
3
Overview of Vitamin D
INTRODUCTION
Vitamin D, first identified as a vitamin early in the 20th century, is now recognized as a prohormone. A unique aspect of vitamin D as a nutrient is that it can be synthesized by the human body through the action of sunlight. These dual sources of vitamin D make it challenging to develop dietary reference intake values.
Vitamin D, also known as calciferol, comprises a group of fat-soluble seco-sterols. The two major forms are vitamin D2 and vitamin D3. Vitamin D2 (ergocalciferol) is largely human-made and added to foods, whereas vitamin D3 (cholecalciferol) is synthesized in the skin of humans from 7-dehydrocholesterol and is also consumed in the diet via the intake of animal-based foods. Both vitamin D3 and vitamin D2 are synthesized commercially and found in dietary supplements or fortified foods. The D2 and D3 forms differ only in their side chain structure. The differences do not affect metabolism (i.e., activation), and both forms function as prohormones. When activated, the D2 and D3 forms have been reported to exhibit identical responses in the body, and the potency related to the ability to cure vitamin D–deficiency rickets is the same (Fieser and Fieser, 1959; Jones et al., 1998; Jurutka et al., 2001). Experimental animal studies have indicated that vitamin D2 is less toxic than vitamin D3, but this has not been demonstrated in humans.
The activation steps involved in converting vitamin D from the diet and cutaneous synthesis are illustrated in Figure 3-1. Vitamin D, in either the D2 or D3 form, is considered biologically inactive until it undergoes two
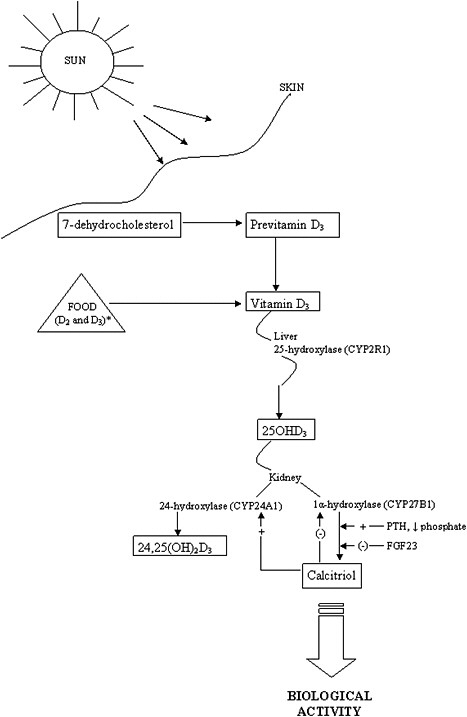
enzymatic hydroxylation reactions. The first takes place in the liver, mediated by the 25-hydroxylase (most likely cytochrome P450 2R1 [CYP2R1]) which forms 25-hydroxyvitamin D (hereafter referred to as 25OHD). The second reaction takes place in the kidney, mediated by 1α-hydroxylase (CYP27B1), which converts 25OHD to the biologically active hormone, calcitriol (1,25-dihydroxyvitamin D). The 1α-hydroxylase gene is also expressed in several extra-renal tissues, but its contribution to calcitriol formation in these tissues is unknown. 25OHD, the precursor of calcitriol, is the major circulating form of vitamin D; it circulates bound to a specific plasma carrier protein, vitamin D binding protein (DBP). DBP also transports vitamin D and calcitriol.
The renal synthesis of calcitriol is tightly regulated by two counter-acting hormones, with up-regulation via parathyroid hormone (PTH) and down-regulation via fibroblast-like growth factor-23 (FGF23) (Galitzer et al., 2008; Bergwitz and Juppner, 2010). Low serum phosphorus levels stimulate calcitriol synthesis, whereas high serum phosphorus levels inhibit it. Following its synthesis in the kidney, calcitriol binds to DBP to be transported to target organs. The biological actions of calcitriol, involve regulation of gene expression at the transcriptional level, and are mediated through binding to a vitamin D receptor (VDR), located primarily in the nuclei of target cells (Jones et al., 1998; Jurutka et al., 2001). Additional hydroxylation reactions, such as that mediated by CYP24A1, as shown in Figure 3-1, result in more polar metabolites with greatly reduced or no apparent biological activity.
The classical actions of vitamin D—which by itself is inactive—are due to the functions of the active metabolite, calcitriol. These actions take the form of the regulation of serum calcium and phosphate homeostasis and, in turn, the development and maintenance of bone health (DeLuca, 1988; Reichel et al., 1989; Jones et al., 1998). Non-classical functions are less well elucidated. VDRs are found fairly ubiquitously throughout the body in tissues not involved with calcium and phosphate homeostasis, and the presence of VDRs in these tissues implies that calcitriol may play a more general role or that ligands other than calcitriol can activate the VDR. Furthermore, the specific vitamin D–responsive elements (VDREs), considered the hallmark of vitamin D action, are present in a large number of human genes involved in a wide range of classical and non-classical roles, such as the regulation of cell proliferation, cell differentiation, and apoptosis. It has been suggested that calcitriol exerts immunomodulatory and anti-proliferative effects through autocrine and paracrine pathways (Adams and Hewison, 2008). These wide-ranging actions of calcitriol have further been hypothesized to play a potential role in preventive or therapeutic action in cancer (Masuda and Jones, 2006) and chronic conditions such
as auto-immune conditions (including type 1 diabetes), cardiovascular disease, and infections (Holick et al., 2007).
Outside of the biological forms of vitamin D, a number of analogues based on the vitamin D structure have been synthesized for use as potential pharmacological agents. These are not, however, dietary or biosynthesized compounds; rather, they are designed for specific applications in research or clinical treatment. Examples of synthetic analogues that have gained importance in clinical medicine are briefly mentioned below.
The term vitamin D is generally used in this report to refer to both the D2 and D3 forms as well as their metabolites, although the two forms are distinguished when necessary for clarification (see Box 3-1 for definitions). Vitamin D levels in the diet—from foods and supplements—are expressed in International Units (IU), but may be expressed elsewhere in micrograms (μg). The biological activity of 1 μg of vitamin D is equivalent to 40 IU. Owing to the frequency with which serum 25OHD levels are included in this report text, the levels are expressed only as nanomoles per liter (nmol/L). As shown in Box 3-1, the nanomoles per liter measure can be converted to nanograms per milliliter (ng/mL) by dividing by a factor of 2.5.
|
BOX 3-1 Terms and Conversions Used in Reference to Vitamin D Terms: Vitamin D—also referred to as calciferol Vitamin D2—also referred to as ergocalciferol Vitamin D3—also referred to as cholecalciferol 25OHD—25-hydroxyvitamin D also referred to as calcidiol or calcifediol; indicates no distinction between D2 and D3 forms. When relevant, forms are distinguished as 25OHD2 and 25OHD3 Calcitriol—1,25-dihydroxyvitamin D3 (Note: Ercalcitriol—refers to 1,25-dihydroxyvitamin D2, but in this report, the term “calcitriol” will be used for both) 24,25(OH)2D—24,25-dihydroxyvitamin D IU = International Unit is a measurement based on biological activity or effect; 1 IU of vitamin D is defined as the activity of 0.025 μg of cholecalciferol in bioassays with rats and chicks. Conversions for Vitamin D3: [sources] 40 IU = 1 μg [serum] 2.5 nmol/L = 1 ng/mL |
SOURCES OF VITAMIN D
Diet
The dietary sources of vitamin D include food and dietary supplements; therefore, “total vitamin D intake” reflects the combined dietary contribution from foods and supplements. There are a few naturally occurring food sources of vitamin D. These include fatty fish, fish liver oil, and egg yolk. Some foods are, however, fortified with vitamin D. After vitamin D was recognized as important for the prevention of rickets in the 1920s (Steenbock and Black, 1924), vitamin D fortification of some foods was initiated on a voluntary basis.
In the United States, fluid milk is voluntarily fortified with 400 IU per quart (or 385 IU/L) of vitamin D (U.S. regulations do not specify the form) (FDA, 2009). In Canada, under the Food and Drug Regulation,1 fortification of fluid milk and margarine with vitamin D is mandatory. Fluid milk must contain 35–45 IU vitamin D per 100 mL and margarine, 530 IU per 100 g. In addition, fortified plant-based beverages must contain vitamin D in an amount equivalent to fluid milk. In analyses conducted in the 1980s and early 1990s, a significant portion of milk samples in the United States were found to contain less than the specified amount of vitamin D (Tanner et al., 1988). Holick et al. (1992) found that 62 percent of milk sampled from five eastern states contained less than 80 percent and 10 percent contained more than 120 percent of the amount of vitamin D stated on the label. Chen et al. (1993) reported similar findings. A more recent report on vitamin D–fortified milk sampled in New York State over a period of 4 years showed that an average of only 47.7 percent of samples fell within the range of acceptable levels of vitamin D fortification (Murphy et al., 2001). However, recent surveys from the U.S. Department of Agriculture (USDA) indicate that these problems have been corrected. In a presentation to this committee, Byrdwell (2009) reported that a USDA survey of milk samples taken in 2007 from 24 locations across the United States showed that most samples had vitamin D levels within the range of 400 to 600 IU/quart.
In Canada, Faulkner et al. (2000) surveyed milk samples and found that 20 percent of skim milk, 40 percent of 2 percent fat milk, and 20 percent of whole milk, contained the recommended level of vitamin D. Samples collected by the Canadian Food Inspection Agency from 1999 through 2009 and analyzed for vitamin D indicated that during the last 4 years of sample collection, 47 to 69 percent were within the range specified by regulation (personal communication, S. Brooks, Health Canada, April
|
1 |
Available online at http://laws.justice.gc.ca/PDF/Regulation/C/C.R.C.,_c._870.pdf (accessed July 23, 2010). |
30, 2010). In addition, over the past 5 years, the average vitamin D content of analyzed milk samples fell within this range. Over time, manufacturers in the United States have added vitamin D to other foods, and the food industry is increasingly marketing foods fortified with vitamin D (Yetley, 2008). Based on data from a U.S. Food and Drug Administration (FDA) survey that provides information on the labels of processed, packaged food products in the United States, Yetley (2008) reported that almost all fluid milks, approximately 75 percent of ready-to-eat breakfast cereals, slightly more than half of all milk substitutes, approximately one-quarter of yogurts, and approximately 8 to 14 percent of cheeses, juices, and spreads are fortified with vitamin D in the U.S. market. Many product labels included in the survey indicated that the form of added vitamin D was vitamin D3. However, some milk substitutes are fortified with vitamin D2. Cereal labels did not specify the form of added vitamin D. Levels of vitamin D ranged from 40 IU per regulatory serving for cereals and cheeses to 60 IU per regulatory serving for spreads and 100 IU per regulatory serving for fluid milk. Several food categories had within-category ranges of 40 to 100 IU of vitamin D per regulatory serving. Serum vitamin D and 25OHD have low penetrance into breast milk, together comprising 40 to 50 IU of antirachitic activity per liter, most of which is contributed by 25OHD (Leerbeck and Sondergaard, 1980; Hollis et al., 1981; Reeve et al., 1982; Specker et al., 1985). Data from the USDA report the vitamin D content of human milk to be 4.3 IU/100 kcal.2 However, the vitamin D biological activity may be higher than the analyzed values, because human milk contains small amounts of 25OHD in addition to vitamin D3 (Reeve et al., 1982); further, the biological activity of 25OHD is approximately 50 percent higher than that of vitamin D (Blunt et al., 1968).
The FDA has established that infant formula must contain 40 to 100 IU of vitamin D per 100 kcal.3 Commercial infant formulas contain approximately 60 IU of vitamin D per 100 kcal, as estimated by the USDA food composition database,4 and Yetley (2008) reported that commercial milk-based infant formulas collected between 2003 and 2006 contained 87
|
2 |
USDA National Nutrient Database for Standard Reference Release 23. NBD No. 01107. Milk, human, mature, fluid. Available online at http://www.ars.usda.gov/Services/docs.htm?docid=8964 (accessed August 3, 2010). |
|
3 |
USDA National Nutrient Database for Standard Reference Release 23. NBD No. 03946. Infant formula, ROSS, SIMILAC LACTOSE FREE ADVANCE, ready-to-feed, with ARA and DHA; and NDB no. 03815. Infant formula, MEAD JOHNSON, ENFAMIL LIPIL, with iron, ready-to-feed, with ARA and DHA. Available online at http://www.ars.usda.gov/main/site_main.htm?modecode=12-35-45-00 (accessed April 28, 2010). |
|
4 |
Available online at http://www.nal.usda.gov/fnic/foodcomp/search/ (accessed March 16, 2010). |
to 184 percent of label declarations. In Canada, infant formula is required by regulation to contain between 40 and 80 IU of vitamin D per 100 kcal.
In recent years, dietary supplements containing vitamin D have become more common and have been more frequently consumed. The form of vitamin D used in supplement products can be either vitamin D2 or vitamin D3. It would appear from informal observations of the market place that manufacturers are increasingly switching from vitamin D2 to vitamin D3, and some are increasing the vitamin D content of their products. Traditionally, many marketed dietary supplements have contained 400 IU per daily dose, but levels in supplements have been increasing. In the United States, vitamin D can now be found in multi-vitamin/multi-mineral formulations as well as a single supplement in a range of dosage levels, including 1,000 to 5,000 IU of vitamin D3 per dose and even up to 50,000 IU of vitamin D2 per dose. In Canada, dosage levels of vitamin D above 1,000 IU are obtainable only with a prescription.
Information about current national survey estimates of the intake of vitamin D from foods and supplements can be found in Chapter 7.
Synthesis in the Skin
Vitamin D3 is synthesized in human skin from 7-dehydrocholesterol following exposure to ultraviolet B (UVB) radiation with wavelength 290 to 320 nm.5 The process of UVB-mediated conversion of 7-dehydrocholesterol to the previtamin D3 form and subsequent thermal isomerization to vitamin D3 occurring in the epidermis is illustrated in Figure 3-2.
The production of vitamin D3 in skin is a function of the amount of UVB radiation reaching the dermis as well as the availability of 7-dehydrocholesterol (Holick, 1995). As such, the level of synthesis is influenced by a number of factors, as described below in the section entitled “Measures Associated with Vitamin D: Serum 25OHD,” including season of the year, skin pigmentation, latitude, use of sunscreen, clothing, and amount of skin exposed. Age is also a factor, in that synthesis of vitamin D declines with increasing age, due in part to a fall in 7-dehydrocholesterol levels and due in part to alterations in skin morphology (MacLaughlin and Holick, 1985).
Toxic levels of vitamin D do not occur from prolonged sun exposure. Thermal activation of previtamin D3 in the skin gives rise to multiple non–vitamin D forms, such as lumisterol, tachysterol and others (Holick et al., 1981; Webb et al., 1989), as illustrated in Figure 3-2; this limits the
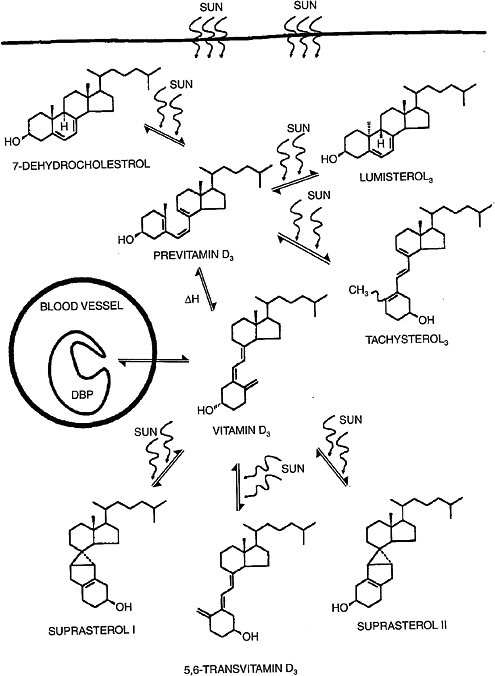
FIGURE 3-2 Photochemical events that lead to the production and regulation of vitamin D3 (cholecalciferol) in the skin.
NOTE: DBP = vitamin D binding protein.
SOURCE: Holick (1994). Reprinted with permission from the American Journal of Clinical Nutrition (1994, volume 60, pages 619-630), American Society for Nutrition.
formation of vitamin D3 itself. Vitamin D3 can also be converted to nonactive forms.
The absolute percentage of circulating 25OHD that arises from cutaneous synthesis versus oral intake of vitamin D in the free-living North American population cannot be clearly specified. Individuals living at Earth’s poles during winter months and submariner crew members with very limited or no measurable UVB exposure have detectable levels of 25OHD in blood, arising from dietary sources and likely from previously synthesized and stored vitamin D. This topic is further explored in the section below that focuses on serum 25OHD.
METABOLISM OF VITAMIN D
Absorption
Owing to its fat-soluble nature, dietary vitamin D (either D2 or D3) is absorbed with other dietary fats in the small intestine (Haddad et al., 1993; Holick, 1995). The efficient absorption of vitamin D is dependent upon the presence of fat in the lumen, which triggers the release of bile acids and pancreatic lipase (Weber, 1981, 1983). In turn, bile acids initiate the emulsification of lipids, pancreatic lipase hydrolyzes the triglycerides into monoglycerides and free fatty acids, and bile acids support the formation of lipid-containing micelles, which diffuse into enterocytes. Early studies demonstrated that radiolabeled vitamin D3 appeared almost exclusively in the lymphatics and in the chylomicron fraction of plasma; as well, subjects with impaired bile acid release or pancreatic insufficiency both demonstrated significantly reduced absorption of vitamin D (Thompson et al., 1966; Blomstrand and Forsgren, 1967; Compston et al., 1981). Subsequently, other clinical and experimental animal studies confirmed that vitamin D is most efficiently absorbed when consumed with foods containing fat (Weber, 1981; Johnson et al., 2005; Mulligan and Licata, 2010) and, conversely, that a weight-loss agent that blocks fat absorption also impairs the absorption of vitamin D (James et al., 1997; McDuffie et al., 2002). The optimal amount of fat required for maximal absorption of vitamin D has not been determined.
Within the intestinal wall, vitamin D, cholesterol, triglycerides, lipoproteins, and other lipids are packaged together into chylomicrons. Importantly, while a fraction of newly absorbed intestinal vitamin D is also transported along with amino acids and carbohydrates into the portal system to reach the liver directly, the main pathway of vitamin D uptake is incorporation into chylomicrons that reach the systemic circulation via the lymphatics. Chylomicron lipids are metabolized in peripheral tissues that express lipoprotein lipase, but particularly in adipose tissue and
skeletal muscle, which are rich in this enzyme. During hydrolysis of the chylomicron triglycerides, a fraction of the vitamin D contained in the chylomicron can be taken up by these tissues. Uptake into adipose tissue and skeletal muscle accounts for the rapid postprandial disappearance of vitamin D from plasma and probably also explains why increased adiposity causes sequestering of vitamin D and is associated with lower 25OHD levels (Jones, 2008). What remains of the original chylomicron after lipolysis is a chylomicron remnant, a cholesterol-enriched, triglyceride-depleted particle that still contains a fraction of its vitamin D content.
Metabolism to the Active Hormonal Form
Vitamin D, regardless of origin, is an inactive prohormone and must first be metabolized to its hormonal form before it can function. Once vitamin D enters the circulation from the skin or from the lymph, it is cleared by the liver or storage tissues within a few hours. The processes that follow are illustrated in Figure 3-3. Vitamin D is converted in the liver to 25OHD, a process carried out by a CYP enzyme that has yet to be fully defined but is likely CYP2R1 (Cheng et al., 2003). The crystal structure of CYP2R1 has been determined with vitamin D in the active site, and the enzyme has been shown to metabolize both vitamin D2 and vitamin D3 equally efficiently (Strushkevich et al., 2008). There is little, if any, feedback regulation of this enzyme. A large genome-wide association study of factors that might be determinants of the circulating 25OHD levels identified the human chromosomal 11p15 locus of CYP2R1 as a significant determinant, whereas the loci of the other enzymes purported to have 25-hydroxylase activity (e.g., CYP27A1 and CYP3A4) were not identified (Wang et al., 2010). The other determinants of serum 25OHD besides CYP2R1 have been reported to be DBP (also known as Gc protein), which has six common phenotypes (Laing and Cooke, 2005) as well as 7-dehydrocholesterol reductase and CYP24A1. Increasing intake of vitamin D results in higher blood levels of 25OHD, although perhaps not in a linear manner (Stamp et al., 1977; Clements et al., 1987).
At this point, 25OHD bound to DBP circulates in the blood stream and, when calcitriol is required due to a lack of calcium (or lack of phosphate), 25OHD is 1α-hydroxylated in the kidney to form calcitriol, the active form, by the 1α-hydroxylase enzyme (also known as CYP27B1) (Tanaka and DeLuca, 1983). This metabolic step is very tightly regulated by blood calcium and phosphate levels through PTH and the phosphaturic hormone, FGF23, and constitutes the basis of the vitamin D endocrine system that is central to maintaining calcium and phosphate homeostasis (see discussion below on functions and physiological actions). FGF23 acts by reducing the expression of renal sodium–phosphate transporters and reducing serum calcitriol levels.
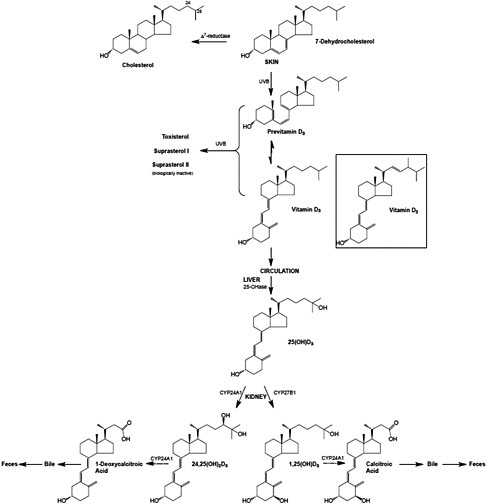
FIGURE 3-3 The metabolism of vitamin D3 from synthesis/intake to formation of metabolites. The process is the same for vitamin D2 once it enters the circulation.
NOTE: CYP = cytochrome P450 (a large and diverse group of enzymes).
SOURCE: Reprinted with permission from Hector DeLuca.
Production of the CYP27B1 enzyme is stimulated by PTH, which is secreted in response to a lack of calcium. It is also stimulated by the hypophosphatemic action of FGF23 on renal phosphate excretion, but to a lesser extent. When PTH is suppressed, or FGF23, produced by osteocytes, is stimulated, 1α-hydroxylation is markedly reduced (Liu et al., 2007; Quarles, 2008). Furthermore, calcitriol can act as a suppressor of CYP27B1, although the mechanism is not fully understood.
Calcitriol has its strongest metabolic activity in inducing its own destruction by stimulating the 24-hydroxylase enzyme (now known as CYP24A1;
Figure 3-1) (Jones et al., 1998). The enzyme CYP24A1 is found in all target tissues and is induced in response to calcitriol interacting with the VDR. CYP24A1 is largely responsible for the metabolic degradation of calcitriol and its precursor, 25OHD, and its deletion in the mouse results in 50 percent lethality at weaning and an inability to efficiently clear the active form of vitamin D (Masuda et al., 2005). CYP24A1 carries out a series of reactions resulting ultimately in production of calcitroic acid from calcitriol and 1-desoxycalcitroic acid from 24,25(OH)2D, the major metabolite of 25OHD. These products are excreted through the bile into the feces (Jones et al., 1998); very little is eliminated through the urine (Kumar et al., 1976). The active forms of vitamin D2 are also catabolized by CYP24A1 into a series of biliary metabolites, somewhat analogous to those of vitamin D3.
As described above, all naturally occurring vitamin D compounds interact with DBP. Calcitriol and vitamin D have significantly lower affinity for this protein than does 25OHD. Whereas vitamin D has an average lifetime in the body of approximately 2 months, 25OHD has a lifetime of 15 days, and calcitriol has a lifetime measured in hours (Jones et al., 1998). Aside from these key elements in vitamin D metabolism, more than 30 other metabolites have been found, including the 3-epi series of vitamin D compounds (DeLuca and Schnoes, 1983; Siu-Caldera et al., 1999). Their importance seems minimal and need not be discussed here.
Although the route of catabolism between 1α,25(OH)2D2 and 1α,25(OH)2D3 differs beyond the initial 24-hydroxylation step, because 24-hydroxylation is primarily a deactivation step (Brommage and DeLuca, 1985; Horst et al., 1986; Lohnes and Jones, 1992; Jones et al., 1998), the rate of this initial step should be the important indicator of the loss of biological action. Comparisons of initial rate kinetics of the 24-hydroxylase enzyme (CYP24A1) activity toward 1α,25(OH)2D2 and 1α,25(OH)2D3 and their precursors suggest that the rates of inactivation by CYP24A1 in vitro are virtually identical (Jones et al., 2009; Urushino et al., 2009). Although side-chain hydroxylation of 1α,25(OH)2D2 represents the primary route of metabolism in the target cell, clearance of the metabolic products in vivo is complicated by additional non-specific liver CYPs (e.g., CYP3A4) (Gupta et al., 2004, 2005) that are inducible by 1α,25(OH)2D3 in certain extra-hepatic tissues (Thompson et al., 2002) and also Phase II enzymes, including uridine diphosphate–glucuronosyl transferases, which are known to subject vitamin D metabolites to glucuronidation (LeVan et al., 1981; Hashizume et al., 2008). The pharmacokinetic consequence of the sum of these catabolic systems, as shown in studies in rats, is a slightly reduced half-life for 1α,25(OH)2D2 compared with 1α,25(OH)2D3 (Knutson et al., 1997).
There are reports that vitamin D2 and vitamin D3 are differentially susceptible to these non-specific inactivating modifications, such as those occurring in the liver in response to a variety of drugs. These enzymes in-
clude the liver and intestinal CYPs that are known to metabolize vitamin D compounds differently, such as CYP27A1, which 25-hydroxylates vitamin D3 and 24-hydroxylates vitamin D2 (Guo et al., 1993), and CYP3A4, which 24- and 25-hydroxylates vitamin D2 substrates more efficiently than vitamin D3 substrates (Gupta et al., 2004, 2005) and 23R- and 24S-hydroxylates 1α,25(OH)2D3 (Xu et al., 2006); the latter enzyme has recently been shown to be selectively induced by 1α,25(OH)2D in the intestine (Thompson et al., 2002; Xu et al., 2006). Both CYP27A1 and CYP3A4 are known to have significantly lower Michaelis-Menten constants (Km values) for 25OHD3 compared with CYP2R1 (Guo et al., 1993; Sawada et al., 2000), in the micromole per liter range; this questions their physiological but not their pharmacological relevance. Recent work (Helvig et al., 2008; Jones et al., 2009) has shown that both human intestinal microsomes and recombinant CYP3A4 protein break down 1α,25(OH)2D2 at a significantly faster rate than 1α,25(OH)2D3, suggesting that this non-specific CYP might limit vitamin D2 action preferentially in target cells, where it is expressed and when the substrate is in the pharmacological dose range. The same type of mechanism involving differential induction of non-specific CYPs may underlie the occasional reports of co-administered drug classes, such as anticonvulsants (Christiansen et al., 1975; Tjellesen et al., 1985; Hosseinpour et al., 2007), causing accelerated degradation of one vitamin D form over the other.
Storage
Adipose tissue stores of vitamin D probably represent “non-specific” stores sequestered because of the hydrophobic nature of vitamin D, but the extent to which the processes of accumulation or mobilization are regulated by normal physiological mechanisms remains unknown at this time. Rosenstreich et al. (1971) first identified adipose tissue as the primary site of vitamin D accumulation from experiments in which radiolabeled vitamin D was administered to vitamin D–deficient rats. Tissue levels of radioactivity measured during vitamin D repletion and during a subsequent period of deprivation showed that adipose tissue acquired the greatest quantity of radioactive compound and had the slowest rate of release. Work by Liel et al. (1988) suggested that there was enhanced uptake and clearance of vitamin D by adipose tissue in obese subjects compared with those of normal weights. Similarly, Wortsman et al. (2000) concluded that in obese subjects, vitamin D was stored in adipose tissue and not released when needed. Finally, Blum et al. (2008) found that, in elderly subjects supplemented with 700 IU of vitamin D per day, for every additional 15 kg of weight above “normal” at baseline, the mean adjusted change in 25OHD level was approximately 10 nmol/L lower after 1 year of supplementation.
The authors estimated that in order for subjects with body mass indexes (BMIs) above the normal range to obtain an increase in serum 25OHD level similar to that of subjects with weight in the normal range, an additional 17 percent increase in vitamin D above the administered dose of 700 IU/day would be needed for every 10 kg increase in body weight above baseline in their study population.
The implication of these studies is that vitamin D deposited in fat tissue is not readily available, and obese individuals may require larger than usual doses of vitamin D supplements to achieve a serum 25OHD level comparable to that of their normal weight counterparts. In support of the hypothesis that vitamin D is stored in adipose tissues, weight reduction studies show that serum 25OHD levels rise when obese individuals lose body fat (Riedt et al., 2005; Zitterman et al., 2009; Tzotzas et al., 2010). Conclusive statements regarding changes in serum 25OHD levels after gastric bypass surgery cannot be made, as a result of confounding factors, such as weight change, possible malabsorption, and diet. There is evidence of a rise in serum 25OHD levels after surgery (Mahdy et al., 2008; Aasheim et al., 2009; Goldner et al., 2009; Bruno et al., 2010), as well as evidence that there is no change after surgery (Riedt et al., 2006; Fleischer et al., 2008; Valderas et al., 2009). Gehrer et al. (2010) indicated that serum 25OHD levels decrease after gastric bypass surgery, although the quality of the methods used is questionable.
Excretion
As described previously, the products of vitamin D metabolism are excreted through the bile into the feces, and very little is eliminated through the urine. This is in part due to renal reuptake of vitamin D metabolites bound to DBP, as mediated by the cubilin–megalin receptor system (Willnow and Nykjaer, 2005).
Excess Intake
Excess intake of vitamin D—but not sun exposure, which is associated with a series of thermal and photoisomerization reactions (see Figure 3-2) —can lead to a state of vitamin D “intoxication” or “hypervitaminosis D.” Chemically synthesized vitamin D became available late in the third decade of the 20th century; reports of vitamin D intoxication were first found from 1928 to 1932 and continued throughout most of the 20th century (DeLuca, 2009). The condition of hypervitaminosis D leads to hypercalcemia and eventually to soft tissue calcification and resultant renal and cardiovascular damage (DeLuca, 1974). In the case of animal models, at necropsy, vitamin D–intoxicated rats show widespread calcification of organs and tissues.
The form of the vitamin implicated in the intoxication is 25OHD (Vieth, 1990; Jones, 2008). In fact, it has been shown in dietary supplementation studies using the CYP27B1 knockout mouse, which is incapable of making calcitriol, sufficiently high concentrations of serum levels of 25OHD can cause changes in vitamin D–dependent general expression even in the absence of calcitriol (Rowling et al., 2007; Fleet et al., 2008).
FUNCTIONS AND PHYSIOLOGICAL ACTIONS OF VITAMIN D
Calcium and Phosphate Homeostasis
The dominant function of vitamin D in its hormonal form (calcitriol or 1,25-dihydroxyvitamin D) is the elevation of plasma calcium and phosphate levels, which are required for mineralization of bone (DeLuca, 1979b; Holick, 1996). Furthermore, the elevation of plasma calcium to normal levels is also required for the functioning of the neuromuscular junction as well as vasodilatation, nerve transmission, and hormonal secretion.
Calcitriol—functioning as part of the endocrine system for maintaining serum calcium levels as outlined in Chapter 2—elevates plasma ionized calcium levels to the normal range by three different mechanisms (see Figure 2-1 in Chapter 2). The first mechanism, which does not require PTH, is the well-established role of calcitriol in stimulating intestinal calcium absorption throughout the entire length of the intestine, although its greatest activity is in the duodenum and jejunum. It is clear that calcitriol directly stimulates intestinal calcium and, independently, phosphate absorption.
In the second mechanism, calcitriol plays an essential role in the mobilization of calcium from bone, a process requiring PTH (Garabedian et al., 1972; Lips, 2006). It induces the formation and activation of the osteoclast to function in the mobilization of calcium from bone, as discussed in Chapter 2. In short, calcitriol facilitates the formation of osteoclasts by stimulating the secretion of a protein called receptor activator for nuclear factor κ B (RANK) ligand, which, in turn, is responsible for osteoclastogenesis and bone resorption (Suda et al., 1992; Yasuda et al., 2005).
In the third mechanism, calcitriol together with PTH stimulates the renal distal tubule reabsorption of calcium, ensuring retention of calcium by the kidney when calcium is needed (Sutton et al., 1976; Yamamoto et al., 1984). These well-known functions dominate vitamin D physiology and many of the functional proteins involved in these processes have been identified, although the exact molecular mechanisms of all of these systems have yet to be elucidated.
Thus, overall, calcitriol acts on the intestine, bone, and kidney as described above, and as illustrated in Figure 2-1 in Chapter 2, to elevate
serum calcium levels, closing the calcium loop. As serum calcium levels rise, PTH secretion drops. If serum calcium levels become too high, the parafollicular cells (“C” cells) of the thyroid secrete calcitonin, which blocks calcium resorption from bone and helps to keep calcium levels in the normal range. Calcitriol, through its receptor, the VDR, suppresses parathyroid gene expression and parathyroid cell proliferation, providing important feedback loops that reinforce the direct action of increased serum calcium levels (Slatopolsky et al., 1984; Silver et al., 1986).
Not shown in Figure 2-1 in Chapter 2 is the mechanism of action of vitamin D in regulating serum phosphorus levels, certain aspects of which remain obscure. What is known is that (1) a deficiency of phosphate stimulates CYP27B1 to produce more calcitriol, which in turn stimulates phosphate absorption in the small intestine; and (2) calcitriol can also induce the secretion of FGF23 by osteocytes in bone, which results in phosphate excretion in the kidney (Liu et al., 2008), as well as feedback on vitamin D metabolism.
Other Actions
It is noteworthy that the VDR is present in the nucleus of many tissues that are not involved in the regulation of calcium and phosphate metabolism. For example, the VDR has been clearly described in epidermal keratinocytes, in activated T cells of the immune system, in antigen-presenting cells, in macrophages and monocytes, and in cytotoxic T cells. Gene array studies in many cells and tissues show that calcitriol regulates several hundred genes throughout the body or as much as 5 percent of the human genome (Pike et al., 2008). However, exactly how calcitriol functions in these tissues and the physiological consequences are not clearly known.
Likewise, the importance of the paracrine or autocrine synthesis of calcitriol under non-disease conditions is unclear. The 1α-hydroxylase (CYP27B1) gene has been reported to be expressed in many extra-renal tissues (Hewison et al., 2007). In some cases, this is based upon in vitro production of calcitriol by cell lines as a consequence of culture conditions, but it also includes detection of the messenger ribonucleic acid (mRNA) transcript or protein for CYP27B1 in tissues in vivo (Hewison et al., 2007). There is no doubt that the kidney is physiologically the overwhelming site of production of calcitriol for the circulation, as chronic kidney disease or nephrectomy results in a significant fall in the serum calcitriol level (Martinez et al., 1995). The contribution of calcitriol to the maternal circulation stemming from production by the placenta is not clearly known; based on a case report for an anephric patient, it appears that the placenta produces calcitriol, but its contribution to the maternal circulation is low (Turner et al., 1988). The pregnancy-related rise in calcitriol is due to up-
regulation of the enzymes in the maternal kidney (Kovacs and Kronenberg, 1997). However, there may be other extra-renal 1α-hydroxylation sites that can act as intracrine systems primarily involved in regulation of cell or tissue growth: skin, gastrointestinal tract, or glandular tissue, such as prostate and breast (Diesing et al., 2006). In mice missing the Vdr gene (Vdr-null), calcitriol and the VDR play a role in lactational physiology; there is accelerated mammary development during pregnancy, but delayed involution of the mammary tissue after lactation (Zinser and Welsh, 2004). Extra-renal CYP27B1 may be up-regulated during inflammation (Ma et al., 2004; Liu et al., 2008) or down-regulated in cancerous tissue proliferation (Bises et al., 2004; Wang et al., 2004). Furthermore, extra-renal production of calcitriol is clearly found in certain pathological diseases, including granulomatous conditions such as sarcoidosis, lymphoma, and tuberculosis (Adams et al., 1989), which can be associated with hypercalcemia. If sarcoidosis is left untreated, the extra-renally produced calcitriol can enter the circulation, resulting in hypercalciuria and eventually hypercalcemia.
There is emerging evidence that calcitriol plays a role in the immune system that has not yet been clearly described. Exogenous calcitriol can suppress autoimmune diseases, but with hypercalcemia as an important side effect (DeLuca and Cantorna, 2001). It has been shown that the local conversion of 25OHD into calcitriol in monocytes or macrophages results in an increase in cellular immunity by stimulating the production of cathelicidin, an anti-microbial peptide capable of killing bacteria, particularly Mycobacterium tuberculosis (Liu et al., 2006). Recently, Stubbs et al. (2010) showed that renal dialysis patients treated with high-dose vitamin D3 develop a population of immune cells with increased CYP27B1, VDR, and cathelicidin expression, although the role of these cells in vivo is unknown. Ironically, calcitriol has an opposite effect on the adaptive immune (B and T cell function) response. Calcitriol generally inhibits T helper cell proliferation and B cell immunoglobulin production. In contrast, calcitriol promotes the proliferation of immunosuppressive regulatory T cells and their accumulation at sites of inflammation (Penna et al., 2007).
A role for vitamin D in carcinogenesis evolved initially from in vitro studies as cell culture approaches became more widely available for the evaluation of the mechanisms of action of vitamin D and its metabolites (Masuda and Jones, 2006). The active hormone, calcitriol, was shown to consistently inhibit the growth of cancer cells and promote differentiation in vitro by regulating multiple pathways (Deeb et al., 2007; Kovalenko et al., 2010). Additional studies documented the presence of the VDR in a wide array of cancer cell types. Vitamin D orchestrates cell cycle progression via alterations in key regulators such as cyclin-dependent kinases, retinoblastoma protein phosphorylation, and repression of the proto-oncogene myc as well as by modulating growth factor receptor-mediated signaling
pathways (Koga et al., 1988; Kawa et al., 1996; Campbell et al., 1997; Xie et al., 1997; Yanagisawa et al., 1999; Sundaram et al., 2000; Gaschott and Stein, 2003; Li et al., 2004). In addition, calcitriol restores or enhances pro-apoptotic effects in cancer cells by several possible pathways, including repression of several pro-survival proteins such as Bc12 and telomerase reverse transcriptase and by activating pro-apoptotic proteins Bax and μ-calpain (James et al., 1996; Diaz et al., 2000; Jiang et al., 2004; Kumagai et al., 2005). Evidence also supports an anti-angiogenic effect of vitamin D. Vascular endothelial growth factor (VEGF) expression by cancer cells is suppressed and endothelial cell responses to VEGF are inhibited by vitamin D, an observation supported by in vivo xenograft studies (Mantell et al., 2000; Bao et al., 2006). The immunoregulatory effects of vitamin D may also have an impact on cancer biology. Inflammation is a critical early step in the carcinogenesis cascade for many cancers, and the ability of vitamin D to exhibit anti-inflammatory effects on cancer cells by down-regulating the pro-inflammatory pathways, such as cyclooxygenase-2, may contribute to cancer inhibition (Moreno et al., 2005). In contrast, the role of vitamin D in cancer immunosurveillance of nascent or established cancers remains to be defined.
The encouraging in vitro findings, tempered with concerns about hypercalcemia, led to the development of many vitamin D analogues in the hope of retaining anti-cancer activity, but without increasing serum calcium, for the pharmacological therapy of cancer, as recently reviewed (Beer and Myrthue, 2004; Masuda and Jones, 2006; Trump et al., 2010).
Vitamin D2Versus Vitamin D3
Vitamins D2 and D3, as described previously, differ only in their side chain structure. Physiological responses to both forms of the vitamin include regulation of calcium and phosphate homeostasis and regulation of cell proliferation and cell differentiation of specific cell types, as described above. Qualitatively, vitamins D2 and D3 exhibit virtually identical biological responses throughout the body (i.e., through gene expression) that are mediated by the VDR (Jones et al., 1998; Jurutka et al., 2001).
Regarding the potency of the two forms of vitamin D, there are reports that certain animals, such as avian species and New World monkeys (Chen and Bosmann, 1964; Drescher et al., 1969), discriminate against vitamin D2. However, it has been assumed for several decades that the two forms are essentially equipotent in humans (Christiansen et al., 1975). Recent reports involving human dietary studies have argued for (Trang et al., 1998; Armas et al., 2004) or against (Holick et al., 2008) a metabolic discrimination against vitamin D2, compared with vitamin D3. Part of the apparent conflict between these different studies (Trang et al., 1998; Armas et al.,
2004; Holick et al., 2008) is almost certainly due to differences in size and frequency of dose (which have ranged from 1,000 IU daily doses to 50,000 IU in a single dose); the differences reported suggest a difference in pharmacokinetic parameters between vitamin D2 and vitamin D3.
This debate runs parallel to the suggestion that vitamin D2 is less toxic than its vitamin D3 counterpart. Experimental animal data from a number of mammalian species ranging from rodents to primates (Roborgh and de Man, 1959, 1960; Hunt et al., 1972; Sjoden et al., 1985; Weber et al., 2001), support the concept that the D2 form is less toxic than D3, but there is no evidence available in humans. Nonetheless, the implication of these diverse studies in several mammalian species is that vitamin D2 compounds may show differences in pharmacokinetics that manifest as lower toxicity from high doses.
There is considerable evidence that most of the steps involved in the metabolism and actions of vitamin D2 and vitamin D3 are identical (Jones et al., 1998). The identification of the series of vitamin D3 metabolites in the late 1960s and early 1970s was followed by the identification of their vitamin D2 counterparts: 25OHD2, 1α,25(OH)2D2, and 24,25(OH)2D2 (Suda et al., 1969; Jones et al., 1975, 1979, 1980a). Noteworthy here is the fact that the structural features unique to the vitamin D2 side chain did not preclude either the 25- or 1α-hydroxylation steps in activation of the molecule or the first step of inactivation, namely 24-hydroxylation. Studies have also shown that the steps in the specific vitamin D signal transduction cascade do not appear to discriminate discernibly between the two vitamin D homologues at the molecular level (e.g., binding to the transport protein, DBP [Hay and Watson, 1977; Jones et al., 1980a] or binding to the receptor, VDR [Jones et al., 1980b; Reinhardt et al., 1989]). Overall, it can be concluded that specific signal transduction systems designed to respond to vitamin D3 respond to physiological doses of vitamin D2 equally well.
At this time, firm conclusions about different effects of the two forms of vitamin D cannot be drawn; however, it would appear that at low doses, D2 and D3 are equivalent, but at high doses, D2 is less effective than D3. In essence, the potency of the two forms (as judged by the dose required to cure rickets) is assumed to be the same (Park, 1940). Differences in toxicity for humans, as judged by the dose to cause hypervitaminosis D, are unclear, but there is evidence from experimental animal data to suggest that D2 is less toxic than D3.
Skeletal Disorders
Vitamin D deficiency results in inadequate mineralization of the skeleton. Commonly referred to as rickets in children and osteomalacia in adults, this disorder has been described in Chapter 2 relative to calcium.
Vitamin D deficiency is characterized by aberrations in the mineralization of the bone. In children, the deficiency results in rickets (see also Chapter 2), in which the cartilage fails to mature and mineralize normally. Rickets is characterized by widening at the end of the long bones, rachitic rosary, deformations in the skeleton, including craniotabes and deformities of the lower limbs, known as bowed legs and knocked knees. In adults, the deficiency of vitamin D leads to osteomalacia in which the newly deposited bone matrix fails to mineralize adequately, and there are wide unmineralized bone matrix (osteoid) seams.
Vitamin D–dependent rickets type I (VDDR I) is an autosomal recessive trait that results in abnormally low calcitriol levels but normal serum 25OHD levels. The mutation in VDDR I affects the 1α-hydroxylase enzyme and leads to impaired intestinal calcium absorption and the resulting rickets (Fraser et al., 1973). VDDR I manifests in the first year after birth and is treated with calcitriol. Supplemental calcium and phosphate are usually not needed. The second disorder is vitamin D–dependent rickets type II (VDDR II), which results in hypocalcemia, tetany, convulsions, alopecia, and rickets. VDDR II is also an autosomal recessive trait, resulting from a mutation in the Vdr gene, which can appear in the second year after birth or go unrecognized until adulthood.
VITAMIN D ACROSS THE LIFE CYCLE
Overall, vitamin D’s role at different life stages is less clearly age-related than that of calcium, and also less well understood, with numerous gaps in basic information. Although some aspects of vitamin D nutrition and physiology have been found to differ with life stage, most of the functions of vitamin D are quite consistent across life stages from infancy and childhood, to adolescence, adulthood, and old age. For all life stages highlighted below, specific studies and conclusions are detailed in Chapter 4.
Infancy
Healthy skeletal development in infancy requires adequate intakes of vitamin D as well as calcium. Inadequate vitamin D intake during periods of growth leads to development of vitamin D deficiency rickets, which when it occurs in North American populations typically manifests around 20 months of age (DeLucia et al., 2003). If rickets is diagnosed early, vitamin D therapy can cure it, but not if skeletal deformities are severe and growth plates have started to mature in puberty (DeLuca, 1979a). Infants at risk for developing rickets include those who are exclusively breast-fed, because vitamin D and 25OHD are normally present at low levels in breast milk (Bachrach et al., 1979; Ward et al., 2007). Health Canada currently
recommends that exclusively breast-fed infants receive a supplement of vitamin D,6 and the American Academy of Pediatrics guidelines support supplementation of breastfeeding infants with vitamin D (Gartner and Greer, 2003). Commercial infant formula contains vitamin D, as discussed previously in this chapter.
Childhood and Adolescence
This life stage is characterized by bone accretion. During the rapid growth phase of adolescence, almost 50 percent of the adult skeletal mass will be accumulated. The onset of puberty stimulates increased metabolism of 25OHD levels to calcitriol (Aksnes and Aarskog, 1982) and subsequent increased calcium intestinal absorption, decreased urinary calcium excretion, and greater calcium deposition into bone (Wastney et al., 1996). Information on relationships between 25OHD levels and optimal intestinal absorption of calcium or risk for rickets or fracture in children and adolescents is lacking, although Abrams et al. (2005) found evidence for an indirect relationship between low serum 25OHD and increased calcium absorption in young adolescents. Although a recent set of systematic reviews (Cranney et al., 2007; Chung et al., 2009), to be discussed in Chapter 4, did not report specifically on bone mass for this age group in relation to vitamin D nutriture, the reviews suggested the possibility of a relationship between serum levels of 25OHD and bone mineral density (BMD) in adolescents. A recent analysis of vitamin D intake and BMD in male and female adolescents and adults ages 13 to 36 years found positive correlations between vitamin D intake and bone density from adolescence into adulthood among male but not female subjects (van Dijk et al., 2009).
Adults
The life stages associated with younger adults, covering several decades, are characterized by a need for adequate nutrition for bone maintenance. The bone is constantly undergoing remodeling, and the maintenance of normal bone density reduces the risk of skeletal disorders ranging from osteomalacia to the onset of osteoporotic fractures later in life. It is also the time of pregnancy and lactation for some female members of this population.
Older adults, especially those characterized as frail, may have poor dairy and vitamin D intake, decreased sun exposure, reduced dermal conversion of 7-dehydrocholesterol to vitamin D3 and secondary hyperparathy-
|
6 |
Available online at http://www.hc-sc.gc.ca/fn-an/nutrition/infant-nourisson/vita_d_supp-eng.php (accessed September 1, 2010). |
roidism, all of which contribute to increased risk for poor bone health and osteoporotic fractures. As discussed below, there is inconsistent evidence as to whether intestinal absorption of vitamin D declines with age. In women, bone loss occurs as a result of the decreased estrogen levels that accompany menopause. As aging continues, both men and women experience age-related bone loss. As is the case for calcium intake, it is not well established whether and to what extent intakes of vitamin D may mitigate the bone loss.
Pregnancy and Lactation
The role of vitamin D in pregnancy and fetal development is the focus of current attention. However, at present the role of vitamin D is not clear, and there are very few data by which to examine the questions surrounding the effect of the nutrient on pregnancy and lactation. Animal studies and inferential human data do not readily elucidate a specific function in fetal development, especially with respect to formation and mineralization of the fetal skeleton. Calcitriol levels increase during pregnancy, but factors other than vitamin D appear to stimulate the increased calcium absorption. Although a number of avenues are still being explored, the bulk of the evidence suggests that calcium is moved from the mother to the fetus without requiring calcitriol.
Breast milk is not normally a significant source of vitamin D for the infant and remains unchanged with supplementation at least up to 2,000 IU/day. Existing evidence suggests that vitamin D nutriture does not appear to affect the maternal processes of bone resorption that occur during lactation, nor its restoration post-lactation.
MEASURES ASSOCIATED WITH VITAMIN D: SERUM 25OHD
Serum 25OHD level is widely considered as a marker of vitamin D nutriture, and consideration of serum 25OHD measures for the purposes of nutrient reference value development has generated notable interest. There is agreement that circulating serum 25OHD levels are currently the best available indicator of the net incoming contributions from cutaneous synthesis and total intake (foods and supplements) (Davis et al., 2007; Brannon et al., 2008; Davis, 2008). Thus, the serum 25OHD level may function as a biomarker of exposure; it is a reflection of the supply of vitamin D to the body and can be a useful adjunct to examining the intake level of vitamin D if the confounders and the measure’s variability depending upon a range of variables are kept in mind. However, what is not clearly established is the extent to which 25OHD levels serve as a biomarker of effect. That is, there is some question as to whether levels of 25OHD relate to
health outcomes via a causal pathway and can serve as predictors of such outcomes.
Research recommendations in the previous Dietary Reference Intake (DRI) review of vitamin D (IOM, 1997), as well as an Institute of Medicine (IOM) workshop on DRI research needs (IOM, 2007), called for studies to evaluate the intake requirements for vitamin D as related to optimal circulating 25OHD concentrations across life stage and race/ethnicity groups of U.S. and Canadian populations, taking into account variability in UVB radiation exposures. The issue of the role of serum 25OHD concentrations was also identified by the sponsors of this current study on vitamin D and calcium DRIs as central to the development of DRIs for vitamin D (Yetley et al., 2009). Much in the way of this information gap for serum 25OHD concentrations has not yet been addressed. Nonetheless, measures of serum 25OHD are important considerations in developing DRI values for vitamin D intake. The sections below highlight factors affecting serum 25OHD level and methodologies for its measurement. It is important to note that these discussions refer to 25OHD, not to calcitriol (i.e., 1,25-dihydroxyvitamin D). Calcitriol, the active hormonal form of the nutrient, has not been used typically as a measure associated with vitamin D nutriture or as an intermediate related to health outcomes. Calcitriol is not useful as such a measure, for several reasons. Its half-life is short (hours), its formation is not directly regulated by vitamin D intake, its levels are regulated by other factors (such as serum PTH), and, even in the presence of severe vitamin D deficiency the calcitriol level may be normal or even elevated as a result of up-regulation of the 1α-hydroxylase enzyme.
Factors Affecting Serum 25OHD Levels
Dietary Intake (Foods and Supplements)
Available literature demonstrates that serum 25OHD levels increase in response to increased vitamin D intake, although overall it can be concluded that the relationship is non-linear rather than linear. Factors that may affect the relationship between vitamin D intake and serum 25OHD levels are not entirely clear, and the reliability of such measures may be less than desirable. Moreover, there remains debate over the equivalence of vitamins D2 and D3 in the diet (Armas et al., 2004; Rapuri et al., 2004; Vieth, 2004), although it has been assumed that they are 25-hydroxylated at similar rates (see previous discussion of functions and physiological actions of vitamin D).
As part of the 2007 Agency for Healthcare Research and Quality (AHRQ) systematic review (Cranney et al., 2007, 2008), referred to hereafter as AHRQ-Ottawa, exploratory meta-regression analysis was conducted
of 16 trials in adults, which suggested an association between vitamin D dose and serum 25OHD concentrations. The analysis found that for each additional 100 IU of vitamin D3, serum 25OHD concentrations rose by 1 to 2 nmol/L. Trials varied in their use of vitamin D2 or vitamin D3. A few of the studies reported different effects of vitamin D2 and vitamin D3 on serum 25OHD levels. Although the later AHRQ-Tufts analysis (Chung et al., 2009) did not identify newer (compared with AHRQ-Ottawa) randomized controlled trials related to intake of vitamin D and serum 25OHD levels, it did graphically evaluate the net changes in serum 25OHD concentrations against the doses of vitamin D supplementation using data from the trials in adults. The analysis confirmed the relationship between increasing doses of vitamin D and increasing net change in serum 25OHD concentrations in both adults and children, but it also concluded that the dose–response relationships differ depending upon study participants’ baseline serum 25OHD levels (≤ 40 vs. > 40 nmol/L) and duration of the supplementation (≤ 3 vs. > 3 months).
In a recent study conducted by Smith et al. (2009), personnel stationed in the Antarctic in winter months (and thus presumed to obtain vitamin D from food and supplements only) were given graded doses of 400, 1,000, or 2,000 IU of vitamin D3 per day for 5 months. Baseline levels of serum 25OHD rose from approximately 44 nmol/L to 57, 63, and 71 nmol/L, respectively, representing a change in 25OHD levels of 13, 19, or 27 nmol/L. Evident in this study is the continuing fall in 25OHD level in the “no pill” group (to 34 nmol/L) of men who were deprived of sunlight and received approximately 250 to 350 IU of vitamin D (which included foods and any non-study supplements) per day. A possible complicating factor in interpreting these data is that the subjects were consuming diets with a vitamin D content that ranged from 241 to 356 IU/day, in addition to the graded doses of vitamin D from administered supplements, although the amounts of vitamin D obtained from foods (or non-study supplements) were not significantly different between treatment groups. Therefore, any effect of these sources of vitamin D on serum levels would have been consistent between treatment groups.
Another study of serum 25OHD response to total intake under conditions of minimal sun exposure used two populations based in Cork, Ireland (51°N), and Coleraine, Northern Ireland (55°N). The study estimated the dose of vitamin D required to maintain 25OHD levels above certain chosen cutoff values (i.e., 25.0, 37.5, 50.0, and 80.0 nmol/L) during the winter months (Cashman et al., 2008). The researchers found that serum 25OHD levels that ranged between 65.7 and 75.9 nmol/L in late fall fell in all groups receiving 200, 400, and 600 IU of vitamin D3 per day, as well as in the placebo group, in winter. The decrease in the 600 IU/day group was minimal, from 75.9 to 69.0 nmol/L. Cashman et al. (2008) went on to
plot the serum 25OHD levels attained after 5 months versus the estimated total vitamin D intake (approximately 0 to 1,400 IU/day) in 215 individuals, as shown in Figure 3-4. They concluded that, in these two populations at 51°N and 55°N, the wintertime intake required to achieve 25OHD cutoff levels of 37.5, 50.0, and 80.0 nmol/L were 796, 1,120, and 1,644 IU/day, respectively.
Importantly, the relationship between vitamin D intake and serum 25OHD response appears not to be linear, given evidence that increasing serum 25OHD level above 50 nmol/L requires more vitamin D intake than does increasing serum 25OHD levels when the starting point is less than 50 nmol/L (Aloia et al., 2008). Factors such as baseline serum 25OHD level in the population may be relevant. Further, there have been reports that the rise in serum 25OHD levels for a given dose tends to stabilize by week 6 (Harris and Dawson-Hughes, 2002; Holick et al., 2008) and that it does
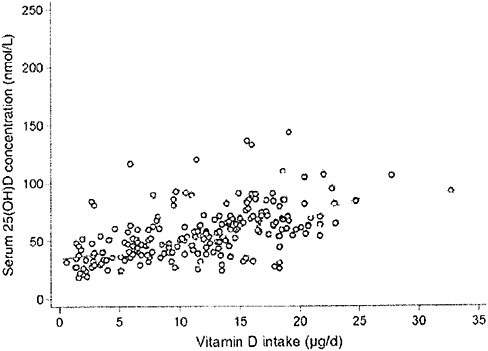
FIGURE 3-4 The relationship between serum 25-hydroxyvitamin D concentrations (in late winter 2007) and total vitamin D intake (dietary and supplemental) in 20- to 40-year-old healthy persons (n = 215) living at northern latitudes (51°N and 55°N).
SOURCE: Cashman et al. (2008). Reprinted with permission from the American Journal of Clinical Nutrition (2008, 88, 1535-42), American Society for Nutrition.
not vary with age at least up to 80 years of age (Harris and Dawson-Hughes, 2002; Cashman et al., 2008, 2009).
Sun Exposure
The cutaneous synthesis of vitamin D, and in turn its contribution to the concentration of serum 25OHD, is initially dependent upon the presence of 7-dehydrocholesterol in the skin. However, many variables can affect the cutaneous synthesis of vitamin D, making it difficult to estimate an average amount of vitamin D and, in turn, serum 25OHD levels that are produced by sun exposure in North America. There is, however, agreement that sun exposure is a significant source of the circulating serum 25OHD in summer for many North Americans, and is notably reduced as a contributor in the winter months. Early work from Webb et al. (1988, 1989) as well as a letter from Holick et al. (1989) have outlined the role of sunlight in regulating cutaneous synthesis of the vitamin and have implicated factors in vitamin D3 synthesis in the skin to include aging, melanin pigmentation, season of the year, latitude, and use of sunscreen. Matsuoka et al. (1992) has discussed the role of clothing in preventing synthesis.
The 2007 AHRQ-Ottawa systematic review (Cranney et al., 2007) noted that the few available randomized clinical trials conducted between 1982 and the time of the analysis, which focused on the effect of UVB radiation on serum 25OHD level revealed little information about the impact of age, ethnicity, skin pigmentation, BMI, or latitude on serum 25OHD levels. However, UVB exposure increased serum 25OHD levels in vitamin D–deficient and –sufficient subjects with mean increases ranging from 15 to 42 nmol/L.
The difference in seasonal contributions to serum 25OHD level from sun exposure is discussed first. Next, factors affecting the synthesis of vitamin D—and, in turn, the levels of 25OHD in serum—are outlined.
Effect of season on circulating serum 25OHD level Sunlight exposure as a source of cutaneous synthesis of vitamin D is subject to a number of limitations. For example, excess exposure can lead to photo-degradation as a regulatory mechanism to avoid toxicity (Chen et al., 2007). In addition, latitude, time of exposure, and season all affect cutaneous synthesis, depending on the ability of UV rays to stimulate vitamin D production. Differences in seasonal exposure can vary by as much as 6 months at extreme northern and southern latitudes (Lucas et al., 2005; Kull et al., 2009).
A number of studies have examined serum 25OHD levels in different seasons. Van der Mei et al. (2007) compared cross–sectional data from men and women less than 60 years of age living in Australia and concluded that season was a strong determinant of serum 25OHD concentrations. Berry
et al. (2009) examined white adults ages 20 to 60 years living in the United Kingdom (UK) at latitude 53°N. In this study, women were found to have higher average serum 25OHD levels than men in both summer and winter (9 and 20 percent higher, respectively). In a study of young adults of diverse ethnic backgrounds living in Toronto, Gozdik et al. (2009) found that in winter, serum 25OHD levels of individuals with East and South Asian backgrounds were significantly lower than those of individuals with European ancestry. Kull et al. (2009) measured seasonal variance of 25OHD levels in adults ages 25 to 70 years living in Estonia in northern Europe, where dairy products are not fortified with vitamin D. During the winter, 73 percent of the study population had 25OHD levels that were below 50 nmol/L, and 8 percent had levels that were below 25 nmol/L, compared with 29 percent and 1 percent, respectively, during the summer. Rapuri et al. (2002) examined white, black, and Hispanic women 65 to 77 years of age in Omaha and reported mean serum 25OHD levels of 68 nmol/L in February and 86 nmol/L in August. These studies, despite variations in age, gender, and ethnicity, all suggest that seasonal change can affect cutaneous vitamin D synthesis. The winter low serum 25OHD concentrations and the summer high serum 25OHD concentrations from these studies are summarized in Table 3-1.
Although there are variations in the available data as well as a number of unknowns that may influence such values, they suggest a seasonal change in serum 25OHD concentrations between the winter nadir and the summer zenith of approximately 25 nmol/L. Free-living individuals in the latitudes studied appear to experience an approximately one-third seasonal increase in their circulating serum 25OHD levels as they moved from the winter months to the summer months.
The 25 nmol/L change in serum 25OHD level would appear to be similar in magnitude to change experienced by subjects given 2,000 IU/day in the Antarctic study (Smith et al., 2009). Although these data suggest that average cutaneous synthesis during the summer in northern latitudes equates to 2,000 IU/day, this may be a questionable conclusion given the many variables that come into play, ranging from feedback mechanisms to skin pigmentation to baseline levels of 25OHD. For example, recent work from Olds et al. (2008) suggested a curvilinear relationship between sun exposure and serum 25OHD levels, as well as variation depending upon initial concentrations of 7-dehydrocholesterol levels. At lower doses, vitamin D3 production rises immediately in response to UV exposure, whereas at higher doses, the rate of production is lower and reaches an earlier plateau.
Effect of skin pigmentation on synthesis The presence of melanin in the epidermal layer is responsible for skin pigmentation. A number of
TABLE 3-1 Winter Low 25OHD Levels and Summer High 25OHD Levels Around the World
|
Location (Latitude) |
Winter Baseline |
Summer High |
|
Toronto, Canadaa 43°N |
35 nmol/L |
50 nmol/Lb |
|
Tasmania, Australiac 43°S |
40 nmol/L |
62 nmol/L |
|
Estonia, northern Europed 59°N |
44 nmol/L |
59 nmol/L |
|
Salford/Manchester, UKe 53°N |
46 nmol/L |
71 nmol/L |
|
Geelong region, Australiac 38°S |
57 nmol/L |
93 nmol/L |
|
Omaha, USAf 41°N |
68 nmol/L |
86 nmol/L |
|
aGozdik et al., 2009. bhigh value recorded in autumn. cvan der Mei et al., 2007. dKull et al., 2009. eBerry et al., 2009. fRapuri et al., 2002. |
||
recent studies have reinforced the relationship of skin pigmentation to the capacity to produce vitamin D3 after UV exposure, but the results are not all consistent. Armas et al. (2007) studied the incremental change in serum 25OHD levels in individuals with average 25OHD baseline levels of 52 nmol/L and with different skin pigmentation who were exposed to different daily doses of 20 to 80 mJ/cm2 of UVB light three times per week for 4 weeks on 90 percent of their skin surface area. The work suggested that for individuals at the lighter pigmentation scale,7 exposed to similar UVB doses (20 to 80 mJ/cm2) resulted in twice the increase in serum 25OHD concentration compared with individuals at the opposite extreme (i.e., 62 vs. 32 nmol/L change) (Armas et al., 2007) (see modeled representation in Figure 3-5). However, other studies have shown that the response to UV dose is non-linear and dependent on genetic factors (Snellman et al., 2009), duration and dose rate of UV exposure, and baseline serum 25OHD levels (Bogh et al., 2010).
Another population study of 237 subjects at a single geographical location (Toronto, 43°N) also explored the effect of skin pigmentation on
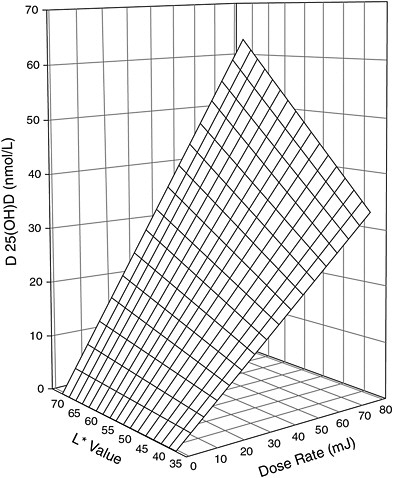
FIGURE 3-5 Three-dimensional scatter-plot of 4-week change in serum 25OHD concentration above baseline expressed as a function of both basic skin lightness (L*) and UVB dose rate. Surface is hyperboloid, plotting equation 1, and was fitted to data using least squares regression methods.
NOTE: Equation 1 is z = b * x * y, where z is the induced increase in serum 25OHD level from baseline (in nmol/L); x is UVB dose (in mJ/cm2 per session); y is skin lightness (expressed as the L* score value from the reflective meter); and b is the sole parameter to be fitted.
SOURCE: Reprinted with permission from Robert Heaney.
25OHD synthesis (Gozdik et al., 2009). The mean serum 25OHD levels in Canadians of European, East Asian, and South Asian ancestry were 71.7, 44.6, and 33.9 nmol/L, respectively, in late fall; and 51.6, 28.1, and 26.5 nmol/L, respectively, in the winter. The authors found that differences between the three subgroups were strongly associated with skin pigmenta-
tion as well as amount of time spent outdoors and total vitamin D intake. A recent study of 182 individuals in Denmark, screened in January and February and selected to reflect wide ranges of baseline 25OHD levels and skin pigmentation, found that the increase in 25OHD levels after UVB exposure was inversely correlated with skin pigmentation as well as with baseline 25OHD (Bogh et al., 2010).
A number of small studies have reported serum 25OHD levels to be consistently lower in persons with darker skin pigmentation, and data from NHANES suggest that serum 25OHD levels are highest in whites, lowest in non-Hispanic blacks, and intermediate in Hispanic groups (Looker et al., 2008). Overall, there is considerable evidence that darker skin pigmentation is associated with a smaller increase in serum 25OHD concentration for a given amount of UVB exposure.
Effect of latitude on synthesis Early on, in vitro methods, such as exposure of sealed vials of 7-dehydrocholesterol to UVB radiation under “idealized conditions” at various geographical locations, were used to assess the effect of latitude, time of day, and season on the rate of vitamin D production (Webb et al., 1988). However, this approach cannot completely simulate the in vivo conditions in the body, where many factors serve to regulate this process. Furthermore, although the measurement of vitamin D concentrations in a mixture of irradiation products is analytically simple, vitamin D3 levels in human serum are rarely used to estimate cutaneous synthesis of the vitamin, in part because of the transient nature of this blood parameter and the difficulty of measuring the low levels in serum (Holick, 1988; Hollis, 2008). Nevertheless, Holick and colleagues used a serum vitamin D3 assay to augment in vitro methodology and suggested that, at latitudes above 43°N, cutaneous synthesis contributes little serum 25OHD to the system in the winter months between October and March in North America (Webb et al., 1988; Matsuoka et al., 1989).
More recent data may call into question current assumptions about the effect of latitude. In fact, Kimlin et al. (2007), using computer modeling, concluded that it may no longer be correct to assume that vitamin D levels in populations follow latitude gradients. Indeed, the relationship between UVB penetration and latitude is complex, as a result of differences in, for example, the height of the atmosphere (50 percent less at the poles), cloud cover (more intense at the equator than at the poles), and ozone cover. The duration of sunlight in summer versus winter is another factor contributing to the complexity of the relationship. Geophysical surveys have shown that UVB penetration over 24 hours, during the summer months at Canadian north latitudes when there are many hours of sunlight, equals or exceeds UVB penetration at the equator (Lubin et al., 1998). Consequently, there is ample opportunity during the spring, summer, and fall months in the far north for humans (as well as animals that serve as food
sources) to form vitamin D3 and store it in liver and fat. These factors may explain why latitude alone does not consistently predict the average serum 25OHD level of a population.
Effect of sunscreen on synthesis Sunscreens are used to protect the skin from ultraviolet A (UVA) and UVB waveband exposure that is associated with deoxyribonucleic acid (DNA) damage—the same UVB exposure that is needed for vitamin D synthesis. Experimental studies suggest that sunscreens can decrease cutaneous vitamin D synthesis (Misra et al., 2008). However, emerging evidence suggests that although sunscreens are effective, many may not actually be blocking UVB because they are improperly or inadequately applied. Thus, sunscreen use may not actually diminish vitamin D synthesis in real world use, although further study is needed to verify its actual impact (Diehl and Chiu, 2010; Springbett et al., 2010).
Other variables affecting synthesis A number of other variables can impede sun exposure and thus inhibit cutaneous vitamin D synthesis. Clothing is an effective barrier to sun exposure and the UVB waveband, but the effectiveness of sun blocking depends on the thickness or weave of the fabric (Diehl and Chiu, 2010). Likewise, ethnic practices, such as extensive skin coverage with clothing, urban environments that reduce or block sunlight, air pollution, and cloud cover that reduces solar penetration can variously reduce sun exposure. In contrast, high altitude reduces the atmospheric protection against UVB waveband and can increase risk for sun damage as well as increase vitamin D synthesis (Misra et al., 2008). There may be a role for measures of physical activity in affecting vitamin D synthesis, although many covariates may be relevant, and some have suggested that genetics can account for some of the differences in synthesis of serum 25OHD.
Confounders Affecting Serum 25OHD Concentrations
Adiposity Interpreting data on serum 25OHD concentrations in obese and overweight persons is particularly challenging. Data from NHANES showed lower circulating levels of 25OHD among young adult obese non-Hispanic white women compared with their leaner counterparts; the relationship appeared to be weaker among non-Hispanic blacks. Differences in physical activity levels partially explain these differences (Looker, 2005, 2007). However, overweight and obese persons in NHANES also reported lower use of dietary supplements than did leaner persons of the same age or gender group (Radimer et al., 2004; Picciano et al., 2007), suggesting that lower dietary exposures could also contribute to the lower serum 25OHD levels in obese and overweight people.
Sequestration of vitamin D into fat likely also plays a significant role in
reducing the amount that can be presented to the liver for 25-hydroxylation. As noted previously, vitamin D is absorbed with fat as part of chylomicrons and is taken up first by peripheral tissues that express lipoprotein lipase, especially adipose tissue and skeletal muscle. This pathway predicts that increased adiposity should lead to lower serum 25OHD levels and, conversely, that weight loss should reduce peripheral sequestration and enable higher 25OHD levels. Consistent with this, not only does increasing adiposity correlate with lower 25OHD levels, but also a few studies of modest weight loss have found the circulating 25OHD levels to increase despite no increased intake of vitamin D from diet or sunlight exposure (Riedt et al., 2005; Reinehr et al., 2007; Zittermann et al., 2009; Tzotzas et al., 2010). The measured increase in serum 25OHD levels in overweight and obese individuals was about 1.5 nmol/L for a 100 IU/day vitamin D intake over 12 months (Sneve et al., 2008; Zittermann et al., 2009). Others found that obese subjects show a lower rise in serum 25OHD levels in response to both oral vitamin D intake and UVB exposure (Wortsman et al., 2000) or in a retrospective analysis in response to 700 IU/day vitamin D3 supplementation (Blum et al., 2008). It is interesting that in severely obese individuals after malabsorptive gastric bypass surgery, vitamin D supplementation resulted in a marked rise in serum 25OHD level of approximately 3 nmol/100 IU intake when the dose was 800 to 2,000 IU/day, but only a 1 nmol/L rise when intake was increased to 5,000 IU/day (Goldner et al., 2009).
African American ancestry Serum 25OHD levels are lower in African Americans compared with light-skinned population groups (Looker et al., 2008), yet the risk for fracture is lower for African Americans than for other ethnic groups (Aloia, 2008). It should be noted, however, that there is a wide range of variability among individuals of any race or ethnicity (Aloia, 2008). Serum 25OHD levels in African Americans and whites have been shown to be similarly responsive to vitamin D supplementation at 40°N latitude (equivalent to Philadelphia or Indianapolis), increasing by 1 to 2 nmol/L per 100 IU/day at a dose of 3,440 IU/day (Aloia et al., 2008), although at doses below 2,000 IU/day, serum 25OHD levels do not increase above 50 nmol/L in African American girls (Talwar et al., 2007). The significance of maintaining a higher serum 25OHD level in African Americans is not understood at this time because of a lack of evidence on extra-skeletal effects of vitamin D.
Size and frequency of dose Dosing of vitamin D daily, weekly, or monthly has been tested, and there are reports of annual dosing as well. The results of a study by Chel et al. (2008) suggested that daily (600 IU/day) and weekly doses of vitamin D will increase serum 25OHD levels more than monthly doses, but Ish-Shalom et al. (2008) using a dose of 1,500 IU/day
found no difference due to timing of the doses. Ish-Shalom et al. (2008) suggested that the attenuated response to monthly dose in the Chel et al. (2008) study may have been due to poor compliance with a powdered monthly supplement compared with pills used for their daily and weekly doses. An alternative explanation is that only a lower (Chel et al., 2008) and not a higher (Ish-Shalom et al., 2008) dose is influenced by timing of the vitamin D supplement. Thus far, studies suggest that weekly and daily dosing give similar serum 25OHD responses.
Assays for Serum 25OHD
Serum 25OHD comprises the sum of 25OHD2 and 25OHD3. Because of the widespread use of both vitamin D2 and vitamin D3 in the United States and Canada, analysts must measure both 25OHD2 and 25OHD3 in order to provide the total 25OHD level in serum. This is in contrast to the situation in Europe where there has been a tradition of using only vitamin D3 and where commercial methods that purport to measure only 25OHD3 are available.
In North America, several assay types are currently in use, each with strengths and weaknesses (Makin et al., 2010). The two most common types of assays are
-
Antibody-based methods, which use a kit or an automated clinical chemistry platform; and
-
Liquid chromatography (LC)-based methods, which use automated equipment featuring either UV or mass spectrometric (MS)-detection.
As discussed below, both these methods are equivalent in terms of measuring the physiologically relevant parameter (total 25OHD level in serum), but there remains controversy over the performance of these assays in clinical and research laboratories. Moreover, reports in the literature for serum 25OHD measures should be interpreted with care, taking into account the type of assay employed, use of automation, year of analysis, and context of the analysis.
Overview of Assay Methodology
Assays for total 25OHD level in serum have existed for four decades since the metabolite was first discovered (Blunt et al., 1968). The earliest assays were competitive protein-binding assays (CPBAs), based upon the ability of either 25OHD2 or 25OHD3 to displace [3H]25OHD3 from the plasma binding protein, DBP (Belsey et al., 1971; Haddad and Hahn,
1973). These assays incorporated extraction, chromatographic purification, and detection steps, but the assays were laborious and not easily simplified, owing to the presence of co-extracted interfering substances. They therefore fell out of favor. Non-chromatographic CPBA assays tended to read erroneously high, and some data from this period (1970s) are viewed as extremely questionable for this reason.
In the late 1970s LC-based assays emerged that used refined extraction and chromatographic steps with some form of fixed- or variable-wavelength UV detector to measure the distinctive native absorbance of the vitamin D metabolites with λmax at 265 nm (Jones, 1978; Horst et al., 1979). These allowed for the separate estimation of 25OHD2 and 25OHD3 from serum samples. This type of assay has undergone much refinement over the years, with improvements in LC, improved automation, or the introduction of diode-array UV detectors that minimize the chance of picking up interfering substances (Lensmeyer et al., 2006). Although these assays require expensive equipment as well as sequential sample analysis, which make them somewhat time-consuming, they are still popular with a minority of analysts.
In the early to late 1980s, antibody-based assays were introduced, which use a proprietary antibody to a vitamin D molecular antigen, usually with a truncated vitamin D side chain. They are therefore devoid of the features that allow the resultant antibody to distinguish between 25OHD2 and 25OHD3 (Hollis and Napoli, 1985); this is of value, but at the expense of detecting other minor metabolites. Consequently, antibody-based methods measure only total 25OHD in serum. Various commercial assays differ because of the nature of the antibody used, some claiming an advantage that they do not discriminate between 25OHD2 and 25OHD3 (Hollis and Napoli, 1985), whereas others in fact do underestimate the 25OHD2 pool and therefore provide correction factors to compensate for high 25OHD2 content. Over the past decade, antibody-based 25OHD assays have become automated into a multiwell plate-format and the increased throughput has made them extremely popular. It is important to note that the majority of the data collected over the past 20 to 30 years have been analyzed using antibody-based assays.
Recently, LC-based assays have also undergone a radical transformation with the replacement of the UV detection step by a “universal detector” in the form of a tandem mass spectrometer (LC-MS/MS) (Jones and Makin, 2000). LC-MS/MS assays allow the analyst to discriminate between 25OHD2 and 25OHD3 and other serum lipids by their unique molecular masses and mass fragments. Accordingly, these methods measure 25OHD2 and 25OHD3, and therefore total 25OHD (Makin et al., 2010). Because these methods use short LC retention times, automated robotic extraction and LC separation steps, and computerized MS systems, they can be made relatively operator-free and provide high throughput. Further, their potential
advantages also include high specificity, high sensitivity, and better reproducibility (< 10 percent). The consensus among analysts is that LC-MS/MS assays will become the “gold standard” for assay performance in the future. Thus, a variety of methods are available to determine serum 25OHD levels, each with its advantages and disadvantages that must be considered in evaluating the data arising from them.
Assay Performance Concerns
The methodologies used over four decades of assaying 25OHD levels in serum have each presented technical problems. These include, first, CPBA assays without chromatography that read high because of poorly defined “interferences” (Morris and Peacock, 1976; Stamp et al., 1976). Second, some LC-MS/MS assays with short LC run times read high because they cannot resolve 3-epi-25OHD3, an isomer found in abundance in neonatal samples (Singh et al., 2006). Third, some antibody-based assays, particularly some of the automated versions, fail to recover or detect, and therefore underestimate 25OHD2 (Carter et al., 2004). Fourth, some antibody-based assays overestimate values by detecting further metabolites of 25OHD (e.g., 24,25[OH]2D and 26,23-lactone) particularly those found in hypervitaminotic D situations (i.e., 25OHD > 250 nmol/L) (Jones et al., 1987); and some antibody-based methods are sensitive to exogenous interferences such as the presence of dilutants (ethanol, non-human serum) used in quality control materials (Phinney, 2009). Thus, no assay is free from problems, and performance must be monitored vigilantly.
Performance has been a concern of analysts and clinicians in the vitamin D field for some time. Inter-laboratory comparisons have been conducted and published for two decades and suggest an unacceptable degree of variability in the different results (Jongen et al., 1984, 1989). This has led to the creation of external quality assurance schemes, such as the Vitamin D External Quality Assurance Scheme or DEQAS8 (Charing Cross Hospital, London, UK), similar to those used in other areas of clinical chemistry. Since its inception in the early 1990s, DEQAS has grown steadily, such that it now serves as a quarterly monitor of performance of analysts and 25OHD analytical methods for approximately 700 laboratories worldwide (Carter et al., 2010). Although initially DEQAS used gas chromatography-mass spectrometry (GC-MS) as its “gold standard,” more recently it has adopted an all-laboratory trimmed mean (ALTM) as the value it uses to judge performance. Using ALTM, DEQAS has served as an early-warning system for method and operator biases that have alerted the commercial kit manufacturers to modify their products or steps in their procedures or withdraw their kits. DEQAS has published performance characteristics (precision,
|
8 |
Available online at http://www.deqas.org/ (accessed March 9, 2010). |
accuracy, and variability) regularly over the past decade (Carter et al., 2004; Carter, 2009; Jones et al., 2009). These publications have been augmented by other independent method comparisons (Glendenning et al., 2006; Lensmeyer et al., 2006; Roth et al., 2008).
In brief, these performance reports suggest some method biases in terms of accuracy and precision as well as variability as high as 15 to 20 percent. However, some skilled analysts can perform better than this with a coefficient of variation less than 10 percent. The recent introduction of the National Institute of Standards and Technology (NIST) reference standards9 calibrated using a “validated” LC-MS/MS method (Phinney, 2009) offers some hope that the variability of all methods can or will be improved in the future. Indeed, recent data suggest that an improvement is already occurring (Carter and Jones, 2009). At this time, however, serum 25OHD data in the literature must be viewed with care based upon the knowledge that they have been acquired using a variety of methods, each with its own shortcomings and subject to high variability.
Assay Shift and Drift: U.S. National Health and Nutrition Examination Survey
Recently, the National Center for Health Statistics (NCHS) posted an analytical note on its NHANES web page informing users about two issues that should be addressed when analyzing and using serum 25OHD data from NHANES.10 The first involved making direct comparisons between serum 25OHD levels from NHANES III (1988 to 1994) and those from the 2000 to 2006 NHANES because of a reformulation of the DiaSorin radioimmunoassay (RIA) kit11 that resulted in shifts in assay results between the two time periods. Second, the note also cautioned that the data from the 2000 to 2006 NHANES were likely affected by drifts in the assay performance (method bias and imprecision) over time. A standard reference material (SRM 972, Vitamin D in Human Serum) and calibration solution (SRM 2972, 25-Hydroxyvitamin D2 and D3 Calibration Solutions) are currently available from the NIST. The NCHS plans to incorporate use of this SRM into future measures of 25OHD in NHANES. The web page also indicates that guidance concerning a correction factor and data interpretation regarding this shift and drift will be forthcoming from the agency. In the case of the Canadian survey of 25OHD levels in the Canadian population, the analytical methods used to determine 25OHD concentrations were the
|
9 |
Available online at http://ts.nist.gov/measurementservices/referencematerials/index.cfm (accessed March 15, 2010). |
|
10 |
Available online at http://www.cdc.gov/nchs/data/nhanes/nhanes3/VitaminD_analyticnote.pdf (accessed March 17, 2010). |
|
11 |
DiaSorin Radio-immunoassay (RIA) (Stillwater, MN). |
Diasorin total 25OHD (Liaison) kit12 (personal communication, S. Brooks, Health Canada, December 18, 2009).
MEASURES ASSOCIATED WITH VITAMIN D: PARATHYROID HORMONE
PTH, which, as described above, plays a role in vitamin D metabolism, is also a known marker for bone resorption, based upon the bone manifestations of secondary hyperparathyroidism—increased bone turnover, increased rates of bone loss, osteoporosis, and increased risk of fractures. Serum PTH level has thus been explored and suggested as a measure indicative of adequate vitamin D nutriture, notably on the basis of the level of serum 25OHD at which serum PTH level rises or, alternatively, the level of serum 25OHD at which serum PTH level no longer declines. This measure is discussed more fully in Chapter 4.
REFERENCES
Aasheim, E. T., S. Bjorkman, T. T. Sovik, M. Engstrom, S. E. Hanvold, T. Mala, T. Olbers and T. Bohmer. 2009. Vitamin status after bariatric surgery: a randomized study of gastric bypass and duodenal switch. American Journal of Clinical Nutrition 90(1): 15-22.
Abrams, S. A., I. J. Griffin, K. M. Hawthorne, S. K. Gunn, C. M. Gundberg and T. O. Carpenter. 2005. Relationships among vitamin D levels, parathyroid hormone, and calcium absorption in young adolescents. Journal of Clinical Endocrinology and Metabolism 90(10): 5576-81.
Adams, J. S., R. L. Modlin, M. M. Diz and P. F. Barnes. 1989. Potentiation of the macrophage 25-hydroxyvitamin D-1-hydroxylation reaction by human tuberculous pleural effusion fluid. Journal of Clinical Endocrinology and Metabolism 69(2): 457-60.
Adams, J. S. and M. Hewison. 2008. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. National Clinical Practice Endocrinology Metabolism 4(2): 80-90.
Aksnes, L. and D. Aarskog. 1982. Plasma concentrations of vitamin D metabolites in puberty: effect of sexual maturation and implications for growth. Journal of Clinical Endocrinology and Metabolism 55(1): 94-101.
Aloia, J. F. 2008. African Americans, 25-hydroxyvitamin D, and osteoporosis: a paradox. American Journal of Clinical Nutrition 88(2): 545S-50S.
Aloia, J. F., M. Patel, R. Dimaano, M. Li-Ng, S. A. Talwar, M. Mikhail, S. Pollack and J. K. Yeh. 2008. Vitamin D intake to attain a desired serum 25-hydroxyvitamin D concentration. American Journal of Clinical Nutrition 87(6): 1952-8.
Armas, L. A., B. W. Hollis and R. P. Heaney. 2004. Vitamin D2 is much less effective than vitamin D3 in humans. Journal of Clinical Endocrinology and Metabolism 89(11): 5387-91.
Armas, L. A., S. Dowell, M. Akhter, S. Duthuluru, C. Huerter, B. W. Hollis, R. Lund and R. P. Heaney. 2007. Ultraviolet-B radiation increases serum 25-hydroxyvitamin D levels: the effect of UVB dose and skin color. Journal of the American Academy of Dermatology 57(4): 588-93.
Bachrach, S., J. Fisher and J. S. Parks. 1979. An outbreak of vitamin D deficiency rickets in a susceptible population. Pediatrics 64(6): 871-7.
Bao, B. Y., J. Yao and Y. F. Lee. 2006. 1alpha, 25-dihydroxyvitamin D3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis 27(9): 1883-93.
Beer, T. M. and A. Myrthue. 2004. Calcitriol in cancer treatment: from the lab to the clinic. Moleecular Cancer Therapy 3(3): 373-81.
Belsey, R., H. F. Deluca and J. T. Potts, Jr. 1971. Competitive binding assay for vitamin D and 25-OH vitamin D. Journal of Clinical Endocrinology and Metabolism 33(3): 554-7.
Bergwitz, C. and H. Juppner. 2010. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annual Review of Medicine 61: 91-104.
Berry, J. L., A. R. Webb, R. Kift, M. Durkin, A. Vail, S. J. O’Brien and L. E. Rhodes. 2009. Should we, in the U.K. be taking vitamin D supplements during the winter? Presented at 14th Workshop on Vitamin D, October 4-8, 2009. Brugge, Belgium.
Bises, G., E. Kallay, T. Weiland, F. Wrba, E. Wenzl, E. Bonner, S. Kriwanek, P. Obrist and H. S. Cross. 2004. 25-hydroxyvitamin D3-1alpha-hydroxylase expression in normal and malignant human colon. Journal of Histochemistry and Cytochemistry 52(7): 985-9.
Blomstrand, R. and L. Forsgren. 1967. Intestinal absorption and esterification of vitamin D3-1,2-3H in man. Acta Chemica Scandinavica 21(6): 1662-3.
Blum, M., G. E. Dallal and B. Dawson-Hughes. 2008. Body size and serum 25 hydroxy vitamin D response to oral supplements in healthy older adults. Journal of the American College of Nutrition 27(2): 274-9.
Blunt, J. W., H. F. DeLuca and H. K. Schnoes. 1968. 25-hydroxycholecalciferol. A biologically active metabolite of vitamin D3. Biochemistry 7(10): 3317-22.
Bogh, M. K., A. V. Schmedes, P. A. Philipsen, E. Thieden and H. C. Wulf. 2010. Vitamin D production after UVB exposure depends on baseline vitamin D and total cholesterol but not on skin pigmentation. Journal of Investigative Dermatology 130(2): 546-53.
Brannon, P. M., E. A. Yetley, R. L. Bailey and M. F. Picciano. 2008. Vitamin D and health in the 21st century: an update. Proceedings of a conference held September 2007 in Bethesda, Maryland, USA. American Journal of Clinical Nutrition 88(2): 483S-592S.
Brommage, R. and H. F. DeLuca. 1985. Evidence that 1,25-dihydroxyvitamin D3 is the physiologically active metabolite of vitamin D3. Endocrine Reviews 6(4): 491-511.
Bruno, C., A. D. Fulford, J. R. Potts, R. McClintock, R. Jones, B. M. Cacucci, C. E. Gupta, M. Peacock and R. V. Considine. 2010. Serum markers of bone turnover are increased at six and 18 months after Roux-en-Y bariatric surgery: correlation with the reduction in leptin. Journal of Clinical Endocrinology and Metabolism 95(1): 159-66.
Byrdwell, W. C. 2009. Analysis of vitamin D in food control materials and fortified foods. Presented at the Committee to Review Dietary Reference Intakes for Vitamin D and Calcium Information-gathering Workshop, March 26, 2009. Washington, DC.
Campbell, M. J., E. Elstner, S. Holden, M. Uskokovic and H. P. Koeffler. 1997. Inhibition of proliferation of prostate cancer cells by a 19-nor-hexafluoride vitamin D3 analogue involves the induction of p21waf1, p27kip1 and E-cadherin. Journal of Molecular Endocrinology 19(1): 15-27.
Carter, G. D., R. Carter, J. Jones and J. Berry. 2004. How accurate are assays for 25-hydroxyvitamin D? Data from the international vitamin D external quality assessment scheme. Clinical Chemistry 50(11): 2195-7.
Carter, G. D. 2009. 25-hydroxyvitamin D assays: the quest for accuracy. Clinical Chemistry 55(7): 1300-2.
Carter, G. D. and J. C. Jones. 2009. Use of a common standard improves the performance of liquid chromatography-tandem mass spectrometry methods for serum 25-hydroxyvitamin-D. Annals of Clinical Biochemistry 46(Pt 1): 79-81.
Carter, G. D., J. L. Berry, E. Gunter, G. Jones, J. C. Jones, H. L. Makin, S. Sufi and M. J. Wheeler. 2010. Proficiency testing of 25-hydroxyvitamin D (25-OHD) assays. Journal of Steroid Biochemistry and Molecular Biology 121(1-2): 176-9.
Cashman, K. D., T. R. Hill, A. J. Lucey, N. Taylor, K. M. Seamans, S. Muldowney, A. P. Fitzgerald, A. Flynn, M. S. Barnes, G. Horigan, M. P. Bonham, E. M. Duffy, J. J. Strain, J. M. Wallace and M. Kiely. 2008. Estimation of the dietary requirement for vitamin D in healthy adults. American Journal of Clinical Nutrition 88(6): 1535-42.
Cashman, K. D., J. M. Wallace, G. Horigan, T. R. Hill, M. S. Barnes, A. J. Lucey, M. P. Bonham, N. Taylor, E. M. Duffy, K. Seamans, S. Muldowney, A. P. Fitzgerald, A. Flynn, J. J. Strain and M. Kiely. 2009. Estimation of the dietary requirement for vitamin D in free-living adults ≥64 y of age. American Journal of Clinical Nutrition 89(5): 1366-74.
Chel, V., H. A. Wijnhoven, J. H. Smit, M. Ooms and P. Lips. 2008. Efficacy of different doses and time intervals of oral vitamin D supplementation with or without calcium in elderly nursing home residents. Osteoporosis International 19(5): 663-71.
Chen, P. S., Jr. and H. B. Bosmann. 1964. Effect of vitamins D2 and D3 on serum calcium and phosphorus in rachitic chicks. Journal of Nutrition 83: 133-9.
Chen, T. C., A. Shao, H. Heath, 3rd and M. F. Holick. 1993. An update on the vitamin D content of fortified milk from the United States and Canada. New England Journal of Medicine 329(20): 1507.
Chen, T. C., F. Chimeh, Z. Lu, J. Mathieu, K. S. Person, A. Zhang, N. Kohn, S. Martinello, R. Berkowitz and M. F. Holick. 2007. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Archives of Biochemistry and Biophysics 460(2): 213-7.
Cheng, J. B., D. L. Motola, D. J. Mangelsdorf and D. W. Russell. 2003. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. Journal of Biological Chemistry 278(39): 38084-93.
Christiansen, C., P. Rodbro and O. Munck. 1975. Actions of vitamins D2 and D3 and 25-OHD3 in anticonvulsant osteomalacia. British Medical Journal 2(5967): 363-5.
Chung M., E. M. Balk, M. Brendel, S. Ip, J. Lau, J. Lee, A. Lichtenstein, K. Patel, G. Raman, A. Tatsioni, T. Terasawa and T. A. Trikalinos. 2009. Vitamin D and calcium: a systematic review of health outcomes. Evidence Report No. 183. (Prepared by the Tufts Evidence-based Practice Center under Contract No. HHSA 290-2007-10055-I.) AHRQ Publication No. 09-E015. Rockville, MD: Agency for Healthcare Research and Quality.
Clements, M. R., M. Davies, D. R. Fraser, G. A. Lumb, E. B. Mawer and P. H. Adams. 1987. Metabolic inactivation of vitamin D is enhanced in primary hyperparathyroidism. Clinical Science (London) 73(6): 659-64.
Compston, J. E., A. L. Merrett, F. G. Hammett and P. Magill. 1981. Comparison of the appearance of radiolabelled vitamin D3 and 25-hydroxy-vitamin D3 in the chylomicron fraction of plasma after oral administration in man. Clinical Science (London) 60(2): 241-3.
Cranney A., T. Horsley, S. O’Donnell, H. A. Weiler, L. Puil, D. S. Ooi, S. A. Atkinson, L. M. Ward, D. Moher, D. A. Hanley, M. Fang, F. Yazdi, C. Garritty, M. Sampson, N. Barrowman, A. Tsertsvadze and V. Mamaladze. 2007. Effectiveness and safety of vitamin D in relation to bone health. Evidence Report/Technology Assessment No. 158 (Prepared by the University of Ottawa Evidence-based Practice Center [UO-EPC] under Contract No. 290-02-0021). AHRQ Publication No. 07-E013. Rockville, MD: Agency for Healthcare Research and Quality.
Cranney, A., H. A. Weiler, S. O’Donnell and L. Puil. 2008. Summary of evidence-based review on vitamin D efficacy and safety in relation to bone health. American Journal of Clinical Nutrition 88(2): 513S-9S.
Davis, C. D., V. Hartmuller, D. M. Freedman, P. Hartge, M. F. Picciano, C. A. Swanson and J. A. Milner. 2007. Vitamin D and cancer: current dilemmas and future needs. Nutrition Reviews 65(8 Pt 2): S71-4.
Davis, C. D. 2008. Vitamin D and cancer: current dilemmas and future research needs. American Journal of Clinical Nutrition 88(2): 565S-9S.
Deeb, K. K., D. L. Trump and C. S. Johnson. 2007. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nature Reviews Cancer 7(9): 684-700.
DeLuca, H. F. 1974. Vitamin D: the vitamin and the hormone. Federation Proceedings 33(11): 2211-9.
Deluca, H. F. 1979a. Vitamin D-resistant rickets. A prototype of nutritional management of a genetic disorder. Current Concepts in Nutrition 8: 3-32.
Deluca, H. F. 1979b. The transformation of a vitamin into a hormone: the vitamin D story. Harvey lectures 75: 333-79.
DeLuca, H. F. and H. K. Schnoes. 1983. Vitamin D: recent advances. Annual Review of Biochemistry 52: 411-39.
DeLuca, H. F. 1988. The vitamin D story: a collaborative effort of basic science and clinical medicine. FASEB Journal 2(3): 224-36.
DeLuca, H. F. and M. T. Cantorna. 2001. Vitamin D: its role and uses in immunology. FASEB Journal 15(14): 2579-85.
Deluca, H. F. 2009. Vitamin D toxicity. Paper prepared for the Committee to Review Dietary Reference Intakes for Vitamin D and Calcium. Washington, DC.
DeLucia, M. C., M. E. Mitnick and T. O. Carpenter. 2003. Nutritional rickets with normal circulating 25-hydroxyvitamin D: a call for reexamining the role of dietary calcium intake in North American infants. Journal of Clinical Endocrinology and Metabolism 88(8): 3539-45.
Diaz, G. D., C. Paraskeva, M. G. Thomas, L. Binderup and A. Hague. 2000. Apoptosis is induced by the active metabolite of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: possible implications for prevention and therapy. Cancer Research 60(8): 2304-12.
Diehl, J. W. and M. W. Chiu. 2010. Effects of ambient sunlight and photoprotection on vitamin D status. Dermatologic Therapy 23(1): 48-60.
Diesing, D., T. Cordes, D. Fischer, K. Diedrich and M. Friedrich. 2006. Vitamin D—metabolism in the human breast cancer cell line MCF-7. Anticancer Research 26(4A): 2755-9.
Drescher, D., H. F. Deluca and M. H. Imrie. 1969. On the site of discrimination of chicks against vitamin D. Archives of Biochemistry and Biophysics 130(1): 657-61.
Faulkner, H., A. Hussein, M. Foran and L. Szijarto. 2000. A survey of vitamin A and D contents of fortified fluid milk in Ontario. Journal of Dairy Science 83(6): 1210-6.
FDA (Food and Drug Administration). 2009. Agency Information Collection Activities; Submission for Office of Management and Budget Review; Comment Request; Food Labeling Regulations. Federal Register 74(201): 53743-6.
Fieser, L. F. and M. Fieser. 1959. Vitamin D. New York: Reinhold. Pp. 90-168.
Fleet, J. C., C. Gliniak, Z. Zhang, Y. Xue, K. B. Smith, R. McReedy and S. A. Adedokon. 2008. Serum metabolite profiles and target tissue gene expression define the effect of cholecalciferol intake on calcium metabolism in rats and mice. Journal of Nutrition 138(6): 1114-20.
Fraser, D., S. W. Kooh, H. P. Kind, M. F. Holick, Y. Tanaka and H. F. DeLuca. 1973. Pathogenesis of hereditary vitamin-D-dependent rickets. An inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1 alpha,25-dihydroxyvitamin D. New England Journal of Medicine 289(16): 817-22.
Galitzer, H., I. Ben-Dov, V. Lavi-Moshayoff, T. Naveh-Many and J. Silver. 2008. Fibroblast growth factor 23 acts on the parathyroid to decrease parathyroid hormone secretion. Current Opinion in Nephrology and Hypertension 17(4): 363-7.
Garabedian, M., M. F. Holick, H. F. Deluca and I. T. Boyle. 1972. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proceedings of the National Academy of Sciences of the United States of America 69(7): 1673-6.
Gartner, L. M. and F. R. Greer. 2003. Prevention of rickets and vitamin D deficiency: new guidelines for vitamin D intake. Pediatrics 111(4 Pt 1): 908-10.
Gaschott, T. and J. Stein. 2003. Short-chain fatty acids and colon cancer cells: the vitamin D receptor—butyrate connection. Recent Results in Cancer Research 164: 247-57.
Gehrer, S., B. Kern, T. Peters, C. Christoffel-Courtin and R. Peterli. 2010. Fewer nutrient deficiencies after laparoscopic sleeve gastrectomy (LSG) than after laparoscopic Roux-Y-gastric bypass (LRYGB)—a prospective study. Obesity Surgery 20(4): 447-53.
Glendenning, P., M. Taranto, J. M. Noble, A. A. Musk, C. Hammond, P. R. Goldswain, W. D. Fraser and S. D. Vasikaran. 2006. Current assays overestimate 25-hydroxyvitamin D3 and underestimate 25-hydroxyvitamin D2 compared with HPLC: need for assay-specific decision limits and metabolite-specific assays. Annals of Clinical Biochemistry 43(Pt 1): 23-30.
Goldner, W. S., J. A. Stoner, E. Lyden, J. Thompson, K. Taylor, L. Larson, J. Erickson and C. McBride. 2009. Finding the optimal dose of vitamin D following Roux-en-Y gastric bypass: a prospective, randomized pilot clinical trial. Obesity Surgery 19(2): 173-9.
Gozdik, A., J. L. Barta, H. Wu, D. Cole, R. Vieth, S. Whiting and E. Parra. 2009. Seasonal changes in vitamin D status in healthy young adults of different ancestry in the greater Toronto area. Presented at the 14th Workshop on Vitamin D. October 4-8, 2009. Brugge, Belgium.
Guo, Y. D., S. Strugnell, D. W. Back and G. Jones. 1993. Substrate specificity of the liver mitochondrial cytochrome P-450, CYP-27, towards vitamin D and its analogs. Proceedings of the National Academy of Sciences of the United States of America 90: 8668-72.
Gupta, R. P., B. W. Hollis, S. B. Patel, K. S. Patrick and N. H. Bell. 2004. CYP3A4 is a human microsomal vitamin D 25-hydroxylase. Journal of Bone and Mineral Research 19(4): 680-8.
Gupta, R. P., Y. A. He, K. S. Patrick, J. R. Halpert and N. H. Bell. 2005. CYP3A4 is a vitamin D-24- and 25-hydroxylase: analysis of structure function by site-directed mutagenesis. Journal of Clinical Endocrinology and Metabolism 90(2): 1210-9.
Haddad, J. G., Jr. and T. J. Hahn. 1973. Natural and synthetic sources of circulating 25-hydroxyvitamin D in man. Nature 244(5417): 515-7.
Haddad, J. G., L. Y. Matsuoka, B. W. Hollis, Y. Z. Hu and J. Wortsman. 1993. Human plasma transport of vitamin D after its endogenous synthesis. Journal of Clinical Investigation 91(6): 2552-5.
Harris, S. S. and B. Dawson-Hughes. 2002. Plasma vitamin D and 25OHD responses of young and old men to supplementation with vitamin D3. Journal of the American College of Nutrition 21(4): 357-62.
Hashizume, T., Y. Xu, M. A. Mohutsky, J. Alberts, C. Hadden, T. F. Kalhorn, N. Isoherranen, M. C. Shuhart and K. E. Thummel. 2008. Identification of human UDP-glucuronosyltransferases catalyzing hepatic 1alpha,25-dihydroxyvitamin D3 conjugation. Biochemical Pharmacology 75(5): 1240-50.
Hay, A. W. and G. Watson. 1977. The binding of 25-hydroxycholecalciferol and 25-hydroxyergocalciferol to receptor proteins in a New World and an Old World primate. Comparative Biochemistry and Physiology. B: Comparative Biochemistry 56(2): 131-4.
Helvig C., D. Cuerrier, A. Kharebov, B. Ireland, J. Kim, K. Ryder and M. Petkovich. 2008. Comparison of 1,25-dihydroxyvitamin D2 and calcitriol effects in an adenine-induced model of CKD reveals differential control over serum calcium and phosphate. Journal of Bone and Mineral Research 23(S357).
Hewison, M., F. Burke, K. N. Evans, D. A. Lammas, D. M. Sansom, P. Liu, R. L. Modlin and J. S. Adams. 2007. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. Journal of Steroid Biochemistry and Molecular Biology 103(3-5): 316-21.
Holick, M. F., J. A. MacLaughlin and S. H. Doppelt. 1981. Regulation of cutaneous previtamin D3 photosynthesis in man: skin pigment is not an essential regulator. Science 211(4482): 590-3.
Holick, M. F. 1988. Skin: site of the synthesis of vitamin D and a target tissue for the active form, 1,25-dihydroxyvitamin D3. Annals of the New York Academy of Sciences 548: 14-26.
Holick, M. F., L. Y. Matsuoka and J. Wortsman. 1989. Age, vitamin D, and solar ultraviolet. Lancet 334(8671): 1104-5.
Holick, M. F., Q. Shao, W. W. Liu and T. C. Chen. 1992. The vitamin D content of fortified milk and infant formula. New England Journal of Medicine 326(18): 1178-81.
Holick, M. 1994. McCollum Award Lecture, 1994: vitamin D—new horizons for the 21st century. American Journal of Clinical Nutrition 60(4): 619-630.
Holick, M. F. 1995. Vitamin D: photobiology, metabolism, and clinical applications. In Endocrinology, 3rd Edition, edited by L. J. DeGroot, M. Besser, H. G. Burger et al. Philadelphia, PA: W. B. Saunders.
Holick, M. F. 1996. Vitamin D: photobiology, metabolism, mechanism of action, and clinical application. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 3rd Edition, edited by M. J. Favus. Philadelphia, PA: Lippincott-Raven. Pp. 74-81.
Holick, M. F. 2008. Vitamin D and sunlight: strategies for cancer prevention and other health benefits. Clinical Journal of the American Society of Nephrology 3(5): 1548-54.
Holick, M. F., R. M. Biancuzzo, T. C. Chen, E. K. Klein, A. Young, D. Bibuld, R. Reitz, W. Salameh, A. Ameri and A. D. Tannenbaum. 2008. Vitamin D2 is as effective as vitamin D3 in maintaining circulating concentrations of 25-hydroxyvitamin D. Journal of Clinical Endocrinology and Metabolism 93(3): 677-81.
Hollis, B. W., B. A. Roos, H. H. Draper and P. W. Lambert. 1981. Vitamin D and its metabolites in human and bovine milk. Journal of Nutrition 111(7): 1240-8.
Hollis, B. W. and J. L. Napoli. 1985. Improved radioimmunoassay for vitamin D and its use in assessing vitamin D status. Clinical Chemistry 31(11): 1815-9.
Hollis, B. W. 2008. Measuring 25-hydroxyvitamin D in a clinical environment: challenges and needs. American Journal of Clinical Nutrition 88(2): 507S-10S.
Horst, R. L., R. M. Shepard, N. A. Jorgensen and H. F. DeLuca. 1979. The determination of the vitamin D metabolites on a single plasma sample: changes during parturition in dairy cows. Archives of Biochemistry and Biophysics 192(2): 512-23.
Horst, R. L., T. A. Reinhardt, C. F. Ramberg, N. J. Koszewski and J. L. Napoli. 1986. 24-hydroxylation of 1,25-dihydroxyergocalciferol. An unambiguous deactivation process. Journal of Biological Chemistry 261(20): 9250-6.
Hosseinpour, F., M. Ellfolk, M. Norlin and K. Wikvall. 2007. Phenobarbital suppresses vitamin D3 25-hydroxylase expression: a potential new mechanism for drug-induced osteomalacia. Biochemical and Biophysical Research Communications 357(3): 603-7.
Hunt, R. D., F. G. Garcia and R. J. Walsh. 1972. A comparison of the toxicity of ergocalciferol and cholecalciferol in rhesus monkeys (Macaca mulatta). Journal of Nutrition 102(8): 975-86.
IOM (Institute of Medicine). 1997. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: National Academy Press.
IOM. 2007. Dietary Reference Intakes Research Synthesis: Workshop Summary. Washington, DC: The National Academies Press.
Ish-Shalom, S., E. Segal, T. Salganik, B. Raz, I. L. Bromberg and R. Vieth. 2008. Comparison of daily, weekly, and monthly vitamin D3 in ethanol dosing protocols for two months in elderly hip fracture patients. Journal of Clinical Endocrinology and Metabolism 93(9): 3430-5.
James, S. Y., A. G. Mackay and K. W. Colston. 1996. Effects of 1,25 dihydroxyvitamin D3 and its analogues on induction of apoptosis in breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology 58(4): 395-401.
James, W. P., A. Avenell, J. Broom and J. Whitehead. 1997. A one-year trial to assess the value of orlistat in the management of obesity. International Journal of Obesity and Related Metabolic Disorders 21(Suppl 3): S24-30.
Jiang, F., J. Bao, P. Li, S. V. Nicosia and W. Bai. 2004. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down-regulation of telomerase. Journal of Biological Chemistry 279(51): 53213-21.
Johnson, J. L., V. V. Mistry, M. D. Vukovich, T. Hogie-Lorenzen, B. W. Hollis and B. L. Specker. 2005. Bioavailability of vitamin D from fortified process cheese and effects on vitamin D status in the elderly. Journal of Dairy Science 88(7): 2295-301.
Jones, G., H. K. Schnoes and H. F. DeLuca. 1975. Isolation and identification of 1,25-dihydroxyvitamin D2. Biochemistry 14(6): 1250-6.
Jones, G. 1978. Assay of vitamins D2 and D3, and 25-hydroxyvitamins D2 and D3 in human plasma by high-performance liquid chromatography. Clinical Chemistry 24(2): 287-98.
Jones, G., A. Rosenthal, D. Segev, Y. Mazur, F. Frolow, Y. Halfon, D. Rabinovich and Z. Shakked. 1979. Isolation and identification of 24,25-dihydroxyvitamin D2 using the perfused rat kidney. Biochemistry 18(6): 1094-11.
Jones, G., B. Byrnes, F. Palma, D. Segev and Y. Mazur. 1980a. Displacement potency of vitamin D2 analogs in competitive protein-binding assays for 25-hydroxyvitamin D3, 24,25-dihydroxyvitamin D3, and 1,25-dihydroxyvitamin D3. Journal of Clinical Endocrinology and Metabolism 50(4): 773-5.
Jones, G., H. K. Schnoes, L. Levan and H. F. Deluca. 1980b. Isolation and identification of 24-hydroxyvitamin D2 and 24,25-dihydroxyvitamin D2. Archives of Biochemistry and Biophysics 202(2): 450-7.
Jones, G., D. Vriezen, D. Lohnes, V. Palda and N. S. Edwards. 1987. Side-chain hydroxylation of vitamin D3 and its physiological implications. Steroids 49(1-3): 29-53.
Jones, G., S. A. Strugnell and H. F. DeLuca. 1998. Current understanding of the molecular actions of vitamin D. Physiological Reviews 78(4): 1193-231.
Jones, G. and H. L. Makin. 2000. Vitamin Ds: metabolites and analogs. In Modern Chromatographic Analysis of Vitamins, 3rd Edition, edited by A. P. de Leenheer, W. E. Lambert and J. F. Van Bocxlaer. New York: Marcel Dekker.
Jones, G. 2008. Pharmacokinetics of vitamin D toxicity. American Journal of Clinical Nutrition 88(2): 582S-6S.
Jones, G., V. Byford, C. Helvig and M. Petkovich. 2009. Differential disposition of vitamin D2 does not involve CYP24A1. Presented at 14th International Vitamin D Workshop, October 4-8, 2009. Brugge, Belgium.
Jongen, M. J., F. C. Van Ginkel, W. J. van der Vijgh, S. Kuiper, J. C. Netelenbos and P. Lips. 1984. An international comparison of vitamin D metabolite measurements. Clinical Chemistry 30(3): 399-403.
Jongen, M., W. J. van der Vijgh, J. C. Netelenbos, G. J. Postma and P. Lips. 1989. Pharmacokinetics of 24,25-dihydroxyvitamin D3 in humans. Hormone and Metabolic Research 21(10): 577-80.
Jurutka, P. W., G. K. Whitfield, J. C. Hsieh, P. D. Thompson, C. A. Haussler and M. R. Haussler. 2001. Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Reviews in Endocrinology and Metabloic Disorders 2(2): 203-16.
Kawa, S., K. Yoshizawa, M. Tokoo, H. Imai, H. Oguchi, K. Kiyosawa, T. Homma, T. Nikaido and K. Furihata. 1996. Inhibitory effect of 220-oxa-1,25-dihydroxyvitamin D3 on the proliferation of pancreatic cancer cell lines. Gastroenterology 110(5): 1605-13.
Kimlin, M. G., W. J. Olds and M. R. Moore. 2007. Location and vitamin D synthesis: is the hypothesis validated by geophysical data? Journal of Photochemistry and Photobiology. B, Biology 86(3): 234-9.
Knutson, J. C., L. W. LeVan, C. R. Valliere and C. W. Bishop. 1997. Pharmacokinetics and systemic effect on calcium homeostasis of 1 alpha,24-dihydroxyvitamin D2 in rats. Comparison with 1 alpha,25-dihydroxyvitamin D2, calcitriol, and calcipotriol. Biochemical Pharmacology 53(6): 829-37.
Koga, M., J. A. Eisman and R. L. Sutherland. 1988. Regulation of epidermal growth factor receptor levels by 1,25-dihydroxyvitamin D3 in human breast cancer cells. Cancer Research 48(10): 2734-9.
Kovacs, C. S. and H. M. Kronenberg. 1997. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocrine Reviews 18(6): 832-72.
Kovalenko, P. L., Z. Zhang, M. Cui, S. K. Clinton and J. C. Fleet. 2010. 1,25 dihydroxyvitamin D-mediated orchestration of anticancer, transcript-level effects in the immortalized, non-transformed prostate epithelial cell line, RWPE1. BMC Genomics 11: 26.
Kull, M., Jr., R. Kallikorm, A. Tamm and M. Lember. 2009. Seasonal variance of 25-(OH) vitamin D in the general population of Estonia, a Northern European country. BMC Public Health 9: 22.
Kumagai, T., L. Y. Shih, S. V. Hughes, J. C. Desmond, J. O’Kelly, M. Hewison and H. P. Koeffler. 2005. 19-Nor-1,25(OH)2D2 (a novel, noncalcemic vitamin D analogue), combined with arsenic trioxide, has potent antitumor activity against myeloid leukemia. Cancer Research 65(6): 2488-97.
Kumar, R., D. Harnden and H. F. DeLuca. 1976. Metabolism of 1,25-dihydroxyvitamin D3: evidence for side-chain oxidation. Biochemistry 15(11): 2420-3.
Laing, C. J. and N. E. Cooke. 2005. Chapter 8: Vitamin D binding protein. In Vitamin D, 2nd Edition, edited by D. Feldman, J. W. Pike and F. H. Glorieux. Burlington, MA: Elsevier Academic Press.
Leerbeck, E. and H. Sondergaard. 1980. The total content of vitamin D in human milk and cow’s milk. British Journal of Nutrition 44(1): 7-12.
Lensmeyer, G. L., D. A. Wiebe, N. Binkley and M. K. Drezner. 2006. HPLC method for 25-hydroxyvitamin D measurement: comparison with contemporary assays. Clinical Chemistry 52(6): 1120-6.
LeVan, L. W., H. K. Schnoes and H. F. DeLuca. 1981. Isolation and identification of 25-hydroxyvitamin D2 25-glucuronide: a biliary metabolite of vitamin D2 in the chick. Biochemistry 20(1): 222-6.
Li, P., C. Li, X. Zhao, X. Zhang, S. V. Nicosia and W. Bai. 2004. p27(Kip1) stabilization and G(1) arrest by 1,25-dihydroxyvitamin D(3) in ovarian cancer cells mediated through down-regulation of cyclin E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2 ubiquitin ligase. Journal of Biological Chemistry 279(24): 25260-7.
Liel, Y., J. Edwards, J. Shary, K. M. Spicer, L. Gordon and N. H. Bell. 1988. The effects of race and body habitus on bone mineral density of the radius, hip, and spine in premenopausal women. Journal of Clinical Endocrinology and Metabolism 66(6): 1247-50.
Lips, P. 2006. Vitamin D physiology. Progress in Biophysics and Molecular Biology 92(1): 4-8.
Liu, P. T., S. Stenger, H. Li, L. Wenzel, B. H. Tan, S. R. Krutzik, M. T. Ochoa, J. Schauber, K. Wu, C. Meinken, D. L. Kamen, M. Wagner, R. Bals, A. Steinmeyer, U. Zugel, R. L. Gallo, D. Eisenberg, M. Hewison, B. W. Hollis, J. S. Adams, B. R. Bloom and R. L. Modlin. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311(5768): 1770-3.
Liu, S., A. Gupta and L. D. Quarles. 2007. Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Current Opinion in Nephrology and Hypertension 16(4): 329-35.
Liu, N., L. Nguyen, R. F. Chun, V. Lagishetty, S. Ren, S. Wu, B. Hollis, H. F. DeLuca, J. S. Adams and M. Hewison. 2008. Altered endocrine and autocrine metabolism of vitamin D in a mouse model of gastrointestinal inflammation. Endocrinology 149(10): 4799-808.
Lohnes, D. and G. Jones. 1992. Further metabolism of 1α,25-dihydroxyvitamin D3 in target cells. Proceedings of the First International Congress on Vitamins and Biofactors in Life Science. Journal Nutritional Science and Vitaminology (Special Issue) 75-78.
Looker, A. C. 2005. Body fat and vitamin D status in black versus white women. Journal of Clinical Endocrinology and Metabolism 90(2): 635-40.
Looker, A. C. 2007. Do body fat and exercise modulate vitamin D status? Nutrition Reviews 65(8 Pt 2): S124-6.
Looker, A. C., C. M. Pfeiffer, D. A. Lacher, R. L. Schleicher, M. F. Picciano and E. A. Yetley. 2008. Serum 25-hydroxyvitamin D status of the US population: 1988-1994 compared with 2000-2004. American Journal of Clinical Nutrition 88(6): 1519-27.
Lubin, D., E. H. Jensen and H. P. Gies. 1998. Global surface ultraviolet radiation climatology from TOMS and ERBE data. Journal of Geophysical Research 103(D20): 26061-91.
Lucas, J. A., M. J. Bolland, A. B. Grey, R. W. Ames, B. H. Mason, A. M. Horne, G. D. Gamble and I. R. Reid. 2005. Determinants of vitamin D status in older women living in a subtropical climate. Osteoporosis International 16(12): 1641-8.
Ma, J. F., L. Nonn, M. J. Campbell, M. Hewison, D. Feldman and D. M. Peehl. 2004. Mechanisms of decreased vitamin D 1alpha-hydroxylase activity in prostate cancer cells. Molecular and Cellular Endocrinology 221(1-2): 67-74.
MacLaughlin, J. and M. F. Holick. 1985. Aging decreases the capacity of human skin to produce vitamin D3. Journal of Clinical Investigation 76(4): 1536-8.
Mahdy, T., S. Atia, M. Farid and A. Adulatif. 2008. Effect of Roux-en Y gastric bypass on bone metabolism in patients with morbid obesity: Mansoura experiences. Obesity Surgery 18(12): 1526-31.
Makin, H. L. J., G. Jones, M. Kaufman and M. J. Calverley. 2010. Chapter 11: Analysis of vitamins D, their metabolites and analogs. In Steroid Analysis, edited by H. L. J. Makin and D. B. Gower. New York: Springer.
Mantell, D. J., P. E. Owens, N. J. Bundred, E. B. Mawer and A. E. Canfield. 2000. 1 Alpha,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circulation Research 87(3): 214-20.
Martinez, M. E., M. T. Del Campo, M. J. Sanchez-Cabezudo, G. Balaguer, A. Rodriguez-Carmona and R. Selgas. 1995. Effect of oral calcidiol treatment on its serum levels and peritoneal losses. Peritoneal Dialysis International 15(1): 65-70.
Masuda, S., V. Byford, A. Arabian, Y. Sakai, M. B. Demay, R. St-Arnaud and G. Jones. 2005. Altered pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology 146(2): 825-34.
Masuda, S. and G. Jones. 2006. Promise of vitamin D analogues in the treatment of hyper-proliferative conditions. Molecular Cancer Therapeutics 5(4): 797-808.
Matsuoka, L. Y., J. Wortsman, J. G. Haddad and B. W. Hollis. 1989. In vivo threshold for cutaneous synthesis of vitamin D3. Journal of Laboratory and Clinical Medicine 114(3): 301-5.
McDuffie, J. R., K. A. Calis, S. L. Booth, G. I. Uwaifo and J. A. Yanovski. 2002. Effects of orlistat on fat-soluble vitamins in obese adolescents. Pharmacotherapy 22(7): 814-22.
Misra, M., D. Pacaud, A. Petryk, P. F. Collett-Solberg and M. Kappy. 2008. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics 122(2): 398-417.
Moreno, J., A. V. Krishnan, S. Swami, L. Nonn, D. M. Peehl and D. Feldman. 2005. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Research 65(17): 7917-25.
Morris, J. F. and M. Peacock. 1976. Assay of plasma 25-hydroxy vitamin D. Clinica Chimica Acta 72(3): 383-91.
Mulligan, G. B. and A. Licata. 2010. Taking vitamin D with the largest meal improves absorption and results in higher serum levels of 25-hydroxyvitamin D. Journal of Bone and Mineral Research 25(4): 928-30.
Murphy, S. C., L. J. Whited, L. C. Rosenberry, B. H. Hammond, D. K. Bandler and K. J. Boor. 2001. Fluid milk vitamin fortification compliance in New York State. Journal of Dairy Science 84(12): 2813-20.
Olds, W. J., A. R. McKinley, M. R. Moore and M. G. Kimlin. 2008. In vitro model of vitamin D3 (cholecalciferol) synthesis by UV radiation: dose–response relationships. Journal of Photochemistry and Photobiology. B, Biology 93(2): 88-93.
Park, E. A. 1940. The therapy of rickets. JAMA 94: 370-9.
Penna, G., S. Amuchastegui, N. Giarratana, K. C. Daniel, M. Vulcano, S. Sozzani and L. Adorini. 2007. 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. Journal of Immunology 178(1): 145-53.
Phinney, K. W. 2009. Methods development and standard reference materials for 25(OH) D. Presented at the Committee to Review Dietary Reference Intakes for Vitamin D and Calcium Information-gathering Workshop, August 4, 2009. Washington, DC.
Picciano, M. F., J. T. Dwyer, K. L. Radimer, D. H. Wilson, K. D. Fisher, P. R. Thomas, E. A. Yetley, A. J. Moshfegh, P. S. Levy, S. J. Nielsen and B. M. Marriott. 2007. Dietary supplement use among infants, children, and adolescents in the United States, 1999-2002. Archives of Pediatrics and Adolescent Medicine 161(10): 978-85.
Pike, J. W., N. K. Shevde, B. W. Hollis, N. E. Cooke and L. A. Zella. 2008. Vitamin D—binding protein influences total circulating levels of 1,25-dihydroxyvitamin D-3 but does not directly modulate the bioactive levels of the hormone in vivo. Endocrinology 149(7): 3656-67.
Quarles, L. D. 2008. Endocrine functions of bone in mineral metabolism regulation. Journal of Clinical Investigation 118(12): 3820-8.
Radimer, K., B. Bindewald, J. Hughes, B. Ervin, C. Swanson and M. F. Picciano. 2004. Dietary supplement use by US adults: data from the National Health and Nutrition Examination Survey, 1999-2000. American Journal of Epidemiology 160(4): 339-49.
Rapuri, P. B., H. K. Kinyamu, J. C. Gallagher and V. Haynatzka. 2002. Seasonal changes in calciotropic hormones, bone markers, and bone mineral density in elderly women. Journal of Clinical Endocrinology and Metabolism 87(5): 2024-32.
Rapuri, P. B., J. C. Gallagher and G. Haynatzki. 2004. Effect of vitamins D2 and D3 supplement use on serum 25OHD concentration in elderly women in summer and winter. Calcified Tissue International 74(2): 150-6.
Reeve, L. E., R. W. Chesney and H. F. DeLuca. 1982. Vitamin D of human milk: identification of biologically active forms. American Journal of Clinical Nutrition 36(1): 122-6.
Reichel, H., H. P. Koeffler and A. W. Norman. 1989. The role of the vitamin D endocrine system in health and disease. New England Journal of Medicine 320(15): 980-91.
Reinehr, T., G. de Sousa, U. Alexy, M. Kersting and W. Andler. 2007. Vitamin D status and parathyroid hormone in obese children before and after weight loss. European Journal of Endocrinology/European Federation of Endocrine Societies 157(2): 225-32.
Reinhardt, T. A., C. F. Ramberg and R. L. Horst. 1989. Comparison of receptor binding, biological activity, and in vivo tracer kinetics for 1,25-dihydroxyvitamin D3, 1,25-dihydroxyvitamin D2, and its 24 epimer. Archives of Biochemistry and Biophysics 273(1): 64-71.
Riedt, C. S., M. Cifuentes, T. Stahl, H. A. Chowdhury, Y. Schlussel and S. A. Shapses. 2005. Overweight postmenopausal women lose bone with moderate weight reduction and 1 g/day calcium intake. Journal of Bone and Mineral Research 20(3): 455-63.
Riedt, C. S., R. E. Brolin, R. M. Sherrell, M. P. Field and S. A. Shapses. 2006. True fractional calcium absorption is decreased after Roux-en-Y gastric bypass surgery. Obesity (Silver Spring) 14(11): 1940-8.
Roborgh, J. R. and T. de Man. 1959. The hypercalcemic activity of dihydrotachysterol-2 and dihydrotachysterol-3 and of the vitamins D2 and D3: comparative experiments on rats. Biochemical Pharmacology 2: 1-6.
Roborgh, J. R. and T. de Man. 1960. The hypercalcemic activity of dihydrotachysterol-2 and dihydrotachysterol-3 and of the vitamins D2 and D3 after intravenous injection of the aqueous preparations. 2. Comparative experiments on rats. Biochemical Pharmacology 3: 277-82.
Rosenstreich, S. J., C. Rich and W. Volwiler. 1971. Deposition in and release of vitamin D3 from body fat: evidence for a storage site in the rat. Journal of Clinical Investigation 50(3): 679-87.
Roth, H. J., H. Schmidt-Gayk, H. Weber and C. Niederau. 2008. Accuracy and clinical implications of seven 25-hydroxyvitamin D methods compared with liquid chromatography-tandem mass spectrometry as a reference. Annals of Clinical Biochemistry 45(Pt 2): 153-9.
Sawada, N., T. Sakaki, M. Ohta and K. Inouye. 2000. Metabolism of vitamin D(3) by human CYP27A1. Biochemical and Biophysical Research Communications 273(3): 977-84.
Silver, J., T. Naveh-Many, H. Mayer, H. J. Schmelzer and M. M. Popovtzer. 1986. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. Journal of Clinical Investigation 78(5): 1296-301.
Singh, R. J., R. L. Taylor, G. S. Reddy and S. K. Grebe. 2006. C-3 epimers can account for a significant proportion of total circulating 25-hydroxyvitamin D in infants, complicating accurate measurement and interpretation of vitamin D status. Journal of Clinical Endocrinology and Metabolism 91(8): 3055-61.
Siu-Caldera, M. L., H. Sekimoto, S. Peleg, C. Nguyen, A. M. Kissmeyer, L. Binderup, A. Weiskopf, P. Vouros, M. R. Uskokovic and G. S. Reddy. 1999. Enhanced biological activity of 1alpha,25-dihydroxy-20-epi-vitamin D3, the C-20 epimer of 1alpha,25-dihydroxyvitamin D3, is in part due to its metabolism into stable intermediary metabolites with significant biological activity. Journal of Steroid Biochemistry and Molecular Biology 71(3-4): 111-21.
Sjoden, G., C. Smith, U. Lindgren and H. F. DeLuca. 1985. 1 alpha-hydroxyvitamin D2 is less toxic than 1 alpha-hydroxyvitamin D3 in the rat. Proceedings of the Society for Experimental Biology and Medicine 178(3): 432-6.
Slatopolsky, E., C. Weerts, J. Thielan, R. Horst, H. Harter and K. J. Martin. 1984. Marked suppression of secondary hyperparathyroidism by intravenous administration of 1,25-dihydroxy-cholecalciferol in uremic patients. Journal of Clinical Investigation 74(6): 2136-43.
Smith, S. M., K. K. Gardner, J. Locke and S. R. Zwart. 2009. Vitamin D supplementation during Antarctic winter. American Journal of Clinical Nutrition 89(4): 1092-8.
Snellman, G., H. Melhus, R. Gedeborg, S. Olofsson, A. Wolk, N. L. Pedersen and K. Michaelsson. 2009. Seasonal genetic influence on serum 25-hydroxyvitamin D levels: a twin study. PLoS ONE [Electronic Resource] 4(11): e7747.
Sneve, M., Y. Figenschau and R. Jorde. 2008. Supplementation with cholecalciferol does not result in weight reduction in overweight and obese subjects. European Journal of Endocrinology/European Federation of Endocrine Societies 159(6): 675-84.
Specker, B. L., B. Valanis, V. Hertzberg, N. Edwards and R. C. Tsang. 1985. Sunshine exposure and serum 25-hydroxyvitamin D concentrations in exclusively breast-fed infants. Journal of Pediatrics 107(3): 372-6.
Springbett, P., S. Buglass and A. R. Young. 2010. Photoprotection and vitamin D status. Journal of Photochemistry and Photobiology B: Biology 101(2):160-8.
Stamp, T. C., J. G. Haddad, Jr., A. N. Exton-Smith, A. Reuben and C. A. Twigg. 1976. Assay of vitamin D and its metabolites. Annals of Clinical Biochemistry 13(6): 571-7.
Stamp, T. C., J. G. Haddad and C. A. Twigg. 1977. Comparison of oral 25-hydroxycholecalciferol, vitamin D, and ultraviolet light as determinants of circulating 25-hydroxyvitamin D. Lancet 1(8026): 1341-3.
Steenbock, H. and A. Black. 1924. Fat-soluble vitamins. Journal of Biological Chemistry 61(2): 405-22.
Strushkevich, N., S. A. Usanov, A. N. Plotnikov, G. Jones and H. W. Park. 2008. Structural analysis of CYP2R1 in complex with vitamin D3. Journal of Molecular Biology 380(1): 95-106.
Stubbs, J. R., A. Idiculla, J. Slusser, R. Menard and L. D. Quarles. 2010. Cholecalciferol supplementation alters calcitriol-responsive monocyte proteins and decreases inflammatory cytokines in ESRD. Journal of the American Society of Nephrology 21(2): 353-61.
Suda, T., H. F. DeLuca, H. K. Schnoes and J. W. Blunt. 1969. The isolation and identification of 25-hydroxyergocalciferol. Biochemistry 8(9): 3515-20.
Suda, T., N. Takahashi and E. Abe. 1992. Role of vitamin D in bone resorption. Journal of Cellular Biochemistry 49(1): 53-8.
Sundaram, S., M. Chaudhry, D. Reardon, M. Gupta and D. A. Gewirtz. 2000. The vitamin D3 analog EB 1089 enhances the antiproliferative and apoptotic effects of adriamycin in MCF-7 breast tumor cells. Breast Cancer Research and Treatment 63(1): 1-10.
Sutton, R. A., N. L. Wong and J. H. Dirks. 1976. Effects of parathyroid hormone on sodium and calcium transport in the dog nephron. Clinical Science and Molecular Medicine 51(4): 345-51.
Talwar, S. A., J. F. Aloia, S. Pollack and J. K. Yeh. 2007. Dose response to vitamin D supplementation among postmenopausal African American women. American Journal of Clinical Nutrition 86(6): 1657-62.
Tanaka, Y. and H. F. DeLuca. 1983. Stimulation of 1,25-dihydroxyvitamin D3 production by 1,25-dihydroxyvitamin D3 in the hypocalcaemic rat. Biochemical Journal 214(3): 893-7.
Tanner, J. T., J. Smith, P. Defibaugh, G. Angyal, M. Villalobos, M. P. Bueno, E. T. McGarrahan, H. M. Wehr, J. F. Muniz, B. W. Hollis et al. 1988. Survey of vitamin content of fortified milk. Journal—Association of Official Analytical Chemists 71(3): 607-10.
Thompson, G. R., B. Lewis and C. C. Booth. 1966. Absorption of vitamin D3-3H in control subjects and patients with intestinal malabsorption. Journal of Clinical Investigation 45(1): 94-102.
Thompson, P. D., P. W. Jurutka, G. K. Whitfield, S. M. Myskowski, K. R. Eichhorst, C. E. Dominguez, C. A. Haussler and M. R. Haussler. 2002. Liganded VDR induces CYP3A4 in small intestinal and colon cancer cells via DR3 and ER6 vitamin D responsive elements. Biochemical and Biophysical Research Communications 299(5): 730-8.
Tjellesen, L., A. Gotfredsen and C. Christiansen. 1985. Different actions of vitamin D2 and D3 on bone metabolism in patients treated with phenobarbitone/phenytoin. Calcified Tissue International 37(3): 218-22.
Trang, H. M., D. E. Cole, L. A. Rubin, A. Pierratos, S. Siu and R. Vieth. 1998. Evidence that vitamin D3 increases serum 25-hydroxyvitamin D more efficiently than does vitamin D2. American Journal of Clinical Nutrition 68(4): 854-8.
Trump, D. L., K. K. Deeb and C. S. Johnson. 2010. Vitamin D: considerations in the continued development as an agent for cancer prevention and therapy. Cancer Journal 16(1): 1-9.
Turner, M., P. E. Barre, A. Benjamin, D. Goltzman and M. Gascon-Barre. 1988. Does the maternal kidney contribute to the increased circulating 1,25-dihydroxyvitamin D concentrations during pregnancy? Mineral and Electrolyte Metabolism 14(4): 246-52.
Tzotzas, T., F. G. Papadopoulou, K. Tziomalos, S. Karras, K. Gastaris, P. Perros and G. E. Krassas. 2010. Rising serum 25-hydroxy-vitamin D levels after weight loss in obese women correlate with improvement in insulin resistance. Journal of Clinical Endocrinology and Metabolism 95(9): 4251-7.
Urushino, N., K. Yasuda, S. Ikushiro, M. Kamakura, M. Ohta and T. Sakaki. 2009. Metabolism of 1alpha,25-dihydroxyvitamin D2 by human CYP24A1. Biochemical and Biophysical Research Communications 384(2): 144-8.
Valderas, J. P., S. Velasco, S. Solari, Y. Liberona, P. Viviani, A. Maiz, A. Escalona and G. Gonzalez. 2009. Increase of bone resorption and the parathyroid hormone in post-menopausal women in the long-term after Roux-en-Y gastric bypass. Obesity Surgery 19(8): 1132-8.
van der Mei, I. A., A. L. Ponsonby, O. Engelsen, J. A. Pasco, J. J. McGrath, D. W. Eyles, L. Blizzard, T. Dwyer, R. Lucas and G. Jones. 2007. The high prevalence of vitamin D insufficiency across Australian populations is only partly explained by season and latitude. Environmental Health Perspectives 115(8): 1132-9.
van Dijk, C. E., M. R. de Boer, L. L. Koppes, J. C. Roos, P. Lips and J. W. Twisk. 2009. Positive association between the course of vitamin D intake and bone mineral density at 36 years in men. Bone 44(3): 437-41.
Vieth, R. 1990. The mechanisms of vitamin D toxicity. Bone and Mineral 11(3): 267-72.
Vieth, R. 2004. Why the optimal requirement for vitamin D3 is probably much higher than what is officially recommended for adults. Journal of Steroid Biochemistry and Molecular Biology 89-90(1-5): 575-9.
Wang, L., J. N. Flanagan, L. W. Whitlatch, D. P. Jamieson, M. F. Holick and T. C. Chen. 2004. Regulation of 25-hydroxyvitamin D-1alpha-hydroxylase by epidermal growth factor in prostate cells. Journal of Steroid Biochemistry and Molecular Biology 89-90(1-5): 127-30.
Wang, T. J., F. Zhang, J. B. Richards, B. Kestenbaum, J. B. van Meurs, D. Berry, D. P. Kiel, E. A. Streeten, C. Ohlsson, D. L. Koller, L. Peltonen, J. D. Cooper, P. F. O’Reilly, D. K. Houston, N. L. Glazer, L. Vandenput, M. Peacock, J. Shi, F. Rivadeneira, M. I. McCarthy, P. Anneli, I. H. de Boer, M. Mangino, B. Kato, D. J. Smyth, S. L. Booth, P. F. Jacques, G. L. Burke, M. Goodarzi, C. L. Cheung, M. Wolf, K. Rice, D. Goltzman, N. Hidiroglou, M. Ladouceur, N. J. Wareham, L. J. Hocking, D. Hart, N. K. Arden, C. Cooper, S. Malik, W. D. Fraser, A. L. Hartikainen, G. Zhai, H. M. Macdonald, N. G. Forouhi, R. J. Loos, D. M. Reid, A. Hakim, E. Dennison, Y. Liu, C. Power, H. E. Stevens, L. Jaana, R. S. Vasan, N. Soranzo, J. Bojunga, B. M. Psaty, M. Lorentzon, T. Foroud, T. B. Harris, A. Hofman, J. O. Jansson, J. A. Cauley, A. G. Uitterlinden, Q. Gibson, M. R. Jarvelin, D. Karasik, D. S. Siscovick, M. J. Econs, S. B. Kritchevsky, J. C. Florez, J. A. Todd, J. Dupuis, E. Hypponen and T. D. Spector. 2010. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet 376(9736): 180-8.
Ward, L. M., I. Gaboury, M. Ladhani and S. Zlotkin. 2007. Vitamin D-deficiency rickets among children in Canada. Canadian Medical Association Journal 177(2): 161-6.
Wastney, M. E., J. Ng, D. Smith, B. R. Martin, M. Peacock and C. M. Weaver. 1996. Differences in calcium kinetics between adolescent girls and young women. American Journal of Physiology 271(1 Pt 2): R208-16.
Webb, A. R., L. Kline and M. F. Holick. 1988. Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. Journal of Clinical Endocrinology and Metabolism 67(2): 373-8.
Webb, A. R., B. R. DeCosta and M. F. Holick. 1989. Sunlight regulates the cutaneous production of vitamin D3 by causing its photodegradation. Journal of Clinical Endocrinology and Metabolism 68(5): 882-7.
Weber, F. 1981. Absorption mechanisms for fat-soluble vitamins and the effect of other food constituents. Progress in Clinical and Biological Research 77: 119-35.
Weber, F. 1983. Absorption of fat-soluble vitamins. International Journal for Vitamin and Nutrition Research (Suppl 25): 55-65.
Weber, K., M. Goldberg, M. Stangassinger and R. G. Erben. 2001. 1Alpha-hydroxyvitamin D2 is less toxic but not bone selective relative to 1alpha-hydroxyvitamin D3 in ovariectomized rats. Journal of Bone and Mineral Research 16(4): 639-51.
Willnow, T. and A. Nykjaer. 2005. Chapter 10: Endocytic pathways for 25-(OH) vitamin D3. In Vitamin D, 2nd Edition, edited by D. Feldman, J. W. Pike and F. Glorieux. Burlington, MA: Elsevier.
Wortsman, J., L. Y. Matsuoka, T. C. Chen, Z. Lu and M. F. Holick. 2000. Decreased bioavailability of vitamin D in obesity. American Journal of Clinical Nutrition 72(3): 690-3.
Xie, S. P., S. Y. James and K. W. Colston. 1997. Vitamin D derivatives inhibit the mitogenic effects of IGF-I on MCF-7 human breast cancer cells. Journal of Endocrinology 154(3): 495-504.
Xu, Y., T. Hashizume, M. C. Shuhart, C. L. Davis, W. L. Nelson, T. Sakaki, T. F. Kalhorn, P. B. Watkins, E. G. Schuetz and K. E. Thummel. 2006. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1alpha,25-dihydroxyvitamin D(3): implications for drug-induced osteomalacia. Molecular Pharmacology 69(1): 56-65.
Yamamoto, M., Y. Kawanobe, H. Takahashi, E. Shimazawa, S. Kimura and E. Ogata. 1984. Vitamin D deficiency and renal calcium transport in the rat. Journal of Clinical Investigation 74(2): 507-13.
Yanagisawa, J., Y. Yanagi, Y. Masuhiro, M. Suzawa, M. Watanabe, K. Kashiwagi, T. Toriyabe, M. Kawabata, K. Miyazono and S. Kato. 1999. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science 283(5406): 1317-21.
Yasuda, H., K. Higashio and T. Suda. 2005. Vitamin D and osteoclastogenesis. In Vitamin D, Volume 1, edited by D. Feldman, J. W. Pike and F. H. Glorieux. San Diego, CA: Elsevier Academic Press. Pp. 665-85.
Yetley, E. A. 2008. Assessing the vitamin D status of the US population. American Journal of Clinical Nutrition 88(2): 558S-64S.
Yetley, E. A., D. Brule, M. C. Cheney, C. D. Davis, K. A. Esslinger, P. W. Fischer, K. E. Friedl, L. S. Greene-Finestone, P. M. Guenther, D. M. Klurfeld, M. R. L’Abbe, K. Y. McMurry, P. E. Starke-Reed and P. R. Trumbo. 2009. Dietary reference intakes for vitamin D: justification for a review of the 1997 values. American Journal of Clinical Nutrition 89(3): 719-27.
Zinser, G. M. and J. Welsh. 2004. Accelerated mammary gland development during pregnancy and delayed postlactational involution in vitamin D3 receptor null mice. Molecular Endocrinology 18(9): 2208-23.
Zittermann, A., S. Frisch, H. K. Berthold, C. Gotting, J. Kuhn, K. Kleesiek, P. Stehle, H. Koertke and R. Koerfer. 2009. Vitamin D supplementation enhances the beneficial effects of weight loss on cardiovascular disease risk markers. American Journal of Clinical Nutrition 89(5): 1321-7.


















































