
2
Challenges of Predicting Pathogenicity from Sequence
INTRODUCTION
Our committee was asked to “identify the scientific advances that would be necessary to permit serious consideration of developing and implementing an oversight system for Select Agents that is based on predicted features and properties encoded by nucleic acids rather than a relatively static list of specific agents and taxonomic definitions.” It is true that the microorganisms and toxins that currently make up the Select Agent list are defined by taxonomy and by their perceived importance to public health and security. They are (or are products of) pathogens, that is, microorganisms capable of causing disease. Most Select Agents are not typical of the common pathogenic microorganisms seen in human or animal medicine, or in agricultural practice. But Select Agents and more commonly encountered pathogenic microorganisms do share a number of biological properties. It is essential to understand that pathogenic microorganisms are not defined by “taxonomy”; it is very common for a given microbial species to have pathogenic and non-pathogenic members. Escherichia coli is found in the colon of virtually all humans and animals and is part of their indigenous flora. They are typically harmless. However, a genetically defined, and more recently sequence-defined, subgroup of E. coli is the most common cause of urinary tract infection in humans and dogs. From a taxonomic standpoint the microorganisms are unequivocally called Escherichia coli; from a genetic and sequence homology standpoint they are distinct categories of E. coli. Similarly, the taxonomic genus Yersinia includes Y. pestis, the causative agent of plague, and other Yersinia species that are certainly enteric pathogens but are not Select Agents, and other Yersinia species that are not known to be pathogens. The pathogens and the non-pathogens are not distinguished by taxonomy but can now be distinguished reasonably well with genetic and molecular analysis.
Is there a potential for developing and implementing an oversight system for Select Agents that is based on features and properties encoded by nucleic acids? The general answer to this question is yes. The committee believes, however, that the entire concept of “predictive” oversight is flawed in that (1) the current Select Agent list has a non-biological as well as a biological basis for existence and (2) functional “prediction” alone cannot provide a level of certainty sufficient to designate a microorganism as a Select Agent, whose possession is legally restricted. Nevertheless, oversight of novel pathogens, whether natural or synthetic, is clearly seen by policy makers and legal experts to be a necessary component of a comprehensive biosecurity strategy. We propose to discuss here only the biological factors relevant to establishing a sequence-based oversight system that is focused on identifying genes and gene products that are likely to be involved in survival and persistence of a microorganism and its interaction with a host. That would include genes of Select Agents, but would also include a far greater number of genes that are associated with pathogenicity (the ability to cause disease) and virulence (the degree of pathogenicity encoded by a given gene or group of genes). Understanding the basis of such an oversight system requires some understanding of the biology of pathogenicity and of the current limitations of genomic analysis.
THE ART OF SEQUENCE-BASED PREDICTION
It is clear that we are immersed in an age of genomics. As of December 27, 2009, the Web site Genomesonline.org (Genomesonline 2009) reported that 3,606 bacterial genomes were being sequenced and that complete DNA sequences of at least 712 distinct bacterial strains were in the public domain. The completed sequences include all the bacterial Select Agents and most common pathogens of humans, animals, and plants. Entrez Genomes contained 3,498 reference sequences for 2,374 viral genomes, including all of the Select Agents and common plant and animal viral pathogens.
The genomes of prokaryotes possess specific and relatively well-understood promoter sequences (signals), such as transcription factor binding sites, that are relatively easy to identify. The gene sequences that code for a protein occur as one contiguous open reading frame (ORF), which is typically many hundreds or thousands of base pairs long. The nucleotide compositions and frequency of use of stop codons (the punctuation between genes) are well known. Furthermore, protein-coding DNA has periodicities of occurrence and other statistical properties. Therefore, recognizing genes in prokaryotic systems is relatively straightforward, and there are well-designed algorithms to do it with high levels of accuracy.
However, identifying a gene and understanding its function are altogether different matters. At least one-fourth of genes that are identified in bacterial genomes, whether large or small, whether from pathogen or non-pathogen,
are “hypothetical” or of unknown function. Considering the long history of biochemical and genetic examination of microorganisms, it is daunting that so much of the “equipment” of microorganisms is still unknown. The hypothetical genes are in two categories: ones that are found in a variety of organisms, and ones that are peculiar to particular lineages. For many genome sequences, the only annotation that will be available for the foreseeable future will be based on computational predictions and comparisons with known functional elements in related microorganisms. Despite the hypothetical genes, it has been pointed out that much of the whole-genome sequence of many microorganisms, when viewed against a backdrop of 100 years or more of biochemistry and microbiology, is not at all a total surprise in that much that has been found was fully expected (Doolittle 1981). Moreover, the full genome itself is a tremendously valuable asset because for a given organism, it provides all the details: The entire “parts list” is on the table—even if we do not know where some of them go or what they do. Seeing an assemblage of parts should not be mistaken for understanding how the parts function. From the standpoint of the present report, for important pathogens, comparisons between strains can pinpoint differences between the virulent and the avirulent microorganisms, and comparisons between species can be informative about host or tissue specificity. Comparisons have become even more useful as we have factored in the complete genomes of the human and other animals that serve as microbial hosts. At the genetic level, genome comparisons begin to reveal the fundamental divergences of microbial life and their evolutionary origins. We have also begun to understand how pathogens got to where they are and, we know to some extent what to look for if we are trying to design a pathogenic microorganism.
The objective of a “predictive oversight” system would be to forecast with a high degree of certainty the pathogenic potential of sequences of
-
Single or small numbers of genes related to Select Agent toxins.
-
Genomes or genomic regions that are closely related to Select Agent pathogens.
-
Genomes or genomic regions of newly identified natural pathogens.
-
Novel genomic sequences that are designed and assembled by synthetic biology.
Sequence prediction in biology is a hierarchy of increasing difficulty that reflects the complexity of the particular system under analysis. The simplest of such predictions would probably be that of a protein, such as a toxin.1 Next
in order of predictive difficulty would be a genetic pathway (a group of co-regulated multiple proteins interacting in concert). The third most problematic set of sequences to evaluate as a means of forecasting function would be those of whole organisms alone in a controlled environment (with multiple pathways interacting in concert). The final and most difficult predictive situation would be one in which two or more organisms interact in their natural environment.2 It is this last level of complexity that gives rise to the key biological attributes of pathogenicity and transmissibility, factors that contribute to the criteria that form the basis of inclusion of an organism on the Select Agent list.
Predicting pathogenicity or transmissibility of a microorganism requires a detailed understanding of multiple attributes of both the pathogen and its host. It is a prediction problem of the greatest complexity. Using a single genomic sequence to predict the potential consequences of the interaction of a microorganism, or a microbial virulence determinant, with a host clearly is not within the bounds of contemporary biology. Current sequence prognostication methods are at best at the level of foretelling the function of an individual protein on the basis of its deduced amino acid sequence. Even with the availability of a high-resolution protein structure, projecting the activity of closely related molecules accurately is not straightforward. There is as yet little work that even attempts to make predictions at the next level, that of genetic or biochemical pathways.
Predicting Biological Function from Sequence
The integration of experimental and computational information suggests that the human genome encodes about 20,000 protein-coding genes and an unknown number of functional RNA molecules; the Bacillus anthracis (anthrax) genome encodes about 6,000 proteins; a large virus, such as the smallpox virus, encodes about 200 proteins; and small positive-strand or negative-strand viruses, such as coronaviruses and influenza viruses, encode 10-30 proteins. As noted above, although these expressed RNAs and proteins can be identified using computational approaches with relative certainty, assignment of function is problematic. Biological experiments are still needed to confirm computational predictions.
The dominant method of function “prediction” uses sequence homology software. The underlying principle of such an approach is that proteins are reused or modified for applications in similar functional systems in different species far more often than entirely new ones are introduced. Most proteins generally fall into a relatively small number of homologous protein families of related structure and usually of at least somewhat related function. For
example, the Pfam protein family database contains about 10,000 protein families that account for about 75 percent of all known proteins. Two proteins that diverged through evolution from a common ancestral sequence (“homologous” sequences) tend to have structural and functional characteristics in common. The sequence that governs the mechanism of action of a particular protein evolves slowly, whereas the sequence that affects how a protein interacts with a binding partner, such as a cellular receptor, evolves rapidly.3 If the function of one protein is known, some aspects of the functional annotation can be inferred for other homologous proteins. Computer programs for sequence-database homology search (such as BLAST, HMMER, and FASTA) are widely used to discern whether a newly annotated protein or RNA sequence is homologous to an already known sequence or sequence family.
Homology offers only a “low-resolution” prediction of function. Sequence-homology analysis can often determine what a protein is likely to do (such as, protein kinase, metalloprotease, or oxidoreductase) but generally will not reveal the biochemical pathway to which its proteins partner(s) belong or the particular residue(s) that will be the target or substrate for it. There are less well-developed computational prediction methods that may occasionally offer clues to help to answer the more detailed questions but, generally, such queries must be addressed directly with controlled laboratory experiments. For example, if a novel influenza-like genome were obtained, sequence analysis would certainly and immediately recognize the homologous parts: the genes that encode hemagglutinin (HA) (Pfam protein family database code PF00509), neuraminidase (NA) (PF00064), nucleoprotein (PF00506), the matrix proteins M1 (PF00598) and M2 (PF00599), the proteins NS1 (PF00600) and NS2 (PF00601), and the RNA-dependent RNA polymerase components PA (PF00603), PB1 (PF00602), and PB2 (PF00604). These families have tens of thousands of examples of sequences; on the basis of the known diversity, the statistical models used in sequence-homology analyses are often capable of recognizing sequences that are separated by hundreds of millions or even billions of years of evolution. The computational approaches would identify the general kinds of “parts” in a genome and would be able to determine whether an expected part were present, missing, or unexpectedly quite different from a currently known virus sequence component. We can recognize some molecular signatures that may be essential for maintaining effective pathogen-host inter-
action,4 replication efficiency and pathogenesis outcomes in natural, but not necessarily, alternative hosts. The identification of genera or species of pathogenic bacteria or viruses is likewise easily accomplished with DNA homology approaches. Those methods are used every day in clinical laboratories all over the world.
What could not be readily foretold from the sequence-homology analyses described above is whether the influenza-like virus is highly pathogenic for humans and other mammals, or whether a particular vaccine will protect against it. Those traits depend on a small number of genetic changes that evolve rapidly in ways that are not well understood; even subtle changes may have a profound biological effect. Those features change so rapidly, they do not correlate well with evolutionary history. Thus, sequence-homology analysis is less informative for such viral characteristics than for simply identifying genome parts components of an influenza or related virus. For example, there is a strong correlation between high pathogenicity and trypsin-independent cleavage of the influenza virus hemagglutinin. This is not a perfect means of prediction, however, inasmuch as the 1918 influenza virus, which was associated with 50 million deaths worldwide, has a cleavage site that appears from sequence analysis to be associated with low pathogenic potential (Box 2.1). Such poor predictive power from sequence analysis is likely to be common for many if not most, microbial virulence determinants because virulence is typically mutifactorial and is affected by details of molecular interactions between a microorganism and a specific target in a specific host. (See also Appendix G)
Protein Structure Prediction
Another possible route to prediction of function from sequence is to predict the folded 3D structure of a protein from its sequence and then use features of that structure (which evolve much more slowly than sequence) to infer catalysis, binding partners, or other functional properties. Function prediction based on structure has been one of the “Grand Challenge” problems in science for the last 50 years, since Anfinsen showed that the information to determine protein 3D structure is encoded in the linear amino-acid sequence (Haber and Anfinsen 1962).
Pure de novo structure prediction was essentially impossible until recently and is only occasionally successful even now. Progress has come mostly from the growing database of experimentally determined structures (the Protein
|
BOX 2.1 Influenza—Hemagglutinin Cleavage One of the most important sequence features of influenza virus A pathogenesis is a protease cleavage site in hemagglutinin (HA). Cleavage is required for HA to catalyze membrane fusion, a necessary step for viral infectivity. In viruses of low pathogenicity, this essential cleavage step tends to be catalyzed by a host-encoded protease (trypsin, in the human respiratory tract), so viral infectivity is limited by tissue distribution of the host protease. Conversely, an essential feature of highly pathogenic influenza viruses is the presence of mutations in the gene for HA that leads to trypsin-independent cleavage of the protein; such mutations enable the virus to infect a broader range of tissues. The HA protease cleavage site is a sequence of only about seven to 10 residues, and the sites that contain small insertions of a few basic residues (lysine and arginine) tend to be associated with trypsin independence. With such changes, HA becomes cleavable by widely distributed subtilisin-like proteases that have a consensus recognition sequence. Yet, the correlation of high pathogenicity and trypsin independent cleavage of HA is not perfect; the 1918 influenza virus, which was associated with 50 million deaths worldwide, has a cleavage site that appears from sequence analysis to be of the low pathogenic. |
Data Bank contains over 60,000; http://www.rcsb.org/pdb), which enable the modeling of new sequences on the basis of homology to known, related structures. Recent achievements have been impressive, as demonstrated by the Critical Assessment of protein Structure Prediction (CASP) competitions (Moult 2005). Two rather distinct current approaches achieve good predictions fairly often: One is inspired by protein evolution and can recognize and piece together distant sequence relationships (Zhang, Wang et al.), and the other is inspired by protein folding and uses a combination of physics and empirical data to construct a model (Raman, Vernon et al. 2009). Homology models are usually approximately correct (Keedy, Williams et al. 2009), and even de novo predictions sometimes succeed (Box 2.2).
Further prediction of binding or catalytic sites from successfully modeled 3D structures (López, Ezkurdia et al. 2009) or prediction of protein/protein binding modes by docking known components, as in the CAPRI (Critical Assessment of PRedicted Interaction) competition (Janin 2005), are also still successful only sometimes and only partially. Approximately correct homology models can enhance the power of purely sequence-based comparisons considerably, especially when they show that known functional residues are brought together into the right 3D relationships. Often, however, the critical biological details hinge on structural details that confer a difference in specificity or in regulation and that are exactly the most difficult places to achieve accurate prediction.
Overall, this route of prediction based on 3D structure is well worth encouraging for both practical and intellectual benefits, but its utility in a robust system of predicting Select Agents (for legal oversight) is still extremely far off.
Gene Regulation
If an organism’s virulence depends on specialized gene products, it must be able to use them when they are needed but not squander its metabolic energy in producing them aimlessly or risk having them detected and prematurely neutralized by host defenses. Consequently, regulating the expression of virulence factors is an additional essential complication of a pathogenic microorganism’s life. The host presents an array of conditions strikingly distinct from those
|
BOX 2.3 Ricin Ricin is a plant toxin isolated from the seeds of the castor bean plant, Ricinus communis. It inhibits protein synthesis in affected cells by modifying the ribosome; this leads to ribotoxic stress and eventually cell death. Ricin represents a family of toxins known as Ribosome Inactivating Proteins (RIPs) that are found throughout the plant and bacterial kingdoms. Despite their very different sequences and sometimes quite different structures, these toxins share three highly conserved amino acids that are responsible for their catalytic activity. One cautionary tale for the prediction of toxin activity from gene sequence comes from the work of Frankel and Robertus (Frankel, Welsh et al. 1990). They genetically mutated ricin amino acid glutamate-177 and predicted that the protein would be inactivated because this side chain is highly conserved and central to the catalytic activity of the toxin. Although more conservative mutations were inactive, when the glutamate was mutated down to an alanine residue, the enzyme still retained about 5 percent of the activity of the wild-type sequence—enough to slow growth of yeast cells sensitive to the toxin. Based on the structure of ricin, the researchers predicted that the nearby glutamate residue at position 208 was able to move and substitute in the reaction. They produced an inactivated ricin A chain only after mutating both glutamates 177 and 208; a crystal structure of the Ala-177 mutant showed that the carboxyl of Glu-208 did indeed move into the former position of catalytic Glu-177 (Kim, Misna et al. 1992). The genes encoding ricin (rtx genes) of the various castor bean cultivars have only one or two nucleotide differences in regions that do not affect protein structure or function. The R. communis genome would not be sequenced to determine the virulence of castor beans, but instead it would be assumed that unmodified beans contain active ricin toxin. Several research groups have genetically altered one or more of the conserved catalytic residues to produce inactive ricin A chain expressed recombinantly in E. coli in an attempt to produce a vaccine to protect against deliberate ricin poisoning (Munishkin and Wool 1995). |
of the outside environment, conditions that are not easily reproduced in the laboratory. In fact, laboratory culture conditions bias our understanding of microbial adaptation to natural environments. Vibrio cholerae, for example, is thought to persist without expression of virulence factors in brackish estuaries and other saline aquatic environments, sometimes in association with the chitinous exoskeletons of various marine organisms. Transition from that milieu to the contrasting environment of the human small intestinal lumen is accompanied by substantial genetic regulatory events.
The microbial cell is relatively simple, yet it possesses the means to detect, often simultaneously, changes in temperature, ionic conditions, oxygen concentration, pH, and calcium, iron, and other metal concentrations that might appear to be subtle signals but are essential for the precise mobilization of virulence determinants. Similarly, environmental regulatory signals prepare a microorganism for its transition from an extracellular to an intracellular state. For example, iron is a critical component of many cell metabolic processes; therefore, it is not surprising that animals rely on high-affinity iron-binding and iron-storage proteins to deprive microorganisms access to this nutrient, especially at the mucosal surface. In turn, most pathogens sense iron availability and induce or repress various iron acquisition systems accordingly. Moreover, many microorganisms produce toxins that are regulated by iron in such a way that low iron concentrations trigger toxin biosynthesis. Reversible regulation of the expression of virulence genes by temperature is common to many pathogens. Thus, a microorganism like E. coli that may be deposited in feces and live for long periods under conditions of nutrient depletion and low temperature, mobilizes its colonization-specific genes when it is returned to the warm mammalian body. The regulatory machinery used to accomplish that is an important feature of many pathogens, including Y. pestis and B. anthracis.
The number of well-characterized virulence regulatory systems is rapidly increasing, in part because of the development of rapid methods for screening gene expression on a genome-wide basis (for example, with the use of DNA microarrays). But, relatively little is known about either the specific environmental signals to which these systems respond and or the exact role of the responses in the course of human infection. One common mechanism of bacterial transduction of environmental signals involves two-component regulatory systems that act on gene expression, usually at the transcriptional level. Such systems make use of similar pairs of proteins: one protein of the pair spans the cytoplasmic membrane, contains a transmitter domain, and may act as a sensor of environmental stimuli; the other is a cytoplasmic protein (a “response regulator”) with a receiver domain and regulates responsive genes or proteins. Those regulatory systems are common both in pathogens and non-pathogens, so their detection by sequence analysis cannot be used as a reliable predictor of whether a microorganism is pathogenic.
The coordinated control of pathogenicity incorporates a regulon; a group
of operons or individual genes controlled by a common regulator, usually a protein activator or repressor. A regulon provides a means by which many genes can respond in concert to a particular stimulus. At other times, the same genes may respond to other signals independently. Global regulatory networks are a common feature of microbial virulence and basic microbial physiology, so their sequences, although often essential for a pathogen, are not reliable predictors of virulence. The apparent complexity of virulence regulation in a single microbial pathogen is magnified by the coexistence of multiple interacting (“cross-talking”) systems and by regulons within regulons.
Thus, the inherent pathogenicity of a microorganism can be greatly altered through regulation of virulence genes. It is extremely difficult to predict how even a single nucleotide change will affect regulation and thereby alter pathogenesis or the viability of the microorganism. (Additional detailed examples of the important role of regulation in allowing pathogens to respond to the environment of the human host are given in Appendix J.)
THE NATURE OF INFECTIOUS DISEASE AND THE ART OF PREDICTING PATHOGENICITY
The preceding sections have shown that several computational approaches have promise for predicting biological function from sequence. Can they be applied effectively to predict pathogenicity? If not, what is required to develop a predictive method that would suffice? To address this issue, we will first discuss the nature of infectious disease.
Infectious diseases affect all living things, from the smallest amoeba to insects, plants, and the largest mammals. The co-existence and co-evolution of microorganisms with their hosts is a dynamic equilibrium ranging from one extreme of mutualism in which both partners benefit from the interaction (for example, bacterial production of organic nitrogen for plants or of vitamin K for the human), to a relationship of commensalism in which one organism benefits but the other is unaffected, to another extreme of parasitism that benefits one partner to the detriment of the other.
Microorganisms are constant companions of plants and animals. Humans carry a vast indigenous microbial flora from shortly after birth until death, and the role of this human microbiome in human health and disease is the subject of considerable interest and recent investigation. Although it is biologically correct to say that most microorganisms that inhabit this planet are harmless to humans or may even benefit humankind, it is also true that humans have a prejudicial view of microorganisms and direct their focus to microorganisms as agents of disease. The biological reality is that most microbial infections are relatively benign and that symptoms of disease are sometimes the result of the human immune system’s response to infection rather than the product of the infecting microorganism (Box 2.4).
|
BOX 2.4 Categories of Microorganisms For humans, and to a lesser extent other animal and plant hosts, interactions with microorganisms are complex and are governed by the health status of the host and by the environment. On the basis of differences in such interactions, microorganisms can be categorized as follows:
|
Recognition of an organism as a pathogen is not always simple, because the interplay between genetic expression of the microorganism and the host is ever-changing. However, in the biological sense, pathogens possess the ability to cross anatomic barriers or breach other host defenses that limit the survival or replication of other microorganisms. The more complicated and relevant question, however, is why some microorganisms are pathogenic to humans or other
animals, yet other closely related microorganisms are not. That question can be approached from the point of view of the host or the microorganism:
-
What specific defense mechanisms of the host allow it to effectively suppress infection (entry, attachment, invasion, and replication) by certain microbes and not others?
-
What are the differences between microbial agents that cause disease and those that do not?
Pathogenicity depends on the biological context, so pathogenic traits (virulence factors) need to be defined in terms of their potential to be associated with infection and disease in a particular host. For example, the human gastrointestinal epithelium is exquisitely susceptible to cholera toxin when delivered during infection by Vibrio cholerae. Although the gut epithelium of most animals is also susceptible to the action of the toxin, the disease cholera is seen only in humans: This finding suggests that additional host-specific virulence factors are involved. The vulnerabilities of the mammalian immune defense mechanisms to factors used by microorganisms to exploit or overcome a specific host defense strategy are of critical importance in defining what is and what is not a microbial virulence factor. To restate the point simply, the nature of microbial virulence factors cannot be understood unless the factors are evaluated in the context of the biology of the host.
Most genes encoded by a pathogen are necessary for its replication and for “general housekeeping”5 and genes are in general shared by most microorganisms. Virulence genes can be considered to be specialized genes essential for survival of a particular organism in a particular environment, usually on or in a host. Even by that limited operational definition, most pathogenic and non-pathogenic bacteria and large viruses that inhabit animals and plants have large numbers of virulence genes. For example, the variola virus (smallpox virus) virulence gene family consists of 64 genes (of a genomic total of 193); however, the presence of a large number of virulence genes does not a priori guarantee pathogenicity. Camelpox and taterapox viruses are the closest orthopoxvirus relatives of variola virus and have similar number of virulence genes, but neither virus causes human disease. Cowpox virus has a larger genome than does variola virus and encodes a greater number of virulence genes, but it causes only a localized lesion in humans and was used by Jenner in the late 1700s to vaccinate against variola virus. Those observations illustrate that variola virus
pathogenicity is a complex trait, possibly due in part to subtle activity differences between the virulence genes of viruses that are pathogenic for humans and the genes of viruses that are not pathogenic for humans. Some host specific pathogenicity can result even from the loss of function (see Appendix J). In the case of bacteria, the picture is a little clearer inasmuch as bacterial pathogens usually have virulence genes that are not present in their non-pathogenic relatives, and this distribution suggests that bacteria evolve to become pathogens by acquiring virulence determinants. For example, Salmonella and E. coli evolved from a common ancestor, but Salmonella acquired genes distinct from those of E. coli that permitted it to cross the mucosal barrier of the gastrointestinal tract. In contrast, Yersinia pestis adapted to life in a flea and in rodents and has retained most of the genes contributed by its most recent ancestor Y. pseudotuberculosis, which is an enteric pathogen. Some of the Y. pseudotuberculosis genes have been inactivated; they are not necessary for Y. pestis survival or they interfered with its new “lifestyle.” In parallel, microbe–host interactions play a critical role in regulating disease severity and distribution in human, animal, and plant populations. It is now generally accepted that pathogenic microorganisms have shaped the genetic population structure of humans and vice versa (see Appendix H).
What Is the Origin of Bacterial Pathogenicity? What Makes a Pathogen?
Microbial evolution continues to challenge the state of human health in part because of the size of the microbial universe; macroscale changes in human, animal, and plant interactions; and the dynamic nature of genomic alterations that result from active gene flow between microorganisms. The net effect of evolution on an organism is a balance between selective pressure in the environment of that organism and the generation of relevant changes in the microorganism through mutation, gene duplication, and horizontal gene transfer.
Most medically important microorganisms have pathogenic and non-pathogenic strains, and virulence factors were well known and characterized long before the advent of the genome projects. However, what we have learned about pathogenicity from the examination of microbial chromosomes has been surprising and useful. One of the most important findings both from the perspective of understanding the biology of pathogenicity and from the standpoint this committee’s task, is that pathogenic bacteria often contain clusters of genes, called pathogenicity islands (PAIs), that are not present in related non-pathogenic bacteria. Those acquired genes have several features (for example, the G+C content of their DNA and other molecular signatures) that indicate that they were once associated with mobile genetic elements, such as bacterial plasmids or bacteriophage (viruses that infect bacteria). Thus, it is clear that although Salmonella and E. coli evolved from a common ancestor, Salmonella
has acquired genes distinct from E. coli that permit it to invade gut epithelial cells. As noted, E. coli strains vary greatly in that some commonly cause disease whereas the majority of E. coli strains are universally restricted to the commensal flora and never cause harm in an immunocompetent host. However, E. coli that “routinely” cause infections, such as urinary tract infections, and the notorious E. coli O157, have genes, indeed large clusters of genes, that are associated with their pathogenicity and are not found in non-pathogens. We now understand that uropathogenic, enterohemorrhagic, and extraintestinal types of E. coli all display mosaic genome structure, with hundreds of gene islands distinct to each type, that makes up as much as 40 percent of the overall gene content in each of these strains. Pathotypes are as distinct from one other as each is distinct from a nonpathogenic laboratory strain of E. coli. E. coli has an “open” pan-genome: with every new genome sequence, a new set of about 300 unique genes is discovered; this suggests continuing evolution of this species by gene acquisition. The O157 case is a good example of how pathogenicity requires biological context; this microorganism is asymptomatically carried by animals, but it can cause diseases ranging from simple, barely noticeable diarrhea to devastating colitis and death in humans.
In contrast, B. anthracis and other pathogens that have restricted environmental habitats display a “closed” pan-genome and a much greater fraction of shared genes. Nevertheless, B. anthracis contains two large plasmids, one of which has a 44.5-kb pathogenicity island that contains genes for (among other things) the toxin that can be lethal to humans. It also contains a sequence signature that suggests a history of gene shuffling and exchange. It is therefore not too surprising to learn that the sequence of the main chromosome of B. anthracis is remarkably similar to those of B. cereus and B. thuringiensis, which also inhabit the soil. B. cereus strains carrying part of the pathogenicity apparatus of B. anthracis are occasionally encountered and present a problem for those attempting to draw a line separating Select Agents from non-Select Agents. Although it may be tempting to think of microorganisms in a clear-cut pathogen and non-pathogen manner, the biological reality is much more complex. Far from black and white, microbial genomes are better described as a multicolor patchwork of sequences, see Figure 2.1 (Read, Peterson et al. 2003). It is also instructive to see that B. thuringiensis produces a toxin fatal to lepidopteran caterpillars; thus, there is a biological theme at play that involves microorganisms, their hosts, and the common mechanisms that microorganisms use to ensure their survival. The distinction of B. anthracis from related soil microorganisms, such as B. thuringiensis and B. cereus, is not always clear in a sequence homology context, whereas the sequence information makes good sense in a biological context. For a more in-depth discussion of pathogenic mechanisms and virulence genes with examples of Select Agents, see Appendix I.
It is perhaps difficult for non-specialists to appreciate that the smallest free-living organisms on the planet engage in a kind of primitive “sex life” that
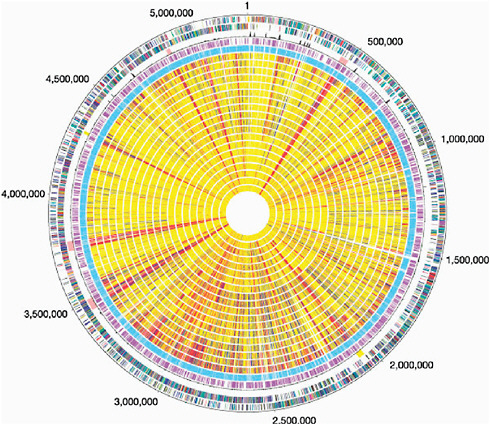
FIGURE 2.1 The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Outer circle, predicted coding regions on the plus strand colour-coded by role categories. Circle 2, predicted coding regions on the minus strand colour-coded by role categories. Circle 3, atypical nucleotide composition curve. Salmon colour, phage regions; yellow, other unique regions located around positions 2.0 and 4.3 Mb. Circle 4, genes not represented on the array. Circle 5, genes present on the array. Genes were classified into three groups: genes present in the query strain (shown yellow), genes absent in the query strain (red), and diverged genes (blue). Missing data are in grey. B. cereus group strains are displayed following the phylogeny of Fig. 2.1 (circle number, strain number): 6, B.c. 874; 7, B.c. 535; 8, B.c. 612; 9, B.w. 1143; 10, B.t. 248; 11, B.t. 442; 12, B.c. 14579; 13, B.t. 775; 14, B.c. 259; 15, B.t. 1031; 16, B.t. 251; 17, B.c. 607; 18, B.c. ATCC 10987; 19, B.c. 812; 20, B.c. 819; 21, B.c. 831; 22, B.t. 840; 23, B.c. 1123; 24, B.c. 816. Here we use B.c, B.t. and B.w to indicate B. cereus, B. thuringiensis and Bacillus weihenstephanensis, respectively (Read, Peterson, et al. 2003).
involves a rather promiscuous exchange of genetic information called lateral (or horizontal) gene transfer (see Box 2.5). The public is already aware that the emergence of antibiotic resistance in microorganisms is a serious health issue, and the mass media have reported that this emergence of resistance is effected by gene exchange among bacteria in which resistance is “donated” to previously susceptible microorganisms. The same is true of pathogenicity, although the transfer of genes associated with pathogenicity is not a single event. Rather, the genomic analysis of pathogenic microorganisms reveals that there have been multiple such events over a long period and that many other genetic selection events have occurred to finely hone only a relatively few microorganisms to emerge as “pathogens.”
A discussion of the current understanding of the emergence of pathogens based on the study of their genomes is beyond the scope of this report, although it can be readily found elsewhere by an interested reader. For present purposes,
it is sufficient to state that it is sometimes possible to recognize genes that arose by lateral gene transfer by simply examining genome sequences. The evolution of pathogenicity is a continuing process that has in some cases been aided by unintentional changes caused by human activity. Moreover, what we have viewed as progress in medicine and agriculture has provided new opportunities for microorganisms to adapt to humans and other organisms and to cause substantial morbidity and mortality (for example, overuse of antimicrobials leads to the selection and transfer of resistance).
A fundamental evolutionary push to bacterial pathogenicity results from gene acquisition. That is not simply a mechanism whereby microorganisms become pathogenic but a general strategy for specialization and success in some environmental niches that are highly competitive. What is the origin of the inherited genes that go on to be important for pathogenicity? The answer is not clear; microorganisms do not leave a fossil trail. However, what is important to our task is that to some extent their history can be read in their sequences. Bacterial adaption of the tactic of horizontal gene transfer to maximize diversity and to increase the opportunity for continuing evolution is most likely a reflection of their haploid state; that is, bacteria and viruses have only a single chromosome, so any genetic change, either beneficial or detrimental, is immediately apparent. Microorganisms have a difficult balancing act. They have the need to conserve fundamental characteristics while being able to try new combinations of genes. The sharing of genes among seemingly disparate microorganisms that occupy the same niche provides these microorganisms with an endless number of combinations of genes for evolutionary experimentation. If we trace the kinds of genes that appear to be peculiar to pathogens, we see that similar kinds of genes encode toxins or invasins found in organisms that infect plants, people and organisms in between. To some extent, we find that virulence genes evolved to prevent predation. For example, there are similarities between a microorganism being devoured by a free-living amoeba, which may have happened early in evolution, and being devoured by a macrophage in the alveolus of a mammalian lung, which evolved much later. In both cases, some organisms’ formation of capsules on their surfaces prevented this event; indeed, the complete genome sequences of many pathogens reveal that clusters of genes in the pneumocococcus, the meningococcus, and some SA are needed for capsule synthesis. That does not mean that capsular genes in all bacteria are suspect, but contemporary sequence analysis can tell us whether the capsular genes come from a pathogen or a non-pathogen. We can also trace how microorganisms evolved to be able to live within phagocytic cells. Legionella pneumophila prefers to parasitize and live in amoebae in nature but, under the right circumstances, can survive and replicate in the phagocytic cells of a human lung. Mycobacterium tuberculosis parasitizes human alveolar macrophages preferentially and can live and persist in these cells for a human lifetime. Yet, Legionella and mycobacteria utilize recognizably similar general mechanisms
for survival in the lung; these mechanisms are revealed in part in the genome sequences of the two organisms. Similarly, we find that many other genetically controlled characteristics can confer virulence on bacteria, including factors that enable the bacteria to attach to and disrupt host cells.
Genomic analysis also led to the discovery that many pathogenic bacteria use similar machinery for injecting proteins into the cytoplasm of the host’s cells. That common feature of pathogens of plants and humans follows, in a sense, the same kind of features found in viruses. While the nature of the proteins injected into animal and plant cells (effector proteins) may vary, there is often surprising homology among injection apparati across diverse pathogenic species. Perhaps even more striking was the finding that the sophisticated apparatus, which is akin to an assembled bacterial hypodermic syringe, is often found as part of a pathogenicity island in various bacterial pathogens in phylo-genetically distinct taxa. For example, the type III secretion system, has been identified in a variety of fully sequenced bacteria ranging from the obligate intracellular parasite Chlamydia trachomatis to the plague bacillus Y. pestis. Again, not all type III secretory apparatus genes are associated with pathogenicity, but the combination of a type III secretion system (or similar distinct secretory machinery) and the signature for effector proteins provides an immediate clue that one may be dealing with a pathogen. For more discussion and examples of how microorganisms become pathogens, see Appendix J.
Thus, bacteria have evolved a kind of biological network to exchange their genetic knowledge. That is as important for the evolution of the organisms that we call pathogens as it is for the organisms that fix nitrogen in the soil. The specialization of bacteria to live at the expense of other, more highly evolved organisms is a reflection of shared experiences. Genomic sequencing has revealed that human DNA contains traces of similar kinds of gene exchanges mediated by viruses and other mobile genetic elements. The novelty of this biology is impressive and shows that the road to bacterial, viral and parasitic specialization has ancient roots. The commonality that is still present in microbial sequences makes the goal of finding unique predictive sequences difficult. But it constitutes the most practical means of identifying pathogenic potential today, and it probably will lead to a much greater predictive capacity in the future.
The Evolution of Bacterial Host Specificity
If the acquisition of genes by horizontal gene transfer seems to be a driving force for bacterial pathogenicity, one might assume that gene loss plays only a minor role in pathogenicity. However, that is not always the case. The fine tuning of pathogenicity, especially with respect to host specificity, appears often to involve gene loss or gene rearrangement (see section on “Gene Loss” in Apendix J). Gene loss can occur by simple mutation (change or deletion of a single or a few nucleotides), which is often reversible. Irreversible gene
|
BOX 2.6
|
loss—the permanent loss of a part of, all of, or many of the genes from the chromosome—is common. For example, as noted above, Salmonella spp. have a number of pathogenicity islands that were acquired by horizontal gene transfer. The Salmonella group of pathogens is notable for its division into specific types that have a preference for a particular host (animal, bird or reptile). A core of identical genes are found in all salmonellae. However, genome comparisons of host-restricted or host-adapted Salmonella spp. (and indeed of other pathogens such as poxviruses) indicate that loss of gene function may be a common evolutionary mechanism through which host adaptation occurs. Gene loss appears to restrict the potential pathogenicity in the host to a more limited or specialized set of interactions, often through specific targeting to particular cell types or organs. For example, long term asymptomatic carriage of Salmonella, which are shed periodically and act as a reservoir of infection for susceptible hosts, occurs because the organisms establish a niche in the gall bladder, an organ that is immunologically protected. S. enterica serovar Typhimurium causes gastroenteritis in humans (typically food poisoning) but causes a chronic infection of mice where the organism can be carried and shed for the life of the animal. Conversely, S. enterica serovar Typhi infects humans exclusively, targets the cells of the reticuloendothelial system, and likewise can be shed asymptomatically for a lifetime (as in the case of “typhoid Mary”). The comparison of S. enterica Typhimurium with S. enterica Typhi shows that Typhi (and other host-restricted Salmonella) harbors a significantly higher proportion of genetic events that are associated with the loss of functional genetic sequences. Modification or loss of some effector proteins can affect whether a Salmonella strain will preferentially be successful in infecting and persisting in a human, a chicken, or a rodent.
In addition, genomic analysis has revealed that gene loss suffered by host-adapted pathogens is often reflected by the appearance of pseudogenes, which
are sequences of bases in the DNA that clearly resemble the sequences of known genes but differ from them in some crucial respect and have no function. Pseudogenes generally do not regain their function except by extensive recombination and their reversion might not lead to a change in pathogenicity. As the pattern of gene gain through horizontal gene transfer and subsequent gene loss through adaptation has been analyzed, it appears that gene loss may be a mechanism of targeting the invading pathogen preferentially to particular tissues or host cells and avoiding the potential stimulation of non-specific inflammation. For example, in both Salmonella and Yersinia, gene loss may be involved in the adaptation from a gastrointestinal to a systemic “lifestyle.” Similarly, genomic analysis seems to refute the popular belief that M. tuberculosis evolved from M. bovis through the adaptation of a bovine strain to the human host. In fact, M. tuberculosis contains chromosomal segments that have been deleted from M. bovis, which raises the converse possibility that humans transmitted tubercle bacilli to animals and those bacilli subsequently evolved into M. bovis.
This is not simply interesting basic science and evolutionary biology. As genomic analysis has been broadly applied to pathogenic microorganisms, analysis of irreversible genetic events—such as chromosomal deletions (large sequence polymorphisms), single nucleotide polymorphisms (SNPs), and direct repeat content (“spoligotype”) patterns—has permitted us to decipher the phylogeny that has occurred during the evolution of pathogenicity. On one hand, we have begun to uncover and understand the mechanism used by nature to “design” a pathogen; this mechanism becomes a blueprint that may be followed by those who would wish to design therapeutics, and by those with more sinister intent who would wish to design novel pathogens. The loss of genetic information, as well as the gain of genetic information, can be important, and this adds to the present uncertainty of predicting pathogenicity on the basis of sequence. On the other hand, as we learn more about pathogen evolution from our studies of nature, we also learn the rules for eventually detecting a pattern of sequences that could be used for monitoring organisms from the wild or those synthesized in the laboratory (see Box 2.7).
The Parallels in the Evolution of Pathogenicity in the Large Viruses
A virus’ genes are associated with its ability to replicate and persist in a specific host cell. However, viral pathogenicity is as dependent on the biological context of the host as it is on the viral agent. Rabies can be carried in a bat harmlessly, but is almost always lethal for an untreated, infected human. An infected human is a biological dead end for both the virus and the host. The evolution of pathogenicity in bacteria, viruses, and eukaryotic pathogens involved horizontal gene transfer. For example, the evolution of the large nuclear and cytoplasmic DNA viruses that gave rise to poxviruses was dominated by
|
BOX 2.7
|
expansion of paralogous gene families and acquisition—by horizontal gene transfer—of numerous genes from early eukaryotic hosts, other viruses and, rarely, bacteria. Comparative genomic studies of poxviruses support a monophyletic origin from a distant ancestral virus on the basis of the presence of 41 homologous genes common to 16 distantly related viruses that have with large DNA genomes (about 150 to 900-kbp predicted protein-coding genes) (Iyer, Balaji et al. 2006). Those 41 genes are a subset of 90 highly conserved core genes (about 100-kbp) common to all poxviruses, which are at the center of the genome, which encode proteins necessary for replication, transcription, assembly of capsid and the acquisition of a membrane (Upton, Slack et al. 2003; Lefkowitz, Wang et al. 2006). The genes at the flanks of the genome (about 60-100 genes) evolve at a rate distinct from that of the essential genes of the central core, and are more often involved with the mitigation or modulation of cellular processes and whole organism responses to infection, (such as apoptosis, ubiquitin signaling, and cytokine signaling) (Moss 2007). Although the progenitor of poxviruses evolved by gene gain from an ancestor common to large nuclear and cytoplasmic DNA viruses, poxvirus species adapted to new hosts through gene loss, keeping the genes that were necessary to parasitize one particular environmental niche successfully (Upton, Slack et al. 2003; Odom, Curtis Hendrickson et al. 2009). In the case of poxvirus pathogens, variola and molluscum contagiosum viruses, this resulted in a narrowing of the host to only humans. As noted above, evolution through gene loss is also carried out by free-living or facultatively parasitic bacteria that adapt to a more dependent association with a host. One study suggests that variola virus diverged from an ancestral poxvirus of African rodents either about 16,000 or 68,000 years ago, depending on the historical record used to calibrate the molecular clock (Li,
Carroll et al. 2007). During that process, the genome maintained its conserved, core gene complement but lost a unique pattern of virulence genes through accumulated mutations or gene deletions to reach its current size of about 186-kbp (Esposito, Sammons et al. 2006). A similar process occurred in the other orthopoxviruses; each species differed in the number and types of virulence genes that were lost except for cowpox viruses (Lefkowitz, Wang et al. 2006). Of the known orthopoxviruses, cowpox virus has the largest genome (224-kbp) and has orthologues of all other genes found in all other orthopoxvirus species (Gubser, Hue et al. 2004; Lefkowitz, Wang et al. 2006). An ancestral poxvirus, similar in size to cowpox virus, is hypothesized as the progenitor of orthopoxviruses (Gubser, Hue et al. 2004). Given that evolutionary history, it is not clear whether the loss of genetic information was a consequence of the lack of requirement of a gene for propagation in a new host, a necessary prerequisite for propagation in a new host, or both. However, we have available the gene sequences from this entire array of possibilities and we can identify those genes that are part of a common core of the poxviruses and those with more specificity for a particular host.
Nature provides an interesting lesson about the value of sequence identity for predicting pathogenicity. The poxviruses, molluscum contagiosum, and variola, have evolved to become successful human pathogens, but in dramatically different ways. Variola virus is a respiratory pathogen that causes a systemic disease with high mortality rate in humans regardless of age or sex. Molluscum contagiosum (caused by Molluscipoxvirus genera) is a common, benign infection of the skin of children and sexually active adults, but it can be a frequent and serious opportunistic infection of immunosupressed patients. [Molluscum contagiosum viruses are more highly restricted in its tissue tropism than variola virus and replicates only in the human keratinocyte (Buller and Palumbo 1991).] Molluscum contagiosum and variola virus share a similar genome structure with a central, conserved core of orthologous genes, and a unique pattern of flanking genes. The evolution of two distinct poxvirus pathogens that cause dissimilar human disease from a common ancestor through gene loss suggests that there can be multiple genetic pathways to becoming a pathogen for the same host. Furthermore, inasmuch as the genomes of molluscum contagiosum and variola viruses are distantly related with 57 percent nucleotide identity (on the basis of DNA polymerase), the evolutionary path to a human poxvirus pathogen does not necessarily demand a high degree of genome sequence identity. Even with the closely related orthopoxviruses that share 96 percent nucleotide sequence identity, phylogenetic analysis cannot predict human pathogenic potential (see Box 2.8).
Evolution of Plant Pathogens in Human-Managed Ecosystems
In evolutionary terms, human-managed artificial agricultural ecosystems are a recent agricultural practice that is highly vulnerable to disease outbreaks.
|
BOX 2.8 Evolutionary relationships, and therefore biological potential, may be inferred through the phylogenetic analysis of orthopoxvirus genomes because the genomic sequence is ultimately the primary genetic map of the species. Examination of phylogenetic predictions based on multiple nucleic acid sequence alignments of the conserved, core genomic regions of isolates of representative orthopoxvirus species found that monkeypox, ectromelia and cowpox viruses (strain Brighton Red) do not group closely with any other orthopoxvirus species, whereas variola, camelpox and taterapox viruses form a more closely related subgroup (Gubser, Hue et al. 2004; Esposito, Sammons et al. 2006). On the basis of that phylogenetic analysis, it would be predicted that if any orthopoxvirus shared biological properties with variola virus and caused human disease, it would be camelpox virus or taterapox virus. Instead, the more distantly related monkeypox virus causes severe human disease that is almost undistinguishable from smallpox, whereas taterapox and camelpox viruses have not been documented to cause any human disease (Damon 2007). Monkeypox and variola viruses together have at least 33 orthopoxvirus virulence genes where a function has been determined for the actual gene or for an orthologue in another orthopoxvirus species. By sequence comparison, both viruses appear to have 21 functional genes in common; of the remaining 12 genes, variola virus lacks 9 and monkeypox lacks 3. Those data suggest that are at least two and probably more unique genetic backgrounds are capable of producing a poxvirus that is lethal for humans. The identity of additional human poxvirus pathogens will probably be an experiment of nature and not a result of phylogenetic genomic comparisons. |
Agricultural ecosystems are artificial and contain a high density of a genetically uniform plant population, which creates a local atmospheric zone where the climate differs from that of the surrounding area, and promotes a high rate of pathogen reproduction and dispersal (Stukenbrock and McDonald 2008). Such agricultural practice allows virulent pathogen genotypes to adapt to a particular host genotype and to increase rapidly, and thereby generates a degree of host specificity in a short period that rarely occurs in natural ecosystems (see Box 2.9). Examples highlighting the major evolutionary mechanisms by which plant pathogens have emerged as threats to agricultural ecosystems are provided in detail in Appendix K. Those mechanisms take place over varied time scales ranging from short to thousands of years.
Interactions of Infectious Agents with the Host
The interplay of expression of genomes of a host and a microorganism dictate the outcome of the interaction. Microbial and host interactions can be characterized as stable, dead-end, evolving, or resistant. The most common
|
BOX 2.9 Evolutionary Mechanism of Plant Pathogen Emergence Host-tracking Host-tracking refers to co-evolution of a pathogen with its host during the process of host domestication, which includes the formation of a specific agro-ecological system. It includes the selection and cultivation of desirable host genotypes, simultaneous selection for pathogen genotypes that are adapted to the selected individuals and for the agro-ecological conditions at the time of the process. The process takes about ten thousand years, and pathogen and host share the same center of origin. Host jump Host jump is a process through which a new pathogen emerges in a host species that is genetically distant from the original plant host (for example, from another class or order). The geographic origin of the host does not always correspond with the geographic origin of the pathogen as observed in host shift. Host shift Host shift is a process in which a new pathogen emerges by adaptation to a new host that is a close relative of the former host (for example, shifting from a wild crop to the new domesticated selection or variety of the crop). The process may take less than 500 years or as much as several thousand years and the pathogen and the host do not always originate in the same center of origin (Stukenbrock and McDonald 2008). |
interaction is the resistant host-microbe interaction that leads to no or minimal amplification of the infectious agent. An evolving microbe-host relationship characterizes the spread of a microorganism from the same or a closely related species (as in the West Nile virus introduction into the Americas in 1999). A stable microbe-host interaction results in survival of both the microorganism and host on a population basis (for example, variola virus, polio virus or M. tuberculosis in humans and plum pox virus in some stone fruit species). In such cases, the host species acts as a reservoir host and it is necessary and sufficient for completion of the natural life cycle of the infectious agent. The resulting disease can be of varied severity as long as transmissibility of the pathogen is ensured. In general, those interactions evolve toward a state of less pathogenicity in the host, while preserving transmissibility. Stable interactions can also include infection of more than one host species with the same microorganism. For example, influenza A virus, Rift Valley fever virus, B. anthracis, tomato spotted wilt virus, cucumber mosaic virus, and turnip mosaic virus are all capable of propagation in a variety of species, and some viruses, such as Rift Valley
fever virus, can replicate efficiently in insects as well as mammals. Dead-end interactions often result in severe, fulminant disease involving infections of an incidental host species that is not needed for maintenance of the natural life cycle of the pathogen (for example, B. anthracis, dengue virus, Nipah virus, and Ebola virus infections of humans, and citrus tristeza virus and Phytophthora infestans infections in citrus grafted onto sour orange and potatoes, respectively). The infectious agents originate in other vertebrate species or are carried by arthropods that cycle between insects and vertebrates or between insects and plants. In some circumstances, a “dead-end” infection can give rise to an emerging infection as was the case for HIV, Rift Valley fever virus, and SARS-CoV in humans and citrus tristeza virus and P. infectans infections mentioned above. The majority of the human pathogens found on the Select Agent list cause dead-end interactions.
The outcome of a microbe-host encounter is based on interactions at the molecular and cellular level that take place over time. For certain viruses, a productive infection is determined by specific receptors that need to be engaged for virus binding and entry (for example, sialic acid and angiotensin-converting enzyme 2 for influenza A virus and SARS-CoV, respectively) the availability of intracellular complementing factors needed for efficient replication and the ability to manipulate intracellular antiviral signaling pathways (for example, interferon, pattern recognition receptors, apoptosis, and autophagy), and the adaptive immune response. In the case of Gram-negative bacteria, a productive infection may be initiated by adhesion through fimbriae (for example, enterotoxigenic E. coli); in the case of Gram-positive bacteria, it may be initiated through cell wall-anchored proteins (for example, microbial surface components recognizing adhesive matrix molecules). Additional virulence factors are needed to counter the innate and adaptive immune response. In the case of plant pathogens, a productive infection is determined in part by success in bridging the plant basal defense or innate immunity system though the expression of countermeasures.
Appendix K, presents examples of various factors that affect microbe-host interactions, and our lack of understanding of the basis of a pathogen’s ability to infect a host or multiple hosts even if we know the genomic sequence of the pathogen and, in the case of human infections, the host. A key factor in the outcome of the microbe-host interaction is the effectiveness of the host innate and adaptive immune response in the face of sophisticated and redundant microbial countermeasures, some of which are conserved in bacterial pathogens that infect plants and animals. Another important factor is selection pressure, which can be manifested in physiological, epigenetic and/or genetic changes in a pathogen in response to the innate or adaptive immune system, the absence of an adaptive immune response, or changes in the microbiome (Box 2.10).
We are just beginning to understand the significance of the microbiome for human health. Microbial interactions may determine whether a would-be pathogen acquires increased virulence or transmissibility, and whether an infec-
|
BOX 2.10 Bacterial Super Infection Following Influenza A Virus Infection Infection of the upper respiratory tract with such a virus as Influenza A virus predisposes the host to superinfection with bacterial pathogens, including Staphylococcus aureus and Streptococcus pneumoniae. The massive host innate response to Influenza A is focused on elimination of the virus from the respiratory tract. The overwhelming inflammatory response directed against the intracellular viral pathogen provides a perfect opportunity for a member of the normal respiratory flora (such as S. aureus or S. pneumoniae) to establish itself deeper in the respiratory tract, where it can cause bronchitis and/or pneumonia. The signals in the bacteria that cause them to transition from commensal organism to pathogen are not entirely known although the complex regulatory networks are slowly being identified and characterized. Once the bacteria migrate to their new niche, a plethora of virulence factors are produced. The host innate response to the bacteria is impaired because the overwhelming response to the virus depletes the neutrophils, macrophages, and dendritic cells and the antimicrobial factors produced by them. As a result, the secondary bacterial infection is often far more life-threatening than the initial viral infection. S. pneumoniae has emerged as the most common cause of secondary bacterial pneumonia in the current H1N1 Influenza A outbreak, although S. aureus, both methicillin-resistant and methicillin-sensitive, is the second most commonly isolated organism in post-influenza bacterial pneumonia. |
tion will result in disease. Thus, the individual microorganism, microbe-host interactions, and the environmental context must be considered in assessing an organism or a gene sequence as a potential threat to human health.
THE SPECIAL CASE OF SYNTHETIC BIOLOGY
Innovations in the chemical synthesis of DNA have led to dramatic improvements—the DNA can be longer, of higher quality, and less expensive per base pair—since the synthesis of the first copy of the 75-base pair tRNAAla in the early 1960s (Agarwal, Buchi et al. 1970). DNA synthesis on solid supports combined with phosphoramidite nucleosides allowed the synthesis of 2.7 kb plasmid DNA, an infectious 7.5 kb poliovirus genome, a 32 kb bat coronavirus (HKU3) that was the precursor to the SARS-CoV epidemic, and the first complete synthesis of a 582 kb artificial bacterial genome. Most recently, a synthetic bacterial genome has been “booted”6 into an autonomous life-form, so the artificial bacterial genomes are self-perpetuating (Gibson, Glass et al. 2010).
It is clear that the price of DNA synthesis has steadily decreased and a cursory survey of commercial suppliers show a cost of about $0.39-0.50/base—a dramatic reduction from synthesis costs commonly seen in the early history, when industry costs of about $5-10.00/base in the early 2000s were common. New optical deprotection chemistries and microfluidic technologies that allow programmable synthesis of hundreds of thousands of oligomers in parallel with fairly high fidelity seem poised to revolutionize inexpensive synthesis. With those and current multiplex technologies, it seems likely that future costs will approach $0.03/base (commercial costs); additional costs will be associated with gene assembly, quality control and other manufacturing issues. In the near future, as gene synthesis approaches $0.10-0.20/base, synthesis will replace most traditional recombinant DNA methods and allow the ready design and synthesis of new gene circuits and biological processes.
The design and testing of artificial biological systems and understanding of functional interactions are key objectives of synthetic biology. The benefits and broad availability of affordable gene synthesis are expected to foster rapid response platform technologies for producing candidate vaccines and therapeutics to address biothreat agents and newly emerging infectious diseases. It will allow more effective diagnostic platform design and basic inquiry into fundamental biological mechanisms, including pathogenesis and pathogenhost interactions. Gene synthesis will assist in designing and testing complex biological systems to fulfill specific purposes ranging from biofuel and petrochemical production, to genetically engineered foods, virus batteries, solar cells and energy systems, and the manufacture of new medicines. Thus, when considering steps that aim to prevent the misuse of the technology, we should also recognize the dramatic impact and potential that synthetic biology offers to the future economic growth, competitiveness and viability of the U.S. biotechnology industry.
In general, the discipline uses either natural cellular components and systems to construct new biological processes (the top-down approach) or the generation of unique biological and/or chemical systems that have novel properties and are designed to mimic living systems (the bottom-up approach). Most investigators have used the former approach because foundational understanding for de novo biological design (for example, protein structure, protein-protein interaction, genetic regulation) is in complete. One controversy is that unanticipated outcomes may occur when engineered organisms reproduce, evolve, and interact with the environment. Another concern is the deliberate misuse of the technology to design and construct new pathogens, either by engineering in components that resist current vaccine or therapeutic interventions, or by altering pathogenesis by blending in virulence genes from alterative pathogens, or by the de novo design of new pathogens. Understanding of the complex genetic and protein networks that regulate replication and disease is substantially lim-
ited, so for the near future, synthesis can for the most part only copy, emulate, or recreate existing gene sets that have been designed by nature.
Top-Down Approach
Since 1980, a standard set of recombinant DNA techniques has been developed that allows the cloning of full length DNA copies of most DNA and RNA virus genomes. In parallel, highly efficient strategies have been developed for “booting” infectivity from the DNA genomes and then recover infectious virus, including recombinant viruses that contain design modifications (for example, mutations, new regulatory networks, and gene insertions or deletions). There are proven strategies exist for reconstituting most of the viral Select Agent and category A-C biodefense pathogens from full-length DNA genomes; however, infectious clones have not been constructed for many Select Agent viruses on these lists. Developing infectious full-length DNAs is traditionally a time-consuming and uncertain process; single nucleotide changes can destroy genome infectivity. In general, genome size is directly proportional to the difficulty of generating an infectious molecular clone because of issues associated with genome and vector stability, sequence accuracy, and technical challenges in manipulating and recovering large genomes. For example, substantial technical sophistication, targeted expertise, and practice are required to reproducibly “boot” genome infectivity of large RNA genomes (for example, coronavirus, Ebola virus and influenza virus), DNA genomes (such as poxvirus) and bacterial genomes. In contrast, small RNA and DNA genomes are much easier to manipulate and recover. The number of people capable of working with these systems is increasing on a daily basis. For example, students at Johns Hopkins University recently worked collaboratively and designed, synthesized and recovered a 280 kb yeast chromosome (Dymond, Scheifele et al. 2009).
Synthetic biology will alter the standard approaches for reconstructing full length infectious DNA genomes of most viruses and computer based genome design will probably become the norm in the near future. It is clear that gene and genome synthesis will allow for synthetic reconstruction of many highly pathogenic human, animal and plant virus genomes, and thereby, removing one major limit to biological warfare and terrorism: availability. There are similar concerns regarding synthetic bacterial genomes, which are on a longer time horizon. Assuming that a cost of $0.10/base is achievable in the near future, the synthesis of the genomes of most “agents of concern” will be readily affordable (RNA genome: about 7.32kb for $700.00-3200.00; DNA genome: about 150-300kb for $15,000-20,000), while half a million base pair bacterial genomes will cost about $50,000 U.S. dollars. Not only is the instrumentation affordable, highly portable, and globally available, but there are commercial vendors on every continent, so it would be difficult or impossible to track and
police nefarious intent. Thus, it is possible that no virus (or microorganism) can ever be considered extinct (for example, poliovirus, 1918 influenza, smallpox, reconstructed extinct retroviruses, wooly mammoth viruses, etc.) as long as basic sequence information is available to support its synthetic reconstruction. The considerable concern surrounding synthetic DNA technology for “dual-use” potential is understandable (NRC 2004).
In general, dual-use concerns include the use of synthetic biology to deliberately host-shift pathogenic microorganisms, to engineer drug or vaccine resistance, or to alter virulence potentials. As noted throughout this chapter, the list of “virulence genes” that have defined biological properties is growing at a considerable rate, and this fuels concerns that virulence is a readily malleable trait. Limited research has focused on the potential of synthetic genome design to enhance viral pathogenesis. With an existing genome as a chassis (from either a pathogenic or a nonpathogenic virus), it is certain that virulence genes from DNA and RNA viruses can easily be introduced into recombinant genomes in an attempt to alter the pathogenic potential of the chassis genome. The capability of using standard recombinant DNA techniques for that purpose on a more limited scale has existed for about 30 years. Moreover, we can imagine synthetic killer viruses that destroy civilization or that cause significant morbidity and mortality—a common topic in cinema.
We know how to synthesize such imaginary “doomsday scenario viruses,” but how well the blended genomes will perform in human populations is unknown. For example, expressing the influenza virus NS1 type 1 interferon antagonist gene in SARS-CoV is simple with a top-down engineering approach, but the pathogenic properties of the chimera is difficult to predict. That is because SARS-CoV encodes at least six other interferon antagonist genes, and this raises the question of the ability of NS1 to offer considerable improvements in the pathogenic and innate immune antagonism capacity of the coronavirus. Moreover, most viral proteins form complex interaction networks that are essential for regulating efficient virus growth and virulence. Removing or introducing new potential interaction partners will most likely adversely affect virus-virus and virus-host interaction networks and thus influence pathogenesis outcomes in unanticipated ways. In addition, dramatically altering the genome content of most RNA viruses by inserting genes could easily attenuate virulence, probably by affecting global gene expression or by altering basic RNA structure and genome packaging and release. In spite of those limitations, insertion of the IL-4 gene (cloned from mus musculus domesticus, the common mouse) into murine poxviruses (such as ectromelia virus) or insertion of the SARS-CoV ORF6 gene into mouse hepatitis virus enhanced pathogenesis in mice, and the influenza virus NS1 gene enhanced Newcastle Disease virus replication in human cells. It is also clear that synthetically designed chimeric viruses would elicit fear in exposed populations, regardless of the actual pathogenic outcomes associated with its intentional release. It might be prudent to shift the focus
away from preventing dual-use proliferation to preparing for it by developing new platforms for rapid vaccine and therapeutic design and stockpiling these reagents against future bioterrorist attacks.
It is important to note that many “non-Select Agent” human viruses and microorganisms are extremely pathogenic and encode virulence determinants that could be blended into other genome chassis or enhanced by introducing new virulence determinants. Thus, one danger in creating lists is that it focuses resources on a select subset of human pathogens and ignores the broader innovation in genome design and gene function that exists throughout the larger microbial world (see Figure 2.2).
FIGURE 2.2 The universe of potential genes and sequences that could be drawn upon to create a biological weapon involves all biology. From “All Biology,” some pathogens and pathogenic products such as toxins, venoms, and others may be known to cause disease or death in humans, animals or plants. In the context of human health, some are recognized as causes of infectious diseases and are reasonably well characterized. Among all infectious diseases, some are further classified as Category A, B, or C pathogens by CDC or NIH, and they may or may not be assigned a biosafety level of laboratory containment (BSL-1, -2, -3, or -4) in the BMBL. Of these, some are designated Select Agents, and a few are prioritized under the DHS Bioterrorism Risk Assessment.
Bottom-Up Approach
The most commonly cited concern is that synthetic biology will allow the de novo design and creation of new life forms, such as synthetically designed killer viruses and microorganisms, either from scratch (which is extremely unlikely) or through the combination of existing gene sets from multiple viruses (which is more likely). Humans are surrounded by large numbers of intentionally genetically modified organisms (for example, domesticated corn from small grass teosinte and domesticated microorganisms used in beer, wine, cheese, and yogurt production) that were generated by breeding, artificial selection or, most recently, direct genetic engineering. In fact, 30 years of genetic manipulation with recombinant DNA technology in thousands of laboratories around the world has not resulted in any human-targeted super virus or microorganism. Although that does not preclude it in the future, it raises the question of whether the degree of concern is exaggerated. Will synthetic designer genomes be more dangerous to society than other highly pathogenic microorganisms that already exist in nature, such as measles (about 150,000 deaths per year), HIV (about 2 million deaths per year), influenza and RSV, respiratory syncytial virus, (about 0.5-1 million excess deaths per year), organisms that cause diarrheal diseases (more than 2 million deaths per year), or malaria (more than 1 million deaths per year)?
The scientific community does not have sufficient knowledge to create a novel, viable life form, even a virus, from the bottom up. Designing an infectious viral genome de novo by sequence requires the accurate prediction of protein structure and function, the design of protein-protein interactions and protein machines, all of which must produce progeny virions efficiently in an order of magnitude more complex host cell. If we cannot predict protein structure and function on the basis of sequences with any accuracy, how can we design and synthesize novel viruses that will replicate, regardless of their disease potential?
Alternatively, de novo design could focus on existing gene sets by emulating or copying known functions. That is also exceptionally difficult. First, entry requires specific interaction between viral and cellular components, including the deliberate orchestration of a series of sequential conformational changes that mediate docking and entry of the virus genome (particle) into the cell. Regulation of entry often involves co-receptors and other host factors like proteases to regulate the entry process. While the process is clear, the ability to design these highly regulated systems de novo is in its infancy. Second, a functional replication complex is needed to replicate the viral genome. Replication complexes are complex multicomponent protein machines (such as, viral and perhaps cellular proteins) that specifically engage and replicate nucleic acid. The formation of a replication complex includes tailored protein-protein and protein-nucleic acid interaction networks that are not known and cannot
be predicted or engineered with existing technology. Pathogenesis involves a regulated set of virus-host interaction networks that influences host responses that antagonize or potentiate disease; these networks are poorly understood and cannot be designed de novo. Virulence genes work together, and the levels of expression that permit virus persistence or spread and transmission, depending on the replication mechanism, are highly regulated. Finally, efficient virus egress from the cell usually involves discrete cellular and viral constituents. The protein-protein interactions, the regulation of the components involved in efficient release, and the design of de novo systems are beyond the capacity of the scientific community. The level of abstraction required to piece together a new life from defined parts is difficult enough—it is a misconception that a viable de novo microorganism can be designed today directly from sequences and a pool of uncoupled gene parts—it would be even more difficult to predict the virulence of such a microorganism if it were assembled and recovered.
Synthetic Biology—Summary
Synthetic biology offers considerable opportunity to improve human health and solve planetary problems in energy, food production, medicine, and public health. However, dual-use applications are of legitimate concern; new synthetic DNA technology alters the old paradigms that regulate pathogen bioavailability, but it is the traditional top-down approaches7 that present the greatest threat to altering virus virulence and pathogenesis. Bottom-up approaches remain extraordinarily complex, and it seems unlikely that sufficient vision and understanding exist to design and recover a true human pathogen using this approach. The time frame for the realization of the synthetic biology revolution is impossible to predict and will depend on whether it remains unfettered and on the support that it receives; undoubtedly, however, it will be a reality within the lifetime of most who read this document.
WHAT CAN CURRENTLY BE PREDICTED FROM SEQUENCE ABOUT THE IDENTIFICATION OF PATHOGENIC MICROORGANISMS, INCLUDING SELECT AGENTS?
It is abundantly clear that the use of sequence alone to predict a naturally occurring or synthetic pathogenic microorganism accurately is infeasible—sequence cannot provide biological context. Whether synthetic or natural, a sequence by itself has no biological properties. However, a remarkable amount of information can be deduced from a sequence of genes or proteins. To begin with, it is clear that all the microorganisms that we deal with can be divided into relatively well-defined general groups. For example, among the groups of
|
7 |
See Chapter 3 for more discussion of the relative threats presented by these approaches. |
bacteria, viruses, and eukaryotic pathogens a core of conserved genes of known and hypothetical function make up the unique set of properties that permit a specific group of microorganisms to occupy, persist, and replicate in a particular biological niche, perform certain metabolic functions, and interact with other living things. That core of genes, which generally constitute about three-fourths of the genes specific to a microbial group, is the basis of much of contemporary taxonomy. A remaining set of genes is concerned with specialization. For our purposes, the specialization is pathogenicity and virulence. We can confidently “predict” the sequence of most of the important microbial toxins or at least suspect, on the basis of the structure of a protein or the deduced catalytic site decoded from a nucleic acid sequence, that a toxin might be encoded therein.
The common features and themes of many bacterial virulence genes are known. Just as we can tell from the sequence of “core” genes that we are dealing with a bacterium of the genus Yersinia or a poxvirus, we can accurately say whether there is a precise sequence of a known virulence gene associated with the plague bacillus or with smallpox. It might be difficult to identify the precise origin of a microbial sequence to a specific Select Agent, but it is surely less difficult to say with some accuracy that a particular sequence is related to a known virulence determinant and to a class of virulence genes with known function. The fact is that the availability of complete microbial genomes, as well as the genomes of their hosts, has made it possible to combine genetic and molecular methods not only to identify microbial genes that are expressed only in certain animal hosts, including humans, but also to determine genes that are essential for pathogenicity using global screening methods. The methods themselves depend on gene sequencing technology. Such lists of genes exist today for Salmonella, Mycobacteria, Yersinia, and the pneumococcus; we believe that they probably exist for most Select Agents in at least a surrogate animal model. Where such lists do not exist, it is relatively easy to determine them experimentally.
The utility of sequence information depends on a number of factors. In some circumstances, the quality of a sequence may be a factor, for example, if it comes from a small sample from an infected individual. The length of a sequence is, of course, also a factor. If a sequence is relatively short, it may be of no immediate consequence unless it can be shown to be part of a much larger assembly of genes. What would be required to determine that a sequence came from a known Select Agent? On the one hand, if the sequence were from a known virulence gene or from a specific (as opposed to core) region of the genome, one could say with a good deal of confidence that it was from that organism. On the other hand, if the sequence is from a core common to pathogenic and non-pathogenic microorganisms, as is common for enteric bacteria (and others), the information is less useful, although it is of value from a surveillance standpoint to eliminate suspicion. Difficulty would arise if a sequence were from a common set of core genes—let us say a core common to the Bacillus
group—and there were sequences that resembled the plasmid-mediated toxin of B. anthracis but they were clearly distinct from the known anthrax virulence determinant. That might heighten the suspicion. However, the level of concern would depend on the biological context. If the sequence were derived from a patient who had sepsis, one would be appropriately alarmed. If the sample were from the soil, the level of suspicion would be far less because soil is a natural habitat for members of the genus Bacillus and they are harmless bacteria that have plasmids encoding related toxin molecules. If the sample were submitted to a DNA synthesis company, there would be cause to learn more about its source and about the reason the DNA was being synthesized.8 There is no fool-proof method for predicting whether a sequence will have the biological properties of a Select Agent (such as pathogenicity), but the common sense use of the considerable amount of sequence data we now have, combined with advances in understanding of microbe-host interactions, does provide for a mechanism that is practical and sufficiently flexible to provide guidance about the potential biological consequences of a DNA sequence from nature or from the biochemist’s bench.
None of this is meant to trivialize the difficulty of understanding the biological importance and function of microbial determinants of pathogenicity or the complexity of microbe-host interactions. Although we have the complete sequence of many of the most feared microbial pathogens and of their host animals or plants, our attempts to devise novel vaccines or therapeutic agents have been difficult at best. If genomics has taught us anything at all about microbe-host interaction, it is just how intimate and intertwined the biology of the two participants may be. That is not always obvious if one thinks about the ravages of the black death of the 14th century or the horror of Ebola infection. Those are the nightmare of those who deal with bioterror, but the most efficient killers of the world are still infectious agents that have a longer-lasting intimate association with humanity. It is not easy to develop antiviral drugs when the interactions of the viral components and the human host are so closely intertwined that inhibiting the invader may mean poisoning the host. Nor is it easy to develop vaccines against many bacterial pathogens that have evolved to interact in subtle specific ways with the biology of the human immune system. Continued investment in genomics, bioinformatics and basic research on infectious diseases of humans, animals and plants, will help us develop new ways to control diseases that plague our planet.
Nevertheless, it is our judgment that the sequence information now available, when combined with that being obtained on a daily basis and deposited in public data banks, can be effectively used to provide a new approach to identifying and monitoring microorganisms of interest much more effectively than the
construction of rigid lists that reflect biological reality poorly and therefore can provide only limited utility for national security. Thus, in Chapter 3 we discuss how the Select Agent list might be clarified by sequence-based classification to better circumscribe the taxonomic distinctions blurred by natural and synthetic variation and modification. Moreover, we present a “yellow flag” biosafety system; this approach is not regulatory and therefore could provide sequence information relevant to biosecurity in a more dynamic and timely manner than the Select Agent Regulations.



































