that bond might be broken selectively, thus achieving mode-selective chemistry. This was truly a revolutionary idea. Synthetic chemistry is more or less like cooking, relying for the most part on heat and on the relative amounts of the ingredients to determine the outcome of a reaction. If a laser could act as a sculpting tool for chemical change, then new chemistry would be possible. Unfortunately, it was soon realized that the coupling among the molecular vibrations was sufficiently strong that before enough energy could be deposited to break a specific bond, much of it had already flowed into different parts of the molecule. Thus, the original attempts at selective bond breaking using lasers were no more effective than simple heating of the molecule, and the laser “scalpel” was behaving more like a laser “blowtorch.” Nevertheless, efforts were rewarded by our new understanding of how energy flowed among the internal degrees of freedom of isolated molecules and led to significant advances in nonlinear dynamics and chaotic systems in physics and chemistry.
Quantum Interference: A Route to Quantum Control
Following these initial attempts, new ideas soon appeared that introduced the use of multiple pathways to enhance the desired process and minimize undesired ones (Box 5–2). According to quantum theory, if there are multiple paths between the starting point and the target for a process, then one cannot tell even in principle which path was taken; the paths are said to be “indistinguishable.” In such cases, the quantum paths can interfere, much like waves interfering on a beach. Constructive interference means that the quantum waves add, and this leads to an enhancement of the process; destructive interference leads to a diminishment of the process. In principle, if there is sufficient control over the light pulse, it is possible to excite molecules via two or more interfering pathways for which the probability amplitudes interfere constructively for the desired process and destructively for the undesired process. New theoretical methods were developed to discover the optimal combination of pathways for controlling a given process, and from these ideas emerged one of the most effective current methods to achieve this goal: pulse shaping.
|
BOX 5–2 Quantum Interference Interference phenomena are familiar in everyday life. For example, when approaching each other, the waves produced by two pebbles thrown in a quiet pond produce an intricate pattern of crests and troughs, with the remarkable property that the height of the wave is higher than that of the individual waves at the crests, and lower at the troughs. The crests and troughs are the result of constructive and destructive interferences, respectively, between the waves produces by the two pebbles. In contrast to pebbles in a pond, which are described extremely well by the laws of classical physics, atoms and molecules are quantum objects governed by the laws of quantum mechanics. Their dynamics is then expressed in terms of “waves of probability amplitudes.” or wavefunctions. The knowledge of these waves is extremely powerful in that it permits the computation of everything of interest about the system under study—for example, the probability of finding a particle at a certain location at a certain time. While these waves are rather abstract objects that are fundamentally different from the waves produced by pebbles, the key point here is that their mathematical properties are nonetheless quite similar; in particular, they too can conspire to produce constructive or destructive interferences. For instance, if an atom or an electron propagates past a screen in which two tiny holes have been drilled, these two holes act much like the pebbles in the pond and result in two pathways with waves of probability that will combine downstream, constructively in some regions and negatively in others. According to the laws of quantum mechanics, there is then zero probability of finding the particle where the waves interfere destructively, and an enhanced probability of finding them where they interfere constructively. Similarly, quantum mechanics predicts that if there is sufficient control over a light pulse, it is possible to excite molecules via two or more interfering pathways for which the probability amplitudes interfere constructively for the desired process and destructively for the undesired process. |
How Do We Shape an Ultrafast Laser Pulse?
Modern quantum control experiments use a single programmable femtosecond pulse to drive the slowest electrons in the molecule to higher energy orbits. Because these electrons control the bonding in the molecule, control of the electron motion is sufficient to direct the motion of the nuclei. Laser pulse shaping is the key tool needed to carry this out. The pulse is shaped using a physical trick based on the time-bandwidth uncertainty relation: The shorter the pulse, the more colors are contained in it. The pulse is therefore first broken down into its different colors, each of which is then modified by passage through a filter and then recombined to form a complex waveform that excites the orbits of interest. A 10-fs pulse, fast though it is, cannot keep up with the electron motion in real time; it can, however, change the electron orbits and thereby alter the forces that govern the motion of the nuclei, determining the outcome of the chemical process.
How do we choose the appropriate pulse shape to achieve a particular chemical reaction? A sufficiently accurate a priori determination of this pulse shape from knowledge of the quantum structure—that is, the orbits—of the molecule is not possible with current theoretical and computational capabilities for most systems. An elegant solution to this problem was provided by turning the problem around and letting the molecule choose the optimum pulse shape for itself. Sophisticated learning algorithms, such as those inspired by biological evolution, are used to optimize the waveform: The sample is exposed to an evolving series of pulse shapes, while the resulting signal of interest is monitored to determine which waveform maximizes the desired outcome (see Figure 5–3). Because the experiments are per-
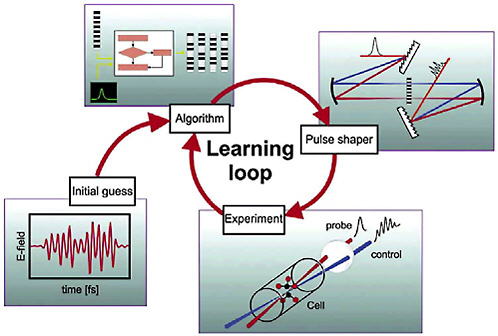
FIGURE 5–3 Letting a molecule solve the problem. A feedback loop can be used to let the experiment choose the optimum shape of the femtosecond laser pulse. In an experiment to optimize the production of a particular product state of a quantum system (e.g., selective fragmentation following the laser-induced breakup of a molecule), the signal from the state is monitored after every laser shot. An algorithm is used to generate a set of initial pulse shapes for the experiment. The femtosecond laser pulse is sent through a pulse shaper to generate the selected waveforms, and these are applied to the quantum system sample. Comparison of the product signal intensities for the different waveforms leads to the selection of a subset of “best” pulses. This subset is then sent through the algorithm again to generate a new, improved set of pulses. The experiment continues around the loop until no further gains can be achieved in the product yield. SOURCE: H.Rabitz, Princeton University, and K.Kompa, Max-Planck-Institut für Quantenoptik.
formed thousands of times per second and the pulse shapes can be modified almost every shot, the molecule can rapidly be exposed to thousands or even millions of different programmed waveforms, and the optimization typically converges on a best pulse shape in seconds to minutes. This learning feedback approach has been used successfully for a number of complex chemical processes, such as maximizing the production of a given quantum state of a molecule, controlling the branching between ionization and dissociation following excitation of a molecule, and controlling the branching between different photofragmentation products. There have even been attempts to control the pathways of energy flow in bacterial lightharvesting proteins.
Quantitative electronic structure and dynamics calculations to predict the optimum laser pulse to control a given process are currently beyond the capabilities of AMO theorists for all but the simplest model systems. Indeed, in general, it is not even a straightforward task for theory to explain why (or if) an experimentally determined pulse is optimal. Such capabilities are ultimately essential to understand quantum control on a fundamental level. In the past decade, tremendous advances in electronic structure theory and computational capabilities have changed the face of molecular physics and chemistry, as recognized by the Nobel prize in chemistry in 1998. Calculations of ground-state equilibrium structures can be exceedingly accurate even for nanoscale systems. In general, however, chemically accurate potential energy surfaces, which describe the energetics of two or more atoms or molecules as they come together in a chemical reaction and rearrange into various products, can only be calculated for systems with a few heavier atoms. Accurate surfaces for excited states of molecules, which are often accessed in quantum control experiments, present even greater challenges. To make matters worse, chemical reactions often involve multiple potential energy surfaces, creating a rapid escalation in the complexity of the problem. Finally, once such surfaces are available, the calculation of the molecular behavior upon them is no simple task.
Nevertheless, theoretical and computational advances in the next decade are expected to lead to major progress on all of these fronts. These advances will not only provide a tremendous boost to the field of quantum control but are also expected to have a broad impact on molecular physics and chemistry and the physical and biological sciences in general.
Aligning Molecules
Selecting the angle of collision between two molecules is yet another way to control chemical reactions. The outcome of a collision depends on the relative orientation and rotation of each collider at the moment of impact. On the molecular scale, the efficiency of chemical reactions and processes like energy transfer
depend on how the molecules are aligned when they come together. Similarly, the interaction of light with molecules depends on the relative orientation of the molecules and the direction of the light’s oscillating electric field. Control of molecular orientation during collisions or interaction with light could provide considerable control over chemical reactions. Controlling the orientation of free molecules is considerably more difficult than controlling the polarization of light. The latter can be done in many ways, including by using Polaroid sunglasses. As molecules in a gas or liquid spin, rotate, and tumble in space, their orientation is constantly changing. Some “polar” molecules, in which one end is slightly positively charged and the other is slightly negatively charged, will tend to line up in an extremely strong DC electric field, but the aligning force is quite weak, and unless the molecules are quite cold—that is, rotating very slowly—to begin with, the applied field will not be sufficient to keep the molecules oriented (see Figure 5–4). New methods for producing polarized ultracold molecules are discussed in Chapter 3.
Recently, a totally different approach to molecular alignment has been developed in which a very short laser pulse is used to kick the molecules. This kick causes each molecule to rotate faster or slower depending on its orientation and its motion at the time of the kick, much like a weathervane spinning after a brief gust of wind. The net effect is that all of the molecules align with each other a short time after the kick, much like setting a clock to 12:00 a.m. aligns all the hands. The molecules rapidly get out of synch as they continue to rotate, just as the clock hands move apart. In the absence of collisions, however, the molecules come back into phase after a known time delay and are again aligned, just as the clock hands become aligned again at 12:00 p.m. Such recurrences of the alignment will continue to occur until collisions or other external effects erase the effects of the initial kick. One advantage of this approach is that there is no laser pulse or external electric field present at the recurrence time, so that the aligned molecules can be studied in their natural state. Collision experiments using aligned samples will provide new
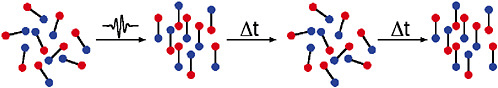
FIGURE 5–4 As described in the text, a random sample of diatomic molecules kicked by a short laser pulse will tend to align themselves right after the pulse. Although this alignment does not distinguish between the two ends of the molecule, methods have been developed to do so. The molecules continue to rotate at different rates and after a few picoseconds dephase into a randomly oriented collection of molecules. However, in the absence of other effects, after a time (tens to hundreds of picoseconds) the molecules will rephase to produce the aligned sample again.
information on the anisotropy of molecular interactions, yielding insight into how reactions actually occur. In addition, the ability to align single molecules will dramatically simplify the analysis of single-shot, and even single-molecule, structure determinations that may become feasible at new x-ray light sources like the Linear Coherent Light Source, currently under construction.
LOOKING TO THE FUTURE: CAN WE SEE AN ELECTRON’S MOTION?
When we peer at an atom or molecule using a femtosecond pulse, the motion of the lighter, rapidly moving electrons is lost in a blur. If we could develop sufficiently short pulses to allow us to strobe the motion of electrons themselves, we would truly enter a new and astonishing realm of science. Two approaches are possible: slowing down the electrons or speeding up the strobes.
Slowing Down the Electrons: Rydberg Electrons
Much insight into the behavior of the electrons comes from the study of a highly excited species known as Rydberg atoms. In these atoms, one or more electrons is excited to a very-high-energy orbit, with just less than the energy necessary for ionization—that is, the energy necessary to escape the influence of the charged core of the atom. This Rydberg electron spends most of its time moving very slowly compared to the electrons in the atomic core: The period of a typical Rydberg electron in an orbit with a radius of ~100 nanometers is 5 picoseconds—200,000 times longer than the 25-as period of an electron in the ground state of a hydrogen atom. Such slow electrons can be filmed in pump-probe experiments using commercially available femtosecond lasers. Studies of Rydberg atoms and molecules have provided enormous insight into the motion of the electrons, and many of their results can be extrapolated to the behavior of the core electrons. For example, time-resolved studies of doubly excited Rydberg atoms—in which two electrons are pumped into highly excited orbits—show how the motions of the two electrons are correlated, as well as how their collisions result in the ejection of an electron or a photon from the atom. Such correlated electron motion drives many of the important processes in chemistry and biology and is often a key factor determining the character of novel complex materials.
SPEEDING UP THE PULSE: ATTOSECOND SCIENCE
The electrons in most atoms and molecules do not move on such a convenient timescale. Yet there is much to be learned from this motion. When sunlight reacts in our skin to help us manufacture vitamin D, how exactly do the electrons permit
the carbon ring to open? Where do the electrons move during this process? Or in chemical physics, can we learn how to make organic solar cells for energy applications? How do x rays damage DNA, and can we learn how to use and control this process for applications in medicine?
Making Attosecond Pulses
To answer questions like these we must find ways to see electron motion directly, and this means that we must shorten the time duration of the strobe. This has led to the birth of “attoscience,” the creation and use of laser pulses less than 1 fs long. To understand this achievement, it helps to realize that green light, for example, has an electric field that oscillates with a period of 1.5 fs. The cycle period becomes shorter as the color moves towards blue, corresponding to higher energy photons. A pulse of light must be longer than one cycle, so attosecond pulses are in the ultraviolet or x-ray region. These attosecond pulses are generated by converting pulses of visible and infrared laser light through a process known as high-harmonic generation, described in Chapter 4. By focusing a high-intensity femtosecond laser into a gas, the laser pulse literally rips the most loosely bound electron from the atom and then smashes it back into the atom. Through this process, a tightly collimated beam of ultraviolet or x-ray radiation is generated. Under the right conditions, these x-ray beams can have durations of a few hundred attoseconds, much shorter then the original laser beams, and can also be directed and laserlike.
Using Attosecond Pulses
Just as important as generating the attosecond pulses are the means to propagate, characterize, and use them productively. New methods and techniques have already been developed to address each of these issues. Box 5–1 showed how these tools have been used together to probe the decay of highly energized atoms of krypton. In the future, we may be able to record the motion of electrons as they undergo transitions between atomic orbitals and even follow the correlated dynamics of multielectron motion in molecules. With attosecond techniques, scientists could capture and manipulate the electrons that serve as the glue that holds atoms and molecules together. This would be a regime where the motion and the interactions of this cloud of electrons swarming around an atom or a molecule can be observed and controlled in real time. To date, simple experiments have observed and manipulated the dynamics of this electronic cloud. In the future, we may be able to follow the correlated motion of entangled multielectron wave packets to understand and to control processes that cannot be accessed using any other type of probe.
HARD PHOTONS AND FAST ELECTRONS
Attosecond science confronts directly the complementarity between photon energy and pulse length: The shorter the pulse, the higher energy the photon must be. In turn, the higher energy the photon, the more it involves interactions with the fastest (innermost) electrons of the atom. Performing new experiments in attosecond physics requires considerable knowledge of the physics of inner-shell phenomena, and one might ask where this information comes from if the timeresolved techniques are only now becoming possible. Much of this information comes from experiments that focus not on time resolution but on energy resolution. For example, experiments can be performed to measure the precise energies of excited states of atoms and molecules and their ions, or to measure the energies and angular distributions of the electrons and other fragments produced when these excited states decay. Experiments can even be performed that determine the spin of the electrons. Synchrotron light sources such as the Advanced Light Source, the Advanced Photon Source, the National Synchrotron Light Source, and the Stanford Synchrotron Radiation Laboratory have enabled such experiments by providing intense sources of far-ultraviolet light and x rays, albeit with relatively long pulse durations. Experiments at these facilities have substantially improved our understanding of complex processes in the inner shells of atoms and molecules and enhanced our ability to characterize materials. This knowledge will continue to grow and is likely to prove invaluable in the development of attosecond science. While the present chapter highlights opportunities in time-domain studies of atoms and molecules, dramatic progress is also being made from the complementary perspective of energy-resolved processes, as described in Box 5–3.
IN REAL LIFE, TIMESCALES OVERLAP
A discussion on the difficulty of quantum calculations for all but the simplest molecules appears earlier in this report. What progress has been made in theory is due to approximations, none more important than the separation of timescales. As was learned above, different types of atomic and molecular motion have characteristic timescales. As a result, it is often possible to separate a complex problem into simplified parts: The fast motion of electrons can be studied independently by assuming that the vibrational and rotational motions of the atoms in a molecule are frozen on the relevant timescales, while slower motions like rotations can be studied by averaging over the much faster electronic and vibrational motions. Such separation of timescales is used as the basis for theoretical approximations that simplify the calculation of molecular properties and dynamics and provide a conceptual basis for understanding the fundamental physics. Nevertheless, many
|
BOX 5–3 Coincidence Measurements In the study of energy-resolved processes, relatively long pulses of light are necessary to define the energies precisely, and such sources of light in the far-ultraviolet and x-ray regions have become available at synchrotron radiation facilities or using harmonics of high-resolution lasers. Because these sources are so much more intense than previously available sources, experiments that were once only a dream have now become routine. Perhaps the most revealing experiments employing these intense tunable light sources are enabled by the concurrent development of new imaging detectors and coincidence techniques, which allow the simultaneous measurement of multiple parameters associated with the fragmentation of the target atoms and molecules. For example, the absorption of a single x-ray photon by an atom often results in the ejection of two or more electrons. The measurement of the energies of these electrons, along with the angular distribution of their velocities with respect to each other and the polarization of the light, can provide enormous insight into the physics of this process, particularly when determined as a function of the photon energy. Inner-shell absorption in molecules can also produce multiply charged ions, and these often fragment into two or more ions. In a diatomic molecule undergoing rapid dissociation, the detection of an ion at a particular angle serves to fix the molecular axis at the moment of ionization, and the coincident detection of the photoelectron energy and direction provides the photoelectron angular distribution in the molecular reference frame (Figure 5–3–1). Such experiments provide an alternative to laser-alignment techniques for recording the spectra of fixed-inspace molecules. These multiply differential measurements, in which the ionization and dissociation dynamics are measured over many dimensions of parameter space, are often essential in differentiating among several possible decay mechanisms and have produced a new appreciation of the limitations of standard theoretical approximations, as well as an improved understanding of correlated electron motion and its role in ionization and dissociation mechanisms. |
important phenomena occur when these approximations break down, providing a great challenge for theorists to develop new approaches and approximations valid in these new regimes.
The separation of timescales for molecular vibration and electron motion results from the vastly different masses of the atomic nuclei and the electrons: The heavier nuclei generally move much more slowly than the lighter electrons. Shortly after the birth of quantum mechanics, this realization led to the development of the Born-Oppenheimer approximation, in which it is assumed that the electron motion within a molecule can follow instantaneously any changes in the positions of the nuclei. This approximation allows calculating the potential energy of the molecule by freezing the geometry and solving the electronic problem.
Important situations arise, however, when the usual separation of timescales breaks down and the electronic and vibrational motion cannot be treated independently. One way in which the Born-Oppenheimer approximation can break down is if the potential energy surfaces of two different electronic states of the molecule cross—that is, if they have the same energy for some geometry of the molecule. If the two states interact, very small changes in the geometry of the molecule can produce large changes in the motion of the electrons (i.e., the electronic structure) as the molecule flips between states. As a result, the electrons cannot instantaneously adapt to the vibrational motion, and the Born-Oppenheimer approximation breaks down. The character and behavior of the molecule can change dramatically as it switches from one surface to the other. In polyatomic molecules, the intersection of
multidimensional potential energy surfaces can lead to intriguing structures such as conical intersections, which play a critical role in how energy deposited into an isolated molecule is redistributed among the electronic and vibrational motions of the molecule and, ultimately, in how the molecule reacts or decomposes. For example, it was only when these nonadiabatic interactions were carefully taken into consideration that one of the simplest chemical reactions—the dissociative recombination of H3+ with an electron—could be properly modeled theoretically. Indeed, nonadiabatic transitions play an important role in many fundamental processes in physics, chemistry, and biology, including photochemistry and nonradiative relaxation, charge transfer and photosynthesis, and solvent caging effects in liquids. There are some environments in which nonadiabatic interactions are inescapable. For example, for particles a few nanometers in size, the electronic-level spacings and vibrational energy spacings are comparable, suggesting that nanoscience will abound with nonadiabatic effects.
Here the ability to sculpt attosecond pulses and control the electronic motion within molecules could prove invaluable. The ability to control the electronic wavefunction will allow scientists to drive molecules through such surface crossings along different trajectories and follow their outcome, providing the means to map out the detailed character of the potential energy surfaces and the electron motion near the crossing and to elucidate mechanisms for the nonadiabatic processes. In such experiments, the motion of the nuclei could be monitored using ultrafast, high-energy x rays, such as those that will be produced at XFELs, discussed in Chapter 4. The advantage of using such x rays is that they excite the innermost electrons of the atoms, which provide the most precise definition of the atomic positions. Together, such capabilities will significantly advance the experimental study of reaction dynamics in molecular physics, chemistry, and biology and provide important clues to understand these dynamics.
Controlling the Ultimate in Timescales
The shortest pulses and most intense electromagnetic fields that have been applied to atoms and molecules are not generated by lasers at all. Rather, they have been obtained by accelerating charged particles to extremely high velocities and crashing them through a sample of the target atoms or molecules. In many ways, the effect of the sharp pulse of charge passing through the sample is similar to an extremely short laser pulse. For decades, the field of atomic collision physics has yielded insights into the response of atoms and molecules to short pulses of electromagnetic radiation. The field of atomic collision physics is sufficiently mature, and its record of accomplishments is so well established in the literature and by applications, that it is largely left to stand on its own powerful historical record
in this report. The generation of high harmonics from short laser pulses is itself a collision phenomenon, involving the recollision of an electron with the parent ion left by the initial laser ionization.
Advances in laser technology allow us to ask new questions about collision physics. Can the element of control that the laser offers be extended to, and united with, this alternative approach to the production of ultrafast pulses? More specifically, can we control the outcome of an impulsive collisional encounter by imposing on the interaction region a laser pulse of sufficient strength and appropriate timing? With the current availability of laser pulses with field strengths that begin to approach those provided in collisions, we can alter the time-dependent fields felt by the colliding components in such a way that the motion of the electrons is changed. For example, theoretical calculations indicate that the likelihood of the transfer of an electron from one colliding atom to another in a moderate-velocity collision can be enhanced by an order of magnitude by applying to the target atom electric fields much weaker than the electric field provided by the atom’s collision partner. The corresponding experiments, while challenging to carry out, are under way in several laboratories around the world. Electron transfer in the interaction between colliding systems plays a key role in a wide range of chemical and biological reactions, and the ability to control this process could have far-reaching consequences. Extensions of this conceptual approach include pump-probe experiments in which a laserprepared (excited, aligned) target is bombarded by a fast ion, as well as heavyparticle diffraction images of molecules and thin solids using picosecond pulses of high-energy, heavy-particle beams.
PROBING TIME-DEPENDENT MOLECULAR STRUCTURE WITH ELECTRONS
Imaging the nuclei in atoms and molecules with matter waves of subatomic particles has its roots in the earliest days of quantum mechanics. Electron microscopes are now used to examine everything from nanostructures in cells to molecules on surfaces, to the inner structure of nuclei and baryons. Until now, however, the pictures have been long-exposure stills, not action-packed movies. Rapid developments of the last decade have added a new element to the mix: time-resolved electron diffraction with pulses only a few hundred femtoseconds in duration. By using femtosecond lasers to generate short electron bursts and high acceleration voltages and small currents to minimize the spreading of the electron bunches, time-resolved electron diffraction can now be used to make molecular movies with a time resolution as short as 500 fs. Beautiful pictures of the evolution of melting in solids and the twisting or isomerization of large molecules have been
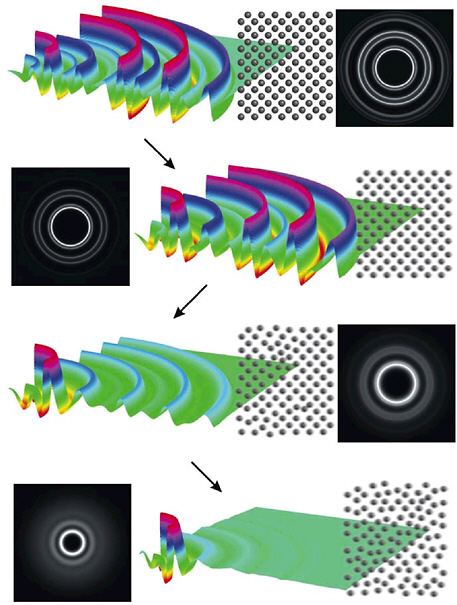
FIGURE 5–5 Ultrafast electron diffraction images, after Fourier filtering high-frequency noise, showing the melting of aluminum, captured in several stages only a few picoseconds apart. Each line shows the diffraction data and the reconstructed image of the aluminum lattice at a particular time as the aluminum melts. Femtosecond time resolution (600 fs) enabled the experiment to determine that the phase transition is thermally driven homogeneous nucleation as opposed to nonthermal electronic mechanisms. SOURCE: R.J.Dwayne Miller, B.J.Siwich, University of Toronto, Canada.
recorded (see Figure 5–5). The faster motions of typical molecular vibrations are still a blur, but the push to shorter timescales is on.
An In Situ Approach to Ultrafast Electron Scattering
In the time-resolved diffraction experiments discussed above, the source of the electron bunches and the sample to be studied are spatially separated. One way to improve the time resolution of the diffraction experiment is to probe the molecule with its own electrons (see Figure 5–6). Here, the ability to control the electronic wavefunction will be particularly valuable. As in the case of atoms, if an intense femtosecond laser is focused onto a molecule, an electron is ripped off the molecule and then slammed back into it. The electron wave packet becomes a short-wavelength replica of the longer-wavelength photon packet and becomes the probe with which the atom or molecule is interrogated. For example, when the returning
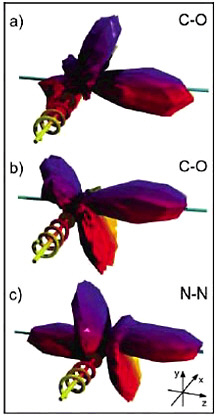
FIGURE 5–6 Molecular tomography. Illustration of an electron in molecular nitrogen as it experiences strong field ionization (top left) and recollision (bottom left) under the driving force of an intense laser beam. The VUV radiation produced in the recombination of the electron with its parent molecule is collected and analyzed to produce a snapshot image of the molecule (top right), which can be compared to a calculation (bottom right). SOURCE: D.Villeneuve, National Research Council of Canada.
electron is captured by the parent ion, the high harmonic light so generated can provide information on the electronic structure of the molecule (see Figure 5–6). In addition, the rescattering electron itself can be diffracted by the molecule and thus also carries detailed information on the molecular structure. Because the recollision is nearly instantaneous—the time interval between each ionization and recollision event is only a fraction of a cycle of visible light, or approximately 3 fs—the electron diffracts from a relatively unchanged molecule, since the positions of the atomic nuclei cannot change appreciably on this timescale. Can illuminating a molecule with its own electron be useful as a probe of the dynamics of the molecule itself? Absolutely! Because the electron wave packet is emitted from, and immediately refocused onto, the molecule to be interrogated, there is little time for the electron wave packet to spread. As a result, the effective intensity of the returning electron wave packet is immense (easily exceeding 100 billion A/cm2). No macroscopic source of electron beams can approach this value. Thus far, theoretical studies of the self-electron-diffraction technique have shown that it is possible to study the motion of the nuclei in diatomic molecules. The future will surely see studies on more complex molecules, and even on molecular reactions. Furthermore, by shaping the laser pulse in time, it will be possible to sculpt the emitted electron wave packet in both time and space to optimize the characteristics of the returning wave. Such advances in the control of electron motion could lead to a new age of electron diffraction and significantly enhance efforts to characterize the structure of complex molecules.
THE FUTURE
As our sophistication with these new experiments grows and new techniques emerge, important questions will arise regarding the use of shaped pulses as reagents in chemical and other processes. These questions will involve the speed and uniqueness of the optimization techniques, the degree of control that is achievable, and the cost of the overall approach relative to other methods. Big questions also remain at the fundamental level. For example, we must ask why a particular pulse shape is optimal. One approach to this question is to take the optimized pulse apart and, through a combination of more traditional pump-probe experiments and detailed quantum chemical calculations, to reconstruct the effect of the pulse on the molecule in a series of steps to see how the result is achieved. While such reverse engineering can help elucidate the control mechanism, it is clear that new insight and modes of understanding are also necessary to make the most of the results.
How fast can we make physical devices? Not faster than the atoms and molecules themselves operate, but perhaps just as fast? It is thus not difficult to see the importance of continuing to press our ability to observe and control matter to this timescale. Is it a stretch to say that in a decade of two we will see computer cycle
times approaching the timescales of electrons in individual molecules? Or that designer molecules for health care will be created and reproduced using molecule-milling machines in which every atom is placed individually? Or that combustion reactions will be made entirely by-product-free by sending the reactants through aligning and exciting laser-preparation instruments on their way to the burn? Perhaps, but then again much of the technology we take for granted today would have seemed completely out of reach 20 years ago. At the heart of this progress in technology is the constant search for faster observation and control. We stand at the threshold of the age when the timescale at hand is in some sense ultimate, namely that of the atoms and molecules which themselves make up matter. Control at this timescale is certain to launch truly revolutionary technology.






















