
3
Strengthening the Scientific Base of the Agency1
The committee that authored the IOM report was charged with reviewing the U.S. drug safety system—primarily the postmarket system—and providing recommendations for how to improve it. While the majority of the report’s recommendations focus on postmarket safety, panelists emphasized that there are opportunities to improve safety throughout the drug development pathway. Gaining a better understanding of the safety profile of a drug and being able to discriminate more precisely among drugs within the same class before clinical testing begins would strengthen the drug safety system before a drug ever reaches the market. Postmarket safety is dependent upon a continuum of knowledge, including understanding of a drug’s mechanism of action, as well as the information gained from clinical trials and epidemiological studies. Salient here are the drug safety recommendations in another (non-IOM) report, Drug Safety and Drug Efficacy: Two Sides of the Same Coin, which was based on feedback from the patient and clinical communities (Young et al., 2007). Key among these recommendations was the advancement of current scientific opportunities through the Critical Path Initiative (see below) in order to create a stronger, safer, science-based FDA. Dr. Sigal argued that “the new science” (e.g., molecular diagnostics and targeting)
will dramatically impact the future of drug safety and that advancing this science will require increased support.
In March 2004, the FDA released a document titled Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products (FDA, 2004). The release of this document marked the launch of the agency’s Critical Path Initiative, designed to revolutionize the drug development pathway. The FDA explained that, despite technological advances, the drug development community is still using the last century’s methods to develop and test new drugs, biological therapies, and medical devices. In fact, a drug entering the Phase I clinical stage of development in 1985 was more likely to reach the market than one entering in 2000 (Lloyd, 2002–2003). During 1995–2000, an average of 1 out of 8 new compounds entering Phase I development reached the market, compared with an average of 1 out of 13 during 2000–2002. Thus between 1995 and 2002, the chance of reaching the market declined from 14 to 8 percent (Gilbert, 2003). One of the primary goals of the Critical Path Initiative is to increase the efficiency of the development process by building safety into products throughout their development life cycle.
RESEARCH NEEDED TO IMPROVE DRUG SAFETY: CURRENT FDA INITIATIVES TO EXPAND RESEARCH CAPACITIES
Dr. Woodcock emphasized the need for safety research and described the FDA’s limited ability to take the lead in the conduct of such research. The type of postapproval research needed to improve drug safety extends well beyond the surveillance activities discussed in Chapter 5. Safety research is lacking in part because there is no particular body or entity charged with conducting it. While some research gaps can be filled through existing consortia, collaborations (e.g., with the National Institutes of Health [NIH] and the Centers for Education and Research on Therapeutics [CERTs]), and other mechanisms, the magnitude of the knowledge gap and the FDA’s currently very limited capacity to fill that gap need to be recognized. The cost of a single large comparative safety study, for example, could exceed the entire appropriated budget of the Center for Drug Evaluation and Research (CDER). Considering that funding for CDER totals approximately $500 million for fiscal year 2007—of which about $240 million is from user fees, $225 million is base appropriations, and $16 million is dedicated for orphan drug research grants—less than a few million dollars remains to fund research (after infrastructure costs, salaries, document and adverse event processing costs, etc. are also subtracted).
Dr. Woodcock briefly outlined a broad spectrum of urgent safety science research needs, most of which focus on mechanisms, not just causal associations:
-
Drug toxicological research—The system needs a way to predict and address drug toxicity questions before a drug reaches the market, as well as to address drug toxicity questions that arise after a drug has been marketed.
-
Predictive safety biomarkers—The suite of biomarkers typically used for tracking during clinical trials, as well as after marketing, is inadequate, and most current safety tests are insensitive and nonspecific. Without good biomarkers, even skilled data mining of medical records will not provide adequate answers. The C-Path Institute’s Predictive Safety Consortium, a group of 16 pharmaceutical firms based in Phoenix, Arizona, that shares and cross-validates new safety assays, could serve as a model for what the IOM report recommended.
-
Individualization of therapy—While tests exist for drug-metabolizing enzymes (i.e., to identify slow metabolizers who are at risk for adverse events), outcome studies are needed to evaluate whether these tests really make drugs safer. Similarly, studies are needed to better understand how individuals vary with respect to target status (e.g., some people may have a slightly different molecular drug target that causes them to experience adverse events).
-
Abuse potential—The animal model–based algorithm used for predicting the drug abuse potential of marketed drugs is very old and needs to be updated with better science.
-
Safety in special populations—The FDA is one of the few organizations with a specific focus addressing many of the safety questions that pertain to special populations (e.g., women with asthma or pulmonary disease who take inhaled medications during pregnancy). Because of liability concerns, there is little involvement of the pharmaceutical companies in these activities.
-
Methodological research on how to use large databases and health records—As discussed in Chapter 5, accessing data does not immediately translate into understanding the data.
-
Effective risk communication—Research is needed to understand how information on adverse events and benefit–risk balance can be communicated effectively in a way that modifies prescriber behavior, and how safety information can be communicated effectively in drug advertisements.
-
Root causes of medication errors—Research is needed to understand the causes of medication errors (e.g., mix-ups involving various strengths).2
-
New risk management programs being initiated by the FDA (and others)—Research is needed to evaluate these programs.
Because of its limited ability to conduct the safety research outlined above, the FDA is promoting collaboration among stakeholders in the development of publicly available scientific and technical tools that all stakeholders could use to design safer and more effective products more efficiently. Dr. FitzGerald encouraged collaboration, arguing that FDA–industry partnerships would serve as a necessary component of an improved drug safety system. An enhanced training ground is needed to develop a workforce of scientists with an integrative understanding of drug safety evaluation, and Dr. FitzGerald proposed a Jet Propulsion Laboratory–style public–private partnership to fund this critical training (see the discussion in the next section). Barbara Alving, Acting Director, National Center for Research Resources, NIH, described the Clinical and Translational Science Award program funded by NIH, and gave several examples of current collaborative safety research efforts (see Chapter 5). For example, the University of Pennsylvania is working with the National Cancer Institute (NCI) to improve the capacity to report adverse events associated with cancer therapies.
BUILDING THE FDA’S CAPACITY FOR SCIENCE-BASED PREMARKET REVIEW
According to Dr. FitzGerald, a decade of revolution in drug discovery has resulted in more rationally selected drug targets and molecules directed against those targets than likely was ever anticipated. However, the number of new drugs emerging through the approval process has decreased (Figure 3-1). The current drug development model is not supporting drug discovery. Factors contributing to this “broken model of drug development” include pervasive concern about drug safety, with the “coxibs”3 serving to illustrate the challenges faced by the U.S. drug safety system in balancing benefit and risk. In light of subsequent experience, it might be said that the traditional epidemiological drug safety approach detected merely “the tip of what turned out to be an iceberg.” That is, while some studies detected a signal from the most selective drug (Vioxx) at a relatively high dose (50 mg/day), virtually all of the epidemiological studies conducted prior to the withdrawal of Vioxx from the market detected a signal at a lower dose (25 mg/day) or at any doses of Celebrex, and none detected a signal from Bextra. Yet placebo-controlled clinical tri-
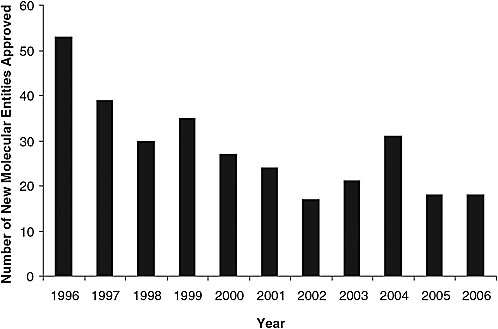
FIGURE 3-1 Number of new drugs approved in the last 10 years (1996–2006).
SOURCE: Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery (Owens, 2006 drug approvals: finding the niche), copyright 2007.
als designed to support new indications were able to detect safety signals for all three of these drugs. This example illustrates the importance of understanding the limitations of each scientific approach—human pharmacology, proof of principle in model systems, observational studies, and randomized trials—when drawing conclusions.
Dr. FitzGerald noted several lessons to be learned from the coxib experience:
-
Industry had no incentive to invest in mechanistic research once the drugs had attained Investigational New Drug (IND) status.
-
While the FDA may have detected and appreciated the relevance of the signals, limited resources made the agency poorly equipped to pursue them.
-
The scientific results from both animals and humans poorly informed the epidemiological studies in terms of both design and analysis.
-
Investigators did not appreciate the limitations of their respective approaches (i.e., epidemiological versus other types of approaches).
-
The placebo-controlled trials that provided the final, compelling evidence were not designed to find that evidence, but to identify new indications.
In light of these lessons, Dr. FitzGerald suggested the need for “a radical new form of science that impinges on drug development in a way that is just as revolutionary as the changes that have occurred in drug discovery.” This new science should have the capacity to (1) develop and project mechanism-based quantitative biomarkers from model systems to humans, (2) evoke phenotypic responses in humans to guide individualization in rational dose selection, and (3) harness unbiased technologies to select among molecules directed against a single target.
Dr. FitzGerald noted that in addition to the deficit of epidemiologists mentioned by Steve Galson, Director, CDER, FDA, and Hugh Tilson, Clinical Professor, University of North Carolina School of Public Health, there is a lack of experts with the integrated skill sets needed to pursue this new type of science (e.g., researchers capable of integrating molecular mechanistic science with clinical science and systems biology). The proposed Jet Propulsion Laboratory–style FDA–industry partnership could be used to address this deficit in human capital. Sites external to the FDA would serve as loci for mechanistic studies and the testing of proposed drug action hypotheses. One major roadblock to this type of initiative is funding, as neither the FDA nor NIH has money available for such an endeavor;4 however, other organizations may be appropriate to spearhead the effort. The partnership could also leverage Clinical and Translational Science Awards and FDA investments in education and informatics. The integrative research challenges would be great, necessitating three or four external centers focused on major disease areas and extramural investments on the order of $10 million annually. Yet Dr. FitzGerald stressed that the investment would be worthwhile because it holds promise for speeding the development of safe and effective medicines, accelerating understanding of the factors involved in personalized medicine, and generally enhancing the benefit provided by the drug development industry.
Many of the symposium panelists expressed similar views on this issue. Gail Cassell, Vice President, Scientific Affairs, and Distinguished Lilly Research Scholar for Infectious Diseases, Eli Lilly and Company, encouraged the audience to consider additional ways of increasing the availability of and interest in epidemiology training programs, perhaps in
collaboration with the FDA and other organizations. Ellis Unger, CDER, FDA, said that the FDA would be interested in having a fellowship program similar to that of NIH. Finally, Dr. Tilson emphasized that before embarking on these programs, it will be important first to understand what competencies the scientists being trained need to have and then develop the appropriate training accordingly.







