
3
The Urgent Need for Regulatory Science
While the world of drug discovery and development has undergone revolutionary change, shifting from cellular to molecular and gene-based approaches, FDA’s evaluation methods have remained largely unchanged over the last half century.
FDA Science Board, 2007
CHALLENGES FACED BY FDA
Congresswoman Rosa DeLauro, keynote speaker for the workshop, explained that the focus on regulatory science is a natural outcome of the drug safety issues that have surfaced in recent years. As the government investigates the origins and causes of these issues, such as contaminated heparin supplies and postmarket adverse events, one area of focus is the extent to which breaches are forming along the drug development path. Specifically, DeLauro cited “most fundamentally, [the] sheer lack of resources at the agency’s disposal.” She also expressed concern that initiatives aimed at accelerating approval could omit safety steps in an effort to speed up patients’ access to new therapies. In addition, observed DeLauro, in 2009 the Government Accountability Office (GAO) released a report (GAO, 2009) alerting FDA to a loophole whereby Class III medical devices (e.g., pacemakers) were being approved without certain essential safety measures and in noncompliance with the premarket safety steps mandated by the Safe Medical Devices Act of 1990.1
According to DeLauro, despite progress made at FDA since 2007 and the enactment of the Food and Drug Administration Amendments Act of 2007, the ad hoc nature of the problems faced by the agency, such as continually emerging safety recalls, forces the agency to act reactively to issues as they arise instead of assuming a leadership role and proactively address-
|
BOX 3-1 Potential Contributions of Regulatory Science to Cancer Therapy Ellen Sigal, Chair and Founder, Friends of Cancer Research, suggested key areas in cancer care that stand to benefit from increased regulatory science capacity at FDA:
|
ing regulatory needs. DeLauro suggested that this characterization of the agency reflects a common public sentiment.
Although FDA has unique opportunities to improve the public health through its access to a diversified workforce and a wealth of data, accomplishing this goal is a daunting task. According to Drazen, a key challenge is that the agency is often forced to “take limited data … based on small numbers of people’s response to a given therapeutic approach—and determine what will happen when this therapy is unleashed to very large numbers of people.”
Another challenge faced by FDA is the rapid emergence of new technologies. A theme among the speakers was that the agency currently is not supported sufficiently to deal with the masses of data that come from large investments in such areas as genomics and health information technology. At the same time, emerging technologies cannot meet the demand for new therapies without coordinated effort from regulatory bodies. The presentations summarized below focused on specific areas of emerging technology and the scientific gaps caused by the lack of strong regulatory science. Box 3-1 lists some ways in which regulatory science could contribute to the development of therapies in the specific area of cancer treatment.
THE NEED FOR REGULATORY SCIENCE IN PREDICTING OR ADDRESSING RARE ADVERSE REACTIONS2
The current safety-focused environment can serve to hinder innovation, as drug companies often are averse to risking investment in the development of new drugs not yet proven safe. The hope is that regulatory science can mitigate this problem by improving risk detection and creating rewards for discovery.
To illustrate this point, Watkins referred to a recent case involving FDA’s review of a New Drug Application (NDA). In this case, FDA supported the NDA sponsor’s conclusion about the drug’s effectiveness; however, 2 of the 4,000 patients treated in Phase III clinical trials developed elevations in liver chemistry. In light of this finding of possible liver toxicity in the 2 patients, the sponsor was required to conduct a new safety study that involved treating 20,000 patients with the drug or a comparator for a full year.
Given the current model of drug development, in which the drug sponsor is responsible for the bulk of clinical testing, requiring such follow-up based on limited experience will reduce the drug’s patent life by approximately 3 years. Together, moreover, the cost of conducting the trials and the lost profits from the drug’s shortened patent life will cost the company millions of dollars, which will ultimately be passed on to the consumer.
The core issue in Watkins’ example is the lack of understanding of idiosyncratic reactions, or serious adverse events (SAEs), in rare individuals for drugs that are otherwise proven safe. These idiosyncratic reactions are time-consuming and costly to address through the current regulatory system, and can result in the failure of effective drugs with the potential to reach previously untreated patients. Initiatives have been undertaken to improve the scientific knowledge surrounding these idiosyncratic reactions, such as the Serious Adverse Events Consortium3 and the National Institutes of Health (NIH)-funded Drug-Induced Liver Injury Network.4 Another such initiative, the Hamner Institute’s study of inbred mice panels, is described in Box 3-2. Nonetheless, the problem persists, as there has
|
BOX 3-2 Hamner Institute’s Study of Inbred Mice Panels In an attempt to identify mechanisms of idiosyncratic toxicities, the Hamner Institute created panels of inbred, genetically engineered mice to recreate the genetic heterogeneity found in patient populations. By injecting a single high dose of acetaminophen into 36 different inbred mice strains, the institute was able to show the effect of the injection among various genetic strains (see Figure 3-1 below). Following analysis, specific genetic variants pointed to high susceptibility to acetaminophen—resulting in severe liver damage—whereas other genetic variants showed no effects from the injection. This work led to the discovery of a new risk factor called CD44, which was then shown to be an indicator for mild acetaminophen liver toxicity in healthy human volunteers. CD44 is present on the surface of white blood cells, and is also found on liver progenitor cells and may play a role in repair. Following its success with inbred mice panels, the Hamner Institute is now partnering with the pharmaceutical industry to study proprietary drugs that have shown success in animal safety and preclinical studies but failed at various stages of the drug’s life cycle due to severe toxicities. According to Watkins, such studies utilize academia’s existing resources and access patient populations not available to FDA. 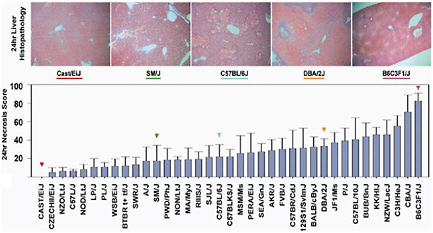 FIGURE 3-1 Graphic presentation of the effect of acetaminophen on various mouse gene strains. The grey bars indicate high liver injury. SOURCE: Harrill et al., 2009. |
been a general inability to access these resources for designing better trials or preventing drug failures in the clinical stages of drug development.
Watkins suggested that a regulatory science infrastructure that would partner the discovery science of academia with the regulatory efforts of FDA could mitigate the challenge to drug development posed by rare SAEs. While academic studies may generate a great deal of data, the ultimate value of the data in improving the public health will come from a regulatory agency’s ability to synthesize that information into a usable and useful form.
THE NEED FOR REGULATORY SCIENCE IN GENOMICS5
In the past 10 years, the field of genomics has made great strides in better understanding of the genetic mapping of organisms. These advances have opened up new possibilities for personalized medicine and the discovery of new drugs. Despite increased funding for research and an extensive literature on the human genome and genomics, however, there is a dearth of new medicines on the market. Treatments with genetic-based side effects are still used widely, and little remains known about how to translate advances in genomics into reliable diagnostics or guidance for practitioners and patients. Roses suggested that regulatory science can help fill these gaps by:
-
enhancing product development by minimizing the likelihood of imperfect data; and
-
making it possible to analyze and interpret data in regulatory submissions taking into account all products of the genome, all genomes, integrative biology, constructive pharmacology, and translational analyses.
There is a great deal of new science in the genomics arena, and FDA must move quickly to adjust its review processes and determinations accordingly. Roses referred to his own experience in discovery of the translocase of outer mitochondrial membrane 40 homolog (TOMM40) gene that was found to greatly increase precise prediction in the estimation of age of late-onset Alzheimer’s Disease for carriers of the 4 allele of the apolipoprotein E (APOE) gene, which is considered the most highly replicated genetic factor for risk and age of the disease (Roses et al., 2009). He cited the subsequent process to gain approval from FDA for conducting a clinical trial as one of success because the agency utilized its capac-
ity for regulatory science to nimbly analyze the scientific data and reach a decision. In particular, Roses highlighted the key factors of success in his experience.
Roses urged for the future establishment of regulatory science for the following reasons:
-
FDA review teams have access to the specific scientific expertise required to produce sound judgments on the safety and efficacy of the products they review; and
-
FDA reviewers of pharmacogenetics and outcome studies are able to balance retrospective and prospective data, benefits and risks, agnostic and hypothesis-driven approaches, clinical validity and epidemiological strengths, and validation and replication.
A regulatory science infrastructure is necessary to address these complex issues and develop consistent standards tailored to the science of genomics, said Roses.
Roses noted that it will be necessary to develop genetic diagnostics with clearly defined clinical parameters as well as reproducible methodologies, informed by an overarching concern for the safety and efficacy of products. Since every individual inherits a single strand of DNA from each parent, regulatory bodies must look to individuals’ genetics to develop predictive data, rather than to the genome-wide association studies that are commonly discussed in the existing literature, according to Roses. An analogy is the approach taken by the typical physician to make an accurate treatment prediction by tailoring the analysis to the specific patient instead of considering some percentage of the clinical population that suffers an adverse event.
Influenza vaccines and human immunodeficiency virus (HIV) mutation are two examples of the successful use of regulatory science at FDA. In the case of influenza vaccines, phylogenetic mapping previously conducted by academic laboratories had prepared FDA for the incoming data, and the agency was quickly able to acquire the necessary expertise to perform due diligence in an efficient regulatory support process.
Regulatory science at FDA, warned Roses, may not be the same as that at NIH: “[Genomics] is very different from finding adverse events. This is about making drugs. This is about discovering which ones work, with fewer people, so you can do trials that are faster and safer.” Therefore, Roses said, FDA’s regulatory science in genomics must be able to balance efficacy determination with the identification of safety issues arising from adverse events.
Conversely, the use of genomic information can enhance the development of the discipline of regulatory science by showing how best to design
clinical trials and evaluate targeted therapies and assays for use in such therapies. Genomics could also help identify optimal analytic approaches for determining genomic characteristics in response to therapy and aid in the development of more efficient strategies for evaluating combination regimens aimed at molecular targets.
THE NEED FOR REGULATORY SCIENCE IN STATISTICAL DESIGN AND ANALYSIS6
Pressing issues in biostatistics stem from the lack of a regulatory science infrastructure in the field. Ellenberg called on her experience as head of the Office of Biostatistics and Epidemiology in FDA’s Center for Biologics Evaluation and Research (CBER) to emphasize the importance of on-the-job regulatory training that can be attained only by working at the agency for a period of time. To fully understand statistical problems in a regulatory setting, said Ellenberg, one must be an FDA statistician, an industry statistician who interacts frequently with FDA, or an academic statistician who has served on FDA advisory committees or consulted frequently for industry. Unfortunately, noted Ellenberg, most statisticians—including epidemiologists, computational biologists, and informaticians—do not seek FDA reviewer positions.
Ellenberg suggested further that, despite expectations for improved quantitative approaches, they will not eliminate the need for sufficiently large populations for safety assessment, adequate duration of follow-up for documentation of sustained efficacy and long-term safety, or long-term data to validate the use of surrogate endpoints. Due to the constant tension between efficiency in getting products to market and the adequacy of safety assessments in regulatory decision making, systematic approaches are needed to transform statistical data into educated action quickly and effectively. Ellenberg described a number of possible innovations in such approaches and associated areas of need.
Bayesian Methods
Adoption of Bayesian methods—a form of meta-analysis using evidence to update beliefs—is one way to build a regulatory science capability at FDA. Ellenberg described FDA staff as taking their responsibilities very seriously, being well versed in potential biases and distortions of traditional analytical approaches, and concerned with potential biases and distortions in newer or less familiar designs and analytical approaches.
The latter concern can often lead to extreme caution in accepting new approaches, such as Bayesian methods, which in turn may impede the agency’s fulfillment of its responsibilities in the long run.
Postmarket Safety Surveillance
Ellenberg remarked that postmarket safety surveillance is the topic with “the greatest likelihood of getting onto the front page of a newspaper.” It is a crucial area for regulatory decision making and a new area for quantitative methodology. Ellenberg urged the use of incentives, such as grants, to draw more statisticians to focus on postmarket surveillance methodology. In addition, she emphasized the importance of involvement by both premarket and postmarket FDA scientists in monitoring spontaneous data, particularly because the Sentinel Initiative is likely to generate a great deal of data in the future; conducting and evaluating meta-analyses of completed trials and improving understanding of such analyses; and designing and analyzing postmarket observational studies and clinical trials.
Assessment of Multiple Related Outcomes
The current regulatory approach to a new product is to require that a sponsor identify a single primary endpoint to avoid concerns about multiple comparisons of its products and the possibility of false-positive errors. However, drugs often have multiple benefits, which are likely to be highly correlated. Ellenberg described the sponsor’s frustration in being forced to “arbitrarily choose one outcome for submission to FDA.” Because existing statistical methods, including global methods,7 do not account for multiple comparisons, new regulatory methods that can calibrate the extent of correlation among the outcome variables are needed.
Adaptive Designs
The hope for adaptive designs is to telescope clinical trials into smaller, more efficient versions of themselves. Although many new approaches to adaptive designs have surfaced in the past 10 years, and some adaptation has already been built into traditional trials, many questions remain about how the adaptive designs will work:
-
How much can efficiency be increased by these designs?
-
How reliable are these designs?
-
Will the designs introduce more biases?
-
Will increasing the efficiency of answering questions about efficacy compromise safety, and in effect lead to the wrong answers more rapidly?
These are legitimate concerns that FDA will need to evaluate fully and systematically, as the agency is uniquely situated with access to a large body of diversified data and knowledge, noted Ellenberg. As a regulatory and public health agency, FDA has an opportunity to ensure that increased efficiencies in trial design can be achieved without compromising safety.
Comparative Effectiveness Research
Ellenberg acknowledged that both the value of comparisons of widely used treatments and the complexities of interpreting results in comparative effectiveness studies have long been appreciated at FDA. This is the case because the results of comparative effectiveness research are ultimately derived in the context of FDA’s supplemental applications and label changes. Thus, Ellenberg believes, the involvement of regulatory scientists in studying and developing an optimal design for comparative effectiveness research is critical.
Other Areas
Ellenberg cited a number of other areas in emerging statistical technologies that warrant a standardized, science-based system of regulatory decision making:
-
developing regulatory pathways for biosimilars;8
-
improving Phase I trial designs beyond cancer trials;
-
developing pediatric indications for drugs already studied in adults;
-
developing therapies for rare diseases;
-
identifying optimal dosage levels in Phase II and III studies; and
-
identifying safety signals during the translation phase from animal to first-in-human studies.
FDA statisticians, observed Ellenberg, have little discretionary time for methodological research. Conversely, research statisticians may not be informed of the regulatory constraints or pitfalls commonly known to regulatory scientists. Therefore, some approaches recommended by research scientists in published journals go unnoticed by agency scientists.
To make progress, said Ellenberg, two components are necessary: first, FDA statisticians who are adept at using newly developed approaches must be empowered to judge whether methods should be applied based on their appropriate scientific value; second, research statisticians must be knowledgeable about the regulatory environment so the advances created by their research will be relevant to, and take into account, issues faced by FDA. A regulatory science infrastructure can provide the mechanism to fill the gap between these two bodies of knowledge that otherwise delays innovation.










