
3
Non–Food and Drug Administration Sources of Adverse Event Data
The Food and Drug Administration (FDA) partners with external groups to collect and analyze postmarket surveillance data. The committee requested information from some of FDA’s partners as well as other groups that collect adverse event information related to medical devices. The committee was presented with overviews of four non-FDA sources of adverse-event data with a focus on the collection of information about specific clinical activities or specific kinds of devices.
THE NATIONAL CARDIOVASCULAR DATA REGISTRY: OPPORTUNITIES AND CHALLENGES IN POSTMARKET SURVEILLANCE
“We are awash in data,” began Frederick A. Masoudi, associate professor of medicine at the Denver Health Medical Center and the University of Colorado and senior medical officer of the American College of Cardiology National Cardiovascular Data Registry (NCDR). It is not necessarily a lack of data that is the problem, he said, but the lack of an ideal data source.
An ideal data source for postmarket device surveillance, he suggested, is one that includes
-
Clinical data (avoiding administrative data whenever possible).
-
Standardized definitions (collection of the same data elements for the same event).
-
Detailed phenotyping of patients (collection of a wide array of clinical characteristics that could be used for risk adjustment).
-
Real-world populations (that is, not in the context of a clinical trial).
-
A population denominator.
-
Followup for adverse events.
The mission of the NCDR is “to improve the quality of cardiovascular patient care by providing information, knowledge, and tools; implementing quality initiatives; and supporting research that improves patient care and outcomes.” Although the NCDR was not set up primarily for postmarket surveillance, Masoudi said, it is one of its benefits. The NCDR is actually a suite of cardiovascular-disease registries (see Box 3-1). Several of the NCDR registry programs are focused on procedures and devices, Masoudi noted.
The data-collection platform is similar in all the NCDR registries, using electronic data collection to capture a wide array of detailed demographic and clinical characteristics for each patient, details of procedures that are performed, and inhospital complications. The data include detailed information on the devices used for each procedure.
Each institution that submits data to the NCDR receives quality benchmark reports that can be used to support self-assessment and quality improvement by the institution.
|
BOX 3-1 NCDR Registries CathPCI Registry—for diagnostic catheterization and percutaneous coronary intervention (PCI) procedures. Includes data since 1998 from over 1,100 hospitals on more than 9 million patients. ICD Registry—tracking implantable cardioverter defibrillators (ICDs). Includes data from 1,445 hospitals on more than 250,000 patients. CARE Registry—for carotid artery revascularization and endartectomy procedures. ACTION-GWTG Registry (Get With The Guidelines)—acute–myocardial-infarction patients. PINNACLE Registry (Practice Innovation and Clinical Excellence)—an ambulatory care, practice-based registry. IMPACT Registry (Improving Pediatric and Adult Congenital Treatment)—tracking cath procedures for congenital heart conditions. |
Postmarket Surveillance with the National Cardiovascular Data Registry
As an example of postmarket device surveillance with the NCDR, Masoudi cited a study led by Paul Varosy looking at complications in implantable cardioverter defibrillators (ICD) patients (Dewland et al, 2008). It has been suggested that dual-lead ICDs may be superior to single-lead devices, but, Masoudi said, there are concerns that the use of dual-lead devices when there is not a clear need may be associated with higher rates of complications.
On the basis of the registry, 206,000 patients who underwent ICD implantation were identified. Patients who received biventricular ICDs or had clear indications for dual-chamber devices were excluded from the analysis. Of the remaining patients who had no clear reason to receive a dual-lead device, about half had received single-lead devices and half dual-lead devices (“discretionary” dual-lead ICD placement).
The analysis showed a substantially higher rate of major inhospital complications and a higher risk of death associated with dual-chamber devices than with single-chamber devices. Both findings were statistically significant, Masoudi noted, even after risk adjustment based on a wide array of patient characteristics that are collected in the registry.
Another example Masoudi cited used the CathPCI Registry to assess the rates of bleeding complications associated with different closure devices used at the groin site after angiography and percutaneous coronary intervention (PCI) (Travis et al., 2005). After adjustment for various characteristics among the different patient categories, the analysis showed that the VasoSeal device was associated with a significantly higher risk of adverse outcomes after angiography than other hemostasis devices. As a result of the analysis, Masoudi said, use of the VasoSeal device was removed from the market.
Several postmarket surveillance collaborations between the NCDR and FDA are going on, Masoudi said. FDA task orders include assessment of device use in carotid revascularization, ICD lead safety, risk factors for ICD malfunction, data elements and metrics for an atrial fibrillation (AF) ablation registry, and a dataset for the National Congenital Heart Disease Registry. The NCDR and FDA are also drafting a white paper discussing the value of registries, such as the NCDR, for postmarket surveillance.
Several challenges to device surveillance using the NCDR, Masoudi said, include followup, data quality, and integration of data collection into care. Cost is an overriding concern; registries are extremely expensive to develop and to maintain.
Strategies for Longitudinal Followup
A number of strategies have been used to address the challenge of longer-term surveillance using registries which are primarily hospital based,
Masoudi said. For example, NCDR data have been linked to administrative data, such as those from the Centers for Medicare and Medicaid Services (CMS), to provide followup of clinical events (for example, later hospitalizations, death, and complications that might be coded in administrative datasets). In some cases, that has been done probabilistically: instead of exact patient identifiers, several aspects of patients’ episodes of care are used to find a match with the Medicare data. Although it is probably not adequate for the purposes of device-based postmarket surveillance, Masoudi said, the probabilistic matching approach (as opposed to direct patient matching) has been shown to be a valid method of data analysis. This is particularly relevant in light of the limitations on patient identifiers as a result of the Health Insurance Portability and Accountability Act privacy rule.
Masoudi referred to a recent report from the Institute of Medicine that concluded that the privacy rule does not do enough to protect patient privacy in all situations and substantially impedes research or efforts to improve public health. The privacy rule is vague in many respects, he said, and it is often assumed that the most conservative approach is the most appropriate one, particularly because the penalties for nonadherence to the privacy rule are very high. As a result, efforts to comply with the rule interfere with important research, in this case in identifying linkages between NCDR data and direct Medicare data.
Another strategy for longitudinal followup is collaboration with health systems that own all their administrative data, such as Kaiser Permanente and the Veterans Health Administration. That allows direct connection between patients in the registries and followup in the system instead of a probabilistic matching strategy.
A final strategy described by Masoudi involves forming a relationship between inpatient registries, such as the CathPCI Registry and the ICD Registry, and outpatient registries, such as the new PINNACLE Registry. The fragmentation of the health-care system creates some challenges to this approach, Masoudi said, for example, how to ensure that the same patients who are followed in a hospital with the ICD Registry are then followed in an outpatient setting with the PINNACLE Registry or another outpatient registry.
The NCDR has a number of data-quality procedures in place as part of a larger quality program, for example, data-quality checks where sites are given green-light, yellow-light, or red-light status on the basis of the quality of the data that they submit to the registries (completeness of the data and range checks of the different data fields).
A limited audit is also performed on a clinical level by the NCDR, but because of insufficient resources, this is done for relatively small numbers of charts and clinical records, Masoudi said. A data-quality program that
ensures the fidelity of data, particularly if it involves clinical-data abstraction, requires substantial resources.
When a physician or other clinical professional enters into a patient chart that, for example, the patient has class III angina, that information does not automatically get into the NCDR. It needs to be re-entered in many cases, and this adds to the challenge of fostering participation in the registries.
One very successful approach to address this, Masoudi said, has been instituted in the Department of Veterans Affairs (VA) health system. In the Clinical Assessment, Reporting, and Tracking System for Cardiac Catheterization Laboratories (CART-CL) program, for example, data collected in the process of routine clinical care are tailored to the NCDR data fields and then entered into the program for device surveillance.
THE DEPARTMENT OF VETERANS AFFAIRS CARDIOVASCULAR ASSESSMENT, REPORTING, AND TRACKING PROGRAM: INTEGRATION OF REAL-TIME DATA COLLECTION INTO THE PROCESS OF CLINICAL CARE
Paul D. Varosy, director of cardiac electrophysiology in the VA Eastern Colorado Health Care System and assistant professor of medicine at the University of Colorado Denver, provided an overview of the VA CART program, describing it as a new paradigm for cardiovascular-disease surveillance and a potential model for medical-device surveillance.
Data Resources
The VA-wide electronic health record, known as the Computerized Patient Record System (CPRS), is an outgrowth of the national VA medical-record system in place since the 1970s. CPRS was developed in the middle 1990s as a rich graphical user interface for health-care data and incorporates text notes and reports, laboratory data, electronic order entry, pharmacy records, and images (such as electrocardiograms and radiology records). It is organized and managed at the regional level by the 23 Veterans Integrative Service Networks, which are all linked to a single nationwide network.
Clinical and administrative data from the VA facilities are warehoused at the Austin Information Technology Center. Data are aggregated and processed for multiple potential uses, including workflow tracking, quality of care and quality assessment, and health-services research. Varosy added that access controls prevent breeches in data security.
Data are also obtained from the VA Office of Patient Care Services, which has clinical oversight over cardiovascular services. The office has specific programs for quality monitoring and improvement and national
programs, such as the pacemaker and ICD surveillance programs and the CART program.
Limitations of Administrative Data
There are some important concerns about administrative data, such as data that are aggregated into the VA datasets, Varosy said. First, most parts of the clinical record are not entirely field-specific. There are text notes and reports that need to be abstracted, either by manually going through the individual records (which is labor-intensive) or by some complex natural-language processing extraction that is cumbersome and difficult to interpret. There can also be a “loss in translation” of information and a lack of clinical granularity of the data.
A second concern is lack of standardization. With regard to heart function, for example, left ventricular ejection fractions can be measured in various ways, including echocardiography, radionuclide ejection fraction measurement, radionuclide myocardial perfusion study, cardiac catheterization, and magnetic resonance imaging. Results can be reported in different places in accordance with different standards, such as a specific number of an ejection fraction, for example, 36.7% as measured with one of the various methods; an estimate of 35–40%; or a qualitative estimate, such as “moderately depressed left ventricular function.” The need to collate information from different sources based on different studies and different ways of codifying the results makes it difficult to interpret the data.
Finally, Varosy said, dependence on administrative coding is problematic in a system where coding is not tied to reimbursement. For example, in the VA system there is relatively little incentive to ensure correct coding according to Current Procedural Terminology (CPT) and International Classification of Diseases 9th Revision (ICD-9) when there is no reimbursement on the basis of these codes. However, analyses of the VA database often use the codes for disease surveillance and quality assessment.
As a result, abstraction of data after care is necessary because data collection is generally not integrated directly into the process of clinical care.
Offering an analogy, Varosy suggested that using administrative data, such as ICD-9 codes or CPT codes, for disease surveillance is like trying to monitor air traffic by reviewing jet-fuel receipts (if a plane refueled in Omaha and then again in Newark, it must have flown from Omaha to Newark). The air-traffic control system is instead designed to see where planes are flying in real time. That is not the case for health-care data.
Department of Veterans Affairs Patient-Care Services Clinical Programs
For nearly 30 years, VA has been a leader in remote pacemaker monitoring. The VA Pacemaker Surveillance Program includes remote followup of pacemaker function, administrative tracking of clinical and administrative cohorts, and support of clinicians and voluntarily enrolled patients. Patients without easy access to a VA hospital are monitored by telephone every 3 months to assess all pertinent characteristics (such as battery life) remotely. The program is administered from two sites: Washington, DC, and San Francisco.
The VA National ICD Surveillance Center (VANISC) was established in 2003 on the basis of the successful pacemaker surveillance program. VANISC monitors voluntarily enrolled patients who have ICDs remotely for arrhythmia episodes and reports results directly to the patients’ providers. It also facilitates disease surveillance and research studies.
To provide a sense of the scope, Varosy said that the western pacemaker surveillance program and the ICD program combined (both using secure data servers based in San Francisco) remotely monitor more than 18,000 veterans, including more than 12,000 veterans who have implantable ICDs. In FY 2009, the staff of 13 reviewed and provided support for over 32,000 pacemaker transmissions and over 40,000 ICD transmissions.
The remote monitoring programs have limitations, Varosy noted. Enrollment in the programs is voluntary. Linkage of remote monitoring programs to electronic health records is problematic, and there is a lack of infrastructure to connect remote and in-clinic device followup. Most important, he said, the ascertainment of long-term clinical outcomes is challenging (outcomes data are necessary for quality improvement, device performance and surveillance, and health-services research).
The Cardiovascular Assessment, Recording, and Tracking Program: A New Paradigm for Care
CART is a clinical tool that improves the efficiency of care by integrating data collection with electronic health records and facilitating report generation. The user interface, designed with clinicians in mind, incorporates VA-wide standardization and allows completion of reports in real time, often, Varosy said, before a patient is even off the examination table in the cardiac-catheterization laboratory.
The integration of data collection into the transaction of health care, Varosy said, allows transactional quality management, real-time patient-safety monitoring, real-time device surveillance, and nearly real-time health-services research.
Critical to the success of CART, Varosy said, are strategic collaborations with clinical champions in the 77 catheterization laboratories in the VA
system nationwide, the VA Office of Patient Care Services, the VA Quality Enhancement Research Initiative (QUERI), the VA Office of Quality and Performance, the VA Office of Information and Technology, and outside VA (for example, in the NCDR and in FDA). Varosy noted that VA has well-established connections with FDA, including monthly CART conference calls with FDA in which information about signals or unexpected problems with devices reported by clinicians at the point of care are shared.
Using the CART-CL program as an example, Varosy demonstrated the user-friendly CART interface consisting of checklists, drop-down boxes, and the opportunity to add text comments. He noted that many of the fields can be prepopulated directly from electronic medical records for data on such items as medications, allergies, vital signs, laboratory studies, and past medical history. For CART-CL, the coronary-angiography documentation process includes images with intuitive interfaces designed specifically for clinicians, which allow rapid and granular notation of specific lesions observed. Once all the data are entered, a uniform text report is generated that can be copied and pasted directly into the text-reports field in an electronic medical record. At the same time, all the data are captured into a database that resides in a secure server within the VA firewall.
Since 2005, nearly 140,000 total cardiac procedures have been documented with the CART-CL system by nearly 3,000 providers (including cardiology fellows in training and attending cardiologists). For decades, Varosy said, VA has been basing its workflow tracking and administrative information on the data resources from the Austin Information Technology Center. He pointed out, however, that for FY 2008, the CART program recorded 7,972 total cardiology procedures, but on the basis of purely administrative codes within VA the Austin Information Technology Center recorded 4,079 cardiology procedures (slightly more than half the number recorded in the CART system).
Transactional quality management in CART includes immediate e-mail reporting of major complications (for example, inhospital or intraprocedure stroke, death in a laboratory, or the need for emergency cardiac surgery during a cardiac-catheterization procedure). Immediate, secure, encrypted e-mail messages are sent to the chief cardiovascular consultant, CART leadership, and the CART Quality Management Committee chair. Committee review and preliminary recommendations occur within 24–72 hours after an event. Formal root-cause analysis or other interventions necessary to produce systemwide improvements in care may occur later. There are also monthly site quality-assurance reports, monthly and biannual national procedure and adverse-event count reports submitted to the VA central office and the CART Quality Management Committee, and quarterly regional reports submitted to the network administrators and chief medical officers.
Beyond the original CART-CL application, VA is creating modules to
address peripheral arterial intervention (CART-Peripheral), arrhythmia procedures (CART-EP), inhospital cardiac arrest (CART-CPR), and ambulatory care (CART-Ambulatory).
Data elements used are based on well-established data standards, including those of NCDR, and work is under way to construct a data link so that data captured in real time in CART-CL will be sent directly to NCDR. Similar integration efforts are planned for other CART modules and NCDR databases. In implementing CART-EP, VA hopes to provide a clinically useful reporting tool that will integrate with the ICD and pacemaker surveillance programs and allow transactional data collection.
The CART program, Varosy concluded, moves beyond the era of after-the-fact data collection to one of transactional data collection, leveraging real-time data to allow not only clinical care but real-time quality management, real-time workflow tracking, and real-time health-services research.
USE OF REGISTRIES FOR POSTMARKET DEVICE SURVEILLANCE
Eric D. Peterson, professor of medicine and associate director of the Duke University Medical Center and director of cardiovascular research at the Duke Clinical Research Institute, reiterated some of the issues that can arise after a device reaches the market, including rare events not observed in premarket evaluation, downstream safety events and long-term followup of devices that remain in patients for long periods, use in a wider array of high-risk patients, off-label use, device–drug interactions, and device–health-care provider interactions (the “learning curve”).
Among the challenges for FDA, Peterson said, are the rapid evolution of technology, which can make device studies quickly obsolete, lack of incentive or ability of device developers to conduct high-quality premarket and postmarket evaluations, and the fact that the clinical community (including patients) can be very eager for access to new devices and often does not demand clinical evidence or does not support the conduct of clinical studies (enrolling patients in clinical studies is challenging).
A Role for Registries
Can clinical registries address some of those challenges? Peterson asked. A creative sentinel system to track medical devices once on the market could, he said, provide an idea of actual device use (a denominator for analysis), gather data on off-label use, and identify potential device-safety signals.
Peterson provided examples of existing clinical-device registries. A device manufacturer may establish its own device-specific registry, as Medtronic has done with its pacemaker registry. There are also multisponsor device registries, such as the Interagency Registry for Mechanically Assisted Cir-
culatory Support (INTERMACS) database, a collaboration of the National Heart, Lung, and Blood Institute, CMS, and FDA. Professional organizations may house registries, such as the clinical cardiovascular registries established by the Society of Thoracic Surgeons (with over 900 centers registering all bypass, valve, cardiac, and thoracic surgery), and the NCDR (discussed by Masoudi above). Peterson added that relevant data have come from randomized controlled trials, and in the future data may be available directly from electronic health records.
Efforts are now under way to build more in-depth device information capacity into each of those registries progressively and to link data from clinical registries with claims data (particularly Medicare claims) to provide information on longitudinal outcomes, such as rehospitalization and device explantation.
Examples of Food and Drug Administration Partnership with Registries for Purposes of Postmarket Surveillance
Peterson highlighted several examples of the use of clinical registries for postmarket assessment of devices. Drug-eluting stents, Peterson said, are remarkable tools in the hands of interventional cardiologists but presented a substantial challenge to FDA because of the very rapid adoption of the devices for both label and off-label uses. Analyzing data from the NCDR, Peterson and colleagues found that the adverse-event rate appeared to be higher for off-label indications, but it was not clear whether this was attributable to the device or to the patient population in which it was used (Rao et al., 2006). Another study using the data bank from the Duke Heart Center showed that patients who had drug-eluting stents in place and who did not remain on dual antiplatelet therapy for long periods had higher adverse-event rates and mortality (Eisenstein et al., 2007). Those findings and other signals from clinical trials and other larger databases prompted FDA to initiate a public debate on safety issues of drug-eluting stents and to establish an advisory panel. Peterson noted that after those publications and presentations, the use of drug-eluting stents in practice declined.
Ultimately, the Agency for Healthcare Research and Quality (AHRQ) and FDA commissioned a database to examine the comparative effectiveness and safety of drug-eluting stents vs bare-metal stents in a national PCI cohort. The DEcIDE (Developing Evidence to Inform Decisions about Effectiveness) Cardiovascular Consortium linked NCDR data on 262,700 PCI patients from 2004 through 2006 to CMS claims data (based on indirect identifiers) to assess long-term outcomes (up to 3 years after stent placement). Peterson said that the analysis could not confirm a unfavorable safety signal for drug-eluting stents (the results with drug-eluting stents were equal to or better than those with bare-metal stents).
As another example, Peterson described an early initiative in which the Duke Clinical Research Institute partnered with FDA and the Society of Thoracic Surgeons to look at the safety and use patterns of transmyocardial revascularization (TMR). Diffusion of the technology into clinical practice was rapid and included off-label use of TMR combined with coronary arterial bypass graft procedures (Peterson et al., 2003). A followup study is under way with a retrospective cohort to compare long-term clinical outcomes of the use of two existing laser devices.
Other examples that Peterson cited included a large FDA-sponsored partnership with the Society of Thoracic Surgeons to compare clinical outcomes of the use of two marketed endoscopic vein-harvesting devices and a series of studies being conducted by the DEcIDE Consortium, supported by AHRQ in partnership with FDA and the Society of Thoracic Surgeons, to evaluate the clinical effectiveness and safety of marketed biologic vs mechanical aortic valve prostheses in older patients.
Those organizations are now working together to develop standardized nomenclature to allow linking of data across datasets, Peterson said. There is also interest in using a registry as a backbone to carry out large clinical trials.
In conclusion, Peterson said that postmarket device surveillance is and will remain an important issue for health care. Clinicians need to be actively involved and to demand better device information and identify device issues. Ideally, clinical registries can be used to provide novel solutions for effective and efficient postmarket surveillance.
AUTOMATED POSTMARKET SAFETY SURVEILLANCE: THE DELTA SURVEILLANCE PROJECT
Medical-device safety surveillance today is primarily passive, said Frederic S. Resnic, director of the Cardiac Catheterization Laboratory at Brigham and Women’s Hospital and assistant professor of medicine at Harvard Medical School. Automating prospective postmarket surveillance involves integration of high-quality data sources, appropriate safety expectations, monitoring systems, and secure data exchange.
Challenges
Resnic concurred with previous speakers regarding the array of challenges associated with device safety monitoring. Completeness and level of granularity of current datasets is a primary challenge. The lack of unique identifiers for devices affects the utility of clinical registries for understanding who has been exposed to which devices. Surveillance is affected by how quickly data become available, and appropriate and comprehensive
outcome ascertainment remains challenging. There are also the issues of data ownership, data security, and patient privacy.
Another major concern is signal detection and methods. Appropriate expectations need to be set, and appropriate comparators chosen. For an active, automated surveillance system to function, information needs to be converted into an alert that could notify regulators when a device appears to be heading out of bounds relative to performance expectations.
Given the availability of high-quality data and the ability to detect a signal, the next challenge is signal interpretation. Any alerts generated in observational surveillance must be verified through detailed clinical and statistical exploration. Device–operator, device–patient, device–drug, and device–device interactions all come into play.
Idealized Safety-Monitoring System
In one approach to surveillance, multiple data sources (such as hospitals) submit information to a centralized database or data owner, and the combined dataset is monitored. For reasons of data security and privacy, however, some data owners may be unable or unwilling to deposit their data into a central database. Rather, they share their data in a virtual fashion. Thus, a monitoring system must also be able to handle distributed datasets.
Resnic described an ideal monitoring system as one that is continuously updated, with data provided in as close to real time as possible, and that has an array of statistical analytic options. Systems should be able to handle multiple prospective analyses simultaneously, he said, and such analyses would be running in the background with various alerting thresholds.
The DELTA Surveillance System
In an effort to address some of those challenges, Resnic and colleagues are developing the Data Extraction and Longitudinal Time Analysis (DELTA) System, a Web-based platform designed to perform automated, real-time monitoring of device postmarket safety. DELTA has a matrix of analytic options (see Figure 3-1) that support exploration of potential safety events and draw inferences from both a frequentist perspective and a Bayesian perspective for uniform, stratified, and risk-adjusted expectations. The system generates and e-mails alerts as appropriate. DELTA is designed to run continuously in the background, much like industrial process-control systems in manufacturing plants, Resnic noted.
For the development and validation of DELTA, Resnic used historical data from the Massachusetts state registry. In 2002, he explained, the Massachusetts Department of Public Health implemented mandatory clinical-outcomes registries for invasive cardiac services, which required all
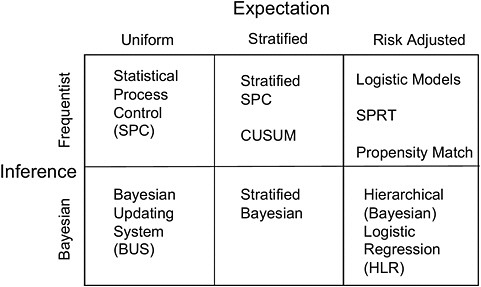
FIGURE 3-1 Statistical methods used in the DELTA System.
NOTE: CUSUM, cumulative sum control chart; SPRT, sequential probability ratio test.
hospitals in the state to submit data to the NCDR CathPCI Registry and the Society of Thoracic Surgery database and to harvest and report those data quarterly to the state registry. At the state level, reports are rigorously adjudicated and audited, and outcomes are linked to vital statistics and inpatient claims data (Resnic noted that this is a direct link, not a probabilistic link). For the retrospective-surveillance demonstration study, 74,000 cases of coronary intervention performed from the launch of the state registry in 2003 through 2007 were used. Devices that were first marketed within 6 months of the launch of the state registry were evaluated, and patients who received such devices were compared with propensity-matched patients who received competing devices of the same class (for example, a drug-eluting–stent patient under study was matched to another drug-eluting–stent patient according to about 35 variables).
A multicenter study that will test the prospective surveillance functionality of DELTA is under way (see Figure 3-2). For the purposes of the study, each participating hospital has a local DELTA system. Through secure data transmissions, the local DELTA agent communicates de-identified, encrypted data to the central DELTA collecting server. Only the data necessary to facilitate the analysis are sent.
To address concerns about facility disclosure of data to a central repository, in this case DELTA, the study will test three levels of data access: case-level data aggregation to the central database, with fully de-identified
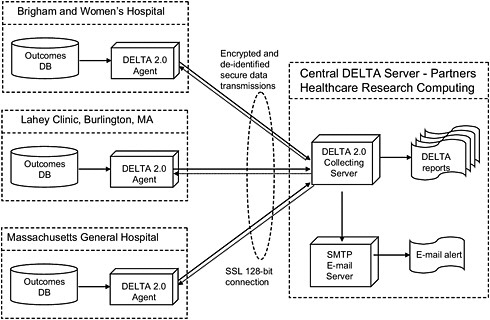
FIGURE 3-2 Prospective DELTA Network Study.
NOTE: DB, database; SMTP, simple mail transfer protocol.
data submitted; case-level outcome aggregation, in which only encrypted case identification, outcomes, and predicted outcomes are sent to the central server; and aggregated results of analyses performed at the local level, with no case-level information.
Costs, Resnic added, are primarily for collecting the data. In Massachusetts, a single implementation of DELTA costs around $25,000–30,000 for the hardware and the software license. The cost of collecting data is borne by the hospitals. However, because of other regulatory requirements for quality management within the hospitals and because many institutions in Massachusetts have adopted the CART-CL model, in which data are collected in real time as part of the clinical process, it is hard to tease out the actual cost of data collection, which has become part of the routine process.
Summary
Detection of low-frequency postmarket safety signals for medical devices challenges traditional methods of statistical surveillance, Resnic concluded. The ideal surveillance system is a time-efficient, high-sensitivity alert system that is designed to trigger detailed investigations of potential safety concerns. Such systems clearly require accurate, granular outcomes data and
device-specific identifiers. Resnic noted that the Massachusetts mandated cardiac registry provides such information.
The DELTA system is a prospective approach to surveillance. It provides flexible statistical and risk-adjustment methods for multiple simultaneous analyses and, Resnic said, meets the design requirements for many of the features of an automated safety surveillance system. Resnic noted that alerts based on analyses must be considered hypothesis-generating and require epidemiologic confirmation. In a future in which registries exist for many products, Resnic opined, systems like DELTA could be used to target regulatory resources and focus efforts on device pairs in which there is an outcomes signal.
Continuing testing of DELTA in a multicenter network study will provide an opportunity to evaluate the applicability and potential role of automated surveillance as a complement to existing methods and as a component of overall active surveillance strategies for new medical devices, Resnic said.
There is plenty of opportunity for further study in existing high-quality registries, Resnic noted. Future DELTA studies include a more in-depth exploration of the Massachusetts cardiac quality data focused on longitudinal outcomes, pilot studies with the VA CART-CL and the NCDR CathPCI Registry, an Orthopedic Implant Registry study, and a cardiac surgical valve safety-surveillance pilot study.
REFERENCES
Dewland, T. A., C. N. Pellegrini, Y. Wang, G. M. Marcus, and P. D. Varosy. 2008. Abstract 4127: Dual chamber ICD selection is associated with racial and socioeconomic disparities and increased complication rates among patients enrolled in the NCDR ICD Registry. Circulation 118:S834-S835.
Eisenstein, E. L., K. L. Anstrom, D. F. Kong, L. K. Shaw, R. H. Tuttle, D. B. Mark, J. M. Kramer, R. A. Harrington, D. B. Matchar, D. E. Kandzari, E. D. Peterson, K. A. Schulman, and R. M. Califf. 2007. Clopidogrel use and long-term clinical outcomes after drug-eluting stent implantation. Journal of the American Medical Association 297(2):159-168.
Peterson, E. D., P. Kaul, R. G. Kaczmarek, B. G. Hammill, P. W. Armstrong, C. R. Bridges, and T. B. Ferguson Jr. 2003. From controlled trials to clinical practice: monitoring transmyocardial revascularization use and outcomes. Journal of the American College of Cardiology 42(9):1611-1616.
Rao, S. V., R. E. Shaw, R. G. Brindis, L. W. Klein, W. S. Weintraub, and E. D. Peterson. 2006. On- versus off-label use of drug-eluting coronary stents in clinical practice (report from the American College of Cardiology National Cardiovascular Data Registry [NCDR]). American Journal of Cardiology 97(10):1478-1481.
Travis, D. R., S. Dey, B. Albrecht-Gallauresi, R. G. Brindis, R. Shaw, W. Weintraub, and K. Mitchel. 2005. Risk of local adverse events following cardiac catheterization by hemostasis device use—phase II. Journal of Invasive Cardiology 17(12):644-650.
















