
2
A New Light on Biology
At the dawn of the 21st century, scientific understanding of microbes is uneven—sophisticated in some ways, primitive in others. Decades of genetic, molecular, and biochemical dissection of microbial life have revealed the detailed structure and inner workings of several bacteria and archaea. Although there is much more to learn even about model organisms, such as E. coli, many individual pathways for nutrient cycling, gene regulation, and reproduction are understood at a satisfying level of precision. But these processes in the majority of microbes remain unknown and knowledge of the evolution and ecology of microbial communities lags far behind cellular microbiology. Basic ideas that organize biologists’ understanding of the living world may need refinement in the face of greater understanding of community function. New concepts of genomes, species, evolution, and ecosystem robustness will have effects beyond the specific field of microbiology. The questions that must be asked are “deep” ones, but answers will in all cases inform and guide the work of putting increased knowledge of microbial communities to practical use. This chapter focuses on some of the more fundamental questions raised by the study of microbial communities that can be addressed through metagenomics.
WHAT IS A GENOME?
When the first microbial genome sequence was completed in 1995, informed opinion was that a few dozen more genomes, chosen to be appropriately diverse, would exhaust the range of variability in how genes could be assembled to make microbes. But as the number of fully sequenced
genomes approaches 500, there seems to be no end to the ways in which genes can be arranged—on linear chromosomes or circular, on one or many, tightly compacted or (in many eukaryotic microbes) separated by “junk” DNA 10 times their length. The number of genes in the genome of a free-living bacterium ranges from 500 to 10,000 or more; the largest bacterial genomes are more than twice the size of the smallest eukaryotic genomes. In contrast, the genomes of many parasitic or symbiotic microbes are highly reduced, with not nearly enough genes to support them independently of their hosts.
Even within a single clonal culture established from a single cell, there will probably be multiple forms of the genome. Many bacteria, especially pathogens, have elaborate mechanisms for rearranging their genes. The mechanisms serve as mutational switches, ensuring that as the microbe’s environment changes due to shifts in chemical, physical, or biological conditions, there will be variants in the population that can flourish. For example, no matter what defenses a host’s immune system mounts against the pathogen, there will be some resistant variants in the pathogen’s population. Variability is also achieved by exchange between genomes: recombination (similar to the genetic exchange that occurs in sexual reproduction) constantly reshuffles the variants (alleles) of genes in the population, generating new adaptive combinations. Plasmids, small and often self-transmissible packets of genes that encode environmentally relevant functions, are rife.
It is, however, the pervasiveness of lateral gene transfer between species that most profoundly challenges the notion that a single bacterial species has a single genome. Several natural processes—transport by viruses, bacterial “mating,” and the direct uptake of DNA from the environment—carry genetic information from one species to another. These processes are regulated and evolutionarily preserved; they are turned on when they are most likely to result in gene transfer, and genes that must function together are often transferred together, forming genomic “islands” (pathogenicity islands, symbiosis islands, or biodegradation islands). Genomic plasticity is an evolved strategy. No single sequence can be said to be the genome sequence of the bacterial species Escherichia coli. And the variations are decidedly not like the trivial differences that account for much of the 0.1% sequence variation among humans. When genomes of multiple strains of the same species (like E. coli K12, O157:H7, and another dozen now available) are compared, they differ up to 25% in genome size and in the number and kinds of genes they carry. Indeed, the genes that are shared by all sequenced E. coli strains amount to less than 40% of the genes present in the species as a whole (see Figure 2-1). Microbial genomicists have started to think in terms of microbial “species genomes” or pangenomes, which comprise a core of genes shared by all strains of a species and a library,
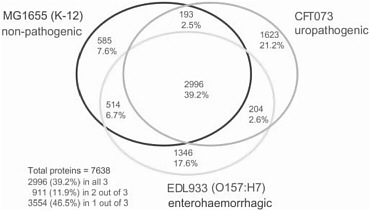
FIGURE 2-1 A Venn diagram showing strain-specific and shared genes for the genomes of three E. coli strains. SOURCE: Welch et al. (2002) PNAS 99: 17020-24. Copyright 2002 National Academy of Sciences, U.S.A.
perhaps much larger, of auxiliary genes that are in some members of the species but not all (Fraser-Liggett 2005).
Probing the extent of genomic diversity is an enormous task best carried out by metagenomic approaches. With suitable experimental and computational methods, environmental gene sequences can be binned (statistically grouped) into provisional pangenomes on the basis of compositional characteristics and site of recovery. As more data accumulate, the definition of what constitutes a microbial genome will be better informed and underlying principles governing genomic plasticity in microbes may emerge. If having a more flexible and dynamic genome structure is a fundamental life-strategy difference between bacteria and archaea, on the one hand, and eukaryotes, on the other, what are its advantages and limits? Can understanding the phenomenon help to explain the emergence of multicellular organisms that have more fixed genomes?
WHAT IS A SPECIES?
In many eukaryotes (especially animals and higher plants), a species contains individuals that can breed and produce fertile offspring together. There is no equivalent definition for bacteria and archaea, because they usually reproduce by binary fission, which does not require sexual compatibility. Moreover, as discussed above, their sexual lives are not limited by relatedness: bacteria and archaea transfer DNA to organisms that are distantly related, even in different phyla, thereby providing no indication of an orderly classification. Traditional bacterial classification has (partly for
that reason) been based on cell appearance, motility, and physiology. The field of bacteriology developed around these classification methods, and most of the names used for common bacteria today are relics of that system. For many purposes, such traditional classification remains useful. But its connection to genomic information is problematic: as discussed above, the very nature of the microbial genome—a fluid entity subject to invasion by large segments of alien DNA—is the problem.
Although molecular methods, in particular the use of rRNA gene sequences (see Box 1-4, page 26), have transformed bacterial classification in the field and in the clinical laboratory, they have not provided entirely satisfying or conclusive answers. Bacteria or archaea that carry similar or even identical rRNA genes can have deeply diverging genomic structure and content because of horizontal gene transfer. Conventions that enable a standard for assigning species names have been established, such as the rule that no examples of the same species should vary in their 16S rRNA gene sequence by more than 3% (Gevers et al. 2005). Those conventions are convenient and informative, but they are controversial, conceptually ungrounded, and thus somewhat arbitrary. Some “species” defined by the above convention contain highly similar members, for example, and others exhibit remarkable differences in gene content and important features of phenotype (see Box 2-1).
Such concerns are not purely academic. What does it mean, for example, for regulatory standards to require that products be free of particular species (for example, E. coli in food products) if the definition of species is uncertain? Medical diagnosis of an infectious disease usually requires determining the species of the pathogen. How closely related to a particular pathogen does an organism need to be to be considered that pathogen? Registering products that contain live microbes, such as those used to control pests and pathogens of crops, requires naming the organisms in the product. How can entities that cannot be clearly defined be dealt with in patent applications? What if the biocontrol agent is in the same species as a human pathogen but does not appear to be pathogenic? On what basis can the organisms be deemed sufficiently safe (see Box 2-1)? How can microbial evidence be used effectively in a court proceeding or policy decision if the microbes cannot be fully and precisely identified and linked to a source with confidence and certainty?
Metagenomics will steer microbiology closer to a more realistic, flexible, and predictive classification scheme and a more rational (if possibly more pluralistic) species concept. The power of such an approach is that it will be predicated on a far more extensive dataset than the one that has informed past attempts at classification and will make use of new computational strategies to cluster and split groups of organisms in ways not predictable with today’s limited information. The definition of species is
|
BOX 2-1 What’s in a Name? Names carry important legal and regulatory implications. A glaring example is the “Bacillus cereus group,” which contains B. cereus, B. thuringiensis, and B. anthracis (Priest et al. 2004). The first species in the group contains strains that induce food poisoning, strains that prevent plant disease, and others that produce unusual antibiotics. Some B. cereus strains can perform more than one of these activities. B. thuringiensis is the most widely used bioinsecticide in the world; it produces crystal proteins that are toxic to certain insect pests. Some B. thuringiensis strains also produce the toxins associated with food poisoning in humans caused by B. cereus, but this was not recognized when B. thuringiensis was first registered for use in 1961. B. anthracis is the causal agent of anthrax, a disease deadly to both cattle and people. Modern phylogeny and genomics indicate that the three species probably make up a single species with a few dramatic phenotypic differences due to a small number of genes. If we call them all by the same name, how will regulatory agencies, the courts, and the public respond to the idea of spraying trees and crops with an organism that has the same name as the anthrax pathogen? If the similarities of the species had been recognized earlier, might the production of the human enterotoxins by B. thuringiensis have been recognized sooner and prevented registration of this bacterium? What we call bacteria does make a difference. |
less important than intelligent and flexible application of species concepts so that estimates of species richness or organisms’ names imply a similar degree of relatedness across groups and can be of genuine utility in the development of ecological theory and environmental applications.
WHAT IS THE ROLE OF MICROBES IN MAINTAINING THE HEALTH OF THEIR HOSTS?
Most multicellular organisms have a closely associated microbial community that provides a variety of functions, from digestion to defense against pathogens. All plants and animals, including humans, can be considered superorganisms composed of many species—animal, bacterial, archaeal, and viral. Historically, the study of physiology has not focused on these host-associated microbial communities; metagenomics offers an opportunity to understand their physiological role and evolutionary significance.
Using the human as an example, the human “metagenome” might be considered an amalgamation of the genes contained in the Homo sapiens genome and in the microbial communities that colonize the body inside
and out. The organisms within these communities are collectively known as the human “microbiome.” The metagenome of these communities encodes physiological traits that humans have not had to evolve, including the ability to harvest nutrients and energy from food that would otherwise be lost because we lack the necessary digestive enzymes (see Figure 2-2). Recent studies suggest that the gut microbiome may play a role in obesity (Turnbaugh et al. 2006). Without understanding the inhabitants of the human microbiome and the mutualistic human-microbial interactions that it supports, our portrait of human biology will remain incomplete.
Metagenomics will enable us to address a number of fundamental questions about ourselves. Is there an identifiable core microbiome shared by all humans? How is each individual’s microbiome selected? What is the role of host genotype? Should differences in each individual’s microbiome be viewed, with the immune and nervous systems, as features of our biology that are profoundly affected by individual environmental exposures? How is the human microbiome evolving (within and between individuals) over different time scales as a function of changing diets, lifestyle, and biosphere? What are the functional correlates of diversity in the membership of a microbiome, and how does this diversity affect the robustness of a community and the host’s ability to respond to various physiological or pathophysiological states? How redundant or how modular are the contributions of individual microbial constituents to community function and to host biology? How should such constituents be defined given that mutualists, like pathogens, do not have a single genomic structure but rather have pangenomes with various degrees of openness to acquisition of genes from other microbes? How can this knowledge be used to manipulate microbial communities to optimize their performance in a person or in a population? Most obviously, how does the microbiome affect health, and vice versa? When we know more, previously unrecognized microbial involvement with disease states will be uncovered. Many host physiological states with primary genetic or biochemical causation will affect the microbiome in ways that may aid in diagnosis. Of course, these questions do not apply only to humans—study of host-associated microbial communities will contribute to understanding of the physiology of all organisms.
HOW DIVERSE IS LIFE?
“How many?” is a fundamentally human question. How many people are there on Earth? How many grains of sand on the beach? How many planets in the universe? Defining the dimensions and limits of an entity is often the first quest of scientific discovery. But as suggested above, the question “How many species of bacteria are there on Earth?” is far from simple. Metagenomics is likely to contribute to a more flexible and useful
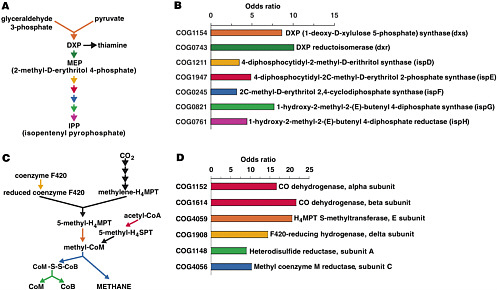
FIGURE 2-2 Some of the metabolism on our own distal gut (isoprenoid synthesis and methane formation) that is the responsibility of genes encoded in the genomes of our microbiota. Results of the metagenomic survey of Gill et al. “Odds ratios” indicate extent to which genes for the indicated biochemical reaction are over-represented in the gut microbiota. SOURCE: Gill et al. (2006) Science 312: 1355-9. Reprinted with permission from AAAS.
definition of microbial species, and no matter how microbial “species” are defined, metagenomics will aid in describing the extent of microbial diversity. In some cases, it may be important to know how many different species—however defined—are present. For other purposes, it may be that what is important is the overall genetic content of an environment, not the number of species it contains. The degree to which genetic diversity and species composition affect the capabilities and stability of a microbial community is another fundamental conceptual question to which metagenomics can contribute.
Soil, for example, is estimated to contain a few hundred species per gram on the basis of culturing, a few thousand per gram on the basis of 16S rRNA gene sequencing and mathematical modeling, and a few million per gram on the basis of DNA-DNA reassociation kinetics and kinetic modeling (Schloss and Handelsman 2006). Although with current tools and knowledge the number of “species” in soil cannot be counted with any confidence, with molecular methods soil’s complexity can be compared with that of other habitats at the gene level.
Molecular and, in particular, high-throughput metagenomic methods that sample all classes of gene, not just phylogenetic markers like 16S rRNA, will guide many aspects of basic and applied microbiology (see Figure 2-3). They will inform the design of experiments that are directed toward capturing or describing diversity. For example, knowing the extent of diversity in a particular habitat will aid in estimating sample sizes required to draw robust conclusions, and knowing the diversity in a biological grouping may determine the choice of habitat for particular types of study. Searches for antibiotics might focus on environments that contain a high diversity of Actinobacteria, the phylum that has yielded the most antibiotic-producing cultured organisms. Metagenomics will provide information about microbial diversity that is intrinsically linked to information about functional attributes of members of microbial communities and that will aid researchers in making strategic choices.
At a more conceptual level, metagenomics will enable us to begin to explore the reasons for the observed genetic diversity. Do communities with extensive genetic diversity also have more functional diversity? Do they respond differently to environmental change? Does genetic diversity correlate with environmental stability or resource availability, or is it a matter of chance and history? Such questions will be addressable through metagenomic approaches.
HOW DO MICROBIAL COMMUNITIES WORK?
Generally speaking, biological community interactions are as important to evolutionary and ecological processes as is the surrounding physical and
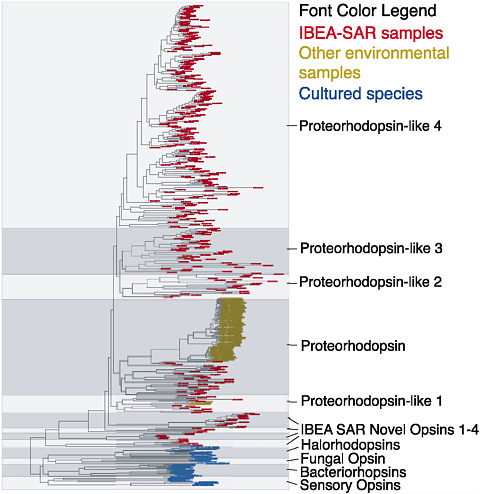
FIGURE 2-3 Diversity of proteorhodopsin sequences in the Sargasso Sea. Proteorhodopsin, a light-driven proton pump, is encoded in many bacterial genomes. Proteorhodopsin genes show a distribution characteristic of lateral gene transfer. As with many other genes, environmental surveys reveal vast and hitherto unexpected numbers and variations of gene sequences. The Sargasso Sea project revealed not only many new gene sequences but whole new classes of sequences. Only those indicated in blue were previously known from cultured organisms. SOURCE: Venter et al. (2004) Science 304: 66-74. Reprinted with permission from AAAS.
chemical environment, and community interactions can shape the properties of the surrounding environment as much as the environment shapes the community. A good example is the influence of microbial communities on the oxidation-reduction potential in their surrounding environment (producing anoxic conditions in sediments, for instance), which in turn shapes the spatial organization of the associated communities. Similarly, community interactions mold biological properties. Lateral gene transfer, cell-cell communication, metabolic complementarity, trophic interactions, interspecies competition and predation, and biogeochemical cycling are all results of community processes.
Macroscopic plant and animal communities have been studied for a long time, but parallel description of natural microbial communities has been more challenging. The typical approach has been to dissect microbial communities into their various components, often by isolating individual microbial strains in pure culture. Even if they were readily cultivated, it is impossible with standard cultivation methods to characterize the hundreds and thousands of microbial strains that make up any single community. Furthermore, the vast intraspecies diversity and variability typically seen in natural microbial populations is usually not examined with most culture-based approaches, which focus on clonal populations. And microbial interactions that in part define community structure and functional relevance (competition, predation, lateral gene exchange, metabolic complementation and syntrophy, and allelopathy) are not readily modeled in most laboratory settings.
Metagenomics promises a new view of microbial-community genomic structure, functional properties, and potential interactions. By mitigating many analytical constraints and using high-resolution community-wide genomic information, we can describe the composition, function, and emergent properties of integrated microbial communities more accurately. The effects of microbial community activities span enormous ranges of time and space, from nanoscale molecular interactions to global-scale biogeochemical cycles. Metagenomic data provide a foundational microbial-community database from which the properties and dynamics of biological organization at a variety of levels (genes, genomes, proteins, metabolic pathways, cells, organisms, populations, and communities) can be inferred. For example, metagenomic datasets provide information about the structure, type, and organization of genes in individual genomes, about within-population allelic variability, and about the patterns of organismal and gene occurrence. Reading metagenomic information may make it possible to infer emergent properties and dynamics of interacting genomes and the relationship of the interactions to the functionality of natural microbial ecosystems.
As metagenomic techniques begin to be applied in natural settings, a
number of fundamental questions about microbial communities can be better addressed. For example, there is evidence that microbial communities may self-assemble in nonrandom ways (Crump and Hobbie 2005; Fuhrman et al. 2006). What are the genetic and physiological drivers? If genetic instructions in part encode species interactions and community assembly, what are the “assembly rules”? Do founder effects influence the nature of spatially structured microbial ecosystems? What are the differences in community organization and interspecies interactions in biofilms vs planktonic communities? Is substantial functional redundancy built into all microbial communities? Does intrapopulation allelic variation have functional significance? All these questions—whose answers have important consequences for understanding evolutionary, ecological, and environmental processes— can potentially be addressed by metagenomics.
HOW DO MICROBIAL COMMUNITIES REACT TO CHANGE?
Robustness is defined as resistance to and recovery from change. It has implications for fundamental understanding of communities and for their management to bring about beneficial outcomes. Many communities maintain their structures across space and time despite continually changing biological and physical conditions, whereas others are more easily destabilized by external disturbance, introduction of new members, or internal processes. Little is known, however, about the basis of robustness or vulnerability to change. Communities are dynamic assemblages governed by dependence and antagonisms among the members, so robustness is likely to depend, in part, on interactions among the members. But this is surmised, not evidence-based; in few communities are the factors that influence robustness known (see Box 2-2).
Community robustness is critical for stability of natural ecosystems. When communities are vulnerable to change, vagaries in weather, seismic activity, or human activity can lead to collapse of a community or the ecosystem in which it resides. The practical implications of community vulnerability are enormous. Managed communities that perform services for humans, such as those in soil or sewage sludge, need to be predictable and steady in their behavior (see Box 2-3). Agricultural productivity depends on the soil community’s protecting plants from disease, transforming minerals, and decomposing organic matter. Similarly, human health is shaped by the robustness of the microbial shield that prevents pathogen invasion of the skin, mouth, and gut.
Metagenomics will add tremendously to our currently limited understanding of robustness by providing large datasets that will facilitate the identification of functional traits or groups of traits that are correlated with robustness or vulnerability to change. Comparing metagenomic data-
|
BOX 2-2 Robustness and the Gut Community The human gut illustrates some key applications of the principle of community robustness. In some situations, robustness of the gut community is desirable. When a person takes antibiotics that alter the gut community, robustness is depended on to return the community to its original structure and function. The inverse of robustness is vulnerability to invasion, and the success of gut pathogens depends on their invasive ability; this highlights another implication of robustness and suggests processes that could be managed better if we understood it. Sometimes, invasion of the gut community is desirable, and robustness interferes with the desired outcome. For example, probiotics, (treatments containing live organisms, such as lactobacilli and bifidobacteria) might be more effective if they survived and colonized the gut. Most healthy gut communities are highly resistant to invasion, providing “colonization resistance” that maintains gut community integrity. Little is known about what makes a gut community resistant to or able to recover from invasion, so there is little rational basis for the design or choice of successful invaders for probiotics. Moreover, there are no predictive models to explain why some people’s gut communities are more robust than others. |
bases for many communities, both robust and vulnerable, over time and space, and applying analytical mathematical tools that can extract patterns will reveal how membership, community structure, specific functions, and functional redundancy and complexity influence robustness. Such analyses might distinguish the characteristics that are associated with all robust communities from those that are specialized or unique to certain habitats or functional units.
HOW DO MICROBES EVOLVE?
Microbial genome variation and the practical and theoretical problems that gene exchange poses for defining species suggest that microbial evolution differs in tempo and mode from the evolution of animals and plants. Current understanding of evolution in general is built on eukaryotes, so a more broadly synthetic evolutionary theory is needed to reconstruct the history of microbial life, to model microbial ecology, and to integrate microbial with eukaryotic evolutionary theory.
The microbial evolutionary model that dominated until recently emphasized clonality and periodic selection. In this model, bacteria are primarily asexual beings. Their populations comprise clones—descendants of a single progenitor cell. Adaptation and divergence are the consequences of favorable mutations in clonal populations that confer advantage on the genomes
|
BOX 2-3 Community Robustness: The Case of Sludge The removal of phosphorus from wastewater by microbes by a process known as enhanced biological phosphorus removal (EBPR) depends upon the stability and robustness of the microbial community responsible for phosphorus accumulation (Levantesi et al. 2002; Garcia Martin et al. 2006). A single organism, Candidatus Accumulibacter phosphatis, supplies all the required biochemical functions to remove phosphorus in many systems. However, although A. phosphatis can be enriched to high numbers in laboratory scale bioreactors, the organisms remain recalcitrant to growth in pure culture, and this suggests a role for additional community members in their maintenance. Although EBPR is generally stable and was first used in full-scale waste-water treatment facilities over thirty years ago, these facilities must continue to maintain backup chemical phosphorus-removal systems to respond to periodic crashes of the biological systems. The cause of crashes is not well understood, but they are hypothesized to result from particular biological and environmental perturbations that destabilize the phosphorus-accumulating microbial community. In laboratory-scale reactors that mimic the wastewater treatment plant cycling, small perturbations in pH and the type of carbon supplied can stimulate the growth of competitors of the phosphorus accumulators and result in less efficient or completely abolished phosphorus removal. In addition, homogeneity of the population of A. phosphatis may leave the community vulnerable to infection by bacteriophage. Greater understanding of the interactions sustaining the EBPR microbial community will lead to more reliable phosphorus-removal systems. |
in which they occur and on the cells that harbor them. Episodes of selection of favored mutants periodically purge populations of genetic and genomic diversity and maintain the cohesiveness (genome-to-genome and cell-to-cell similarity) that allows us to recognize and define species.
However, discoveries of the last decade indicate that gene transfer between similar but nonidentical genomes is, at least in some bacteria, more often the cause of genetic diversity than are new mutations in clones. Indeed, recombination may well be the principal generator of evolutionary novelty in such groups and has parallels to the role of sex in the evolution of animal species. But in other respects there are important differences between microbes and animals: the boundaries of cross-species homologous recombination may be much less distinct, and lateral gene transfer, almost by definition a transgressor of species boundaries, clearly is an important cause of divergence and adaptation in bacteria.
Debate will continue to rage over the frequency and evolutionary importance of such cross-species transfer. Metagenomics, by focusing on genes in an environmental rather than an organismal context, will recast the terms of the debate, as it will of the question “What is a species?” Under-
standing the genetic and ecological processes that determine the structure and function of microbial metagenomes cannot but lead to new ways of describing patterns in Nature and could lead to the emergence of new theories integrating microevoutionary and macroevolutionary principles.
WHAT ECOLOGICAL AND EVOLUTIONARY ROLES DO VIRUSES PLAY?
Viruses are important not only as pathogens, but also as agents of lateral gene transfer and catalysts that generate tremendous genetic variation in their specific hosts. Viral activity also has important consequences for turnover of the elements, for example, in carbon cycling in aquatic systems. It has only recently been recognized that virus particle numbers are enormous, often exceeding those of co-occurring cellular life. For example, seawater contains 10 times more bacteriophage than cellular microbes. Estimates suggest the biosphere harbors perhaps as many as 1031 viral particles (Edwards and Rohwer 2005). Given these vast numbers, the influence of viruses on biodiversity and evolutionary catalysis, and their role in biogeochemical cycling, there is considerable interest in characterizing naturally occurring virus populations. Metagenomics has recently provided an important avenue for exploring these ubiquitous and biologically important entities.
Of special interest is the recent evidence that viruses infecting marine cyanobacteria carry genes involved in photosynthesis (Lindell et al. 2004). Presumably that prolongs the lives of infected hosts (and thus increases virus yields), but another effect is to serve as a genetic bridge between different host species, coupling their evolution, at least as far as such genes are concerned.
Viruses present several unique and interesting opportunities and challenges for metagenomic analyses. Their numbers are large, their genomes are small, and their diversity is impressive. Viruses typically evolve rapidly, so gene sequence conservation is typically much less than that in cellular organisms. Practically speaking, although their numbers are great, their biomass is small, and cloning of viral genes has sometimes been problematic because of modified nucleotides and the cellular toxicity of some of their genes. Metagenomic methods, especially newer sequencing technologies that do not require cloning, may mitigate some of these problems.














