
5
Enabling Technologies and Tools for Research
INTRODUCTION
One could argue that it has been the tools of the physicist and the chemist that have driven the life sciences forward at an ever-increasing rate. From the invention of X-ray crystallography to the invention of the gene chip, new technologies and tools have allowed us to look deeply into biology at increasing depth and breadth. These tools have enabled the study of the structures and dynamics that drive biological systems, and the progress has been spectacular. However, there still is much to be learned at all length scales of biological systems, from nanosized organisms to global ecosystems, and suitable tools and technologies will be critically important in studying those systems over the next 20 years.
As we ask increasingly probing questions, often guided by theory, no doubt the truly transforming ones will be those that are least expected. This chapter is not a laundry list of all the latest and greatest technologies transferring from the physical sciences to the life sciences—there is not room enough, and such a list would inevitably be skewed toward the fields of the writer. Rather, the chapter highlights areas of promising research at different size scales and seeks to identify the new technologies most urgently needed to make advances in these fields.1
PHYSICAL BASIS OF MOLECULAR RECOGNITION
Molecular recognition is arguably the single-most-important molecular process. It is the key to the structure-specific association of a macromolecule (protein or nucleic acid) with another molecule and is the basis for a number of subcellular activities. These include protein-ligand binding, catalysis, the action of receptors, the formation and operation of mechanical structures in the cell, the generation of energy and vectorial movement of charge, and sensing. The processes involved in molecular recognition are the same as those in the folding of proteins or nucleic acids and, somewhat more loosely, in the formation of lipid bilayers. Arguably, molecular recognition is more fundamental than any other single process in the cell—that is, more fundamental than replication and translation of DNA, synthesis of proteins, or the operation of signaling networks—since it is the basis of all of them. And, astonishingly, it still is not well understood at the molecular level.
Chemistry and biophysics have led to a picture of molecular recognition, a metaphor for which is a lock and key. In this metaphor, two molecules associate when they have complementary shapes. Complementary shapes maximize van der Waals interactions and make it possible to associate complementary electrostatic charges. Much of molecular recognition is ascribed to the hydrophobic effect, which is the association of nonpolar surfaces in water. The problem with this attractively simple metaphor is that, like many metaphors, it gives a distorted picture. Close complementary fit between associating molecules may improve the enthalpy of interaction, but it is unfavorable entropically. A better metaphor is now believed to be a cow in a tent—that is, a loose fit between molecules that minimizes the Gibbs free energy of association. What is needed for good binding of molecules to one another is the right kind of sloppy fit, but the meaning of “right kind” is not clear.
The problem of molecular association has been clearly posed for 50 years but there is still no resolution. For example, it still is not possible to rationalize quantitatively the Gibbs free energy of the binding of ligand–potein pairs or to predict the structure of new ligands for a protein, even if one has detailed knowledge of the structure of the binding site. One thing that has made the problem so difficult is that while water is clearly a necessary component of molecular recognition, the role of solvent in biology is sufficiently inconvenient that it has been ignored for the most part. For example, it is incorrect to express the molecular recognition problem in terms of protein and ligand. Instead, it must be expressed in terms of protein, ligand, water, and, perhaps, other components of biological media as well.
Understanding the interactions between water and proteins is a problem that will require high-resolution structural methods; new theoretical methods, including new methods in statistical mechanics that can handle the large numbers of particles involved; and thermodynamic analysis. Each type of information will need to be supported by physical tools such as high-resolution X-ray and neutron
sources, fast computers, quantum-mechanically based potential functions, and statistical methods for single-molecule analysis.
These tools and technologies reflect a top-down approach to probing living systems: designing and building technologies using macroscale techniques and then using those technologies to examine biological systems. No doubt top-down approaches to designing new tools will continue to be extraordinarily useful, but the scope of what can be accomplished through such tools is limited. Recently, bottom-up technology—whereby self-assembly of nanostructures can be used to create new materials and to perform functions that can probe biological systems—has started to allow collecting much more useful data for understanding these and other complex biological issues (Whitesides and Grzybowski, 2002).
Until recently it was not possible to control the molecules and assemblies of molecules from which the bottom-up-designed devices are composed. However, such control is now becoming possible, and although technological hurdles still must be overcome, not only will this control allow for the design and manipulation of bottom-up technologies, but also a new array of techniques developed from such technologies should provide a substantially different perspective on a given problem. For example, the effect of a controlled-design molecular assembly on the behavior of protein-ligand binding could provide immensely useful information about the mechanisms behind molecular recognition.
STRUCTURES AND DYNAMICS WITHIN CELLS
Cellular Environment
Moving beyond the fundamentals of molecular recognition, new tools and techniques will be needed to study molecules within cells, the function of cells and assemblies of cells, and larger biological systems. The complexity of the biological milieu of a single cell, and the fundamental challenges it poses for a technology that tries to probe it, cannot be overstated. Several characteristics of the subcellular milieu are particularly relevant:
-
High concentration of macromolecules,
-
Extreme heterogeneity of components,
-
Highly organized components, from the nanoscale and on up,
-
Local protein densities that may approach the densities of closely packed spheres,
-
Dynamic and directed transport of components coupled with diffusion,
-
Two phases of water: one behaves like bulk solvent and the other, presumably water of hydration, has very different physical properties, and
-
Rapid (subsecond) and specific variation of metabolic components on the nanoscale.
All functions within a cell occur within a medium called the cytosol. This medium fills the cell, contains ca. 300 g/L of organic material, has an ionic strength of approximately 1 mole, and is extraordinarily complex. For many years, the study of biological materials took place not in a cytosol-like medium but in water or dilute buffer solutions. Biochemical and biophysical studies have concentrated on the average (ensemble average) behavior of purified protein molecules in these dilute aqueous solutions, but the unique properties of the interiors of cells dictate that the in vivo behavior of these molecules depends on their interactions, not only with water and small molecular solutes, but also with macromolecules in a very crowded neighborhood (Minton, 2006). Although enormous strides have been made in the development of single-molecule dynamics tools, driven by optical tweezers and/or single fluorophore techniques, except for a few rare exceptions these tools are still employed in dilute solutions, not in living cells.
Dilute aqueous solutions were historically used as a model medium because the tools to do single-cell studies simply did not exist originally. When molecular biology began, proteins were very difficult to obtain in pure form, and analytical systems were crude. The introduction of the Beckman Model D spectrophotometer was a revolutionary event: it enabled, for almost the first time, quantitative physical measurements in biological systems, but its sensitivity was low. Unfortunately, the media used in those early days (very dilute solutions, low ionic strengths, buffers that did not absorb in the ultraviolet), were chosen for compatibility with the crude instrumentation of that day, not for their relevance to biology or the cell. As data began to accumulate, it was convenient to continue to use these simple media, since they provided a basis for comparing data. The result now is an immense body of data collected over the last 50 years in media known to be dissimilar in almost every respect to the media filling the cell.
It is thus an active area of research to determine the relevance (and the limitations) of this ex vivo data accumulated by molecular biologists, protein chemists, and enzymologists in understanding the processes that occur inside a cell (see Rivas et al., 2004). In the future, to ensure that ex vivo experiments are relevant to the real problems of biology, chemists, physicists, and biologists need to collaborate to define a model intracellular medium and then to develop tools to probe such a complex medium. Mapping the mass of historical data to gain such increased understanding of the cytosol will require comparing the properties of relevant biological molecules and processes in the model medium and in the cytosol and then defining an adequate cytosol model system. The model system will likely vary with the particular system being studied because the cytosol in a red blood cell is markedly different from the cytosol in a lymphocyte.
Meeting this challenge will require both bottom-up and top-down tools, as described in the preceding subsection, because the modeling of the cytosol cannot be improved without the development of techniques capable of in vivo measurements such as NMR, fluorescence resonance energy transfer (FRET), and other techniques sufficiently sensitive to study single molecules. Part of this effort would require analyzing existing information to determine what are the most pertinent characteristics of the cytosol, such as its polarity, ionic strength, dielectric constant, viscosity, polarizability, free volume, and compressibility. Another part would involve determining how to model these characteristics in a simpler fluid. The involved communities would have to agree that this simplified fluid is a valid model substitute for the cytosol. Tools both molecular (hence bottom-up) and top-down in origin would be needed to physically test and then study the model cytosol. For example, confocal-imaging coherent technologies could be used to model the dynamics of the local densities of components of the cytosol as they are transported throughout the cell.
Interactions Within Cells
Understanding the cytosol is one part of the puzzle, but understanding the internal mechanisms of cells requires functional imaging of the space- and time-resolved metabolic components and their interactions. Remarkably, demands principally arising in the physical sciences are driving the development of tools that can characterize this microecology in a spatially resolved way (Yu et al., 2006). In chemistry, probes are being developed that can be used as highly specific labels, while in physics laboratories tools are being created that can separate and detect at the single molecule level different components of a single cell. One example is the matrix-assisted laser desorption/ionization (MALDI) technique: Coupled with time-of-flight mass spectrometry it can function as a rapid and sensitive top-down analytical tool (Tsuyama et al., 2008). It has the potential to obtain molecular weights of peptides and proteins from single-cell samples and to perform in situ peptide sequencing and can map peptides in cells and tissues directly. Mass spectrometry is being used in other systems-level analyses of cellular metabolism as well, and those studies are beginning to reveal how the concentrations and fluxes of small-molecule metabolite levels in cells are controlled.
Within (as well as beyond) a single cell, biology depends on macromolecular assemblies. This dependence is apparent in the molecular mechanisms underlying gene expression, signal transduction, cell migration, cell organization, and cell division. To understand and manipulate specific biological processes, it would be advantageous to be able to design and then to generate defined molecular assemblies. While scientists are adept at devising chemical syntheses of specific compounds, their ability to design specific molecular assemblies is rudimentary,
and the interface between the two technologies is still poorly understood (Bertozzi and Kiessling, 2001). Synthetic macromolecules that mimic the features and functions of naturally occurring biomolecules can illuminate fundamental principles and control biological responses. For example, synthetic oligomeric compounds have been generated that, like proteins, can adopt a specific conformation such as folding on themselves like proteins. Moreover, compounds have been devised that mimic the light-harvesting properties of the photosynthetic reaction center, and agents of this type could become new sources of energy. Macromolecules have been generated to serve as agents for drug delivery, as scaffolds for cell growth or differentiation, and as therapeutic agents. The potential of synthetic macromolecules to manipulate cellular responses by other means is great and has not been explored widely.
One strategy for creating synthetic functional assemblies is to use the molecular reaction processes employed by nature. The recognition properties of nucleic acids, for instance, are being used to generate nonnatural structures and assemblies, as is shown in Figure 5-1. This example shows how understanding biology can lead to new approaches for generating materials that function in an abiotic as well as a biotic realm. Another strategy that can be exploited to generate assemblies is to create multifunctional ligands. Agents that can direct the formation of multiprotein complexes and/or control the localization of multiple proteins within a cell would be valuable. Such ligands, which can be used like small molecules for temporal control, could illuminate how proteins assemble or how protein localization controls cellular responses. For example, multifunctional ligands could serve as scaffolds to effect signaling pathways not known to exist in nature or could endow cells with unexpected plasticity. Because modular protein assemblies are essential cellular control elements, myriad possibilities exist for using multifunctional ligands to manipulate cellular responses.
Examining Structures Within Cells
There are many underexplored but essential internal structures in the cell that exist between the atomic scale resolution of x-ray crystallography and the diffusion-limited 100-nm-scale resolution of conventional optical microscopy: cytoskeleton components, moving chromosomes, lipid rafts, folding membranes, etc. While great strides have been made in breaking the 100-nm resolution length scale of conventional microscopy, many of the techniques are quite slow (a minimum of 1 minute scanning time on a fixed sample) and require fluorescent labeling techniques, which can be difficult to implement. For example, one of the great challenges at this size range is determining how nanoscale molecular motors work. Fluorescence probes such as FRET have given us some insight into the conformational motions of these wonderful motors, but at the cost of bulky and difficult
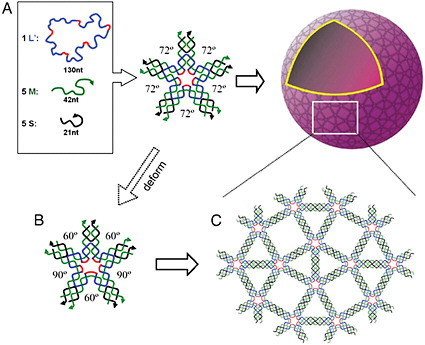
FIGURE 5-1
Synthetic Functional Assemblies
A recently proposed assembly process for creating three-dimensional nanosized objects. (A) At high DNA concentrations, five-point-star tiles can assemble, arranged into tetragonal two dimensional crystals. (B) Three-dimensional spheres are capable of self-assembling by taking advantage of the propensity of such tiles to have out-of-plane bending and asymmetrical bends in the molecular plane. (C) In the assembled structure, the angles between two neighboring branches varies (three 60-degree and two 90-degree angles), and all are different from the 72-degree angle in the free tiles. In these kinds of three-dimensional structures the conformational flexibility of the molecules is a critical part of the energetics of self-assembly. The structures created through such self-assembly processes are not only interesting in and of themselves but could also serve as encapsulation agents, nanoreactors, or organizational scaffolds. SOURCE: C. Zhang et al., 2008, courtesy of the Proceedings of the National Academy of Sciences.
probes that are chemically attached to the protein. Optical tweezers have allowed us to monitor the motion of these with remarkable precision, but little information is gained about how the motion actually proceeds. Molecular dynamics is greatly limited in its time range and is forced to greatly simplify the interactions between the atoms. We need tools that can see, at the single-molecule level—that is, at the sub-nanometer scale—how biological molecules proceed in functional activities, without the use of probes.
Super-resolution techniques are being developed that overcome some of the time constraints, allowing researchers to map the trajectories of individual molecules and organelles in live cells. These techniques typically involve illuminating the sample with patterned light, collecting the low-resolution image that contains moiré-fringed patterns of the sample, and then drawing from those fringed patterns high-resolution information about the sample. Different combinations of lenses, light sources, and modulated patterns have produced a plethora of acronymed microscopies, such as photo-activated localization microscopy (PALM) and stimulated emission depletion microscopy (STED), which now are allowing the mapping of trajectories of individual molecules and organelles in live cells.
Coherent, soft x-ray light sources also show great promise for studying biological systems and will probably provide high-resolution imaging in the middle of these underexplored but biologically critical length scales (Gibson et al., 2003). Furthermore, a rapid temporal sequence of images of a single cell would for the first time provide direct visualization of the complex dynamics that occur on the 10- to 200-nm length scale. Contrast mechanisms exist for high specific imaging modalities because of the narrow bandwidth of the coherent beam and its continuous tunability. Element-specific imaging (by tuning to different element L-edges and/or by using XANES/EXAFS spectroscopy in the imaging, which give the chemical state and the nearest neighbor distances and coordination, respectively) will greatly aid in the interpretation of the images. For example, we could examine how bacteria segregate their chromosomes during bacterial cell division or ask if there are specific highways for transport of mRNA molecules from the eukaryotic nucleus to the cytoplasm. Biological membranes are exceedingly important, and detailed knowledge of membrane features at the 10- to 100-nm length scale is critical. Ion channels in membranes could be imaged directly using resonances associated with specific ions. Imaging the membrane and cellular trafficking through the membrane may be the single most important application. Because over 50 percent of drug targets are G-coupled receptors or ion channels that are associated with cell signaling pathways and transport across the membrane, the pharmaceutical industry will benefit greatly if we can image receptor targets inside live, wet cells, by using specific ions at their L-edges.
Temporal or spatial relationships among individual molecules as they move within the cell cannot be captured by examining isolated static structures in vitro or by analyzing indirect biochemical or genetic data. Imaging organelle structure in frozen or red cells gives information about cellular context but is limited by its static snapshot view. Dynamic imaging of molecules in vivo is required to track structural changes over time and to obtain direct information about native structures within the cell. Despite the increasing demand to image cellular processes, however, the tools and reagents are not well developed. The linear accelerator coherent light source (LCLS) under construction and the proposed energy-recovery
linear accelerator light sources (ERL) will produce coherent hard X rays, offering stroboscopic atomic-scale imaging. These light sources will revolutionize X-ray imaging and related coherent applications, including probing complex materials dynamics by X-ray photon correlation spectroscopy (XPCS). In particular, the X-ray free-electron laser (XFEL) under construction will access dynamical processes at 1 to 0.001 picoseconds including solvent and vibrational relaxations as well as energy transfer during photosynthesis. Figure 5-2 shows how the new ultrabright free electron laser sources might provide breakthroughs in this area.
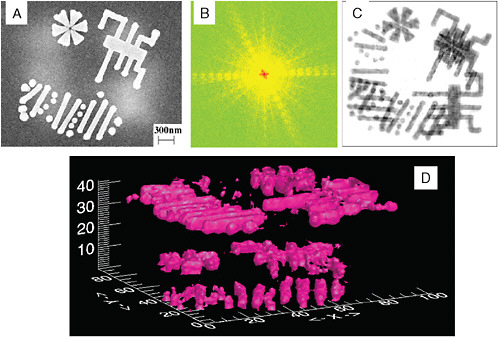
FIGURE 5-2
New Light Source Imaging
Imaging is an area where the ultrahigh brilliance and time structure of deep-UV lasers to soft X-ray, free electron lasers can have an important impact. The coherence of the source opens up imaging possibilities, including quantitative phase contrast imaging. The time structure allows for very rapid exposures over timescales below the characteristic damage time structure of some samples. This has implications for both condensed matter and biological samples. (A) A scanning electron microscope (SEM) image of a three-dimensional nanoscale specimen, which shows the surface structures but not the internal structures; (B) a coherent X-ray diffraction pattern from (A); (C) an image reconstructed from (B), showing both the surface and internal structures; (D) iso-surface rendering of a three-dimensional image reconstructed from 31 two-dimensional diffraction patterns. SOURCE: Courtesy of John Miao, University of California at Los Angeles and the California NanoSystems Initiative.
There is urgent need for a new form of system engineering at the interface between the nanoworld and the macro world. While we can now create some self-assembling nanostructures over which we have some design control, interfacing these nanoconstructs to the external macro world is difficult. The bottom-up approach might be able to give rise to massively parallel, heterogeneous, nanoscale self-assembled components, but integration of these nanoscale components into higher order structures and devices that resemble what living systems routinely accomplish is as yet out of our grasp. The integration of the two approaches, termed “hybrid top-down bottom-up” (HTBP), lies at the present cutting edge of technology development (http://www.sinam.org/). To quote a recent report from the Center for Scalable and Integrated Nano-Manufacturing (SINAM) at UCLA: “HTBP combines the best aspects of top-down and bottom-up techniques for massively parallel integration of heterogeneous nano-components into higher-order structures and devices. HTBP assembles by pick-and-place the nanoscale functional components, namely nano-LEGOs, into a defined pattern (a top-down approach); then the functional molecules attached to the nano-LEGOs can start to glue the adjacent nano-LEGOs by self-assembly, thus forming a stable structure (a bottom-up approach). Depending on designed functionalities, the nano-LEGOs can be in the form of nano-wire, quantum dots, DNA, protein, and other functional entities” (Zhang et al., 2004, pp. 126-127). Figure 5-3 shows some of the progress being made in interfacing bottom-up with top-down technologies.
THEORY AND SIMULATIONS
The diverse sizes and compositions of the heterogeneous molecules synthesized in biological environments generate a multitude of correlated phenomena on time and length scales that cannot be described with present analytical and numerical techniques. The understanding of biological processes requires the development of new theoretical approaches, modeling algorithms, and accurate effective potentials that bridge these scales. Biomolecules within the cell—in chromosomes, for example—are assembled into strong structures at short length scales but organized into soft networks at large length scales (Marko, 2008). Networks of fibers give mechanical integrity permitting interactions between molecules, possibly via compositional gradients and other long-range fields. In principle, compositional gradients result from the competition of entropy, which favors homogeneous mixing of the various components, and specific and nonspecific interactions among the molecules, which favor the formation of dense systems. The understanding of these entities requires calculations of entropy in systems with long-range interactions, transport in heterogeneous media, and interactions in inhomogeneous fluctuating environments. Moreover, since the functionality of biological organizations is dictated in part by the symmetries or lack of symmetries of their components, it is imperative to understand how symmetries are generated and broken in biologi-
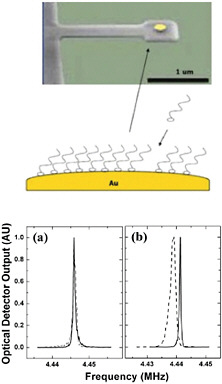
FIGURE 5-3
Interfacing Technologies
Microelectronicmechanical systems (MEMS), a top-down technology, can be combined with self-assembling monolayers, a bottom-up technology, to create extremely sensitive, label-free biosensors. A typical sensor consists of a cantilevered high-Q resonant MEMS on which the self-assembled monolayers are affixed. Here, a gold dot is placed on the cantilever to achieve adhesion for a self-assembled thiol array (top). When a molecule of the species to be detected attaches to a member of the thiol array, the resonant frequency of the MEMS shifts (bottom), thereby generating a measurable signal. Several methods for monitoring the resonant shifts and transferring the signal to external circuitry have been proposed, including direct eloectromechanical activity, wireless signals, and all-optical systems where the signal is measured using waveguide gradient forces. SOURCE: top, Harold Craigshead, Cornell University; bottom, Ilic et al., 2004, reprinted with permission from Journal of Applied Physics 95: 33694-33370, copyright 2004, American Institute of Physics.
cal media. Figure 5-4 shows some examples of symmetries found in biology that resemble symmetries found in assemblies of charged molecules. Finally, concepts developed in condensed matter theory to describe how emergent phenomena arise should have much to contribute to any question where interactions between many constituents is important.
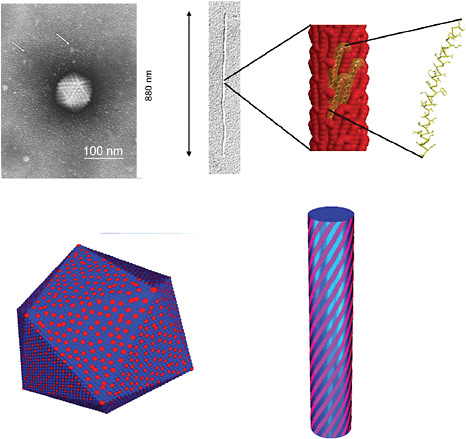
FIGURE 5-4
Symmetries in Nanostructures and Computational Challenges
The emergence and breaking of symmetries, such as rotational, translational, mirror symmetry, and chirality are known to be essential for generating functionalities at the molecular level. For instance, a conjecture has been proposed that Coulomb interactions are a means by which broken symmetries, in particular chirality, can arise at the nanoscale from the ordering of charged molecules on the surface of fibers. This may be particularly relevant in biology since in aqueous media the electrostatic energy is of the same order as the thermal energy. The figures show how basic structures and transitions induced by changes in ionic strength of co-assemblies of charged molecules, such as viruses and DNA-protein complexes, can be explored with statistical mechanics. Examples include virus structures, such as shown in the upper left, that have similar structural arrangements as obtained in models by the faceting of ionic shells into icosahedra, as seen in the lower left. Virians that assemble at bacterial membranes, as shown in the upper right, show structural similarities to the results of models computing the optimal arrangement of charged stripes over a cylindrical fiber, shown in the lower right. SOURCES: upper left, Biel, 2006; upper right, Marvin et al., 2006, reprinted with permission from Elsevier; lower left, Vernizzi and de la Cruz, 2007; lower right, Vernizzi et al., 2009, courtesy of the Proceedings of the National Academy of Sciences.
One approach for exploring collective behavior in physical and biochemical systems is by using complex network theory (Newman, 2008). Graphic-theoretic and statistical physics tools integrate the increasingly available information about the components of biological systems, such as genes and proteins in a cell, into a framework that can describe system-level properties. Previous network paradigms have addressed structural properties, including the determination of correlations between hierarchical structures and global properties, of complex systems. These theories can be extended to include nonlinear dynamical effects. For example, system-level analysis capable of describing and modeling the integrated functional behavior of complex systems can provide a network-based approach to control and recover metabolic function in faulty or suboptimally operating cells (Motter et al., 2008). This approach is based on identifying local modifications of the underlying network structure that can drive the system to a desired global functional behavior and can be used, for example, to identify synthetic rescues—gene pairs in which the deletion of one gene is lethal but the concurrent deletion of a second gene rescues cell viability. Besides its implications for the transformation of materials and the discovery of multitarget drugs, this approach is extremely versatile and promises to deepen our understanding of the interplay between network structure and dynamics in a variety of systems. Additional efforts that promise to lead to further advances in this field include exploratory network analysis and its variants.
The understanding of biological processes via computational methods requires force field development and coarse-grained approaches that include solvent effects within molecular dynamics, which could be developed by using physicalchemical models extended to accurately bridge length and timescales. For example, one of the main problems in computational protein folding is the question of timescales. The dynamics of individual amino acids is, in general, several orders of magnitude faster than the entire folding process of the protein. Most molecular dynamics simulations are plagued by this problem, and although preliminary efforts in this direction exist (see Sega et al., 2007), more needs to be done. Other issues in this field include the need to improve conformational search strategies for large biomolecules and a more accurate treatment of polar interactions that are crucial for identifying enzyme active sites and general properties of the protein surface. Furthermore, the role played by electrostatic interactions in shaping the folding landscape (and therefore the thermodynamics and kinetics of the folding) is far from being understood. The problem stems from the computational challenge in simulating electrostatic effects in complex environments (different dielectrics, boundary conditions, polar molecules) correctly and efficiently. The problem is still waiting for some new, and probably revolutionary, progress. Electrostatics is also crucial in the study of large structures. For studies of processes at large timescales, algorithms that accurately treat rigid-body dynamics and combine molecular dynamics with continuum boundary conditions methods may be needed. Theory is essential to construct
both these and other algorithms required to describe complex biological processes and environments. Modeling will aid our understanding only if the algorithms are developed using appropriate physical arguments and mathematics.
COLLECTIVE DYNAMICS
As has been emphasized in previous chapters, the behavior of an individual cell does not determine the behavior of a collection of cells. Studying the dynamics of single cells underlies studying the collective dynamics of tissue, the extraordinarily complex assembly of cells and connective components that results in the creation of high-order plants and animals. Certainly no structure is more complex and mysterious than the human brain. Just the interconnect complexity of the human brain is truly staggering and dwarfs any foreseeable implementation of the Internet. Since each neuron has on the order of 104 interconnects, and there are on the order of 1012 neurons, the number of neuronal interconnects in the brain, which is a three-dimensional system, is on the order of 251014, a staggering number. The dynamics of these interconnections presumably results in the phenomena of consciousness, yet we have precious few tools to probe noninvasively deep into tissue with high spatial resolution. What is the enabling technology that can probe at the micron scale in centimeter depths within tissue?
In the interest of brevity, only two potentially enabling technologies are discussed here: (1) two-photon fluorescence imaging and (2) magnetic resonance imaging (MRI). Both technologies are top-down techniques and have their strengths and weaknesses. Two-photon imaging’s greatest strength lies in its potential spatial resolution of submicrons within tissue. However, two-photon imaging requires the use of externally applied fluorescent probes or engineered cells that express fluorescent proteins. Because such visible light scatters, it limits penetration depths to well under a centimeter under optimal conditions. MRI’s great promise is that it can do whole-tissue three-dimensional imaging, yet it is limited in its spatial resolution to, at best, the millimeter length scale. This constraint is due to Gibbs ringing, which is an inevitable artifact in MRI caused by truncating k-space. A corollary to MRI is functional MRI (fMRI), which uses changes in the metabolic state, such as oxygen concentration within tissue, to develop a spatial image of metabolic activity in whole tissue. This is a truly transformative technology, with applications from oncology to brain activity, and it is the closest thing we have to a technology that allows deep imaging of tissue. However, it is not yet good enough; the sensitivity of MRI is notoriously low due to the small magnetic moment of the nuclei; the spatial resolution is coarse; and the present cost of superconducting MRI magnets is prohibitive.
These technologies are still at the development stage, with complementary strengths and weaknesses, and in some sense all have fatal technology flaws that prevent them from achieving the spatial and temporal information we need from
whole tissue analysis. It is difficult at this point in technology development to see what future technologies could be possibly developed to provide the answers we seek. The committee believes there is a powerful need to push for new enabling technologies for deep-imaging of tissue connectivity, metabolism, and dynamics at the 10-cm depth scale and at the 100-micron (at least) length scale.
Figure 5-5 shows an example of the remarkable three-dimensional imaging now possible using diffusion tensor imaging (DTI).
COMPLEX COMMUNITY SIGNALS AND SHARED RESOURCES AT LARGE LENGTH SCALES
So far, this chapter has only discussed the tools available for studying biology at the subcellular, cellular, and organism levels. Biological communities exist at many different length scales, however, from the local interactions between cells in tissue all the way up to the massive forests and plains that cover our land masses and the marine communities that exist in the oceans. These communities are in constant chemical communication with one another and reacting to the signals being sent and the production and consumption of metabolites, yet we lack the exquisitely sensitive and selective tools needed to understand the flow of chemical information among their inhabitants. These signals have profound importance: Organisms release metabolites in order to live, and they not only signal for cooperation but also present potential targets of opportunity for predators and parasites. The connection to evolution and fitness is clear, as is the connection to bioterrorism of a natural kind caused by humans.
For example, the coastal marine waters that encircle the continents are an extraordinarily important and imperiled ecology. Fully 60 percent of the world’s population lives within the coastal zone (100 km from the coastline), and about 20 percent of the world’s food comes from the sea. In spite of the fact that 75 percent of the Earth’s surface is covered by water, most of the ocean resources come from the far smaller coastal waters.
The coastal marine ecosystem is under increasing stress that is out of proportion to the stress experienced by other critical ecosystems as the world’s population continues to grow. Developing nations have historically drawn many of their resources from the sea and the coastal marine environment. This exploitation of the sea is increasing as the population grows, predominantly in large coastal urban areas. Developed nations, such as the United States, find themselves looking increasingly to the coastal marine environment for resources. Yet, in spite of this ever-increasing emphasis on a small resource and its exploitation, we are also seeing dramatic changes in the coastal marine ecosystems because of our failure to understand and protect this region. There is an urgent need to develop satellite imaging technologies that give us detailed temperature, cell density, metabolite concentrations, dissolved oxygen levels,
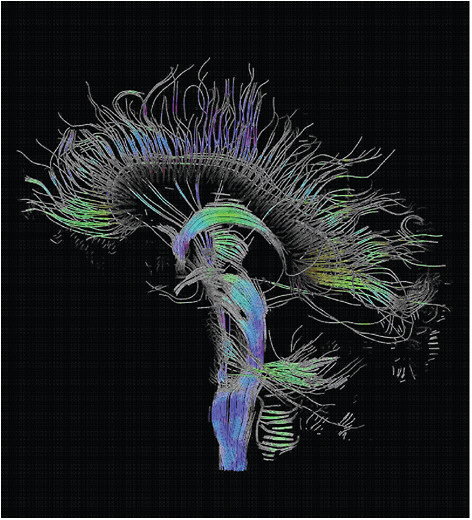
FIGURE 5-5
Three-Dimensional Diffusion-Tensor Imaging
Diffusion tensor imaging (DTI) is an MRI method that takes advantage of the ease with which water diffuses in various types of tissue, directly reflecting the internal fibrous structure of that tissue. By compiling many MRI measurements of the polarized water in a biological sample and extracting information from those images about the rates at which water is diffusing and its preferred directional paths, tensor mappings of the water diffusion can be developed and used to generate very detailed images of the tissue being measured. The figure visualizes a DTI measurement of the human brain. Depicted are reconstructed fiber tracts that run through the midsagittal plane. Especially prominent are the U-shaped fibers that connect the two hemispheres through the corpus callosum (the fibers come out of the image plane and consequently bend towards the top) and the fiber tracts that descend toward the spine (blue, within the image plane). SOURCE: T. Schultz, University of Chicago.
and the like of organisms and organism interactions in the marine environment. These top-down active and passive imaging technologies are increasingly powerful but still probe only a small range of the key processes and locations. Figure 5-6 shows satellite imaging of biological dead zones in the Gulf of Mexico. These zones are the result of an overabundance of nutrients from fertilizer run-off, followed by hypoxia, the condition in which bottom water oxygen concentrations are less than 2 mg/L, causing what is known as eutrophication.
Researchers have many tools and techniques at their disposal with which to study biological systems. These tools allow for the study of cells, organisms, and
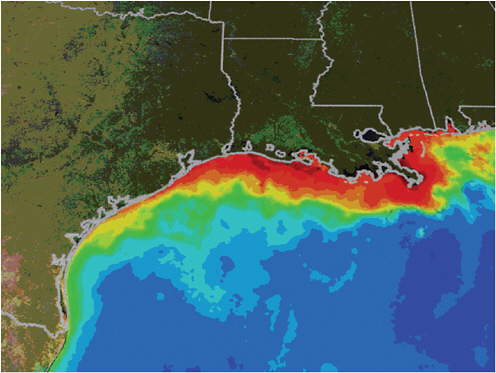
FIGURE 5-6
Satellite Imagery of Eutrophication along the U.S. Gulf Coast
Summertime satellite observations of ocean color from MODIS/Aqua show very turbid waters, which may include large blooms of phytoplankton extending from the mouth of the Mississippi River all the way to the Texas coast. When these blooms die and sink to the bottom, bacterial decomposition strips oxygen from the surrounding water, creating an environment in which it is very difficult for marine life to survive. Reds and oranges represent high concentrations of phytoplankton and river sediment. Image taken by NASA and provided courtesy of the NASA Mississippi Dead Zone Web site. Available at http://serc.carleton.edu/microbelife/topics/deadzone/general.html.
ecosystems in great detail. It is obvious, however, that new techniques must be developed to study interactions at small and large scales. The top-down approach to instrument design and technique development will continue to be important for research; indeed, for some systems and size scales, it will likely be the only path available. On the molecular level, however, bottom-up technologies promise to make the direct study and control of subcellular interactions possible.
REFERENCES
Bertozzi, C.R., and L.L. Kiessling, 2001. Chemical glycobiology, Science 291: 2357.
Biel, S., 2006. Adenoviridae: Human adenovirus C. ICTVdB Management 00.001.0.01.001. Human adenovirus C. In ICTVdB —The Universal Virus Database, version 4. C. Büchen-Osmond, ed. New York, N.Y.: Columbia University.
Gibson, E. A., A. Paul, N. Wagner, R. Tobey, D. Gaudiosi, S. Backus, I. Christov, A. Aquila, E.M. Gullikson, D.T. Attwood, M.M. Murnane, and H.C. Kapteyn, 2003. Coherent soft x-ray generation in the water window with quasi-phase matching, Science 302: 95.
Ilic, B., H.G. Craighead, S. Krylov, W. Senaratne, C. Ober, and P. Neuzil, 2004. Attogram detection using nano-electromechanical oscillators, Journal of Applied Physics 95: 33694-3703.
Marko, J.F., 2008. Micromechanical studies of mitotic chromosomes, Chromosome Research 16: 469.
Marvin, D.A., L.C. Welsh, M.F. Symmons, W.R.P. Scott, and S.K. Straus, 2006. Molecular structure of fd (f1, M13) filamentous bacteriophage refined with respect to x-ray fibre diffraction and solid-state NMR data supports specific models of phage assembly at the bacterial membrane, Journal of Molecular Biology 355, 294-309.
Minton, A.P., 2006. How can biochemical reactions within cells differ from those in test tubes? Journal of Cell Science 119: 2863.
Motter, A.E., N. Gulbahce, N. Almaas, and A.L. Barabasi, 2008. Predicting synthetic rescues in metabolic networks, Molecular Systems Biology 4: 168.
National Research Council, 2009. A New Biology for the 21st Century. Washington, D.C.: The National Academies Press.
Newman, M., 2008. The physics of networks, Physics Today 61: 33.
Rivas, G., F. Ferrone, and J. Herzfeld, 2004. Life in a crowded world, EMBO Reports 5: 23.
Sayre, D., 1980. Imaging Processes and Coherence in Physics. Springer Lecture Notes in Physics, Vol. 112, M. Schlenker et al., eds. Berlin: Springer Vertag pp. 229-235.
Sega, M., P. Faccioli, F. Pederiva, G. Garberoglio, and H. Orland, 2007. Quantitative protein dynamics from dominant folding pathways, Physical Review Letters 99: 118102.
Tsuyama, N., H. Mizuno, E. Tokunaga, and T. Masujima, 2008. Live single-cell molecular analysis by video-mass spectrometry, Analytical Sciences 24: 559.
Vernizzi, G., and M.O. de la Cruz, 2007. Faceting ionic shells into icosahedra via electrostatics, Proceedings of the National Academy of Sciences 104: 18382.
Vernizzi, G., K.L. Kohlstedt, and M.O. de la Cruz, 2009. The electrostatic origin of chiral patterns on nanofibers, Soft Matter 5: 736.
Whitesides, G. M., and B. Grzybowski, 2002. Self-assembly at all scales, Science 295: 2418.
Yu, J., J. Xiao, X.J. Ren, K.Q. Lao, and X.S. Xie, 2006. Probing gene expression in live cells, one protein molecule at a time, Science 311: 1600.
Zhang, C., M. Su, Y. He, X. Zhao, P. Fang, A.E. Ribbe, W. Jiang, and C. Mao, 2008. Conformational flexibility facilitates self-assembly of complex DNA nanostructures, Proceedings of the National Academy of Sciences of the United States of America 105: 10665.
Zhang, X., C. Sun, and N. Fang, 2004. Manufacturing at nanoscale: Top-down, bottom-up and system engineering, Journal of Nanoparticle Research 6:125.


















