
Acceptability
Have You Determined How to Address the Following Challenges to Implementing the Strategies?
-
How are public concerns affecting the community? What can you do to empower personal responsibility for protective actions?
-
Will the community support the strategies under consideration? What can you do to increase support?
-
What secondary effects (for example, child nutrition, job security, financial support, health service access, and educational progress) might result from the strategies under consideration? Can you get the message out to businesses and employers that they need to have flexible leave policies that align with public health recommendations?
-
Can these secondary effects be mitigated? Which community entities and organizations can help reduce the secondary effects?
-
What can be done to increase community buy-in?
Preparing for the Flu: A Communication Toolkit for Schools (Grades K-12)16
A2
PREDICTING EMERGING DISEASES IN THE TWENTY-FIRST CENTURY: THE CASE OF ZOONOTIC INFLUENZA
Kurt J. Vandegrift, Parviez Hosseini, Ph.D., and Peter Daszak, Ph.D.
Wildlife Trust17
In this paper, we would like to pose a simple question: What have we learned from past experiences with emerging diseases that could help us understand the emergence and spread of 2009-H1N1 influenza A and the next pandemic pathogen? We hope to illustrate how, through intensive study and fusion of evolution, ecology, virology, and microbiology, we could be better prepared for, or even predict for, the next emergent pathogen.
Over the past four decades, we have seen the emergence of diseases such as AIDS, methicillin-resistant Staphylococcus aureus (MRSA) infection, and
severe acute respiratory syndrome (SARS). The rate of disease emergence has increased significantly over this time, and there seem to be parallel problems in wildlife and even plants (Daszak et al., 2000; Anderson et al., 2004; Jones et al., 2008). In wildlife, a fungal disease has caused a series of extinctions of amphibian species globally, and a transmissible cancer threatens extinction of the Tasmanian devil (McCallum et al., 2007). In plants, diseases of crops and trees have been linked to anthropogenic spread through trade, climate change, and other factors (Anderson et al., 2004). What are the commonalities among these seemingly disparate groups? Are there patterns to emergence that might allow us to predict and prevent the next emerging disease? We should strike a note of caution at this point. In his 1998 address to the International Congress on Emerging Infectious Diseases, Professor Fred A. Murphy reminded us that predicting the next emerging disease’s origin or impact is a significant challenge (Murphy, 1998). The biggest obstacle is probably the sheer size of the unknown pathogen diversity in wildlife, livestock, and other reservoir species with potential to infect humans, should we make the right type of contact. Later in this paper, we will present our approach to dealing with this unknown, but first we consider the commonalities in the process of emergence and how they lead us to a potential solution. We focus here on the emergence of new zoonotic diseases from other animal reservoirs.
There are undoubtedly factors that influence a pathogen’s potential to spill over from wildlife to humans. As a simple case in point, rodent-borne zoonotic pathogens (e.g., hantaviruses) require the presence of rodent reservoirs and, although these creatures exist throughout the world, there are certain areas where rodent abundance is greater or the contact with humans is higher. Although this does not tell us exactly where a rodent-borne pathogen will emerge, it does provide an indication of where there is higher risk. In the same vein, substantial molecular phylogenetic evidence points to a Central-West African origin of HIV-1 from chimpanzees, a species widely hunted for bush meat there. The origins of SARS and some Ebola virus outbreaks have also been linked to the consumption of wildlife. It follows that patterns of human hunting, butchering, and consumption of bush meat will likely predict patterns of the emergence of some zoonotic infections. Finally, SARS coronavirus spread rapidly from China to the New World via infected people traveling on planes (Hufnagel et al., 2004). If we examine trends in global air travel, surely that will give us significant predictive power in analyzing where the next new pathogen in people is likely to spread to. In a very general sense, it becomes clear as we look at the source of each emerging pathogen that almost every emerging disease (perhaps every single one) was driven to emerge by some type of change in human behavior, demography, or anthropogenic environmental change. These emerging diseases are not, after all, “natural” events. If this is true, then it follows that we should be able to predict disease emergence by analyzing trends in demographic, socioeconomic, or environmental changes.
To do this, our group has used a database approach pioneered by Mark Woolhouse’s group in Edinborough and based on the database analyses commonly used in ecological studies of animal life history traits. In this approach, global spatial data on environmental changes (e.g., agricultural land-use change) and the outcomes of these changes (in this case of the occurrence of an emerging disease) are tested for correlation. To do this for disease emergence, we expanded a database of all pathogens known to emerge in people (Woolhouse, 2008). The distribution of the types of newly emerging pathogens offers a glimpse of what sort of pathogens are more likely to cause the next emerging disease. A disproportionate amount of these pathogens are drug-resistant bacteria (e.g., MRSA) and viruses (mainly RNA viruses, e.g., HIV-1, SARS CoV, and Chikungunya virus). This is not entirely surprising because of the recent rise in global use of a diverse array of antibiotics, and because of the mutation rates and lack of copy editing mechanisms in the RNA viruses, which make them better able to produce more diverse strains capable of establishing in new host species. The origins of emerging pathogens are also informative, with the majority being zoonotic (e.g., SARS CoV, the Lyme disease spirochete, and Ebola virus) and these zoonoses include many of the most significant infections to emerge recently. This likely reflects our increasingly close association with animals, a factor that may appear counterintuitive in developed countries where our meat is bought prepackaged in plastic, but is a virtue of the unprecedentedly large global human population and our globalized travel and trade networks. Even as we eat our lunch here at the Institute of Medicine workshop, we may be eating beef produced in Australia, anchovies from Peru, and blackberries grown in Guatemala. Thus, in our database of emerging diseases, we find zoonotic diseases emerging from this complex network of globalized agriculture and trade.
Taking the database of emerging diseases, we surveyed the literature for the most accurate information available on the geographic origin of the first known outbreaks for each pathogen. In plotting out the origin of each of the more than 400 emerging disease “events,” we find a strong bias toward the developed countries of Europe, North America, and the Far East. This likely reflects the increased ability of these richer countries to identify emerging disease outbreaks and is perhaps due to their higher spending on healthcare. To correct for geographic and temporal biases in global reporting, we trawled through every paper published in Journal of Infectious Diseases18 from 1980 to 2002, collated each author’s geographic origin and the date of the work, and then incorporated these data into our analyses. Next, we developed a strategy to estimate the global spread of the vast diversity of unknown pathogens. To do this, we used a global database of the distribution of every mammalian species (Jones et al., 2008) and made the simple assumption that every species will carry a roughly equal number of pathogens,
known or unknown (Grenyer et al., 2006). We then used a general linear model (GLM), a multiple regression model, to test for correlation between the risk of an emerging disease and a series of presumed drivers of emergence: rainfall distribution, human population density and growth, and so on. Our results (Jones et al., 2008) show that all groups of emerging diseases (vector-borne, zoonotic diseases from wildlife, zoonotic diseases from other species, and drug-resistant infections) show strong correlation with human population density and growth. We found that zoonotic diseases from wildlife were strongly correlated with human density and mammalian biodiversity, suggesting that it is regions where human populations are coming into close contact with wildlife that are most at risk for the highest impact of future zoonotic emerging infectious diseases (EIDs). Finally, we were able to use the geographic distribution of risk, corrected for biases in reporting, to produce the first ever global maps of the risk of future emerging diseases (Jones et al., 2008). The maps for zoonotic diseases pointed to developing countries in the tropics (Central and West Africa, Mexico, parts of tropical Latin America, South Asia, Southeast Asia) as those places most likely to spawn the next emerging zoonotic pathogen. Importantly, these are also the regions least covered by our global effort to conduct surveillance for new diseases.
This predictive approach has great relevance for new strains of influenza. If we can develop predictive approaches to the emergence and spread of new pathogens, it may be possible to also do this for new strains of influenza. Influenza pandemics have occurred repeatedly in the twentieth century. In 1918, 1957, and 1968, these pandemic strains resulted in 50 million, 1 million, and 0.5 million deaths, respectively (Cox and Subbarao, 2000). The resulting strains circulating annually as seasonal flu cause millions of severe illnesses and approximately 500,000 deaths per year (Cox and Subbarao, 2000). In 1998, with the emergence of H5N1 virus direct from birds and again in 2009, with the emergence of a new strain of 2009-H1N1 influenza A virus, it became evident that there is significant potential for novel strains, to which humans have little or no immunity, to arise and spread as worldwide pandemics. Furthermore, it became clear that these strains could emerge from zoonotic reservoirs (poultry, wild birds, pigs) into the human population. What factors underlie this phenomenon? Influenza viruses are able to evolve into new strains capable of establishing in new host species, specifically their potential for genetic reassortment. This results in a diversity of influenza strains which was illustrated well in the 2009 pandemic, wherein the new strain included segments of avian, human, and swine origin (Smith et al., 2009). Swine-origin H1N1 viruses have circulated in North American pigs for over 80 years (Shope and Lewis, 1931). The precursor to this virus was first detected in commercial swine in the United States and was subsequently labeled as a notifiable disease in 2007. Further mixing and reassortment with other cocirculating viruses (e.g., H3N2 and H1N2) led the 2009-H1N1 influenza A virus to have gene segments from humans, swine, and birds and these segments were associated with three different continents (Smith et al., 2009). Phylogenetic
analysis suggests the strain emerged between 10 and 15 years ago, but, due to a lack of surveillance, the direct ancestors are not known. However, the new gene segments that were not previously known to circulate in North American swine most closely resemble the Eurasian avian-like swine H1N1 (Smith et al., 2009). This suggests that live hog trade between Eurasia and North America could have facilitated the mixing that led the World Health Organization (WHO) to declare the first pandemic of the twenty-first century.
Phylogenetic analysis of 2009-H1N1 influenza A is useful, but it is limited in helping our understanding of the virus’ origin and emergence. For example, it can point to the involvement of swine production, but, due to the incomplete surveillance and availability of global swine influenza sequences for the past two decades, it is currently not possible to trace back the virus spread through the swine trade. Likewise, it is currently impossible to deduce the relative roles of swine production, poultry production, wild bird migration, and human travel in the emergence of the strain. Our group analyzed swine and poultry imports to Mexico in the decade preceding the emergence of 2009-H1N1 influenza A using Food and Agriculture Organization (FAO) data from the UN Comtrade data portal (2009). We found that there was little trade between Mexico and countries other than Canada, the United States, and the United Kingdom, but that the volume of trade with these countries was extremely high (tens of thousands to hundreds of thousands during this period). Likewise, the volume of poultry traded among these countries was in the hundreds of thousands to tens of millions during this period, with multidirectional trade confounding the issue. This supports the phylogenetic findings of evidence for mixing of multiple strains. However, the lack of knowledge of recent evolution of each H1N1 viral gene segment precludes the use of this approach to determine viral origins.
There are very detailed data on human travel capacity, and it is possible to analyze the spread of the strain postemergence and to make some useful predictions. We used data on human air travel capacity from the International Air Transport Association (IATA, 2009) from around the time of the first emergence of 2009-H1N1 influenza A. We found that these data are a good predictor of the early spread of the virus from its origin near La Gloria, Mexico, especially when we included the likely secondary travel of passengers out of connection hubs (e.g., Los Angeles, Houston, and others). However, apparent anomalies in the case load were evident for some countries. For example, our air travel data predict that Brazil and Argentina (two countries traveled to extensively from Mexico) should have had higher caseloads than were reported in the weeks follow ing the outbreak. We predicted that there was a “hidden” caseload due to the likely lower propensity of these countries to report than richer countries such as the United States—a product of less funding available for healthcare (testing and surveillance), less incentives for poorer people to report, and the lower number of testing facilities and doctors per capita. We tested this theory by incorporating measures of gross domestic product (GDP) and money spent on healthcare in
these countries. We found that incorporating national healthcare resource data into our analyses allowed a much greater capacity to predict the international reporting of spread of this virus. In countries with lower healthcare resources, the reporting of 2009-H1N1 influenza A cases was significantly delayed, reflecting a likely lower capacity for testing and reporting, as well as other demographic issues. We concluded that strategies to prevent pandemic influenza virus emergence and spread in the future may include enhanced surveillance for reassortant strains in traded livestock and rapid deployment of control measures in the initial spreading phase to countries where travel data predict spread and where lower healthcare resources predict delays in reporting. Our results highlight the benefits, for all parties, when higher income countries provide additional healthcare resources for lower income countries, particularly those that have high air traffic volumes. The result is the potential for earlier detection of pathogens and reduced impact of pandemics.
What lessons can we learn from these approaches to disease emergence that we can apply to zoonotic influenza viruses? Broadly, we can conclude that predictive approaches to disease emergence require measurement of the capacity of anthropogenic changes to alter dynamics of viruses and their risk of spilling over to people. This has great relevance to highly pathogenic H5N1, which has repeatedly spilled over to people in Asia but so far has not been efficiently transmitted between people. Our group is involved in a new Fogarty International Center-funded initiative to collect the sort of data necessary for developing a predictive model for this pathogen that will be of use in predicting the risk for other zoonotic influenza strains. This project involves collaboration among groups working on the ground in South America, Africa, South Asia, and Southeast Asia. The ultimate goal is to build a mathematical model that describes the risk of influenza virus movement within wild bird populations, and between these and domestic poultry farms, pig farms, and then people. Mathematical models work best when they are underpinned (parameterized) with data on the factors involved in each important stage of emergence. In this case, each group will gather data on wild bird populations (e.g., diversity, abundance, contact with poultry), on poultry and pig populations (e.g., farm size, density, and agricultural practices), and on human populations (e.g., density and cultural practices relating to pigs and poultry). Once the data collection is complete, the spillover rates can be observed and the data used to parameterize a model that hopefully will help us to identify important risk factors a priori.
It seems logical to focus particular interest on farming practices. For example, poultry production has changed tremendously in the past 50 years following the widespread availability of cheap antibiotics to combat coccidiosis, the development of new rapid weight-gain bird varieties, and the growing demand for protein, particularly in Asia. The annual per capita consumption of poultry, pork, and beef in the United States shows a comparatively large increase in poultry compared to pork or beef (Figure A2-1). This increased demand has resulted
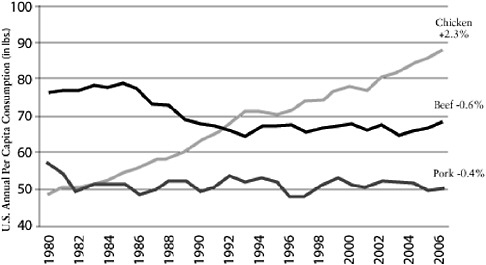
FIGURE A2-1 Chicken: a growth category. Compound annual growth rate 1980-2006. Per capita consumption has grown consistently for 26 years.
SOURCE: Reprinted with permission from Boric (2006).
in a shift from backyard production to vertically integrated commercial facilities that can now generate over 1 million kg of meat per year (MacDonald, 2008). The number of farms has decreased by 50 percent, yet production has gone up 500 percent (MacDonald, 2008). These commercial farms house birds in dense populations and, although there are varying degrees of biosecurity, there is a great deal of transmission potential within-farm and between farms. Live bird markets have also been associated with disease outbreaks (i.e., SARS) in the past and are another complicating factor in the influenza transmission cycle. One of the risk factors here is the mixing and clustering of many different species from vastly different areas. The markets typically house animals very tightly and have unsanitary conditions that may promote transmission. Waterfowl are thought to be the primary reservoir host for influenza viruses; however, nearly 100 species of birds have tested positive. Anseriformes (ducks, swans, and geese) includes the most commonly infected species and the prevalence for influenza viruses normally ranges between 1 and 15 percent (Olsen et al., 2006). H5N1 has also been isolated from a range of sick mammals, and the host diversity of this strain is probably underestimated. A further complexity here is that some species may act as “silent” carriers or reservoirs. For example, an H5N1 isolate that was very lethal in commercial poultry was found to only cause a mild passing illness in juvenile mallard ducks (Sturm-Ramirez et al., 2005) and similar findings have been seen with quail. More experimentation with different subtypes and species needs to be accomplished before we will be able to understand how these patho-
gens impact the wide variety of hosts they can infect. This in turn will influence the potential for viral persistence and potentially spill over into humans.
Of the Anseriformes, the dabbling (or puddle) ducks have the greatest prevalence and mallards in particular have the highest prevalence (Olsen et al., 2006). The age structure of these populations is also important in that juvenile birds are more likely to have an infection than adults, and this likely is influenced by immunological status. Other migratory birds including the Charadriiformes (shorebirds, gulls, terns, and waders) can be infected but typically only at very low levels (Krauss et al., 2004; Olsen et al., 2006). However, this does not mean they are unimportant in the transmission cycle or maintenance of the virus. More intensive long-term data on how these viruses circulate and transmit between these birds are needed.
Finally, the potential for newly reassorted strains to emerge is probably heightened now because of the widespread circulation of 2009-H1N1 influenza A. We hope that our Fogarty International Center-funded program will both help identify the risk of co-infections (e.g., regions with high poultry and hog farm density) and actually find evidence in the testing that our groups will be doing. The recent report of hog farms in Indonesia with high prevalence of H5N1 (Cyranoski, 2005) and two very recent suspected cases of humans passing 2009-H1N1 influenza A onto hogs highlight this risk.
We conclude that there are a growing number of strategies being developed to predict the origin and spread of novel emerging pathogens. These strategies meld ecological, virological, and mathematical approaches to identify high-risk regions, activities, and behaviors, and they have some potential for prevention and control. At the same time, far more detailed and structured studies are needed to truly get to the underlying causes of zoonotic influenza emergence and help prevent the next human-to-human high-pathogenicity pandemic. To do these studies effectively will require some capital investment, likely within the range of a few tens of millions of dollars. However, we believe the potential reduction in pandemic risk would be a wise investment because the predicted pandemic mortality and associated economic costs are within the tens of billions of dollars (Meltzer et al., 1999).
References
Anderson, P. K., A. A. Cunningham, N. G. Patel, F. J. Morales, P. R. Epstein, and P. Daszak. 2004. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends in Ecology and Evolution 19(10):535-544.
Boric, J. 2006. Investing in chicken, http://pennysleuth.com/investing-in-chicken-2/ (accessed March 24, 2010.
Cox, N. J., and K. Subbarao. 2000. Global epidemiology of influenza: past and present. Annual Review of Medicine 51:407-421.
Cyranoski, D. 2005. Bird flu spreads among Java’s pigs. Nature 435(7041):390-391.
Daszak, P., A. A. Cunningham, and A. D. Hyatt. 2000. Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287(5452):443-449.
Grenyer, R., C. D. L. Orme, S. F. Jackson, G. H. Thomas, R. G. Davies, T. J. Davies, K. E. Jones, V. A. Olson, R. S. Ridgely, P. C. Rasmussen, T. S. Ding, P. M. Bennett, T. M. Blackburn, K. J. Gaston, J. L. Gittleman, and I. P. F. Owens. 2006. Global distribution and conservation of rare and threatened vertebrates. Nature 444(7115):93-96.
Hufnagel, L., D. Brockmann, and T. Geisel. 2004. Forecast and control of epidemics in a globalized world. Proceedings of the National Academy of Sciences 101(42):15124-15129.
IATA (International Air Transport Association). 2009. Aviation data for airline planners, http://www.iata.org (accessed November 16, 2009).
Jones, K. E., N. G. Patel, M. A. Levy, A. Storeygard, D. Balk, J. L. Gittleman, and P. Daszak. 2008. Global trends in emerging infectious diseases. Nature 451(7181):990-993.
Krauss, S., D. Walker, S. P. Pryor, L. Niles, C. H. Li, V. S. Hinshaw, and R. G. Webster. 2004. Influenza A viruses of migrating wild aquatic birds in North America. Vector-Borne and Zoonotic Diseases 4(3):177-189.
MacDonald, J. M. 2008. The economic organization of U.S. broiler production, http://www.ers.usda.gov/publications/eib38/eib38.pdf (accessed November 16, 2009).
McCallum, H., D. M. Tompkins, M. Jones, S. Lachish, S. Marvanek, B. Lazenby, G. Hocking, J. Wiersma, and C. E. Hawkins. 2007. Distribution and impacts of Tasmanian devil facial tumor disease. EcoHealth 4(3):318-325.
Meltzer, M. I., N. J. Cox, and K. Fukuda. 1999. The economic impact of pandemic influenza in the United States: priorities for intervention. Emerging Infectious Diseases 5(5):659-671.
Murphy, F. A. 1998. Emerging zoonoses. Emerging Infectious Diseases 4(3):429-435.
Olsen, B., V. J. Munster, A. Wallensten, J. Waldenstrom, A. Osterhaus, and R. A. M. Fouchier. 2006. Global patterns of influenza A virus in wild birds. Science 312(5772):384-388.
Shope, R. E., and P. Lewis. 1931. Swine influenza: experimental transmission and pathology. Journal of Experimental Medicine 54(3):349-359.
Smith, G. J. D., D. Vijaykrishna, J. Bahl, S. J. Lycett, M. Worobey, O. G. Pybus, S. K. Ma, C. L. Cheung, J. Raghwani, S. Bhatt, J. S. M. Peiris, Y. Guan, and A. Rambaut. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459(7250):1122-1125.
Sturm-Ramirez, K. M., D. J. Hulse-Post, E. A. Govorkova, J. Humberd, P. Seiler, P. Puthavathana, C. Buranathai, T. D. Nguyen, A. Chaisingh, H. T. Long, T. S. P. Naipospos, H. Chen, T. M. Ellis, Y. Guan, J. S. M. Peiris, and R. G. Webster. 2005. Are ducks contributing to the endemicity of highly pathogenic H5N1 influenza virus in Asia? Journal of Virology 79(17):11269-11279.
UN Comtrade. 2009. United Nations Commodity Trade Statistics Database, http://comtrade.un.org/ (accessed November 16, 2009).
Woolhouse, M. E. J. 2008. Epidemiology: emerging diseases go global. Nature 451(7181):898-899.









