
C
Workshop Speakers’ Papers
Overview of Traumatic Brain Injury Within the Department of Defense
Kimberly S. Meyer1 and Michael S. Jaffee2
INTRODUCTION
Traumatic brain injury (TBI) has been declared by the media as the signature injury of the current conflicts in Iraq and Afghanistan. The Department of Defense (DoD) defines TBI as a traumatic blow or jolt to the head resulting in an alteration or loss of consciousness (DoD, 2007).
TBI surveillance efforts within DoD began in 2000 and, through the fourth quarter of 2010, 202,281 service members have been diagnosed with TBI.3 The majority of these injuries (77 percent) are classified as mild TBI (mTBI) or concussion. The severity of injury for the remainder of cases is as follows: moderate (16.8 percent), severe (1 percent), and penetrating (1.7 percent). A small proportion (3.5 percent) is of undetermined severity, likely because of coding incongruencies. Since 2000, the frequency of diagnosis has increased each year (Table C-1), which is likely a result of the aggressive pursuit of increased screening efforts instituted by DoD in 2006.
SCREENING
Screening of TBI occurs at various time points following combat activities. During the acute stages, screening takes place in theater. Early efforts regarding TBI screening required
|
1 |
Defense & Veterans Brain Injury Center. |
|
2 |
San Antonio Uniformed Services Health Education Consortium. |
|
3 |
Available online: http://www.dvbic.org/TBI-Numbers.aspx (accessed March 25, 2011). |
TABLE C-1 The Number of DoD Service Members (All Armed Forces) Diagnosed with TBI, 2000–2010a
|
Year |
Number of Service Members |
|
2000 |
10,963 |
|
2001 |
11,830 |
|
2002 |
12,470 |
|
2003 |
12,898 |
|
2004 |
13,312 |
|
2005 |
12,192 |
|
2006 |
16,946 |
|
2007 |
23,160 |
|
2008 |
28,555 |
|
2009 |
29,252 |
|
2010b |
30,703 |
|
Total |
202,281 |
|
aAvailable online: http://www.dvbic.org/TBI-Numbers.aspx (accessed March 25, 2011). bAs of quarter 4 of 2010, as of February 17, 2011. |
|
evaluation when an individual presented to medical care with symptoms concerning for TBI. An in-theater assessment of TBI care sponsored by the Joint Chiefs of Staff, however, found that individuals at risk for TBI failed to seek medical evaluation; consequently, mandatory event-based screening protocols were implemented in July 2010. These protocols include obligatory medical evaluations for all individuals within 50 meters of a blast, those who were located in a building or vehicle damaged by a blast, and those with certain other indications like blunt trauma to the head.
The Military Acute Concussion Evaluation (MACE) is the tool used in acute TBI screening (Figure C-1). The MACE tool was instituted in 2006 and assesses the following four domains: history of the traumatic event, including presence or absence of changes in consciousness; current symptoms; neurological exam; and, if indicated, a brief cognitive appraisal. The MACE tool is based on the Standardized Assessment of Concussion used for sports-related injuries where scores of 25 or less are indicative of cognitive impairment (McCrea et al., 2000). A validation study of MACE use in an austere environment is currently under way. Preliminary evidence suggests cognitive scores slightly less than 25 in a deployed setting may be normal because simple orientation may be affected by lack of differentiation during daily routines. Further validation testing is ongoing to optimize the use of this cognitive evaluation in theater.
Initially, the only documentation required when utilizing MACE was the numeric score associated with the cognitive assessment, which led to incomplete capture of the TBI exposure and an immediate written record in a member’s medical history. Subsequently, documentation requirements were modified and the following were added: cognitive score, neurological assessment, and current symptoms (CNS). Using this additional documentation facilitates the identification of a temporal relationship between the traumatic event and symptom onset in addition to changes in symptom profiles over time. Although MACE is not a definitive diagnostic tool for TBI, positive screens trigger a detailed clinical exam to confirm the diagnosis or determine the differential diagnosis for ongoing symptoms.
Service members evacuated to Landstuhl Regional Medical Center (LRMC) for any injury or illness undergo additional screening using the MACE tool. From May 2006 to October 2008, approximately 18,000 patients (approximately 12,200 inpatients and 5,800

FIGURE C-1 An example of a MACE form.
SOURCE: Available online: http://www.dvbic.org/Providers/TBI-Screening.aspx (accessed April 5, 2011).
outpatients) completed the initial MACE screening at LRMC. Sixteen percent of outpatients screened positively as being at risk for TBI. Additional screening revealed that 78 percent of the positive screens described symptoms associated with TBI. Thirty-one percent of inpatients screened positively as being at risk for TBI. Of those inpatients screening positive, 66 percent reported associated symptoms (Dempsey et al., 2009). Those with significant findings are triaged to a stateside military medical facility with appropriate resources to further evaluate and treat TBI. Increased funding has led to allocation of resources at most military installations, thereby allowing those with mTBI to be treated at their home base. The U.S. Army has credentialed its hospitals based on resources available (Figure C-2). Similarly, the U.S. Navy and U.S. Air Force have declared certain facilities as TBI centers. Those deter-
mined to need specialized services (i.e., severe and penetrating TBI patients), however, are directed to Walter Reed Army Medical Center (WRAMC) or National Naval Medical Center (NNMC) for additional acute and subacute care.
Active-duty service members undergo a final screening during mandatory Post-Deployment Health Assessment (PDHA, DD-Form 2796). This assessment is not limited to TBI and covers symptoms associated with a variety of other medical and psychological conditions. The TBI component consists of the following four questions, which are based on the Brief Traumatic Brain Injury Screen (Schwab et al., 2006):
-
During this deployment, did you experience any of the following events?
-
Blast or explosion
-
Vehicular accident
-
Fragment wound or bullet wound above the shoulders
-
Fall
-
Other event causing injury to the head
-
-
Did any of the following happen to you, or were you told happened to you, IMMEDIATELY after any of the event(s)?
-
Lost consciousness or got “knocked out”
-
Felt dazed, confused, or “saw stars”
-
Didn’t remember the event
-
Had a concussion
-
Had a head injury
-
-
Did any of the following problems begin or get worse after the event(s)?
-
Memory problems or lapses
-
Balance problems or dizziness
-
Ringing in the ears
-
Sensitivity to bright light
-
Irritability
-
Headaches
-
Sleep problems
-
-
In the past week, have you had any of the symptoms you indicated?
-
Memory problems or lapses
-
Balance problems or dizziness
-
Ringing in the ears
-
Sensitivity to bright light
-
Irritability
-
Headaches
-
Sleep problems
-
One study of a returning U.S. Army Combat Brigade Team revealed that 22.8 percent of soldiers who reported injury during assessment with PDHA sustained a clinician-confirmed TBI (Terrio et al., 2009). The majority were mTBIs (i.e., concussions). Although 33.4 percent of this sample reported multiple symptoms immediately following the injury, symptom reporting decreased to 7.5 percent in the postdeployment period. These results suggest that most individuals recover within weeks to months of concussion injury, which is consistent with findings among the civilian population.
Further screening occurs for those entering the Veterans Health Administration (VHA) for care. Affirmative responses on all questions are required to be a positive screen. This
serves to identify those still in need of medical care for TBI-related symptoms but does not enhance current TBI surveillance methods.
NEUROCOGNITIVE ASSESSMENT TESTING
In accordance with the congresional National Defense Authorization Act of 2007, predeployment neurocognitive testing was implemented in 2008. To date, 723,981 service members have completed this testing, which currently uses the Automated Neurocognitive Assessment Metrics (ANAM). This is a brief computerized battery that is designed to detect speed and accuracy of attention, memory, and thinking ability. By completing the ANAM within 12 months of deployment, each service member establishes an individual preinjury baseline; thus, if a TBI is sustained in theater, the ANAM can be repeated to allow for pre- and postinjury comparison and to better inform return-to-duty determinations. Pending inclusion in the electronic medical record, test results are stored in a central repository. Health-care providers can obtain these results from the helpdesk by emailing pertinent demographic information to anam.baselines@amedd.army.mil. To date, there have been 5,820 requests for baseline scores with 1,826 from Afghanistan, 202 from Iraq, and 3,792 from the Continental United States.
TREATMENT
Clinical practice guidelines have been developed to guide the evaluation and management of TBI in both the acute and chronic settings. These guidelines incorporate available evidence and expert consensus. Acute care begins on the battlefield with theater-based guidelines, which incorporate the tactical requirements of the combat setting. Previous guidelines were only useful if the service member sought a medical evaluation. Recent revisions, supported by the Joint Chiefs of Staff, require all service members involved in a traumatic event to undergo screening and a mandatory 24-hour rest period. Those with serious neurologic injury are evacuated to facilities with imaging capabilities. Those with symptoms of concussion, such as headache or dizziness, are managed aggressively with local medical assets. Once symptoms resolve, exertional testing is performed prior to return to duty to ensure that symptoms do not return with physiologic stress. Those with persistent symptoms undergo combat stress evaluations and more detailed medical examinations. Because of concerns that multiple concussions may result in slower recovery or, in more severe cases, chronic traumatic encephalopathy (Guskiewicz et al., 2003; McKee et al., 2009), service members sustaining three or more concussions in a 12-month period are required to undergo a complete neurological and psychological evaluation. The result of this exam may lead to one of the three following dispositions: stateside evacuation, restricted duty, or return to full duty.
Subacute and chronic TBI care is guided by the Department of Veterans Affairs (VA) and DoD Evidence-Based Clinical Practice Guideline for the Management of Concussion/ Mild Traumatic Brain Injury (2009). This document was developed under the auspices of the VHA and DoD with the intent of providing evidence-based recommendations to patients and their providers, reduce practice variability, and provide structure for measurement of patient outcomes. The guidelines include three algorithms: initial presentation, symptom management, and follow-up of persistent symptoms. More detailed guidance is included for the use of pharmacotherapy and management of common physical symptoms associated with concussion.
TELEMEDICINE
Various telemedicine modalities are currently used in the care of TBI patients within the Military Health System (MHS). For example, TBI.consult is an electronic consult service available to deployed providers, and it is staffed by neurologists, neurosurgeons, and nurse practitioners with expertise in TBI. A confidential history and physical record are transmitted to the consult team via email. The team considers local resources and within two to three hours of consult receipt an initial response is provided. The team makes individualized recommendations based on the patient’s medical condition. Resources such as the theater clinical practice guidelines (Brain Trauma Foundation, 2005) and MACE are provided when needed as well as in-theater contacts for specialty care. In some cases, collaboration occurs with other specialty services (i.e., ear, nose, and throat services for hearing loss associated with TBI), with TBI staff in theater, or with LRMC staff to facilitate necessary evacuations. Stateside TBI care in more remote locations is supported by the virtual TBI (vTBI) clinic. The vTBI clinic currently provides symptom management and neuropsychological screening via videoconferencing. Plans are in process to increase capabilities to include mass screening and some components of neurorehabilitation.
The Defense Centers of Excellence for Psychological Health and Traumatic Brain Injury (DCoE) has established a 24/7 Outreach Center. Service members can contact the center via email, live online chat, or telephone for immediate assistance. After immediate concerns are addressed by trained operators, callers are connected with appropriate TBI resources, most often through the Defense and Veterans Brain Injury Center (DVBIC).
CARE COORDINATION
In the acute care phase, service members sustaining severe or penetrating TBI are assigned a federal recovery coordinator to facilitate coordination of care across the health-care continuum. Those with mTBI may also require coordination of services during their recovery. This is accomplished by the DVBIC Care Coordination Program. Once identified with TBI, service members are contacted by a regional care coordinator (RCC), who conducts assessments and identifies patient needs. The RCC works with the individual’s case managers to identify ongoing needs and local TBI resources for up to two years. Data from this program indicate that physical symptom reporting decreases with time while psychological symptom reporting increases. Further work is needed to determine the cause and implications of these findings.
EDUCATION
TBI-related educational efforts within DoD are aimed at two main consumers: providers and patients/families. Three websites have been developed to assist patients and their families with their understanding of TBI.
-
www.traumaticbraininjuryatoz.org
Sponsored by the U.S. Air Force Center of Excellence for Medical Multimedia, this award-winning site provides an overview of TBI by severity, expected courses of recovery, and personal stories of service members with TBI. In addition, there are
-
downloadable components to the congressionally mandated DoD/VA TBI Family Caregiver Curriculum.
-
www.afterdeployment.org (mTBI)
Developed by the DCoE’s National Center for Telehealth and Technology, this website provides an overview of mTBI. Self-assessments are available for many common symptoms or conditions associated with mTBI. A resource library is also included.
-
A service of WETA4 public communications with funding by DVBIC, this website provides information about TBI for patients, families, and providers. Various research topics, TBI experts, and other headlines are presented.
The National Defense Authorization Act of 2007 established a 15-member panel to develop a curriculum to train family caregivers of service members and veterans with TBI. Panel members were appointed by the DoD and the Department of Health and Human Services on March 6, 2008. The members of the panel include professionals from DoD and the VA specializing in TBI, family caregivers, and experts in the development of curricula. The curricula were approved for distribution in April 2010. The curricula are presently being disseminated by recovery coordinators at WRAMC and NNMC to family members of patients with significant TBI. They are also available to the public for download at www.dvbic.org, in addition to other currently approved patient and provider materials.
Many modalities are used to ensure that DoD providers have an adequate understanding of TBI. Since 2007, DVBIC has hosted the annual TBI military training conference. The 2010 event registered more than 850 participants from all branches of service for the two-day conference. Experts from across the country contributed to education through case studies, panel discussions, and podium presentations. The DCoE and DVBIC host regularly scheduled webinars to further facilitate provider education. TBI education is also provided at deployment platforms. The U.S. Army Proponency Office for Rehabilitation and Reintegration convened a panel of subject-matter experts-both military and civilian-to develop a series of TBI materials ranging from a public service announcement (101) to treatment paradigms.
-
TBI 101: Introduction and Awareness-Army
-
TBI 101: Introduction and Awareness-Joint
-
TBI 201: TBI Overview for Healthcare Personnel
-
TBI 401: Primary Care Assessment and Management for Concussion
This program is available for use at deployment platforms and, most recently, has been included on the MHS Learning Portal (MHS Learn), accompanied by appropriate continuing medical education credits. Thirteen other TBI-related lessons are also available on MHS Learn.
RESEARCH
In 2009, more than $40 million were allocated for TBI and psychological health research through the Congressionally Directed Medical Research Program (CDMRP). Funding priorities included cellular regeneration and interconnection strategies for the central
nervous system, evidence-based prevention and rehabilitation strategies, three-dimensional models of blast injury, and advanced diagnostic modalities (e.g., neuroimaging, biomarkers). Figure C-3 depicts an overview of current CDMRP-funded studies.
Key collaborations with academia and industry are likely to provide high-yield information in the upcoming years. A partnership between DVBIC and the Armed Forces Institute of Pathology established a state-of-the-art research lab. This alliance also introduced an organizational structure for the development of a “brain bank” allowing for detailed neuropathological examinations. In addition, the lab has a small animal imaging facility featuring a 7 Tesla horizontal bore imager used in various preclinical research protocols. In conjunction with Massachusetts Institute of Technology and the Institute of Soldier Nanotechnology, DoD scientists have developed the most comprehensive computerized simulation model of the interactions between blast and brain. It is anticipated that this initial effort will lead to further work on the utilization of nanotechnology to protect and improve survivability of wounded service members.
Findings from the U.S. Army Medical Research and Materiel Command Blast Symposium reveal pathological differences in blast and blunt trauma seen on diffusion tensor imaging. Data presented at the symposium from functional Magnetic Resonance Imaging (fMRI) showed statistically significant differences between breacher instructors and students. During their training to be breachers, students are exposed to 50 to 70 blasts of weapons-grade explosives. Finally, animal models suggest axonal, neuronal, and glial damage following blast injury as well as physiologic, histologic, and behavioral differences between blunt and blast injury. Proceedings from this symposium are scheduled to be published in an upcoming special issue of the journal Neuroimage. Other studies still in progress include helmet sensor studies, a 15-year longitudinal study of TBI, and the Head to Head Study of computerized neurocognitive tools. All of these studies will greatly enhance the understanding of TBI and its consequences.
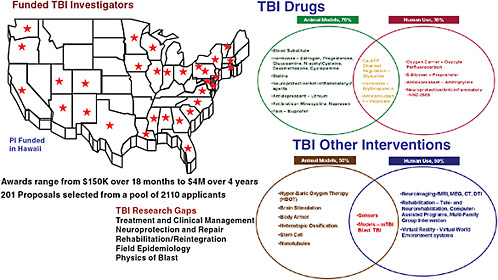
FIGURE C-3 CDMRP-funded studies and their locations.
SOURCE: Jaffee, 2010.
TABLE C-2 Key Combat-Related TBI Scholarly Papers, 2003–2009
|
Category |
Reference |
Title |
|
Screening |
Ivins et al., 2009 |
Performance on the Automated Neuropsychological Assessment Metrics in a nonclinical sample of soldiers screened for mTBI after return returning from Iraq and Afghanistan: A descriptive analysis |
|
|
Terrio et al., 2009 |
Traumatic brain injury screening: Preliminary findings in a U.S. Army Brigade Combat Team |
|
|
Ivins et al., 2003 |
Traumatic brain injury in U.S. Army paratroopers: Prevalence and character |
|
Clinical Findings |
Bell et al., 2009 |
Military traumatic brain and spinal column injury: A 5-year study of the impact blast and other military grade weaponry on the central nervous system |
|
|
Brahm et al., 2009 |
Visual impairment and dysfunction in combat-injured service members with traumatic brain injury |
|
|
Hoge et al., 2008 |
Mild traumatic brain injury in U.S. soldiers returning from Iraq |
|
Blast Neurotrauma |
Ling et al., 2009 |
Explosive blast neurotrauma |
|
|
Moore et al., 2009 |
Computational biology—Modeling of primary blast effects on the central nervous system |
|
|
Dewitt et al., 2009 |
Blast-induced brain injury and posttraumatic hypotension and hypoxemia |
|
TBI & Post-Traumatic Stress Disorder |
Stein and McAllister, 2009 |
Exploring the convergence of posttraumatic stress disorder and mild traumatic brain injury |
|
|
Nelson et al., 2009 |
Relationship between processing speed and executive functioning performance among OEF/OIF veterans: Implications for post deployment rehabilitation |
|
|
Kennedy et al., 2007 |
Posttraumatic stress disorder and posttraumatic stress disorder-like symptoms and mild traumatic brain injury |
|
Imaging |
Moore et al., 2009 |
Diffusion tensor imaging and mTBI—A case-control study of blast (+) in returning service members following OIF and OEF |
|
|
Huang et al., 2009 |
Integrated imaging approach with MEG and DTI to detect mild traumatic brain injury in military and civilian patients |
|
Outcomes |
Bjork and Grant, 2009 |
Does traumatic brain injury increase risk for substance abuse? |
|
|
Han et al., 2009 |
Clinical, cognitive, and genetic predictors of change in job status following traumatic brain injury in a military population |
|
|
Gottshall et al., 2003 |
Objective vestibular tests as outcome measures in head injury patients |
|
|
Drake et al., 2000 |
Factors predicting return to work following mild traumatic brain injury: A discriminant analysis |
|
Category |
Reference |
Title |
|
Rehabilitation |
Lew et al., 2009 |
The potential utility of driving simulators in the cognitive rehabilitation of combat-returnees with traumatic brain injury |
|
|
Vanderploeg et al., 2008 |
Rehabilitation of traumatic brain injury in active-duty military personnel and veterans: Defense and Veterans Brain Injury Center randomized controlled trial of two rehabilitation approaches |
|
|
Trudel et al., 2007 |
Community-integrated brain injury rehabilitation: Treatment models and challenges for civilian, military, and veteran populations |
|
|
Walker et al., 2007 |
Motor impairment after severe traumatic brain injury: A longitudinal multicenter study |
|
Miscellaneous |
Cote et al., 2007 |
A mixed integer programming model to locate traumatic brain injury treatment units in the Department of Veterans Affairs: A case study |
|
|
Warden, 2006 |
Military TBI during the Iraq and Afghanistan wars |
|
|
Ivins et al., 2006 |
Hospital admissions associated with traumatic brain injury in the U.S. Army during peacetime: 1990s trends |
Clinicians continue to contribute to the understanding of combat-related TBI. Table C-2 summarizes recent observations of providers involved in the day-to-day care of those with TBI. These efforts identify some of the consequences associated with TBI and lead to additional scientific inquiry.
SUMMARY
Much work remains to be done in order to fully understand TBI and its long-term consequences. Efforts within DoD to rapidly transform bench science and observed best practice to clinical practice continue. This is most readily evident by the annual revisions of educational materials, clinical practice guidelines, and screening techniques.
REFERENCES
Brain Trauma Foundation. 2005. Guidelines for field management of combat-related head trauma. New York, NY: Brain Trauma Foundation.
Dempsey, K. E., W. C. Dorlac, K. Martin, R. Fang, C. Fox, B. Bennett, K. Williams, and S. Flaherty. 2009. Landstuhl Regional Medical Center: Traumatic brain injury screening program. Journal of Trauma Nursing 16(1):6–12.
DoD (Department of Defense). 2007. Traumatic brain injury: Definition and reporting. Memorandum. HA Policy 07-030. Dated October 1, 2007. Available online at http://mhs.osd.mil/content/docs/pdfs/policies/2007/07-030.pdf (accessed March 25, 2011).
Guskiewicz, K. M., M. McCrea, S. W. Marshall, R. C. Cantu, C. Randolph, W. Barr, J. A. Onate, and J. P. Kelly. 2003. Cumulative effects associated with recurrent concussion in collegiate football players: The NCAA Concussion Study. The Journal of the American Medical Association 290(19):2549–2555.
Jaffee, M. S. 2010. Overview of Combat Related Traumatic Brain Injury and DoD TBI Initiatives. Presented at Institute of Medicine’s Workshop on Nutrition and Neuroprotection in Military Personnel, Washington, DC, June 23, 2010.
McKee, A. C. M., R. C. M. Cantu, C. J. A. Nowinski, E. T. M. Hedley-Whyte, B. E. P. Gavett, A. E. M. Budson, V. E. M. Santini, H.-S. M. Lee, C. A. Kubilus, and R. A. P. Stern. 2009. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. Journal of Neuropathology & Experimental Neurology 68(7):709–735.
Schwab, K. A., G. Baker, B. Ivins, M. Sluss-Tiller, W. Lux, and D. Warden. 2006. The brief traumatic brain injury screen (BTBIS): Investigating the validity of a self-report instrument for detecting traumatic brain injury (TBI) in troops returning from deployment in Afghanistan and Iraq. Neurology 66(5)(Suppl. 2):A235.
Terrio, H., L. A. Brenner, B. J. Ivins, J. M. Cho, K. Helmick, K. Schwab, K. Scally, R. Bretthauer, and D. Warden. 2009. Traumatic brain injury screening: Preliminary findings in a U.S. Army brigade combat team. Journal of Head Trauma Rehabilitation 24(1):14-23.
VA/DoD (Department of Veterans Affairs and Department of Defense). 2009. VA/DoD clinical practice guideline for management of concussion/mild traumatic brain injury. http://www.healthquality.va.gov/mtbi/concussion_mtbi_full_1_0.pdf (accessed January 19, 2011).
Pathophysiology and Mechanisms of Neurotrauma: Blast and Civilian
Mårten Risling5
Traumatic brain injury (TBI) is not only a leading cause of death and disability in young active people, but also a significant health problem in elderly people. It is a fundamental type of trauma in both civilian life and at the battlefield. However, the spectrum of the injuries seems to be different in these two areas, and has also changed over time. For example, the signature TBI in recent military conflicts has evidently changed from penetrating TBI to blast-induced TBI. Below we will first describe a basic classification of TBI. All types of TBI may occur in both the civilian and military setting. However, there are some important considerations for TBI on the battlefield, mostly relating to the extreme energy transfer.
CLASSIFICATION
The term TBI includes a number of different injuries, ranging from mild to severe lesions. Several mechanisms contribute to the induction of the injury. The World Health Organization (WHO) publishes an International Classification of Diseases (ICD), where trauma and head injury are included in the chapter “Injury and poisoning.” The latest edition, ICD-10, lists the injuries as:
SO6 Intracranial injury
SO6.0 Concussion
-
Commotio cerebri
SO6.1 Traumatic cerebral edema
SO6.2 Diffuse brain injury
Cerebral:
-
contusion NOS
-
laceration NOS
-
traumatic compression of brain NOS
SO6.3 Focal brain injury
Focal:
-
cerebral:
-
contusion
-
aceration
-
-
traumatic intracerebral haemorrhage
SO6.4 Epidural hemorrhage
SO6.5 Traumatic subdural hemorrhage (SDH)
SO6.6 Traumatic subarachnoid hemorrhage (SAH)
SO6.7 Intracranial injury with prolonged coma
SO6.8 Other intracranial injuries
SO6.9 Intracranial injury, unspecified
Adoption of ICD-10 seems to be slow in the United States. Since 1999, the ICD-10 (without clinical extensions) was adopted for reporting mortality, but ICD-9-CM is apparently still often used for morbidity, unlike the situation in many European countries. The causes of the injuries have been systematically listed in the International Classification of External Causes of Injury (ICECI). It contains seven modules for the mechanism of injury (objects/substances producing injury, place of occurrence, activity when injured, the role of human intent, use of alcohol, use of psycho-active drugs) and five additional modules referring to special topics (violence, transport, place, sports, occupational injury). The ICECI contains a module V7—TYPE OF CONFLICT, which includes war.
This type of classification is requisite for meaningful statistics and epidemiology. According to data from the U.S. Centers for Disease Control and Prevention (CDC), the annual incidence for TBI in the United States is approximately 1.5 million. Among these, around 50,000 die and a larger number (around 85,000 people) survive with long-term disabilities. Because many of the victims are young at the time of the injury, the accumulated number of people with disabilities caused by TBI is assumed to be more than 5 million. Sport activities, firearms, and road traffic accidents represent common causes of TBI in young people, whereas fall injury is a common cause in the elderly.
MILD TBI
A TBI is often classified as mild (concussion or commotion) if loss of consciousness or confusion is shorter than 30 minutes. Magnetic resonance imaging (MRI) and computerized axial tomography (CAT) scans are usually normal but the patient may have headache and cognitive problems (memory problems, mood disturbance, attention deficits), and the effect on the patient can be devastating. The majority of blast-induced TBI fall into this category, although the pathophysiology is largely unknown. Cerebral concussion is often associated with other types of brain injury.
MODERATE AND SEVERE TBI
These injuries can be divided into closed head injuries and penetrating injuries. The penetrating TBI will always induce a focal injury and often diffuse secondary injuries. Closed head injuries may be both diffuse and focal (Reilly and Bullock, 2005).
DIFFUSE TBI
The most common diffuse injury is the diffuse axonal injury (DAI). With increasing sensitivity in imaging techniques, the proportion of DAIs will probably increase in statistics. DAI is defined as the presence of diffuse damage to axons in the cerebral subcortical parasagittal white matter, corpus callosum, brain stem, and cerebellum (Reilly and Bullock, 2005). Another type of diffuse injury is diffuse vascular injury (DVI) (Reilly and Bullock, 2005). DAI is often, but not always, also associated with vascular injury. Different parts of
the brain move at different speeds because of their relative density. This can lead to shearing injury and DAI (Anderson and McLean, 2005). Beta-amyloid precursor protein (APP) is a membrane-spanning glycoprotein originating from a gene on chromosome 21. APP, which is transported by fast axoplasmic transport, has been proven to be an excellent marker for axonal injury in histology (Sherriff et al., 1994). Modern imaging techniques, such as MRI with diffusion tensor imaging (DTI), have provided improved possibilities to detect DAI.
FOCAL TBI
Vascular injury can result in intracerebral hemorrhage, subdural hemorrhage, epidural hemorrhage, or subarachnoid hemorrhage. Extradural hematomas (EDH) are the result of bleeding that occurs between the calvarium and the dura mater, most frequently in the temporo-parietal region near the middle meningeal arteries. There are two main types of traumatic acute subdural hematoma (ASDH). In traumatic ASDH related to contusions and lacerations, the hematoma is located adjacent to damaged brain. The second type of ASDH is the result of rupture of the bridging veins (Reilly and Bullock, 2005).
Cerebral contusions are focal injuries. Bleeding from damaged blood vessels is usually the most obvious feature upon macroscopic or microscopic examination. Contrecoup contusions occur opposite the impact site. Fracture contusions occur beneath the site of a fracture. Gliding contusions occur in the parasagittal regions and are often associated with DAI. In a simple contusion the pia–glial membrane is intact. Disruption of this membrane with tearing of the underlying tissue constitutes a laceration. Contusions and lacerations form a continuum of tissue injury.
Lacerations of the brain may be defined as primary disruptions of the brain tissue at the moment of injury. In direct lacerations the tissue disruption is caused by a penetrating injury from various types of missiles or an open depressed fracture of the skull with penetration of the brain by fragments of bone and foreign bodies.
SCREENING AND EXAMINATION AFTER TBI
In the acute phase physical examination, CAT scans and biomarkers are used to differentiate from more severe injuries. The Glasgow Coma Scale (GCS) is a 15-point scale for estimating the acute effect of TBI. The test measures the motor response, verbal response, and eye-opening response. The score is determined by adding the values of these three parameters. Mild TBI should have a score between 13 and 15 (Servadei et al., 2001). Lower scores indicate a more severe injury, that is, a moderate or severe TBI. This test is useful for screening in the emergency room, and repeated tests can be useful for monitoring. However, GCS seems to be less reliable for penetrating injuries and maybe also blast injuries with late onset of symptoms. Therefore, there is an interest for additional tools for screening after TBI, especially in the military setting.
Biomarkers such as the calcium-binding protein S100B can be used as a screening tool. S100B can be detected in serum samples after TBI (Berger et al., 2002; Elting et al., 2000; Pelinka et al., 2003). It has a short half-life in serum, and it has been suggested that repeated samples can be used to monitor the progress of a TBI and make predictions on the outcome. Some guidelines for mild TBI in children advocate the use of S100B instead of CAT scans in order to reduce the exposure to radiation. Several research programs focus on the development of more sensitive and more CNS-specific biomarkers, such as various axonal markers.
Imaging techniques such as CAT scans and MRIs have increased the precision and sensitivity for exact diagnosis after TBI. The resolution and protocols are improving. For
example, DTI can be used to visualize fiber tracts in the brain and enhance the diagnosis of diffuse TBI. Additional techniques such as PET (positron emission tomography) scanning may, for example, add knowledge on metabolic parameters after the injury but are not employed as a screening tool.
In the neurointensive-care unit (NICU) additional techniques can be used to monitor the TBI patients (Matz and Pitts, 1997). Microdialysis is a technique to monitor the chemistry of the extracellular space in the brain (Alessandri et al., 2000). The microdialysis probe consists of a semi-permeable catheter that is constantly perfused with a physiological solution. The perfusate can give information on the metabolic state in the injured brain. Digital electroencephalography (EEG) can provide information on the occurrence of post-TBI seizures or to monitor sedative treatment after the injury. Microdialysis studies in the NICU have revealed that the level of glucose in the brain is difficult to predict from analysis of peripheral blood or samples from fat tissue (Rostami and Bellander, 2011). However, microdialysis is a difficult technique and probably very dependent on the position of the catheter. It is more useful for the monitoring of patients in the NICU than for prediction of secondary events in the injury.
SECONDARY INJURIES
Secondary traumatic brain damage occurs as a complication of the different types of TBI and includes ischemic and hypoxic damage, swelling, raised intracranial pressure, and infection (Figure C-4). Neurochemical alterations, such as excitatory stress, may mediate important components of brain physiology associated with TBI, and such alterations may be responsive to pharmacologic therapy (Miller et al., 1990; Palmer et al., 1993). The secondary TBI is potentially reversible with adequate treatment. A part of the treatment is to reduce the metabolic load by controlled sedation. It should be mentioned that there is some evidence that neurons reduce their metabolic load after injuries in the central nervous system by active removal of excitatory synapses.
Thus, the secondary injuries are extremely important because they represent the components of the injury that could be treated or prevented if the mechanism and threshold have been identified. Becaquse TBI from high-energy incidents may be induced by more than one mechanism (e.g., primary blast + acceleration movements), we can assume to find more than one threshold for these secondary assaults.
The blood-brain barrier (BBB) can be assumed to have a key function in the aftermath
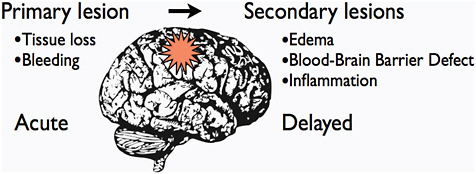
FIGURE C-4 A schematic representation of the relation between primary and secondary traumatic brain damage after TBI. The interval between the primary acute lesion and the secondary lesions can vary from hours to days.
of TBI. The BBB is the result of a complex interaction of astrocyte endfeet and the endothelium of the capillaries in the central nervous system. On the molecular level, the BBB is a complicated interaction of specialized contact proteins at junctions between the cells and active transporters for essential compounds, like the Glut-1 glucose transporter (Stark et al., 2000). The BBB can be rapidly distorted after TBI, resulting in extracellular edema and infiltration with inflammatory cells. The threshold for BBB defects after TBI has not been established, but could possibly be different after different types of injury. The BBB function following TBI is usually restored within four weeks, although more permanent BBB defects have been observed after lesions in the spinal cord (Risling et al., 1989).
Axonal damage in both DAI and focal injuries interferes with axoplasmic transport. Severe traumatic injury results in primary axonal disruption, termed primary axotomy, which can initiate a series of poorly understood events culminating in secondary axonal degeneration or secondary axotomy (Maxwell et al., 1997). Thus, adequate treatment could probably limit the axonal damage.
Edema is an important and variable secondary response to TBI, the causes and consequences of which are only partly understood. Effective treatment is lacking. Impairment of the BBB leads to accumulation of fluid in the extracellular space. The localized edema around contusions and penetrating TBI is mostly vasogenic. Cytotoxic edema occurs in association with hypoxic-ischemic damage where there is a disturbance of ionic gradients leading to an accumulation of intracellular fluid. Energy crisis and mitochondria malfunction can be important components of the cytotoxic edema (Castejon and Castejon, 2000; Castejon and de Castejon, 2004; Clausen et al., 2001; Klatzo, 1987). Severe blast-induced TBI is often complicated by acute edema. The mechanisms for this edema formation have still to be identified. Decompressive craniotomy is often needed (Ling et al., 2009).
COMPLICATIONS WITH LATE ONSET
Epilepsy occurs in many TBI patients. Recent studies indicate an association between TBI and the subsequent development of Alzheimer’s disease (Emmerling et al., 2000; Uryu et al., 2002). Genetic background, such as changes in the APO-E gene can increase the risk for trauma-related Alzheimer dementia, and it has been suggested that the APOE-epsilon4 genotype may result in an earlier onset of the disease, rather than increased incidence (Hartman et al., 2002).
RISK ASSESSMENT
Several measures have been developed in an attempt to quantify the tolerance of the head to impact in terms of the magnitude of both the resulting acceleration of the head and the duration of the impact. The Head Injury Criterion (HIC) is the most widely used. Such values have a definite role for improved car crash safety and body armor. However, although HIC can provide an assessment of the risk for fractures of head it does not seem to give a reliable description of the risk for diffuse injuries, such as DAI (Margulies and Thibault, 1992).
BLAST-INDUCED BRAIN INJURIES—THE GRAND CHALLENGE IN TBI RESEARCH
The U.S. government has initiated the largest coordinated research programs ever in neurotrauma to get insight on mechanisms and to provide better treatment for blast-induced
traumatic brain injuries (Risling, 2010). TBI has been identified as a major health problem in military personnel returning from service. The injuries range from severe multitrauma to a number of mild TBIs that still has to be settled (Jaffee and Meyer, 2009; Ling et al., 2009).
The enormous energy transfer in blast TBI creates a number of specific problems:
-
Propagation of blast waves is very complex. It could involve both direct propagation through the skull and indirect propagation via blood vessels (Cernak and Noble-Haeusslein, 2010). If the latter mechanism is important, we could expect effects from vascular disturbance. Several lines of evidence seem to point in that direction.
-
It is not known whether blast TBI is a specific type of injury that will require specific and new types of treatment, or if, for example, the mild TBI from blast exposure is more like a classical type of concussion injury (Hoge et al., 2009).
-
A reliable borderline between mild blast-induced TBI and posttraumatic stress syndrome (PTSD) has yet to be identified. Many of the symptoms are similar, and many patients might suffer from both TBI and PTSD (Jaffee and Meyer, 2009).
-
There is some debate about whether blast-induced TBI is an entirely new problem. The shellshock syndrome (Anderson, 2008) that was seen after the enormous artillery battles during World War I had similarities to blast-induced TBI and post blast-induced TBI symptoms, but for many years it has been regarded as PTSD rather than physical injuries. The new situation with improvised explosive devices (IEDs) is that the explosive often detonates at short distances, and improved body armor and helmets protect much better against penetration from fragments.
-
Although the epidemiology of blast-induced TBI has been established in terms of approximate numbers of people injured, it is very difficult to assess the injury mechanisms in individual cases. The actual situation during exposure to an IED is usually very complicated because of complex propagation and reflection of the primary supersonic blast wave, effects from acceleration and rotation (Moss et al., 2009), effects from impact of fragments, effects from heating, and effects from emitted gases and electromagnetic waves (Figure C-5). Well-designed experimental models as well as data from acceleration probes and pressure sensors that have been mounted into helmets and body armor are required to increase the knowledge of the critical mechanisms.
EXAMPLE OF ONGOING EXPERIMENTAL RESEARCH
As indicated earlier, it may be assumed that several mechanisms contribute to the injury. This study was an attempt to characterize the presumed components of blast-induced TBI (Risling et al., 2011). Our experimental models included a blast tube in which an anesthetized rat can be exposed to controlled detonations of explosives that result in a pressure wave with a magnitude between 130 and 260 kPa. In this model, the animal is fixed with a metal net to avoid head acceleration forces. The second model is a controlled penetration of a 2-mm thick needle. In the third model, the animal is subjected to a high-speed sagittal rotation angular acceleration.
Immunohistochemical labeling for amyloid precursor protein revealed signs of DAI in the penetration and rotation models. Signs of punctuate inflammation were observed after focal and rotation injury. Exposure in the blast tube did not induce DAI or detectable cell death but did not functional changes. Affymetrix gene arrays showed changes in the expression in a large number of gene families including cell death, inflammation, and neurotransmitters in the hippocampus after both acceleration and penetration injuries. Exposure to the
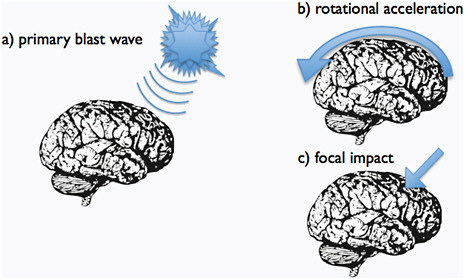
FIGURE C-5 Primary blast wave, rotational acceleration, and focal impact can be assumed to represent key components in the injury mechanism of blast TBI.
primary blast wave induced limited shifts in gene expression in the hippocampus. The most interesting findings were a downregulation of genes involved in neurogenesis and synaptic transmission. These experiments indicate that rotational acceleration may be a critical factor for DAI and other acute changes after blast-TBI. The further exploration of the mechanisms of blast-induced TBI will have to include a search for long-term effects.
The microarray data indicated that the metabolic load and mitochondrial function were very different in the three models (Risling et al., 2011). For example, 47 different mitochondrial genes showed a changed expression in the cortex surrounding the penetrating TBI, whereas no mitochondrial gene showed any sign of significant expression changes in the primary blast model. A number of genes for the metabolic cytochrome p450 (CYP) enzymes also showed significant changes in the cortex subjected to penetrating TBI. It may be assumed that such expression changes could have implications for drug and hormone metabolism. The relatively modest changes in the employed primary blast model could be related to the simple pulse form and short duration of the primary peak that is achieved by a detonation at short distance (1 meter) in this type of blast tube (Clemedson and Criborn, 1955). This may be relevant for some types of IED at short distance but in a protected vehicle, could be very different (Courtney and Courtney, 2011). Preliminary data from experiments employing a rigid body protection for the torso and a higher load of explosive, indicate a rapid induction of GFAP and s-fos, indicators of TBI pathology, but without any signs of DAI. Thus, these data indicate that blast-induced TBI may be the result of a number of simultaneous mechanisms with different thresholds.
REFERENCES
Alessandri, B., M. Reinert, H. F. Young, and R. Bullock. 2000. Low extracellular (ECF) glucose affects the neurochemical profile in severe head-injured patients. Acta Neurochirurgica. Supplement 76:425–430.
Anderson, R. J. 2008. Shell shock: An old injury with new weapons. Molecular Interventions 8(5):204–218.
Anderson, R., and J. McLean. 2005. Biomechanics of closed head injury. In Head injury. pathophysiology and management, edited by P. L. Reilly and R. Bullock. London: Hodder Arnold. Pp. 26–40.
Berger, R. P., M. C. Pierce, S. R. Wisniewski, P. D. Adelson, and P. M. Kochanek. 2002. Serum s100b concentrations are increased after closed head injury in children: A preliminary study. Journal of Neurotrauma 19(11):1405–1409.
Castejon, O. J., and H. V. de Castejon. 2000. Oligodendroglial cell behaviour in traumatic oedematous human cerebral cortex: A light and electron microscopic study. Brain Injury 14(4):303–317.
Castejon, O. J., and H. V. de Castejon. 2004. Structural patterns of injured mitochondria in human oedematous cerebral cortex. Brain Injury 18(11):1107–1126.
Cernak, I., and L. J. Noble-Haeusslein. 2010. Traumatic brain injury: An overview of pathobiology with emphasis on military populations. Journal of Cerebral Blood Flow and Metabolism 30(2):255–266.
Clausen, T., A. Zauner, J. E. Levasseur, A. C. Rice, and R. Bullock. 2001. Induced mitochondrial failure in the feline brain: Implications for understanding acute post-traumatic metabolic events. Brain Research 908(1):35–48.
Clemedson, C. J., and C. O. Criborn. 1955. A detonation chamber for physiological blast research. The Journal of Aviation Medicine 26(5):373–381.
Courtney, M. W., and A. C. Courtney. 2011. Working toward exposure thresholds for blast-induced traumatic brain injury: Thoracic and acceleration mechanisms. Neuroimage 54(Suppl. 1):S55–S61.
Elting, J. W., A. E. de Jager, A. W. Teelken, M. J. Schaaf, N. M. Maurits, J. van der Naalt, C. T. Sibinga, G. A. Sulter, and J. De Keyser. 2000. Comparison of serum s-100 protein levels following stroke and traumatic brain injury. Journal of the Neurological Sciences 181(1–2):104–110.
Emmerling, M. R., M. C. Morganti-Kossmann, T. Kossmann, P. F. Stahel, M. D. Watson, L. M. Evans, P. D. Mehta, K. Spiegel, Y. M. Kuo, A. E. Roher, and C. A. Raby. 2000. Traumatic brain injury elevates the alzheimer’s amyloid peptide a beta 42 in human CSF. A possible role for nerve cell injury. Annals of the New York Academy of Sciences 903:118–122.
Hartman, R. E., H. Laurer, L. Longhi, K. R. Bales, S. M. Paul, T. K. McIntosh, and D. M. Holtzman. 2002. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer’s disease. Journal of Neuroscience 22(23):10083–10087.
Hoge, C. W., H. M. Goldberg, and C. A. Castro. 2009. Care of war veterans with mild traumatic brain injury—flawed perspectives. The New England Journal of Medicine 360(16):1588–1591.
Jaffee, M. S., and K. S. Meyer. 2009. A brief overview of traumatic brain injury (TBI) and post-traumatic stress disorder (PTSD) within the Department of Defense. Clinical Neuropsychologist 23(8):1291–1298.
Klatzo, I. 1987. Pathophysiological aspects of brain edema. Acta Neuropathologica 72(3):236–239.
Ling, G., F. Bandak, R. Armonda, G. Grant, and J. Ecklund. 2009. Explosive blast neurotrauma. Journal of Neurotrauma 26(6):815–825.
Margulies, S. S., and L. E. Thibault. 1992. A proposed tolerance criterion for diffuse axonal injury in man. Journal of Biomechanics 25(8):917–923.
Matz, P. G., and L. Pitts. 1997. Monitoring in traumatic brain injury. Clinical Neurosurgery 44:267–294.
Maxwell, W. L., J. T. Povlishock, and D. L. Graham. 1997. A mechanistic analysis of nondisruptive axonal injury: A review. Journal of Neurotrauma 14(7):419–440.
Miller, L. P., B. G. Lyeth, L. W. Jenkins, L. Oleniak, D. Panchision, R. J. Hamm, L. L. Phillips, C. E. Dixon, G. L. Clifton, and R. L. Hayes. 1990. Excitatory amino acid receptor subtype binding following traumatic brain injury. Brain Research 526(1):103–107.
Moss, W. C., M. J. King, and E. G. Blackman. 2009. Skull flexure from blast waves: A mechanism for brain injury with implications for helmet design. Physical Review Letters 103(10):108702.
Palmer, A. M., D. W. Marion, M. L. Botscheller, P. E. Swedlow, S. D. Styren, and S. T. DeKosky. 1993. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. Journal of Neurochemistry 61(6):2015–2024.
Pelinka, L. E., E. Toegel, W. Mauritz, and H. Redl. 2003. Serum s100b: A marker of brain damage in traumatic brain injury with and without multiple trauma. Shock 19(3):195–200.
Reilly, P. L., and R. Bullock. 2005. Head injury. Pathophysiology & management. 2nd ed.: Hodder Arnold.
Risling, M. 2010. Blast induced brain injuries—a grand challenge in TBI research. Frontiers in Neurotrauma 1(1):1–2.
Risling, M., H. Linda, S. Cullheim, and P. Franson. 1989. A persistent defect in the blood-brain barrier after ventral funiculus lesion in adult cats: Implications for CNS regeneration? Brain Research 494(1):13–21.
Risling, M., S. Plantman, M. Angeria, E. Rostami, B. M. Bellander, M. Kirkegaard, U. Arborelius, and J. Davidsson. 2011. Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage 54(Suppl. 1):S89–S97.
Rostami, E., and B.-M. Bellander. 2011. Monitoring of glucose in brain, adipose tissue and peripheral blood in patients with traumatic brain injury, a microdialysis study. Journal of Diabetes Science and Technology 5(3):596–604.
Servadei, F., G. Teasdale, and G. Merry. 2001. Defining acute mild head injury in adults: A proposal based on prognostic factors, diagnosis, and management. Journal of Neurotrauma 18(7):657–664.
Sherriff, F. E., L. R. Bridges, and S. Sivaloganathan. 1994. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathologica 87(1):55–62.
Stark, B., T. Carlstedt, S. Cullheim, and M. Risling. 2000. Developmental and lesion-induced changes in the distribution of the glucose transporter Glut-1 in the central and peripheral nervous system. Experimental Brain Research 131(1):74–84.
Uryu, K., H. Laurer, T. McIntosh, D. Pratico, D. Martinez, S. Leight, V. M. Lee, and J. Q. Trojanowski. 2002. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. Journal of Neuroscience 22(2):446–454.
Recurrent Sports-Related Traumatic Brain Injury and Tauopathy
Robert A. Stern,6 Brandon E. Gavett,7 Christine Baugh,8 Christopher J. Nowinski,9 Robert C. Cantu,10 and Ann C. McKee11
Dementia pugilistica and related terms (e.g., punch drunk, slug nutty) have evolved over the previous century (Millspaugh, 1937; Parker, 1934). The origin of these terms can be traced to boxers that were perceived by the lay public to have developed serious cognitive and movement abnormalities as a result of being repeatedly punched in the head in their sport (Critchley, 1957). The first neuropathological examination of a “punch drunk” boxer was conducted by Harrison Martland in 1928 (Martland, 1928). One half-century later, a more comprehensive neuropathological study of the brains of boxers was carried out by Corsellis and colleagues, who examined 15 deceased retired boxers and found a characteristic pattern of brain injury that was distinct from other known causes of neurodegeneration (Corsellis et al., 1973).
As research into the link between the long-term effects of repeated head injury grew, it became clear that this condition was not restricted to boxers, but rather, could result from other causes of head injury. Similar neuropathological findings were found in individuals with a common history of head trauma of various causes, including soccer (Geddes et al., 1999), head-banging behavior (Geddes et al., 1999), and domestic abuse (Roberts et al., 1990b). As such, the term chronic traumatic encephalopathy (CTE) became the most commonly used term to describe what had previously been referred to as dementia pugilistica (Miller, 1966). Chronic traumatic encephalopathy is a progressive neurodegenerative disease, similar to Alzheimer’s disease, but with unique features (McKee et al., 2009). It is believed to be caused by repeated trauma to the brain, including mild concussions and subconcussive blows. Its symptoms occur years or decades following head trauma and continue to worsen; these symptoms are distinct from the acute or post-acute (e.g., postconcussion syndrome) effects of a head injury. The early symptoms of CTE are believed to include memory and cognitive difficulties, depression, impulse control problems, and behavior changes. Later on,
movement abnormalities (including Parkinsonism) become more common, and, in many cases, full-blown dementia occurs. CTE is the only preventable cause of dementia.
Recently, our group combined a review of the existing CTE neuropathology literature with a neurological and clinical case series of three athletes (McKee et al., 2009). At the time of the McKee et al. publication, there were 52 cases of neuropathologically verified CTE in the world’s literature (including the three cases in that case series). Since that time, our group has examined more than 35 cases of CTE (well over half of the known cases) and helped to refine its neuropathological characterization.
The microscopic pathology of CTE includes neurofibrillary degeneration in the form of extensive tau-immunoreactive neurofibrillary tangles, glial tangles, and neuropil neurites throughout the brain in a widespread distribution. Within the cerebral cortex, the frontal and temporal lobes are commonly affected, with very dense neuropathology in the medial temporal lobe structures—the amygdala, hippocampus, and entorhinal cortex. Tauimmunoreactive inclusions are also found in the subcortical white matter, thalamus, hypothalamus, mammillary bodies, and brainstem (Corsellis et al., 1973; McKee et al., 2009).
CTE may be similar to Alzheimer’s disease (AD), both in terms of clinical presentation and neuropathological changes. Both are progressive, incurable, neurodegenerative diseases, and both are currently only diagnosed through postmortem examination of brain tissue. However, there are a number of important distinctions between CTE and AD. Age of onset in AD is usually later than in CTE; while sporadic AD most often occurs after age 65, the onset of CTE has been found to range from ages 20 to 50. Neuropathologically, AD is associated with an abundance of both tau-immunoreactive inclusions and significant deposition of neuritic beta-amyloid plaques (Braak and Braak, 1991). In contrast, fewer than half of the neuropathologically documented cases of CTE showed evidence of amyloid deposition, and when amyloid is present, the plaques tend to be very modest and diffuse, as opposed to neuritic (Clinton et al., 1991; Roberts et al., 1990a).
Athletic participation is ubiquitous amongst people of all ages, especially children. The link between sports-related head injuries and CTE is alarming considering that an estimated 1.6 to 3.8 million people suffer sports-related concussions each year (Langlois et al., 2006). This highlights the importance of expanding the study of CTE beyond boxing. Within the past decade, CTE has been documented in a number of professional athletes who were not boxers. The first five of these cases included four professional football players and one professional wrestler. Mike Webster was a retired National Football League (NFL) center who died at age 50 because of a myocardial infarction; in addition to suffering from depression and cognitive problems, he was also unemployed and homeless at the time of his death in 2002. Terry Long was a retired NFL guard who committed suicide in 2005 at the age of 45. Andre Waters was a retired NFL safety who committed suicide in 2007 at the age of 44. Justin Strzelczyk, a retired NFL offensive lineman, was 36 years old at death; his life had begun to slip into a “downward spiral” of depression and behavioral changes. His death in 2004 was caused by erratic behavior leading to a high-speed police chase that ended when he drove his truck into a tractor trailer. Chris Benoit was a 40-year-old professional wrestler when he committed suicide in 2007 after murdering his wife and child (Cajigal, 2007; Omalu et al., 2005, 2006, 2010a, 2010b).
Following the report of these five cases, the Boston University (BU) Center for the Study of Traumatic Encephalopathy (CSTE) was established to further investigate the relationship between head trauma and later neurodegenerative disease. The CSTE (McKee et al., 2009) first reported on John Grimsley, a retired NFL linebacker who died at the age of 45 after a 10-year career with the Houston Oilers and Miami Dolphins. There was no evidence that he used performance-enhancing drugs in his lifetime, and he reportedly suffered at least eight
concussions during his NFL career. He died of a gunshot wound to his chest while cleaning his gun, and his death was ruled an accident. For the five years prior to his death, he reportedly experienced worsening memory and cognitive functioning, as well as an increasingly “short fuse.” Neuropathologically, Mr. Grimsley’s brain was characteristic of CTE and was noteworthy for the dense tau-immunoreactive inclusions in the medial temporal lobes. Tom McHale was a retired offensive lineman who spent 10 years in the NFL with the Tampa Bay Buccaneers. He was a graduate of Cornell University, former restaurateur, husband, and father of three boys. It was reported that, during his playing career, he suffered two to three concussions, but as lineman, he suffered routine subconcussive blows. He died from a drug overdose after a multi-year battle with drug addiction. Of note, Mr. McHale’s drug abuse problems began late in his life, after retiring from the NFL. Neuropathologically, Mr. McHale’s brain was consistent with CTE and contained numerous areas of dense tau-immunoreactivity in the cerebral cortex. The first member of the Professional Football Hall of Fame and the first participant in the NFL’s “88 Plan” to pass away and undergo neuropathological examination for CTE was Lou Creekmur. Mr. Creekmur died at the age of 82. He played 10 seasons as an offensive lineman for the Detroit Lions and was an eight-time Pro Bowler. He was famous for suffering at least 13 broken noses while playing without a facemask. He died from complications of dementia while in a nursing home after a 30-year decline that included cognitive and behavioral issues, memory loss, problems with attention and organization, and angry and aggressive outbursts. His wife referred to him as “punchy” for the last 30 years of his life. Neuropathological findings revealed extensive tau-immunoreactive inclusions throughout the cortex and medial temporal lobes consistent with CTE. Remarkably, there was no neuropathological evidence of AD in this 82-year-old.
In addition to finding evidence of CTE in professional athletes, recent evidence suggests that CTE can occur following shorter athletic careers. Mike Borich played football in college, but did not play football professionally. He died at age 42 after exhibiting a pattern of erratic behavior throughout much of his adult life. His college-playing career included stints with Snow College and Western Illinois University in the 1980s. He was known to have approximately 10 concussions during his college football career with no subsequent concussions or head injuries after college. He worked as a Division I college football coach, and was named Offensive Coordinator of the Year in 2001 while coaching at Brigham Young University. He also coached for the NFL’s Chicago Bears in 1999–2000. He left coaching in 2003 while struggling with overwhelming drug and alcohol addictions, and ultimately died from a drug overdose in February 2009 at the age of 42. Neuropathological examination of Mr. Borich’s brain revealed less pathology overall than many previous cases of confirmed CTE, but was consistent with CTE nonetheless. Of particular salience was the patchy, superficial distribution of tau protein throughout Mr. Borich’s frontal cortex and medial temporal lobes.
Although evidence of CTE has become increasingly common in retired football players, it has been found in other sports as well. The first professional ice hockey player to be examined for CTE was Reggie Fleming, who died at the age of 73 in 2009. Fleming was a defensemen and forward for six National Hockey League (NHL) teams from 1959 to 1971. His 13 seasons and 749 NHL games were part of a storied professional career that lasted more than 20 years. He is remembered today for his hard-nosed play and combative style that led to 108 NHL goals, 1,468 penalty minutes, and a Stanley Cup with the 1961 Chicago Blackhawks. He reportedly exhibited symptoms of CTE for decades; he was diagnosed with “manic depression” in his early 40s because of frequent extreme behavioral outbursts. He was described as “out of control” because of significant problems controlling his eating, drinking, gambling, and temper. He reportedly suffered from significant attention,
concentration, memory, and executive impairment that progressed to frank dementia in his final two years of life. Neuropathologically, examination of Mr. Fleming’s brain was also consistent with CTE, as evidenced by the extensive tau-immunoreactive inclusions distributed throughout the neocortex and medial temporal lobes, particularly at the depths of sulci.
Although much of the proceeding discussion has emphasized the ubiquity of tau protein abnormalities in the pathogenesis of CTE, more recent evidence also suggests the involvement of a second abnormal protein in CTE: Tar-DNA Binding Protein-43 (TDP-43). This abnormal protein has been found in 85 percent of CTE-positive cases (King et al., 2010; McKee et al., 2010). It is also associated with other neurodegenerative diseases like frontotemporal lobar degeneration, and, in some cases, may be associated with motor neuron disease that mimics amyotrophic lateral sclerosis (King et al., 2010; McKee et al., 2010; Tatom et al., 2009).
The prevalence of CTE is currently unknown, but appears to be more common than was previously thought. All 13 of the 13 football players examined by the BU CSTE have had CTE. However, the athletes examined are not representative of the general population or even the population of retired athletes, thus calling into question the denominator used to estimate CTE prevalence. However, assuming that neuropathological findings observed in the next 87 football players to come to autopsy are negative, this would nonetheless suggest a prevalence rate of 13 percent in this population. Clearly, there is a need for longitudinal research with a large, representative sample to more precisely estimate prevalence and identify potential risk and protective factors of CTE.
Such a longitudinal study has recently been implemented through the CSTE’s Brain Donation Registry. Recruitment for this registry began approximately 12 months ago and currently, approximately 300 active or retired athletes have agreed to participate in annual telephone interviews and to donate their brains at death. The goal of the CSTE is to recruit a total of 750 athletes altogether. The annual evaluations consist of telephone interviews regarding cognitive and behavioral symptoms, athletic, concussion, and medical history, and include a brief cognitive assessment. The CSTE is also beginning a new registry for combat veterans, another population at risk for brain trauma due to blast injuries caused by improvised explosive devices.
Many combat veterans exposed to blast injuries, like athletes who may suffer incidental blows to the head, experience repetitive subconcussive trauma. In other words, trauma caused by forces that are not substantial enough to cause symptoms of concussion. It is believed that concussions are just the tip of the iceberg when it comes to repetitive head trauma; the chronic neurodegeneration seen in CTE also may be influenced by a large number of subconcussive blows to the head that in and of themselves are not acutely problematic. It is believed that the long-term progressive tauopathy of CTE is caused by repetitive blows to the head, including mild, non-symptomatic, subconcussive trauma. However, this hypothesis has yet to be tested empirically.
In addition to the contribution of subconcussive blows to the pathogenesis of CTE, there are many other research questions that have yet to be answered. How common is CTE in athletes at all levels? Is CTE found in combat veterans? What are the risk factors of CTE? Is risk influenced by genetics? How does the type of trauma (loss of consciousness, severity of concussion, subconcussive blows) influence CTE? Can the impact of blast injuries (single, repetitive) lead to CTE? Can the frequency and time interval between successive head traumas affect CTE? For athletes, how can the positions and sports played (i.e., “load” of trauma) influence CTE? Does the age of the individual at time of injury(ies) and the duration of exposure affect CTE? How can we detect and diagnose CTE prior to death? What treatments and prevention strategies will be effective? Is CTE triggered by repetitive blast
injury in a similar fashion to repetitive (sub)concussive trauma? What roles do diffuse axonal injury and microhemorrhages play in CTE’s pathogenesis?
Returning to the discussion of blast injuries in a military combat setting, the long-term consequences of repetitive blast injuries are currently unknown. In athletes, the clinical symptoms of CTE begin years or decades following trauma. If the physical mechanics of athletic trauma and blast injuries are both capable of initiating the same metabolic cascade that may initiate CTE pathogenesis (Giza and Hovda, 2001), it is possible that, in several decades, we may see a growing epidemic of progressive dementia in veterans. One confounding factor that must be better understood is the fact that the symptoms of CTE and of posttraumatic stress disorder can manifest similarly. A second confound pertains to the fact that many members of the military also may have had previous and/or concurrent contact sport involvement.
Given that the risk factors for CTE are currently not well characterized, how can the risk of CTE be reduced, especially in young people? Under ideal circumstances, the best method to reduce CTE risk is to avoid head trauma. However, short of banning sports and preventing all accidents, head trauma cannot be completely avoided. Proper management of head trauma then becomes paramount. In sports, the following strategies are often recommended to reduce the overall trauma load suffered by an athlete: limit repetition (e.g., “return to play” guidelines); make changes to helmet technology to better absorb the force of the impact; identify the players who may be at increased risk (e.g., based on position, stature, style of play, etc.); and change rules to reduce head injuries without affecting the overall “feel” of the sport.
CTE research is in its infancy. However, a number of important changes have already been made, including changes to rules, increased public awareness, and increased research participation. It is hoped that these efforts can contribute to the prevention of this fully preventable neurodegenerative disease.
REFERENCES
Braak, H., and E. Braak. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82(4):239–259.
Cajigal, S. 2007. Brain damage may have contributed to former wrestler’s violent demise. Neurology Today 7(18):15–16.
Clinton, J., M. W. Ambler, and G. W. Roberts. 1991. Post-traumatic Alzheimer’s disease: Preponderance of a single plaque type. Neuropathology and Applied Neurobiology 17(1):69–74.
Corsellis, J. A. N., C. J. Bruton, and D. Freeman Browne. 1973. The aftermath of boxing. Psychological Medicine 3(3):270–303.
Critchley, M. 1957. Medical aspects of boxing, particularly from a neurological standpoint. British Medical Journal 1(5015):357–362.
Geddes, J. F., G. H. Vowles, J. A. R. Nicoll, and T. Révész. 1999. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathologica 98(2):171–178.
Giza, C. C., and D. A. Hovda. 2001. The neurometabolic cascade of concussion. Journal of Athletic Training 36(3):228–235.
King, A., F. Sweeney, I. Bodi, C. Troakes, S. Maekawa, and S. Al-Sarraj. 2010. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer’s disease. Neuropathology 30(4):408–419.
Langlois, J. A., W. Rutland-Brown, and M. M. Wald. 2006. The epidemiology and impact of traumatic brain injury: A brief overview. Journal of Head Trauma Rehabilitation 21(5):375–378.
Martland, H. 1928. Punch drunk. Journal of the American Medical Association 91(15):1103–1107.
McKee, A. C., R. C. Cantu, C. J. Nowinski, E. T. Hedley-Whyte, B. E. Gavett, A. E. Budson, V. E. Santini, H. S. Lee, C. A. Kubilus, and R. A. Stern. 2009. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. Journal of Neuropathology and Experimental Neurology 68(7):709–735.
McKee, A. C., B. E. Gavett, R. A. Stern, C. J. Nowinski, R. C. Cantu, N. W. Kowall, D. P. Perl, E. T. Hedley-Whyte, B. Price, C. Sullivan, P. Morin, H. S. Lee, C. A. Kubilus, D. H. Daneshvar, M. Wulff, and A. E. Budson. 2010. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. Journal of Neuropathology and Experimental Neurology 69(9):918–929.
Miller, H. 1966. Mental after-effects of head injury. Proceedings of the Royal Society of Medicine 59(3):257–261.
Millspaugh, J. 1937. Dementia pugilistica. United States Naval Medical Bulletin 35(3):297–303.
Omalu, B. I., S. T. DeKosky, R. L. Minster, M. I. Kamboh, R. L. Hamilton, and C. H. Wecht. 2005. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57(1):128–133.
Omalu, B. I., S. T. DeKosky, R. L. Hamilton, R. L. Minster, M. I. Kamboh, A. M. Shakir, and C. H. Wecht. 2006. Chronic traumatic encephalopathy in a National Football League player: Part II. Neurosurgery 59(5):1086–1092.
Omalu, B. I., J. Bailes, J. L. Hammers, and R. P. Fitzsimmons. 2010a. Chronic traumatic encephalopathy, suicides and parasuicides in professional American athletes: The role of the forensic pathologist. American Journal of Forensic Medicine and Pathology 31(2):130–132.
Omalu, B. I., R. L. Hamilton, M. I. Kamboh, S. T. DeKosky, and J. Bailes. 2010b. Chronic traumatic encephalopathy (CTE) in a National Football League player: Case report and emerging medicolegal practice questions. Journal of Forensic Nursing 6(1):40–46.
Parker, H. 1934. Traumatic encephalopathy (‘punch drunk’) of professional pugilists. Journal of Neurology and Psychopathology 15(57):20–28.
Roberts, G. W., D. Allsop, and C. Bruton. 1990a. The occult aftermath of boxing. Journal of Neurology, Neurosurgery, & Psychiatry 53(5):373–378.
Roberts, G. W., H. L. Whitwell, P. R. Acland, and C. J. Bruton. 1990b. Dementia in a punch-drunk wife. Lancet 335(8694):918–919.
Tatom, J. B., D. B. Wang, R. D. Dayton, O. Skalli, M. L. Hutton, D. W. Dickson, and R. L. Klein. 2009. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig’s disease in rats via TDP-43 overexpression. Molecular Therapy 17(4):607–613.
Standardized Clinical Management of Traumatic Brain Injury by the U.S. Military
Geoffrey S. F. Ling12
INTRODUCTION
Traumatic brain injury (TBI) is a common disorder associated with military service. For Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (Afghanistan) (OEF), TBI has been referred to as the “signature injury of the war” (Warden, 2006).
The exact incidence and prevalence of this disease among OIF and OEF troops is uncertain. There are estimates that as many as 19.5–40.0 percent of those deployed are affected. For civilians, the TBI rates also are uncertain with estimates suggesting that as many as 8 million head injuries occur each year in the United States alone. The reasons for the uncertainty are that many patients receive care by non-medical professionals, do not seek medical attention at all, or are improperly diagnosed. This is particularly true for mild TBI (mTBI), where signs and symptoms may be subtle. For moderate to severe TBI, where diagnosis is more certain, among U.S. civilians there are an estimated 1.7 million new cases per year.
Recently, the U.S. military, in response to this rising TBI problem, has instituted a system-wide standardized approach to diagnosis and clinical management of TBI. Critical elements of this approach are criteria for TBI screening; return to duty; neuroimaging; and, especially important, clinical practice guidelines for prehospital, in-hospital, and chronic treatment. As this is a collaborative effort between the Department of Defense (DoD) and
the Veterans Administration (VA), it is national in scope. This may be the first such large system-wide effort to apply clinical practice guidelines (CPGs) for TBI patients. Importantly, this has been endorsed and mandated at the highest level of command, which ensures its adoption and execution by medical care providers.
SEVERITY OF TRAUMATIC BRAIN INJURY
There are three major TBI categories: mild, moderate, and severe. These are differentiated by the patient’s presenting Glasgow Coma Scale (GCS). A traumatic injury to the brain leading to a GCS of 13–15 is defined as mild. If the GCS is 9–12, it is moderate TBI, and if 8 or less, then it is severe.
Mild TBI is further clarified by the Mild Traumatic Brain Injury Section of the American Congress of Rehabilitation Medicine (1993) as the loss of consciousness, loss of memory preceding or following injury (amnesia), alteration in mental status at time of injury, and/or focal neurological deficit. The American Academy of Neurology’s (AAN’s) Quality Standards Subcommittee (1997) reports that mTBI and concussion is often associated with brief (< 5 min) loss of consciousness or situational awareness where the person suffers a performance decrement within his/her required environmental context.
In clinical practice, mTBI and concussion are used interchangeably. However, they are distinct. Concussion is altered function following injury. Mild TBI is a pathological state of brain resulting from injury.
Concussion has three grades of severity (ANN, 1997). The grades are differentiated by duration of altered mental status and any loss of conscious. Although not part of the original AAN criteria, amnesia is an independent diagnostic indicator of TBI severity, with the loss of memory preceding (retrograde) or following (posttraumatic or anterograde) injury. Grade 1 concussion is defined as injury leading to altered mental status lasting less than 15 minutes without loss of consciousness. Grade 2 concussion is altered mental status lasting more than 15 minutes without loss of consciousness. Grade 3 concussion is any loss of consciousness.
Moderate TBI is usually associated with prolonged loss of consciousness and/or neurological deficit (Geocadin, 2004). These patients require advanced medical care including neurosurgical and neurointensive care. Later, as they recover, they may develop postconcussion syndrome (Jarell et al., 2003).
Severe TBI is when injury causes the patient to be significantly neurologically compromised such as obtundation or coma. Typically, this is associated with significant neurological injury, often with structural brain or skull lesions revealed by neuroimaging, e.g., head computerized tomography (CT) scan revealing skull fracture, intracranial hemorrhage, and early diffuse cerebral edema. Severe TBI patients require advanced medical care even in the pre-hospital setting. After initial resuscitation and stabilization in the field, severe TBI patients should be quickly evacuated directly to the nearest combat support hospital (CSH) with neurosurgical capability. These patients need airway protection, mechanical ventilation, neurosurgical evaluation, neurocritical care, intracranial pressure (ICP) monitoring, and highly skilled nursing in a trauma or neurointensive care unit. For these patients, recovery will be prolonged and often incomplete with many not surviving to 1 year (Ashwal et al., 1994; BTF, 2007; Bullock et al., 2006).
Mild TBI
Diagnosis and therapy begins at the site of injury, whether the battlefield or the playing field. It is now required that all troops at risk of TBI be evaluated as soon as possible after
exposure. During combat operations, at risk is defined as being within a certain distance of an explosive blast. That distance is different depending on circumstances, such as being mounted or dismounted when exposed. Evaluation is done using the military’s standardized MACE13 or military acute concussion evaluation. This is a paper-based tool developed by the Defense and Veterans Brain Injury Center (DVBIC). It begins with obtaining relevant patient information including history, especially exposure details, e.g., blast versus impact, etc. Embedded within the MACE is the standardized assessment of concussion (SAC). The SAC is a neuropsychological clinical test of orientation, concentration, memory, processing, etc. It is based on a similar test used by the National Football League. At present, there are six versions of the SAC so as to minimize the effect of learning the test.
Without a history of altered mental status, if the SAC is normal and the patient does not have any symptoms, he/she does not have TBI and is returned to duty. However, some patients may have symptoms, such as dizziness, for which other non-TBI etiologies are considered such as dehydration. This and other mild symptoms are treated conservatively with simple therapies such as rehydration or sleep hygiene. The patient may need to be referred to an advanced medical care provider such as the battalion surgeon, who is typically a physician.
With a history of change in mental status after injury and abnormal SAC (score < 25) then the patient has suffered mTBI with impairment and is referred to an advanced medical care provider. A more detailed history and neurological exam is performed. If there are any neurological deficits, the patient is evacuated to the nearest CT for neuroimaging. If CT is normal, patients are returned to their unit to be managed. These patients are automatically taken off of combat operation duty (“take a knee”) for a prescribed period of time, on the order of a few days. During this time, patients are treated symptomatically based on the VA/DoD Clinical Practice Guidelines for Management of Concussion/mTBI (2009). These guidelines have both pharmacological and nonpharmacological (such as sleep, physical therapy, etc.) treatment recommendations. Patients are given crossword puzzles, Sudoku and similar cognitive games. Finally, there are daily education sessions to teach each patient about his/her disease, including possible symptoms (headache, dizziness, insomnia, etc.) and expectations of recovery. An important aspect of this care is that each patient is purposely kept with his/her unit. By doing so, the service member has an important support group and is able to maintain a sense of normality and purpose (Bell et al., 2009).
If the SAC remains abnormal or symptoms persist beyond seven days, patients are referred to an in-theater restoration center, which is clinically staffed with a neurologist and/or occupational therapist and is located near a CSH so that more advanced medical care can be rendered. The neurologist is able to perform more detailed neurological and medical assessments, treat with a wider selection of medications, and test with more sophisticated methods. The occupational therapist can provide a more focused nonpharmacologic treatment plan. Typically, patients stay up to 14 days. If they still have not recovered, then they are evacuated out of theater (Linquist et al., 2010).
This approach has been very successful. Most service members are able to stay with their unit throughout treatment. The majority of those that have been referred to the restoration centers have been able to return to their units. The recovery rate exceeds 90 percent (Bell et al., 2009).
If a service member incurs three concussions over the course of his/her deployment, he/she is removed from further combat operations. The service members call this the “three strikes and you are out” rule (Hancock, 2008).
|
13 |
Available online: www.pdhealth.mil/downloads/MACE.pdf (access March 30, 2011). |
Moderate to Severe TBI
It is especially important for medical care for moderate to severe TBI to begin at the site of injury. In 2000, the Brain Trauma Foundation published the first edition of Guidelines for the Prehospital Management of Severe TBI, which is now in its second edition. Recognizing the need for similar CPGs for the battlefield, the Brain Trauma Foundation and the U.S. military developed the Guidelines for the Field Management of Combat-Related Head Trauma. These CPGs are specific treatment recommendations and goals to be used by medics, corpsmen, and other prehospital medical care providers for use on the battlefield. The emphasis is on maintaining optimal physiology to support the injured brain and prevent exacerbation of injury while under the constraints of military operations (Badjatia et al., 2008; Knuth et al., 2005).
Key guidelines are preventing hypoxia, maintaining perfusion (systolic blood pressure > 90 mmH), and avoiding potentially deleterious interventions (prophylactic hyperventilation). This is accomplished by placing an artificial airway if the patient’s GCS ≤ 8, supporting breathing and oxygenation if oxygen saturation < 90 percent, and maintaining systolic blood pressure > 90 mmHg. Unless a patient is actively herniating, hyperventilation and mannitol are to be avoided. They are not effective for prophylaxis. Mannitol also should not be used when maintenance of intravascular volume can be assured. Other guidelines are analgesic and sedation use, which are important to optimize patient safety, particularly during evacuation. Of note, no particular resuscitation fluid is endorsed because none have sufficient evidence to show clear superiority. However, hypertonic fluids are preferred because they may have benefit in maintaining serum osmolality, which could be beneficial for intracranial pressure (ICP) management (Knuth et al., 2005).
The GPG salso include triage and evacuation. As early as reasonably possible, a GCS for each patient should be determined as well as evaluation of pupillary function. Patients with GCS < 13 should be evacuated as early as possible to a CSH with a neurosurgeon.
Military in-hospital care of moderate to severe TBI treatment has benefited from advances previously developed for civilian TBI. The same civilian-developed CPGs are used in OEF and OIF CSHs. These in-hospital CPGs are directed at optimizing the general physiology; avoiding exacerbation of injury; careful clinical monitoring; and preventing conditions that could worsen outcome, such as deep vein thrombosis (DVT). The key guidelines are placing an artificial airway if the GCS ≤ 8, placing an ICP monitor for GCS ≤ 8, developing criteria to obtain neuroimaging, maintaining ICP < 25 mmHg, systolic blood pressure > 90 mmHg, pO2 > 60 mmHg or O2 saturation > 90 percent, normothermia, and hematocrit ≥ 28. Cerebral perfusion pressure (CPP) is kept between 50 to 70 mmHg if fluids or vasoactive agents are needed to achieve this range. If assistive therapy is not required to keep CPP above 50, then it is permitted to be at any level above that. To optimize cerebral venous drainage, the patient’s head is to be keep midline and the head of the bed at 30°. Antiepileptic medications are given for only the first seven days to minimize the occurrence of early posttraumatic seizures. To prevent DVT, anticoagulation begins as soon as hemorrhages are stable. The approach is using sequential compression devices (SCDs) and anticoagulation with low molecular weight heparin (LMWH) or low dose unfractionated heparin (LDUH), with or without mechanical compression devices such as graduated compression stockings and SCDs. If intracerebral hemorrhage is present, then it is recommended that only SCDs are used until the risk of further bleeding decreases, at which time anticoagulation may be started. Also important is early institution of nutritional support and gastrointestinal prophylaxis to prevent stress ulcers (BTF, 2007; Geerts et al., 2008).
In the event of cerebral herniation, mannitol and hyperventilation can be considered.
Again, mannitol can be used so long as the patient’s intravascular volume can be maintained. Hyperventilation should be to pCO2 34–36 mmHg. Neither should be used as prophylaxis against herniation. Because many combat wounded suffer from hemorrhagic shock and/or dehydration, hypertonic saline solutions are often considered, including 3 percent saline infusion or 23 percent saline bolus. Hemicraniectomy is also a therapeutic option frequently used, especially with long evacuation times from theater back to the United States during which managing ICP can be challenging (Ling et al., 2009).
Nutrition is now recognized to be an important component of proper TBI care. The patient should be fed as soon as practical. In moderate to severe TBI, patients usually need nasogastric or orogastric tubes. This is preferred over using parenteral nutrition as enteric feeding more easily allows meeting metabolic needs. Another benefit is that by minimizing free water, enteral feeds can help maintain the intravascular osmolar gradient used in treating intracranial hypertension. Because most TBI patients have some cerebral edema, hyperosmotic feeds are typically used. Because the injured brain is hypermetabolic, TBI patients typically require 140 percent of their basal metabolic caloric needs (BTF, 2007).
A concern is the risk of cerebral vasospasm. Armonda et al. (2006) reported that close to 50 percent of a series of patients with blast-related severe TBI developed cerebral vasospasm that led to symptomatic neurological deterioration. This is diagnosed with neurological examination, transcranial Doppler, and cerebral angiogram. It is often responsible for delayed or late neurological deterioration. Treatment with intra-arterial nicardipine at the site of spasm is effective in reversing this and restoring neurological function (Armonda et al., 2006).
Close neurological monitoring is essential for optimal outcome. While in the acute period, all TBI patients need to be examined neurologically on a regular basis—at least every hour for the first 24 hours and then less often as clinically indicated. Patients with intracranial lesions require continuous ICP and CPP measurements. Typically, the most critical period is during the 48 to 96 hours following injury when cerebral edema is greatest. Thereafter, edema gradually resolves and the patient should improve clinically.
CONCLUSION
Sadly, TBI is a common consequence of armed conflict. Clinical estimates of prevalence are high. In light of this, the military has enacted a comprehensive system-wide program to identify, treat, and rehabilitate TBI wounded service members. It uses evidence-based CPGs. Even though there is not yet a specific “brain rescue” or neuroprotective drug, evidenced-based CPG for treatment and return to duty provide a rational approach to proper management of the TBI patient. It must be emphasized that this is merely a beginning. To be truly effective, the CPGs must be regularly updated. More research and quality assurance follow-up are essential. The system in place is imperfect so there is ample opportunity for improvement. The ultimate goal is that every TBI service member will receive the highest level of medical care—no matter where they are.
DISCLAIMER
The opinions expressed herein belong solely to the author. They do not nor should they be interpreted as representative of or endorsed by the Uniformed Services University of the Health Sciences, Defense Advanced Research Projects Agency, U.S. Army, or Department of Defense.
REFERENCES
AAN (American Academy of Neurology). 1997. Practice parameter: The management of concussion in sports (summary statement). Neurology 48(3):581–585.
American Congress of Rehabilitation Medicine. 1993. Definition of mild traumatic brain injury. Journal of Head Trauma Rehabilitation 8(3):86–87.
Armonda, R. A., R. S. Bell, A. H. Vo, G. Ling, T. J. DeGraba, B. Crandall, J. Ecklund, and W. W. Campbell. 2006. Wartime traumatic cerebral vasospasm: Recent review of combat casualties. Neurosurgery 59(6):1215–1225.
Ashwal, S., R. Cranford, J. L. Bernat, G. Celesia, D. Coulter, H. Eisenberg, E. Myer, F. Plum, M. Walker, C. Watts, and T. Rogstad. 1994. Medical aspects of the persistent vegetative state (first of two parts). The New England Journal of Medicine 330(21):1499–1508.
Badjatia, N., N. Carney, T. J. Crocco, M. E. Fallat, H. M. A. Hennes, A. S. Jagoda, S. Jernigan, P. B. Letarte, E. B. Lerner, T. M. Moriarty, P. T. Pons, S. Sasser, T. Scalea, C. L. Schelein, and D. W. Wright. 2008. Guidelines for prehospital management of traumatic brain injury, 2nd ed. Prehospital Emergency Care 12(Suppl. 1).
Bell, J., K. Nasky, and W. Skipton. 2009. Personal communication from Afghanistan.
Bullock, M. R., R. Chestnut, J. Ghajar, D. Gordon, R. Hartl, D. W. Newell, F. Servadia, B. C. Waters, and J. E. Wilberger. 2006. Guidelines for the surgical management of traumatic brain injury. Neurosurgery 58(3):S2-1–S2-62.
BTF (Brain Trauma Foundation). 2007. Guidelines for the management of severe traumatic brain injury, 3rd ed. Journal of Neurotrauma 24(Suppl. 1):S1–S106.
Geerts, W. H., D. Bergqvist, G. F. Pineo, J. A. Heit, C. M. Samama, M. R. Lassen, C. W. Colwell, and P. American College of Chest. 2008. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th ed.). Chest 133(6 Suppl.):381S–453S.
Geocadin, R. 2004. Traumatic brain injury. In Handbook of neurocritical care, edited by A. Bharwaj, M. A. Mirski and J. A. Ulatowski. Totowa, NJ: Humana Press.
Hancock, J. 2008. Personal communication from Afghanistan.
Jarell, A., J. M. Ecklund, and G. Ling. 2003. Traumatic brain injury. In Combat medicine, edited by G. Tsokos and J. Atkinson. Totowa, NJ: Humana Press.
Knuth, T., P. B. LeTarte, G. S. F. Ling, L. E. Moores, P. Rhee, D. Tauber, and A. Trask. 2005. Guidelines for the field management of combat-related head trauma. New York: Brain Trauma Foundation.
Ling, G., F. Bandak, R. Armonda, G. Grant, and J. Ecklund. 2009. Explosive blast neurotrauma. Journal of Neurotrauma 26(6):815–825.
Linquist, J., K. Radcliff, and M. Wagner. 2010. Personal communication from Afghanistan.
VA/DoD. 2009. The clinical practice guidelines for management of concussion/mild traumatic brain injury. Journal of Rehabilitation Research and Development 46(6):CP1–68.
Warden, D. 2006. Military TBI during the Iraq and Afghan wars. The Journal of Head Trauma Rehabilitation 21(5):398−402
The Perspective of an R.D. Working with Civilian Traumatic Brain Injury
Natalia Bailey14
INTRODUCTION
Traumatic brain injury (TBI) causes a very serious assault to the body, not only because of the primary or secondary injuries, but also because of the effects it has on all of the body’s systems. Brain injury results in a significant increase in metabolism and catabolism that, if left unchecked, can lead to malnutrition. It has been shown that adequate nutrition support can attenuate these metabolic changes that result in muscle loss and therefore positively affect outcomes. This paper will review the current nutrition support standards of care in the moderately to severely brain-injured population, potential routes of nutrient administration, benefit of specific nutrients, and drug-nutrient interactions of concern.
TABLE C-3 Chemical Messengers Effects on Inflammatory Response
|
Messenger |
Function |
Result |
|
Cortisol |
↑ gluconeogenisis ↑ proteolysis |
↑ rate of muscle catabolism |
|
Glucagon |
↑ gluconeogenisis |
↑ rate of muscle catabolism |
|
Catecholamines (epinephrine, norepinephrine) |
↑ insulin resistance |
↑ rate of muscle catabolism |
|
Cytokines (IL-1, IL-6, TNF) |
Activates immune response ↑ RMR |
↑ rate of muscle catabolism |
RATIONALE FOR NUTRITION SUPPORT IN TBI
In the initial acute phase following TBI, the patient is in a hypermetabolic and catabolic state. The literature indicates energy expenditure ranges between 100–200 percent of resting metabolic rate, and this increased energy expenditure can last anywhere from one week to one year following the injury (Deutschman et al., 1986; Loan, 1999; Moore et al., 1989). During the initial acute stage of injury, glycogen stores are quickly depleted. This results in a need to use muscle proteins as a source of required glucose energy, leading to significant lean body mass catabolism (Berg et al., 2006). Non-stressed individuals lose approximately 200–300 grams of muscle per day, whereas TBI patients lose up to 1,000 grams of lean body mass per day (Loan, 1999). The major cause of this highly catabolic state is a postinjury increase in chemical messengers. These messengers include cortisol, glucagon, catecholamines, and cytokines, all of which contribute to breakdown of muscle rather than fat for energy (Table C-3) (Darbar, 2001; Loan, 1999; Moore et al., 1989; Young et al., 1988). In addition to an increase in catabolic chemical messengers postinjury, there is a decrease in anabolic hormones, such as human growth hormone (Demling, 2009). The increase in metabolic rate and resulting muscle catabolism can lead to malnutrition if not attenuated by provision of adequate nutrition support.
Inadequate nutrition support postinjury can lead to significant malnutrition. This can include significant weight loss resulting in poor outcomes including increased mortality and, increased length of hospitalization and rehabilitation. If the TBI patient is fed inadequately for one week, then this may lead to a 10 percent loss of lean body mass. If nutrition needs are not met for two weeks, then this may lead to a 30 percent loss of lean body mass, and increased mortality (Darbar, 2001). Malnourished TBI patients (BMI < 15) entering rehabilitation have a length of stay approximately 28 days longer than those who are not malnourished (Dénes, 2004).
STANDARDS OF CARE
The major principles driving the nutrition care of TBI patients include early nutrition support, provision of adequate calories (kcal) and protein, preference for enteral nutrition, use of nutrition protocols, and ongoing assessment of efficacy of nutrition support.
Evidence for early nutrition support (within the first 24–72 hours post-injury) is limited
in the TBI population; however, there is growing evidence that this practice is beneficial. In one study, early nutrition support (within five days after trauma) was shown to be one of the few therapies that could positively affect two-week mortality in TBI patients (Hartl et al., 2008). Benefits of early nutrition support in other critically ill populations include lower risk of infection, decreased activation and release of inflammatory cytokines, decreased hospital and intensive care unit (ICU) length of stay, and attenuation of catabolism of skeletal muscle (McClave et al., 2009).
Determining calorie requirements is difficult in this population because many factors influence the rate of metabolism. Calorie needs may be decreased with barbiturate coma, propofol infusions, and other sedatives (Frankenfield, 2006; McCall et al., 2003; Moore et al., 1989; Rajpal and Johnston, 2009). Infection, fever, posturing, storming, and presence of other injuries may increase caloric needs (Clifton et al., 1986; Frankenfield, 2006; Moore et al., 1989; Rajpal and Johnston, 2009). Typically the energy needs are calculated by prediction formulas, which predict basal energy expenditure (BEE), such as the Harris-Benedict formula, which also includes an injury factor (Cook et al., 2008). The Brain Trauma Foundation (BTF) recommends a calorie provision of 140 percent of BEE (Bratton et al., 2007). It has been shown that prediction formulas often under- or over-predict calories, and if able, it is desirable to measure energy expenditure via indirect calorimetry. This measurement is considered the “gold standard” in determining calorie needs (Felípez and Sentongo, 2009; McClave et al., 2009; Pepe and Barba, 1999).
Protein needs are elevated in the TBI population, and the BTF guidelines suggest needs that range from 1.5-2.0 g per kg (Bratton et al., 2007). Several studies have shown variable beneficial results in delivering greater than 2 grams per kg. One study showed that when given 2.2 g of protein per kg, TBI patients corrected their negative nitrogen balance at a faster rate than those who were given less protein; however, they had increased urinary nitrogen excretion (Twyman et al., 1985).
Enterally delivered nutrition support is the preferred route of nutrient delivery if the patient’s gastrointestinal (GI) tract is functioning and accessible. Benefits of enteral nutrition (EN) include the following (Artinian et al., 2006; McClave et al., 2009; Taylor et al., 1999):
-
gut barrier maintenance,
-
modulation of stress and immune response,
-
lower risk of infection when compared to parenteral nutrition (PN) administration,
-
reduction in hospital length of stay,
-
lower cost of nutrition support, and
-
quicker return of cognitive function and in neurosurgery patients.
Although EN support is less risky than PN support, there are still some risks associated with it. If enteral feeds are started prior to adequate resuscitation, or if GI perfusion is poor, there is an increased risk of GI tract ischemia. High infusion rates of pressors and/or sedatives, anesthetics, as well as abdominal injuries influence blood perfusion to the GI tract. Additionally, enteral feeding intolerance, such as abdominal pain, nausea, and vomiting have been described in critically ill patients, negatively affecting the rate at which patients achieve their nutritional goals.
PN is another potential route of nutrition support delivery; however, its use can result in more complications than enteral nutrition, and therefore, is not the preferred option. Risks of PN include increased risk of infection, increased cost, and increased risk of mortality (McClave et al., 2009). In the TBI population, however, it has been shown that patients on PN have fewer interruptions in feeding and their nutrition goals are reached more quickly
(Bratton et al., 2007; Young et al., 1987). One study showed that for those with head trauma, EN and PN were equally effective in meeting nutrition goals with similar infection rates and similar cost (Borzotta et al., 1994).
An assessment by a registered dietitian (RD) early in the TBI patient’s admission is essential in determining nutrition goals and the nutrition plan of the TBI patient. It has been shown that implementing the RD’s recommendations is correlated with decreased length of stay, higher serum albumin, and increased weight gains (Braga et al., 2006). In addition to the RD assessment, use of nutrition protocols is useful in enhancing nutrition delivery. In one study, use of the nutrition protocol increased the percentage of calories provided, and was identified as the factor having the greatest impact on successful delivery of EN in the first week of neurocritical illness (Zarbock et al., 2008).
Assessing efficacy of nutrition support is a difficult task in the ICU setting because of the confounding effects of critical illness and treatment on typical nutrition assessment parameters. Patient weights are strongly influenced by clinically induced fluid gains and losses, and are not immediately useful, although they may be more helpful in the later stages of healing. Nutrition laboratory tests are highly inaccurate in the setting of critical illness because the acute-phase response results in re-direction of the synthesis of visceral proteins toward wound healing and the immune response (McClave et al., 2009; Moore et al., 1989; Young et al., 1985, 1988). It is recommended that if albumin or transthyretin (pre-albumin) is used to assess nutrition status, C-reactive protein (CRP) must be checked as well, to determine the patient’s level of inflammatory response. When CRP is elevated, this is an indication that albumin and pre-albumin will be decreased, and their use as a tool for assessment of nutrition status will not be useful. If using visceral proteins to monitor nutrition status, trends in laboratory values are more helpful compared to isolated measurements of these laboratory tests. Other indicators of adequacy of nutrition include wound healing, ability to wean from mechanical ventilation, as well as ability to participate in rehabilitative therapies.
Standards of care regarding nutrition therapy in the TBI patient are based on guidelines from various organizations including the American Dietetic Association, American Society of Parenteral and Enteral Nutrition (ASPEN), Society of Critical Care Medicine (SCCM), and Brain Trauma Foundation (BTF). The most comprehensive nutrition guidelines come from the combined efforts of ASPEN and SCCM; however, these guidelines are not specific to the TBI population. The BTF has published nutrition guidelines as well, though, because of the lack of nutrition studies in the TBI population, these guidelines are not specific. Table C-4 is a comparison of the nutrition guidelines from ASPEN/SCCM and the brain trauma foundation for moderate to severe brain injury, and may be helpful in identifying areas where more research is needed in the TBI patient population.
TABLE C-4 Comparison of ASPEN/SCCM Guidelines and BTF Guidelines
|
Guidelines Pertaining to |
ASPEN/SCCM |
BTF |
|
Calories |
No specific guidelines in TBI; Met cart gold standard |
100–140 percent replacement of resting metabolism |
|
Protein |
1.2–2.0 g/kg in critical illness |
15–20 percent of kcals |
|
Timing of initiating feeds |
24–48 hours following admission |
No recommendation |
|
Dosing of EN |
50–65 percent of goal by day 7 |
100 percent of goal by day 7 |
|
EN vs. PN |
EN preferred, PN only when necessary |
No recommendation |
|
Nutrition protocols |
Should be implemented |
No recommendation |
It should be noted that currently there are no published nutrition guidelines for those with mild TBI and/or concussion.
SPECIFIC DIETS OR NUTRIENTS OF CONCERN
Specific diets or nutrients to be avoided in the TBI population are unknown, although there is some question regarding the safety of the use of the amino acid glutamine during the acute phases of TBI. During critical illness, glutamine becomes conditionally essential, and administration of exogenous glutamine in this patient population has shown some promise (McClave et al., 2009). Benefits include glutamine’s anabolic/anticatabolic properties, use as an antioxidant, and use as a fuel for dividing cells. However, in the TBI population, it has been hypothesized that increased glutamine in the diet leads to increased glutamate in the interstitial fluid. Increased glutamate has been linked to high intracranial pressures and increased cerebral swelling (Cook et al., 2008; Enriquez and Bullock, 2004). Several studies have shown that feeding patients additional glutamine does increase glutamate levels in the interstitial fluid; however, more studies are needed to determine the benefit of this practice in the brain-injured population (Berg et al., 2006).
IMPORTANCE OF NUTRITIONAL FORMULATIONS
The use of immune-enhancing formulas in the critically ill population is of growing interest, and studies have shown better outcomes with the use of these formulas. Immune-modulating formulas may include higher ratio of n-3 vs. n-6 fatty acids, and/or increased provision of antioxidants including vitamins E and C, zinc, and selenium. ASPEN and SCCM recommend the use of immune-enhancing formulas in surgical and medical ICU patients, but suggest using standard formulas in populations in which there is little evidence that use of enhanced formulas improve outcomes (McClave et al., 2009). In the TBI population, evidence is lacking that these formulas provide additional benefits compared to those receiving standard formulas (Martindale et al., 2009). One study looked at early nutrition along with using an immune-enhancing formula in head injury patients, and no additional benefit was found (Minard et al., 2000). Another small study (n = 13) looked at feeding an immune-enhancing enteral formula to trauma patients, and this did not affect outcomes or levels of pro-inflammatory cytokines (Jeevanandam et al., 1999). Because of the positive results seen in other populations, larger, randomized control trials are warranted in the TBI population.
DRUG AND NUTRIENT INTERACTIONS
When devising a nutrition plan, drug-nutrient interactions should be taken into account. It is possible that calorie needs will be lower with certain medications, such as propofol, which provides 1.1 kcals of fat with each milliliter infused in addition to decreasing metabolic rate. Other medications may cause increased need for nutrients, such as vitamin D and folic acid for those on seizure prophylaxis (Felípez and Sentongo, 2009). The effect that any drug has on the GI tract should also be considered. For instance, vasopressors decrease gut perfusion; therefore, one should be cautious about aggressive nutrition regimens. Table C-5 shows a list of common drug-nutrient interactions in the TBI population. This is not a comprehensive list of all interactions; however, these are most common in the neurocritical care unit.
TABLE C-5 Common Drug/Nutrient Interactions in TBI
|
Drug |
Nutrient Interaction |
|
Barbiturates |
Decreased metabolic rate |
|
Propofol |
|
|
Seizure prophylaxis |
Increased need for vitamin D, folic acid |
|
Dilantin |
Carbohydrates interfere with absorption |
|
Vasopressors |
Decreased gut perfusion |
CONCLUSION
Traumatic brain injury can cause a very dramatic increase in metabolism and catabolism, resulting in extensive loss of lean body mass if adequate nutrition support is not provided. It has been shown that adequate and timely nutrition therapy can lead to positive outcomes for those with brain injury. Standards of care for nutrition support of TBI patients include early enteral nutrition if possible, measurement using indirect calorimetry rather than prediction of calories to be provided, and the use of nutrition protocols to deliver support. Because of the need for nutrition research in this population, the benefit of specific nutrients is unknown.
REFERENCES
Artinian, V., H. Krayem, and B. DiGiovine. 2006. Effects of early enteral feeding on the outcome of critically ill mechanically ventilated medical patients. Chest 129(4):960-967.
Berg, A., B. M. Bellander, M. Wanecek, L. Gamrin, Å. Elving, O. Rooyackers, U. Ungerstedt, and J. Wernerman. 2006. Intravenous glutamine supplementation to head trauma patients leaves cerebral glutamate concentration unaffected. Intensive Care Medicine 32(11):1741-1746.
Borzotta, A. P., J. Pennings, B. Papasadero, J. Paxton, S. Mardesic, R. Borzotta, A. Parrott, and F. Bledsoe. 1994. Enteral versus parenteral nutrition after severe closed head injury. Journal of Trauma 37(3):459-468.
Braga, J. M., A. Hunt, J. Pope, and E. Molaison. 2006. Implementation of dietitian recommendations for enteral nutrition results in improved outcomes. Journal of the American Dietetic Association 106(2):281-284.
Bratton, S. L., R. M. Chestnut, J. Ghajar, F. F. McConnell Hammond, O. A. Harris, R. Hartl, G. T. Manley, A. Nemecek, D. W. Newell, G. Rosenthal, J. Schouten, L. Shutter, S. D. Timmons, J. S. Ullman, W. Videtta, J. E. Wilberger, and D. W. Wright. 2007. Guidelines for the management of severe traumatic brain injury. XII. Nutrition. Journal of Neurotrauma 24(Suppl. 1):S77-S82.
Clifton, G. L., C. S. Robertson, and S. C. Choi. 1986. Assessment of nutritional requirements of head-injured patients. Journal of Neurosurgery 64(6):895-901.
Cook, A. M., A. Peppard, and B. Magnuson. 2008. Nutrition considerations in traumatic brain injury. Nutrition in Clinical Practice 23(6):608-620.
Darbar, A. 2001. Nutritional requirements in severe head injury. Nutrition 17(1):71-72.
Demling, R. 2009. Nutrition, anabolism, and the wound healing process: An overview. Open Access Journal of Plastic Surgery 9:65-88.
Dénes, Z. 2004. The influence of severe malnutrition on rehabilitation in patients with severe head injury. Disability and Rehabilitation 26(19):1163-1165.
Deutschman, C. S., F. N. Konstantinides, S. Raup, P. Thienprasit, and F. B. Cerra. 1986. Physiological and metabolic response to isolated closed-head injury. Part 1: Basal metabolic state: Correlations of metabolic and physiological parameters with fasting and stressed controls. Journal of Neurosurgery 64(1):89-98.
Enriquez, P., and R. Bullock. 2004. Molecular and cellular mechanism in the pathophysiology of severe head injury. Current Pharmaceutical Design 10(18):2131-2143.
Felípez, L., and T. A. Sentongo. 2009. Drug-induced nutrient deficiencies. Pediatric Clinics of North America 56(5):1211-1224.
Frankenfield, D. 2006. Energy expenditure and protein requirements after traumatic injury. Nutrition in Clinical Practice 21(5):430-437.
Hartl, R., L. M. Gerber, Q. Ni, and J. Ghajar. 2008. Effect of early nutrition on deaths due to severe traumatic brain injury. Journal of Neurosurgery 109(1):50–56.
Jeevanandam, M., L. M. Shahbazian, and S. R. Petersen. 1999. Proinflammatory cytokine production by mitogen-stimulated peripheral blood mononuclear cells (PBMCs) in trauma patients fed immune-enhancing enteral diets. Nutrition 15(11–12):842–847.
Loan, T. 1999. Metabolic/nutritional alterations of traumatic brain injury. Nutrition 15(10):809–812.
Martindale, R. G., S. A. McClave, V. W. Vanek, M. McCarthy, P. Roberts, B. Taylor, J. B. Ochoa, L. Napolitano, and G. Cresci. 2009. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine and American Society for Parenteral and Enteral Nutrition: Executive summary. Critical Care Medicine 37(5):1757–1761.
McCall, M., K. Jeejeebhoy, P. Pencharz, and R. Moulton. 2003. Effect of neuromuscular blockade on energy expenditure in patients with severe head injury. Journal of Parenteral and Enteral Nutrition 27(1):27–35.
McClave, S. A., R. G. Martindale, V. W. Vanek, M. McCarthy, P. Roberts, B. Taylor, J. B. Ochoa, L. Napolitano, and G. Cresci. 2009. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.). Journal of Parenteral and Enteral Nutrition 33(3):277–316.
Minard, G., K. A. Kudsk, S. Melton, J. H. Patton, and E. A. Tolley. 2000. Early versus delayed feeding with an immune-enhancing diet in patients with severe head injuries. Journal of Parenteral and Enteral Nutrition 24(3):145–149.
Moore, R., M. P. Najarian, and C. W. Konvolinka. 1989. Measured energy expenditure in severe head trauma. Journal of Trauma 29(12):1633–1636.
Pepe, J. L., and C. A. Barba. 1999. The metabolic response to acute traumatic brain injury and implications for nutritional support. Journal of Head Trauma Rehabilitation 14(5):462–474.
Rajpal, V., and J. Johnston. 2009. Nutrition management of traumatic brain injury patients. Support Line 31:10–19.
Taylor, S. J., S. B. Fettes, C. Jewkes, and R. J. Nelson. 1999. Prospective, randomized, controlled trial to determine the effect of early enhanced enteral nutrition on clinical outcome in mechanically ventilated patients suffering head injury. Critical Care Medicine 27(11):2525–2531.
Twyman, D., A. B. Young, and L. Ott. 1985. High protein enteral feedings: A means of achieving positive nitrogen balance in head injured patients. Journal of Parenteral and Enteral Nutrition 9(6):679–684.
Young, A. B., L. G. Ott, D. Beard, R. J. Dempsey, P. A. Tibbs, and C. J. McClain. 1988. The acute-phase response of the brain-injured patient. Journal of Neurosurgery 69(3):375–380.
Young, B., L. Ott, and J. Norton. 1985. Metabolic and nutritional sequelae in the non-steroid treated head injury patient. Neurosurgery 17(5):784–791.
Young, B., L. Ott, D. Twyman, J. Norton, R. Rapp, P. Tibbs, D. Haack, B. Brivins, and R. Dempsey. 1987. The effect of nutritional support on outcome from severe head injury. Journal of Neurosurgery 67(5):668–676.
Zarbock, S. D., D. Steinke, J. Hatton, B. Magnuson, K. M. Smith, and A. M. Cook. 2008. Successful enteral nutritional support in the neurocritical care unit. Neurocritical Care 9(2):210–216.
Nutritional Considerations in Clinical Treatment: The Perspective of a Neurosurgeon
Jamshid Ghajar,15 Roger Härtl,16 Linda M. Gerber,17 and Jane E. McCormack18
INTRODUCTION
Traumatic brain injury (TBI) remains a highly lethal injury with mortality ranging from 20–50 percent (Bulger et al., 2002; Demetriades et al., 2004; Jiang et al., 2002). Approximately 52,000 patients die from TBI each year (Sosin et al., 1995; Thurman et al., 1999) with approximately 85 percent of the deaths occurring within the first two weeks (Roberts
et al., 2004). Pharmaceutical trials of TBI have failed to demonstrate any efficacy in reducing deaths (Narayan et al., 2002). Proper trauma transport systems and maintenance of cerebral perfusion and oxygenation by avoidance of hypoxemia, arterial hypotension, and intracranial hypertension reduce mortality and improves outcome (Brain Trauma Foundation, 2000; Chesnut et al., 1993; Härtl et al., 2006; Sampalis et al., 1999; Smith et al., 1990).
Currently, the metabolic status and nutritional needs of TBI patients are less of a priority than maintaining cerebral perfusion. However, TBI results in a hypermetabolic and catabolic state that increases systemic and cerebral energy requirements (Clifton et al., 1986; Deutschman et al., 1986; Hovda et al., 1995; Weekes and Elia, 1996). A recent review from the Cochrane Collaboration states that early feeding may be associated with a trend towards better outcomes after TBI (Perel et al., 2006). The Guidelines for the Management of Severe Traumatic Brain Injury recommend that the patient’s feeding requirements should be met by the end of the first week after TBI (Brain Trauma Foundation, 2000). These recommendations were based on two small, randomized trials (Rapp et al., 1983; Taylor et al., 1999). There are no studies on the relationship of mortality to the amount and frequency of feeding in TBI patients. In the few studies done, none controlled for factors known to affect mortality from TBI, such as hypotension, age, pupillary status, and computed tomography (CT) scan findings.
The Brain Trauma Foundation (BTF) prospectively collects data on pre- and in-hospital TBI management in 20 Level I and 2 Level II trauma centers in New York state as part of a TBI quality improvement program. An analysis was conducted examining the effect of timing and quantity of nutritional support on early mortality. Early onset of nutritional support and amount of nutritional support was hypothesized to be associated with a reduced mortality at two weeks. In addition, a feeding compliance implementation program was undertaken at one of the participating hospitals to increase the net caloric intake of patients.
METHODS
The BTF designed and implemented a quality improvement initiative in New York state to improve severe TBI acute care and outcome. The program is funded by the New York State Department of Health, Division of Healthcare Financing and Acute and Primary Care Reimbursement. This program tracks pre- and in-hospital severe TBI data through an online Internet database called TBI-trac®. The database consists of clinical information from the prehospital environment, emergency department, the first 10 days of intensive care unit (ICU) care, and two-week mortality. When the study began in 2000, enrollment was limited to five Level I trauma centers, and this number increased to a total of 24 trauma centers in 2005, 22 of which were Level I trauma centers and 2 of which were Level II centers. This report is based on patients treated at these trauma centers between June 6, 2000 and December 31, 2005.
RESULTS
Data for 1,818 patients were entered in the database from June 6, 2000, through December 31, 2005. Patients were excluded if they had a Glasgow Coma Scale (GCS) greater than or equal to 9 on day 1 (92 patients), or a GCS motor score of 6 on day 1 (16 patients). Patients also were excluded if they had a GCS score of 3 with pupils bilaterally fixed and dilated and were not pharmacologically paralyzed (152 patients). In addition, patients were excluded for a GCS greater than or equal to 4 with pupils bilaterally fixed and dilated or missing pupillary information (93 patients), or with missing outcome assessment (51 pa
tients). Because nutritional requirements for pediatric and adult patients are different, 153 pediatric patients less than 16 years of age were excluded. After these exclusion criteria were applied, a total of 1,261 patients were eligible for analysis.
In order to examine the effect of nutritional support within the first week after admission, only patients who had at least seven days of inpatient data (were alive for at least seven days) were analyzed. A total of 464 patients had less than seven days of data resulting in a final sample of 797 patients. Patients who had less than seven days of data were older (41.9 vs. 39.0 years, p < 0.01) and were more likely to be hypotensive on day 1 (17.6 percent vs. 13.3 percent p < 0.04) than patients with greater than seven days of data. A greater proportion of their CT scans were abnormal (84.3 percent) compared to those with greater than seven days of follow-up (74.4 percent, p < 0.001). Two-week mortality was also significantly higher when compared to those with seven or more days of records (42.0 percent vs. 9.9 percent, p < 0.0001).
Administration of feeding began in 61 percent of patients during days 1 through 3; however, 5 percent of patients were not fed during the seven-day period, and the majority (62 percent) of patients never reached 25 kcal/kg/day within seven days. No differences were found in patient characteristics or severity of illness by nutrition level achieved within the first five days of treatment. Two-week mortality by nutrition status was significantly higher among patients never fed within 5 (p = 0.0008) or seven (p < 0.0001) days. Mortality significantly decreased with increasing nutritional level such that the rate was 6.3 percent and 7.6 percent among patients fed more than 25 kcal/kg/day within five and seven days, respectively. Older age and having a high intracranial pressure (ICP) were also significantly associated with two-week mortality, while CT scan status was marginally related. The lack of correlation between hypotension and pupillary status and mortality may be explained by the specific exclusion criteria in this study; patients with bilaterally fixed and dilated pupils and GCS scores of 3 were excluded, as were patients who were not alive by day seven.
Nutrition level continued to predict two-week mortality after controlling for age, hypotension, pupillary status, initial GCS, and CT scan status. Patients not fed within five days had 2.1 times the risk of two-week mortality, while those not fed within seven days had 4.1 times the risk of two-week mortality. The amount of nutritional support given within five and seven days also contributed significantly to mortality risk. Every 10 kcal/kg decrease in nutritional support administered within five and seven days resulted in 30–40 percent increased risk of mortality (Figure C-6).
Further analysis of the relationship between ICP monitoring and early nutrition reveals that nutrition had a significant impact in patients with elevated ICP. In the first five days, patients with high ICP and without nutritional support, had a significantly increased mortality when compared to patients with intracranial hypertension who were fed (25.7 percent vs. 12.9 percent, respectively, p = 0.04). In patients who did not undergo ICP monitoring the lack of early nutritional support had an even more pronounced impact on mortality (25.8 percent vs. 6.3 percent mortality, p = 0.0004).
DISCUSSION
Current Findings
The present study adds several significant findings to the existing literature. It is the largest database that has used prospectively collected data to address the relationship between nutrition and early mortality after TBI. The main findings follow. First, any nutrition within the first five days after TBI is associated with reduced mortality. Second, there is a significant
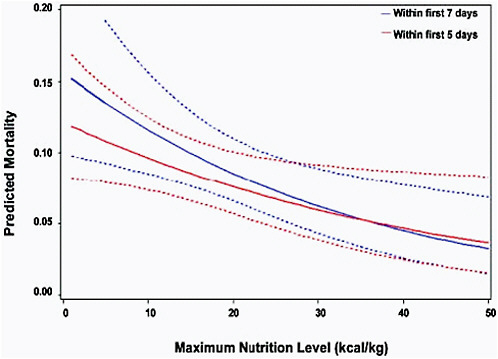
FIGURE C-6 Line graph shows the predicted mortality as a function of maximum nutrition level. Regression models were adjusted for age, hypotension status on day 1, pupil status (normal or abnormal) on day 1, initial GCS score, and CT scan findings. Solid lines represent the maximum amount of nutritional support, and dashed lines indicate the 95 percent confidence interval.
SOURCE: Härtl et al., 2008.
relationship between the maximum level of nutrition reached and mortality; every 10 kcal/kg decrease in caloric intake is associated with a 30–40 percent increase in mortality. Third, early nutrition within five days after TBI emerges as an independent factor affecting mortality even after controlling for known predictors of mortality such as arterial hypotension, age, CT diagnosis, GCS, and pupillary status. Patients who were not fed within five or seven days after TBI had a two- and four-fold increased likelihood of mortality, respectively. In addition, patients with elevated ICPs or patients who did not undergo ICP monitoring had a significantly increased mortality if they were not fed within five days after trauma when compared to patients who received nutrition. These findings demonstrate that feeding is as significant an intervention as avoidance of early arterial hypotension and hypoxia in reducing mortality from severe TBI.
How Does Feeding Affect Mortality?
Generally, nutritional support has emerged as a significant factor in improving the outcome of critically ill patients. Early initiation of enteral nutrition is associated with a lower incidence of infections, reduced length of hospital stay and possibly improved outcome in critically ill patients in surgical and medical ICUs (Marik and Zaloga, 2001). However, the mechanism by which nutrition affects outcome is unclear. One possibility is that it may provide important nutrients during a critical time period when demand exceeds available resources.
Studies have shown a rise in energy expenditure after TBI, even in paralyzed patients (Clifton et al., 1986). This hypermetabolic state after TBI may be due to systemic factors
such as infection and a posttraumatic stress response, but there appears to be a cerebral component as well. There is an increase in cerebral metabolic rate for glucose in TBI possibly as a result of mitochondrial dysfunction (Merenda and Bullock, 2006). Studies in humans indicate that this increase in glucose utilization may last up to five to seven days after TBI (Bergsneider et al., 1997; Hovda et al., 1995). The significance of this cerebral hypermetabolic state is illustrated by the therapeutic effect of interventions that suppress cerebral metabolism such as barbiturate coma, hypothermia and interventions that improve blood flow and supply of nutrients such as cerebral perfusion pressure management. One of the beneficial effects of early aggressive nutritional support may be the steady supply of glucose when the brain depends on increased glucose metabolism to maintain metabolic energy balance.
Another effect of early nutrition may be its attenuation of the posttraumatic stress response and improvement of early immunological function (Bastian et al., 1998; Rovlias and Kotsou, 2000). This could result in an indirect effect on outcome mediated by a lower infection rate. A meta-analysis that compared early (within 36 hours) to delayed initiation of enteral nutrition in critically ill patients (not only TBI) demonstrated a 55 percent reduction in infection rate in patients who received early nutritional support (Marik and Zaloga, 2001). As infection rate was not collected as part of the TBI-trac® database, this relationship could not be examined. It is unlikely, however, that infection rate would have such a significant impact on two-week mortality.
Nutrition also could have an impact on the posttraumatic stress response that is associated with adverse outcome from TBI (Rovlias and Kotsou, 2000). The posttraumatic stress response is characterized by increased blood levels of glucose, lactate, catecholamines, and cortisol. In the blunt trauma population, however, early feeding within 24 hours after injury had no effect on the metabolic stress response (Eyer et al., 1993).
Arterial hypotension doubles mortality from TBI (Chesnut et al., 1993). The relationship between nutritional support and hypotension was examined based on the hypothesis that the fluid volume given with nutrition improves the patient’s hemodynamic status and prevents arterial hypotension. There was, however, no relationship between arterial hypotension and nutritional support within the first five days after TBI.
Another finding in this study was that early nutritional support may have a protective effect in patients with intracranial hypertension. Results indicate that patients with high ICPs who are fed have a significantly lower mortality when compared to patients who do not receive nutritional support (12.9 percent vs. 25.7 percent, respectively). Nutritional support may protect the brain by providing large amounts of energy substrates, during a critical time period when hyperglycolysis and hyperemia are present, in an effort to maintain energy balance and cerebral ionic hemostasis (Bergsneider et al., 1997).
Why Were Patients Not Fed?
Our analysis showed that the lack of nutritional support was not related to the severity of the injury or other factors associated with outcome from TBI. Beyond this, it is difficult to determine what affected the decision not to feed patients early on. Possibilities include that patients did not tolerate enteral nutrition early after TBI and that early nutritional support may not have been given a priority by the treating physicians. Other factors that interfere with feeding and that were not registered in this database include patient transport within the hospital and enteral administration of phenytoin.
Program in Participating Trauma Center to Improve Compliance
Achieving adequate nutritional intake in this patient population is difficult, and nutritional therapy in many ICUs is suboptimal (Cahill et al., 2010). Despite best intentions many trauma centers fail to achieve adequate caloric intake. One Level 1 trauma center was consistently well below the goal of 25 kcal/kg/day, and a Quality Improvement (QI) initiative was undertaken.Its process and results are described below.
Methods
Nurse registars collect kcal/kg per patient per day into an online data base (TBI-trac®) as part of a quality initiative through the BTF. Quarterly data review demonstrated that the mean kcal/kg/day was well below goal. The QI team reviewed patient records, developed protocols, obtained attending physician consensus, monitored compliance, and revised the nutrition protocol as needed. The Nutrition Protocol was revised several times as results were analyzed. The current protocol calls for enteral feeding over 20 hours of the day, with a 4-hour time period built in for “catch up.”
Results
The percentage of patients receiving nutrition by day 2 increased from 29 percent in 2007 to 50 percent in 2010. Average kcal/kg on hospital day 5 increased from a mean of 14.4 in 2007 to 28.8 in 2010 (p = 0.006, Figure C-7). The percentage of patients who received at least 25 kcal/kg on day 5 increased from 19.4 percent in 2007 to 75 percent in 2010 (p = 0.018). The percentage of patients who achieved 25 kcal/kg/day on any day within the first 5 days postinjury increased from 25.8 percent in 2007 to 91.7 percent in 2010 (p = 0.001).
Findings
A successful feeding protocol must address the unique needs of the TBI patient. Our ICU found success by building into the protocol time each day to make up or catch up if feeds were held for any reason. With this protocol we have been successful in achieving adequate nutritional intake in the severe TBI patient.Figure C-7.
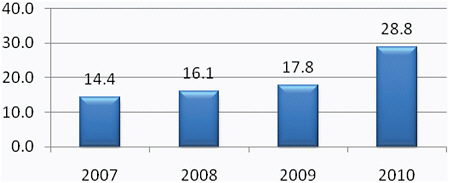
FIGURE C-7 Average kcal/kg received by patients in a Level I trauma center on hospital day 5 significantly increases from 2007 to 2010.
SOURCE: Ghajar, 2010.
CONCLUSIONS
In this severe TBI prospective database, nutritional support initiated within five days after trauma is associated with a significant reduction in two-week mortality. Furthermore, the amount of nutrition is related to mortality. These results held after controlling for other parameters known to affect mortality such as arterial hypotension, age, pupillary status, initial GCS, and CT scan findings. Thus, nutrition may be an independent predictor of mortality. A prospective, randomized trial would be necessary to confirm this finding to generate class I evidence. It is doubtful, however, that such a trial comparing nutrition vs. no nutrition will be done given the ethical implications. Together with arterial hypotension, hypoxia and intracranial hypertension, early nutritional support is one of the few therapeutic interventions that can directly affect outcome.
ACKNOWLEDGMENT
Permission obtained from Journal of Neurosurgery for the following article: Härtl, R., L. M. Gerber, Q. Ni, and J. Ghajar. 2008. Effect of early nutrition on deaths due to severe traumatic brain injury. Journal of Neurosurgery 109:50–56.
REFERENCES
Bastian, L., A. Weimann, W. Bischoff, P. N. Meier, M. Grotz, C. Stan, and G. Regel. 1998. Clinical effects of supplemental enteral nutrition solution in severe polytrauma. Der Unfallchirurg 101(2):105–114.
Bergsneider, M., D. A. Hovda, E. Shalmon, D. F. Kelly, P. M. Vespa, N. A. Martin, M. E. Phelps, D. L. McArthur, M. J. Caron, J. F. Kraus, and D. P. Becker. 1997. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. Journal of Neurosurgery 86(2):241–251.
Brain Trauma Foundation. 2000. Part 1: Guidelines for the management of severe traumatic brain injury. Journal of Neurotrauma 17(6–7):499–553.
Bulger, E. M., A. B. Nathens, F. P. Rivara, M. Moore, E. J. MacKenzie, and G. J. Jurkovich. 2002. Management of severe head injury: Institutional variations in care and effect on outcome. Critical Care Medicine 30(8):1870–1876.
Cahill, N. E., R. Dhaliwal, A. G. Day, X. Jiang, and D. K. Heyland. 2010. Nutrition therapy in the critical care setting: What is “best achievable” practice? An international multicenter observational study. Critical Care Medicine 38(2):395–401.
Chesnut, R. M., L. F. Marshall, M. R. Klauber, B. A. Blunt, N. Baldwin, H. M. Eisenberg, J. A. Jane, A. Marmarou, and M. A. Foulkes. 1993. The role of secondary brain injury in determining outcome from severe head injury. Journal of Trauma 34(2):216–222.
Clifton, G. L., C. S. Robertson, and S. C. Choi. 1986. Assessment of nutritional requirements of head-injured patients. Journal of Neurosurgery 64(6):895–901.
Demetriades, D., E. Kuncir, J. Murray, G. C. Velmahos, P. Rhee, and L. Chan. 2004. Mortality prediction of head abbreviated injury score and Glasgow Coma Scale: Analysis of 7,764 head injuries. Journal of the American College of Surgeons 199(2):216–222.
Deutschman, C. S., F. N. Konstantinides, S. Raup, P. Thienprasit, and F. B. Cerra. 1986. Physiological and metabolic response to isolated closed-head injury. Part 1: Basal metabolic state: Correlations of metabolic and physiological parameters with fasting and stressed controls. Journal of Neurosurgery 64(1):89–98.
Eyer, S. D., L. T. Micon, F. N. Konstantinides, D. A. Edlund, K. A. Rooney, M. G. Luxenberg, and F. B. Cerra. 1993. Early enteral feeding does not attenuate metabolic response after blunt trauma. Journal of Trauma 34(5):639–644.
Ghajar, J. 2010. The perspective of a neurosurgeon. Presented at Institute of Medicine’s Workshop on Nutrition and Neuroprotection in Military Personnel, Washington, DC, June 23, 2010.
Härtl, R., L. M. Gerber, L. Iacono, Q. Ni, K. Lyons, and J. Ghajar. 2006. Direct transport within an organized state trauma system reduces mortality in patients with severe traumatic brain injury. Journal of Trauma—Injury, Infection and Critical Care 60(6):1250–1256.
Hovda, D. A., S. M. Lee, M. L. Smith, S. Von Stuck, M. Bergsneider, D. Kelly, E. Shalmon, N. Martin, M. Caron, J. Mazziotta, M. Phelps, and D. P. Becker. 1995. The neurochemical and metabolic cascade following brain injury: Moving from animal models to man. Journal of Neurotrauma 12(5):903–906.
Jiang, J. Y., G. Y. Gao, W. P. Li, M. K. Yu, and C. Zhu. 2002. Early indicators of prognosis in 846 cases of severe traumatic brain injury. Journal of Neurotrauma 19(7):869–874.
Marik, P. E., and G. P. Zaloga. 2001. Early enteral nutrition in acutely ill patients: A systematic review. Critical Care Medicine 29(12):2264–2270.
Merenda, A., and R. Bullock. 2006. Clinical treatments for mitochondrial dysfunctions after brain injury. Current Opinion in Critical Care 12(2):90–96.
Narayan, R. K., M. E. Michel, B. Ansell, A. Baethmann, A. Biegon, M. B. Bracken, M. R. Bullock, S. C. Choi, G. L. Clifton, C. F. Contant, W. M. Coplin, W. D. Dietrich, J. Ghajar, S. M. Grady, R. G. Grossman, E. D. Hall, W. Heetderks, D. A. Hovda, J. Jallo, R. L. Katz, N. Knoller, P. M. Kochanek, A. I. Maas, J. Majde, D. W. Marion, A. Marmarou, L. F. Marshall, T. K. McIntosh, E. Miller, N. Mohberg, J. P. Muizelaar, L. H. Pitts, P. Quinn, G. Riesenfeld, C. S. Robertson, K. I. Strauss, G. Teasdale, N. Temkin, R. Tuma, C. Wade, M. D. Walker, M. Weinrich, J. Whyte, J. Wilberger, A. B. Young, and L. Yurkewicz. 2002. Clinical trials in head injury. Journal of Neurotrauma 19(5):503–557.
Perel, P., T. Yanagawa, F. Bunn, I. Roberts, R. Wentz, and A. Pierro. 2006. Nutritional support for head-injured patients. Cochrane Database of Systematic Reviews (4):CD001530..
Rapp, R. P., D. B. Young, and D. Twyman. 1983. The favorable effect of early parenteral feeding on survival in head-injured patients. Journal of Neurosurgery 58(6):906–912.
Roberts, I., D. Yates, P. Sandercock, B. Farrell, J. Wasserberg, G. Lomas, R. Cottingham, P. Svoboda, N. Brayley, G. Mazairac, V. Laloe, A. Munoz-Sanchez, M. Arango, B. Hartzenberg, H. Khamis, S. Yutthakasemsunt, E. Komolafe, F. Olldashi, Y. Yadav, F. Murillo-Cabezas, H. Shakur, and P. Edwards. 2004. Effect of intravenous corticosteroids on death within 14 days in 10,008 adults with clinically significant head injury (MRC Crash Trial): Randomised placebo-controlled trial. Lancet 364(9442):1321–1328.
Rovlias, A., and S. Kotsou. 2000. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery 46(2):335–343.
Sampalis, J. S., R. Denis, A. Lavoie, P. Fréchette, S. Boukas, A. Nikolis, D. Benoit, D. Fleiszer, R. Brown, M. Churchill-Smith, and D. Mulder. 1999. Trauma care regionalization: A process-outcome evaluation. Journal of Trauma—Injury, Infection and Critical Care 46(4):565–581.
Smith, J. S., L. F. Martin, W. W. Young, and D. P. Macioce. 1990. Do trauma centers improve outcome over non-trauma centers: The evaluation of regional trauma care using discharge abstract data and patient management categories. Journal of Trauma 30(12):1533–1538.
Sosin, D. M., J. E. Sniezek, and R. J. Waxweiler. 1995. Trends in death associated with traumatic brain injury, 1979 through 1992: Success and failure. Journal of the American Medical Association 273(22):1778–1780.
Taylor, S. J., S. B. Fettes, C. Jewkes, and R. J. Nelson. 1999. Prospective, randomized, controlled trial to determine the effect of early enhanced enteral nutrition on clinical outcome in mechanically ventilated patients suffering head injury. Critical Care Medicine 27(11):2525–2531.
Thurman, D. J., C. Alverson, K. A. Dunn, J. Guerrero, and J. E. Sniezek. 1999. Traumatic brain injury in the United States: A public health perspective. Journal of Head Trauma Rehabilitation 14(6):602–615.
Weekes, E., and M. Elia. 1996. Observations on the patterns of 24-hour energy expenditure changes in body composition and gastric emptying in head-injured patients receiving nasogastric tube feeding. Journal of Parenteral and Enteral Nutrition 20(1):31–37.
Nutrition Therapy for Patients with Traumatic Brain Injury in the Military
Kelli M. Metzger19
NUTRITION STATUS PRIOR TO TRAUMATIC BRAIN INJURY
Nutrition status of military service members prior to injury may vary greatly depending on the location to which they are deployed. During my deployment from December 2008
through November 2009, I was primarily in central Iraq. The Central Operating Bases (COBs) and Forward Operating Bases (FOBs) where I lived and those I visited in central and southern Iraq contained at least one, and sometimes several, contract-operated dining facilities. Most of these dining facilities provided four meals in a 24-hour period to serve both day- and night-shift workers. Meals at the dining facilities typically included two to four protein sources, two to four starch choices, two to three hot vegetables, salad bar, sandwich bar, specialty bar, fresh fruit, and desserts. Beverages available included water, juices, coffee, tea, sodas, ultra-high temperature (UHT) milk, and soy milk. In addition to the dining facilities, most COBs and FOBs contained at least one Army/Air Force Exchange where service members could purchase snack foods and beverages; some COBs and FOBs also housed local and American chain restaurants, which provide even more dining variety to those stationed there. Of course, many service members also received food items sent from friends and family members. Although service members at these locations have opportunities to make more healthful food choices, not all do on a regular basis. Many may be calorically nourished but lack important nutrients, particularly those associated with improved cognitive function, in their diets. In addition, some service members may be assigned to or routinely travel to more remote locations where the food choices are more limited and thus have less opportunity to obtain adequate nutrients.
MEDICAL TREATMENT OF INJURED MILITARY SERVICE MEMBERS IN THEATER AND THE UNITED STATES
Medical treatment for injured U.S. service members occurs at four echelons of care. Level 1 occurs at the unit level and includes the Battalion Aid Station and Combat Medic (Borden Institute, 2008). The combat medic provides initial treatment on the battlefield with the goal of casualty evacuation in less than an hour to at least the Level II echelon of care. Treatment in the field may consist of airway stabilization, fluid resuscitation, pain management, and brain-specific therapies. Airway management is critical in the TBI patient because of the risk of loss of consciousness impacting the ability to protect one’s airway (Girard, 2007). Level II care often includes a Forward Surgical Team (FST) comprised of one orthopedic surgeon, three general surgeons, two nurse anesthetists, one critical care nurse, and technicians (Nessen, 2008). Based on location, service members evacuated from Level I care may go to either the Level II or Level III echelon of care. Level III care is provided at Army Combat Support Hospitals (CSHs), Air Force theater hospitals, and Navy ships. Level III care involves triage; resuscitation; transfusion; initial, definitive, and reconstructive surgery; postoperative care; intensive care; and patient holding capacity (Borden Institute, 2008). Depending on the length of time to stabilize a patient and flying conditions, service members may be evacuated from theater the same day they are wounded or at the latest within 72 hours. Level IV echelon of care is provided at Landstuhl Regional Medical Center (LRMC) in Landstuhl, Germany. Almost 100 percent of U.S. service members evacuated from Afghanistan and Iraq pass through LRMC for general and specialized medical and surgical care (Borden Institute, 2008). The average length of stay at LRMC is less than four days. Research conducted by the RAND Corporation (Tanielian and Jaycox, 2008) found approximately 19.5 percent of the casualties admitted to military health-care facilities in the first five years of the war in Iraq suffered from TBI. Dr. Louis French, a neuropsychiatrist at Walter Reed Army Medical Center (WRAMC), estimates that approximately 35 percent of wounded warriors entering WRAMC in the past two years suffered from some type of TBI.
INITIAL NUTRITION GOALS
A dietitian’s first goal is ensuring patients receive adequate nutrition to prevent malnutrition and promote healing. A study (Härtl et al., 2008) based on information collected by the Brain Trauma Foundation in 20 Level 1 and 2 Level 2 trauma centers in New York state, found TBI patients who were not fed within five days of their injury had 2.1 times the risk of two-week mortality, while those not fed within seven days suffered 4.1 times the risk. Medical staff at LRMC typically place a nasogastric (NG) tube and initiate early enteral feeding within 24–48 hours of admission. In certain cases, such as hemodynamic instability or abdominal injury, this procedure may not be followed. The dietitians at Brooke Army Medical Center (BAMC) feed TBI patients with 2 Cal HN supplemented with glutamine, vitamin C, and selenium. According to a description by Abbott Nutrition, 2 Cal HN is a nutritionally complete, high-calorie liquid food designed to meet the increased protein and calorie needs of stressed patients and patients requiring low-volume feedings. This formula provides two calories per milliliter with 43 percent of calories from carbohydrate, 40 percent from fat, and 17 percent from protein. Dietitians at National Naval Medical Center (NNMC) also use 2 Cal HN with additional protein, glutamine, multivitamin, and Vitamin C, providing patients with 30–35 calories/kg body weight and close to 2 g/kg of protein. At WRAMC, dietitians aim for a similar calorie and protein level as NNMC, but feed with Impact Glutamine, specialized medical nutrition for surgical and trauma patients by Nestle Nutrition. Impact Glutamine contains a blend of glutamine, arginine, n-3 fatty acids, and nucleic acids providing 1.3 calories per milliliter; 46 percent of the calories are from carbohydrate, 30 percent are from fat, and 24 percent are from protein.
NUTRITION CONSIDERATIONS
In selecting the best feeding method and formulation, dietitians must consider other injuries and conditions as well as medications the patient is taking. The TBI or concurrent injuries may cause damage to the gut structure, interfering with the digestion process. If the gastrointestinal tract cannot be used to achieve nutritional goals within three days, total parenteral nutrition (TPN) should be started within 24–48 hours with a goal of reaching nutritional needs by the third or fourth day (Escott-Stump, 2008). Although parenteral nutrition may be easier than obtaining adequate enteral access, enteral nutrition has fewer incidence of complication and lower cost than parenteral with no significant differences in measured nutritional parameters (Kirby et al., 2007). Enteral feedings are preferred because they stimulate blood flow to the gastric lining mucosa and provide a comprehensive mix of macro and micronutrients. A greater number of patients tolerate jejunal feedings better than gastric feeding within 72 hours after injury (Bratton et al., 2007). A jejunal feeding also can be used to reduce gastric intolerance and residuals found in gastric feeding as well as the use of intravenous catheters required in TPN (Bratton et al., 2007). Enteral feedings should begin as soon as the patient is hemodynamically stable.
DETERMINING AND MAINTAINING ADEQUATE CALORIE LEVELS
According to research by Wilson et al. published in 2001, the nutrition goals should include attempting to reach 35–45 kcal/kg and a protein intake of 2–2.5 g/kg on day 1 or as soon as possible (Escott-Stump, 2008). Adequate calories are important to prevent malnutrition and to promote healing and recovery. The brain’s function as the regulator for metabolic activity leads to a complex milieu of metabolic alterations in TBI consisting of hormonal
changes, aberrant cellular metabolism, and a vigorous cerebral and systemic inflammatory response in an effort to liberate substrate for injured cell metabolism. The degree of this hypermetabolic state is proportional to the severity of injury and motor dysfunction (Fruin et al., 1986). Indirect calorimetry is the gold standard for determining the calorie needs of the patient.
NUTRIENT-DRUG INTERACTIONS
Dietitians need to keep in mind, however, that medications may affect calorie needs and/or digestion by either their effect on metabolism or the way they are packaged. For example, pentobarbital, used to induce a pharmacologic coma, reduces calorie needs to as low as 76–86 percent of predicted energy needs. Protein requirements also may be less, as reflected by a 40 percent decrease in urinary nitrogen excretion (Cook et al., 2008). Propofol, a short-acting sedative, is delivered in a lipid emulsion, and 1 mL of propofol contains approximately 0.1 g of fat (1.1 kcal). Because energy contributed from propofol may provide as much as 50 to 80 percent of resting energy expenditure (REE), nutritional requirements of patients receiving propofol over an extended period should be adjusted accordingly (Rajpal and Johnston, 2009). In addition, narcotics and neuromuscular blocking agents may slow peristalsis resulting in nausea, vomiting, gastroparesis, and ileus. Metoclopramide, erythromycin, or raglan may be used to promote gastric emptying. Until normal peristalsis resumes, however, high fiber enteral formulas should be avoided.
NUTRIENTS FOR CONSIDERATION
Once an adequate calorie level is determined and a formula is selected, the dietitian may consider adding nutrients to improve outcomes. Glutamine is an immune-enhancing nutrient that has been tested and found beneficial. Glutamine is used as a source of energy for cells of the intestinal epithelium and immune system (Falcão De Arruda and De Aguilar-Nascimento, 2004). Glutamine also supplies nitrogen for purine and pyrimidine synthesis, which are essential for cells in mitosis. The use of glutamine seems to be able to decrease the occurrence of bacterial translocation and inflammatory response, reducing the possibility of events such as systemic inflammatory response syndrome and sepsis. In a study in a hospital in Brazil (Falcão De Arruda and De Aguilar-Nascimento, 2004), enhancing enteral nutrition with glutamine and probiotics significantly reduced the incidence of infection in head trauma patients. Another potential benefit of early enteral nutrition enriched with probiotics and glutamine reduction is the period of time in the intensive care unit and the number of days requiring mechanical ventilation (Falcão De Arruda and De Aguilar-Nascimento, 2004).
Zinc is another nutrient of interest for the TBI patient. Zinc is an important co-factor for substrate metabolism, immune function, and N-methyl-D-aspartate (NMDA) receptor function (Cook et al., 2008). Because of zinc losses through the gastrointestinal tract and its role in wound healing, it may be prudent to supplement zinc in acutely injured patients (Winker and Malone, 2010). Supplementation of zinc appears to improve protein metabolism and neurologic outcome at one month after TBI (Young et al., 1996). Magnesium may also be neuroprotective because of activity at the NMDA receptor and modulation of cellular energy production and calcium influx, but supplementation of magnesium in humans has yet to yield definitive benefits (McKee et al., 2005).
Choline also may be an important nutrient for patients with TBI. Choline, a B-complex vitamin found in eggs, meat, fish, nuts, legumes, and soy, is a component of the neurotransmitter acetylcholine (Hecht, 2007). Doses of choline as high as 2,500 mg twice per day may
improve memory in adults. Every cell membrane requires phosphatidylcholine; nerve and brain cells especially need large quantities for repair and maintenance (Hecht, 2007). Additional studies have found doses of choline at 100 mg and 400 mg/kg significantly reduce brain edema and breakdown of the blood-brain barrier following TBI (Hecht, 2007).
Arginine is a conditionally essential amino acid. Under usual conditions, arginine is synthesized endogenously; in stressful periods, however, endogenous synthesis is insufficient (Kirby et al., 2007). Arginine is used for protein synthesis; as part of the urea cycle; as a precursor to glutamate, proline, and polyamines; and as a substrate for creatine and nitric oxide production (Sy et al., 2006). When pharmacologic doses are given, arginine stimulates the pituitary growth hormone, insulin-like growth factor, prolactin, insulin, and other hormones, resulting in a net positive effect on wound healing and immune functions (Alexander, 1993). Arginine also is a precursor for nitrates, nitrites, and nitric oxide. Nitric oxide is important as a vasodilator, but also participates in immunologic reactions which include the ability of macrophages to kill tumor cells and bacteria. Studies have demonstrated T-cell function suppressed following major surgery or trauma. Daly and colleagues found that patients receiving 30 g/day of arginine demonstrated a quicker return to normal T-cell level compared to post-surgical patients receiving placebo (Daly et al., 1988).
NUTRITION CONCERNS IN OUTPATIENT REHABILITATION
Although initial nutrition concerns include adequate calories and supplementation of nutrients, nutrition concerns continue into rehabilitation. Patients with mild TBI may experience memory problems and difficulty concentrating, which affect their ability to perform daily activities and return to work (Miele and Bailes, 2009). Patients who sustained a moderate TBI have highly variable outcomes. Moderate brain injury survivors may suffer from cognitive or behavioral impairments that disrupt relationships, employment, or psychological well-being (Timmons and Winestone, 2009). Ninety percent of TBI patients who had good outcomes experienced memory difficulties, and 87 percent had problems performing activities of daily living (Timmons and Winestone, 2009). Despite intensive intervention, long-term disability occurs in a large portion of the survivors of severe head injury (Remig, 2010). A significant percentage of TBI patients admitted to long-term rehabilitation centers or sent home with skilled nursing support are markedly disabled and physically dependent on others for care. Many of these patients have cognitive and motor dysfunction; less than 33 percent are able to eat independently, and about 37 percent require either enteral or parenteral nutrition support (Cook et al., 2008).
Although some TBI patients require assistance with eating, most TBI patients regain their independence in oral feeding within six months after injury (Cook et al., 2008). Those with dental or facial fractures or a need for prolonged cervical immobilization with a hard cervical collar may experience a delay in initiation of an oral diet. Dysphagia can affect 25 to 60 percent of TBI patients and as many as 42 percent suffer from frank aspiration (McNamee et al., 2009). Speech pathologists can assess a patient’s endoscopic instrumental swallowing evaluations. Based on these results, patients may require modified food and liquid consistencies for their safety (Cook et al., 2008). The overall goal is to find the least restrictive diet that promotes safe swallowing and maintains nutritional status (Kirby et al., 2007). In addition to swallowing evaluation, patients should be assessed for readiness to feed. Barriers to self-feeding include cognitive and motor planning behaviors, including impulsivity, distractibility, inability to stay on task, poor sequencing skills, lack of initiation, and inability to motor plan self-feeding (Kirby et al., 2007). Patients with other injuries also may experience difficulty eating. If limb weakness, paralysis, or amputation occur on
the dominant side of the body, poor coordination resulting from a new reliance on the non-dominant side may make eating difficult and unpleasant. Small frequent feedings can help if fatigue or early satiety is a problem (Remig, 2010).
The role of the dietitian is important in the care of patients with TBI. Dietitians are needed to ensure the patient receives adequate nutrition immediately following the injury to prevent malnutrition and promote recovery. They also have a role to play as patients transition to rehabilitation centers and outpatient status. Dietitians can work with other team members to ensure the patient understands the foods they can best tolerate and help patients determine an appropriate calorie and nutrient level as they recover.
REFERENCES
Alexander, J. W. 1993. Immunoenhancement via enteral nutrition. Archives of Surgery 128(11):1242–1245.
Borden Institute. 2008. Trauma system development and medical evacuation in the combat theater. In War surgery in Afghanistan and Iraq: A series of cases, 2003–2007, edited by S. C. Nessen, D. E. Lounsbury and S. P. Hetz. Washington, DC: Office of the Surgeon General, U.S. Army.
Bratton, S., D. Chestnut, J. Ghajar, F. Hammond, O. Harris, R. Hartl, J. Schouten, L. Shutter, S. Timmons, J. Ullman, W. Videtta, J. Wilberger, and D. Wright. 2007. Nutrition. Journal of Neurotrauma 24(Suppl. 1):S77–S82.
Cook, A. M., A. Peppard, and B. Magnuson. 2008. Nutrition considerations in traumatic brain injury. Nutrition in Clinical Practice 23(6):608–620.
Daly, J. M., J. Reynolds, A. Thom, L. Kinsley, M. Dietrick-Gallagher, J. Shou, and B. Ruggieri. 1988. Immune and metabolic effects of arginine in the surgical patient. Annals of Surgery 208(4):512–523.
Escott-Stump, S. 2008. Nutrition and diagnosis-related care. 6th ed. Baltimore, MD: Wolters Kluwer Health/Lippincott Williams & Wilkins.
Falcão De Arruda, I. S., and J. E. De Aguilar-Nascimento. 2004. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clinical Science 106(3):287–292.
Fruin, A. H., C. Taylon, and M. S. Pettis. 1986. Caloric requirements in patients with severe head injuries. Surgical Neurology 25(1):25–28.
Girard, P. 2007. Military and VA telemedicine systems for patients with traumatic brain injury. Journal of Rehabilitation Research and Development 44(7):1017–1026.
Härtl, R., L. M. Gerber, Q. Ni, and J. Ghajar. 2008. Effect of early nutrition on deaths due to severe traumatic brain injury. Journal of Neurosurgery 109(1):50–56.
Hecht, J. 2007. Nutraceuticals. In Brain injury medicine: Principles and practice, edited by N. Zasler, D. Katz, and R. Zafonte. New York: Demos Medical Publishing. Pp. 1037–1047.
Kirby, D., L. Creasy, and S. Abou-Assi. 2007. Gastrointestinal and nutritional issues. In Brain injury medicine: Principles and practice, edited by N. Zasler, D. Katz and R. Zafonte. New York, NY: Demos Medical Publishing. Pp. 657–671.
McKee, J. A., R. P. Brewer, G. E. Macy, B. Phillips-Bute, K. A. Campbell, C. O. Borel, J. D. Reynolds, and D. S. Warner. 2005. Analysis of the brain bioavailability of peripherally administered magnesium sulfate: A study in humans with acute brain injury undergoing prolonged induced hypermagnesemia. Critical Care Medicine 33(3):661–666.
McNamee, S., T. Pickett, S. Benedict, and D. Cifu. 2009. Rehabilitation. In Neurotrauma and critical care of the brain, edited by J. Jallo and C. Loftus. New York: Thieme. Pp. 385–403.
Miele, V., and J. Bailes. 2009. Mild brain injury. In Neurotrauma and critical care of the brain, edited by J. Jallo and C. Loftus. New York: Thieme. Pp. 175–207.
Nessen, S. C., D. R. Cronk, J. Edens, B. J. Eastridge, T. R. Little, J. Windsor, L. H. Blackbourne, and J. B. Holcomb. 2008. U.S. Army two-surgeon teams operating in remote Afghanistan—An evaluation of split-based forward surgical team operations. The Journal of Trauma 66(4 Suppl.):S37-47.
Rajpal, V., and J. Johnston. 2009. Nutrition management of traumatic brain injury patients. Support Line 31(1):10–19.
Remig, V. 2010. Medical nutrition therapy for neurologic disorders. In Krause’s food and nutrition therapy. 12th ed., edited by L. Mahan and S. Escott-Stump. St. Louis, MO: Saunders, Elsevier. Pp. 1067–1101.
Sy, B., E. Dweik, and R. Sweik. 2006. Arginine and nitric oxide. In Modern nutrition in health and disease. 10th ed., edited by M. Shils, M. Shike, A. Ross, B. Coballero, and R. Cousins. Baltimore, MD: Lippincott, Williams, and Wilkins. Pp. 571–581.
Tanielian, T., and L. Jaycox. 2008. Invisible wounds of war: Psychological and cognitive injuries, their consequences, and services to assist recovery. Santa Monica, CA: RAND Corporation.
Timmons, S., and J. Winestone. 2009. Moderate brain injury. In Neurotrauma and critical care of the brain, edited by J. Jallo and C. Loftus. New York: Thieme. Pp. 208–219.
Winker, M., and A. Malone. 2010. Medical nutrition therapy for metabolic stress: Sepsis, trauma, burns, and surgery. In Krause’s food and nutrition therapy. 12th ed., edited by L. Mahan and S. Escott-Stump. St. Louis, MO: Saunders, Elsevier. Pp. 1021–1041.
Young, B., L. Ott, E. Kasarskis, R. Rapp, K. Moles, R. J. Dempsey, P. A. Tibbs, R. Kryscio, and C. McClain. 1996. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. Journal of Neurotrauma 13(1):25–34.
The Therapeutic Potential of Diet and Exercise to Counteract Brain Dysfunction Following Traumatic Brain Injury
Fernando Gomez-Pinilla20
INTRODUCTION
Traumatic brain injury (TBI) induces a state of vulnerability within neurons that survive the initial insult, and this may result in long-term deficits in higher order cognitive and intellectual functions. Advances in understanding how nutritional factors affect brain function and repair have put forward the interesting possibility that dietary therapy is a realistic strategy to reduce the type of weaknesses encountered in TBI pathology. In particular, recent studies show that the efficacy of nutritional factors after TBI is displayed at the level of processes involved in re-establishing energy homeostasis and providing structural substrates that can foster neuronal signaling. An increasing number of studies indicate that certain types of dietary factors, such as n-3 fatty acids, can positively influence molecular systems that serve synaptic function, while diets rich in saturated fats or high in calories do the opposite.
Dietary interventions have the advantage of being non-invasive, highly efficacious, and borne with a broad spectrum of action. These features provide the confidence that findings in animal models of TBI can be easily translated to human applications directed to reduce TBI pathology. This capacity contrasts with the large amount of unsuccessful clinical trials to assess the therapeutic action of many pharmacological compounds in TBI patients. This implies that the type of broad protection elicited by dietary factors can be particularly suitable to treat brain disorders characterized by multiple components (Figure C-8). For example, it is likely that the diffuse nature of TBI, can compromise fundamental and broad aspects of neuronal signaling events that are required for mental operation. Although cognitive and psychiatric disorders are a common feature in TBI patients, such as posttraumatic stress disorders, their cellular/molecular basis and treatment are poorly understood. The fact that select dietary factors support a large range of molecular mechanisms important for cognitive function and neural repair, strongly suggests that dietary therapy is a suitable strategy to promote mental health and brain plasticity after TBI.
THE METABOLIC DEPRESSION AFTER TBI
Brain metabolic depression is a common stage in the sequence of events occurring after TBI as observed in humans (Vagnozzi et al., 2008) and in animal models (Dietrich et al.,
FIGURE C-8 Synaptic transmission where most signaling events take place requires proper energy and a functional plasma membrane (disrupted by TBI).
SOURCE: Gomez-Pinilla, 2008.
1994; Hovda et al., 1991; Moore et al., 2000). The period of energy depression seems to impose a toll on the ability of the brain to maintain normal function and support repair events. It has been known that during this period, the brain is more vulnerable to a second injury. For example, it has recently been reported in the results of a pilot study performed in a cohort of athletes exposed to a single and a double concussion (Vagnozzi et al., 2008). Brain imaging based on 1H-MR spectroscopy was performed to assess the neuronal metabolic marker N-acetylaspartate (NAA) in the cerebral cortex of athletes, at different time points after concussion. Results showed that the concussion opens a temporal window of brain metabolic imbalance, the ending of which does not necessarily coincide with the im-
provement of clinical symptoms. Remarkably, a second concussive event occurring within the period of brain vulnerability prolonged the time required for NAA normalization by 15 days.
Experiments performed in animal models of concussion have shown similar results to those observed in humans and have revealed some of the mechanisms involved in the period of brain vulnerability. Following experimental concussion or lateral fluid percussion injury in the rat, the brain exhibits a decrease in glucose utilization lasting for up to 10 days after the injury (Ginsberg et al., 1997; Yoshino et al., 1991). During this postinjury period of metabolic depression, cells are unable to respond to physiological levels of stimulation, resulting in subsequent neurobehavioral deficits (Colle et al., 1986; Hovda et al., 1987). The energy depression seems to impose a toll on the molecular machinery that supports activity-dependent plasticity. This means that the reduced capacity for synaptic plasticity can have critical implications for the ability of the brain to start a recovery phase. Therefore, it is critical to develop strategies directed to normalize the period of metabolic depression following TBI and counteract the events that arrest proper neuronal function.
THE FUNCTION OF BRAIN-DERIVED NEUROTROPHIC FACTOR SEEMS CRUCIAL TO UNDERSTAND THE EFFECTS OF FOOD ON THE BRAIN
Brain-derived neurotrophic factor (BDNF) is an important regulator of neuronal growth and survival at different developmental stages, and it can function to enhance synaptic plasticity underlying cognitive function. As discussed in subsequent sections, experimental TBI has been shown to reduce BDNF-mediated synaptic plasticity while the action of several dietary factors on the brain has been associated with restoring the function of BDNF. Clinical studies support the importance of proper BDNF function for maintaining learning and memory capacities in humans (Egan et al., 2003; Hariri et al., 2003), i.e., individuals expressing a specific polymorphism in the BDNF gene exhibit learning impairments (Egan et al., 2003). A new line of investigations shows that the action of BDNF on synaptic plasticity is intimately related to the regulation of energy metabolism. The results of these investigations have opened new avenues to understand the mechanism of action of dietary factors on the brain. For example, it is now known that BDNF influences synaptic plasticity by acting on molecular systems important for the regulation of energy homeostasis in the hippocampus (Gomez-Pinilla et al., 2008; Vaynman et al., 2006). These actions of BDNF seem to have profound consequences for the neural control of body metabolism as animals with genetic deletion of the BDNF gene are hyperphagic and develop obesity (Lyons et al., 1999), while infusion of BDNF has been found to reduce body weight, normalize glucose levels, ameliorate lipid metabolism in diabetic rodents, and increase insulin sensitivity (Tsuchida et al., 2002). It is also notable that BDNF protein is most abundant in brain areas foremost associated with cognitive and neuroendocrine regulation, such as the hippocampus and hypothalamus.
NUTRITIONAL FACTORS WITH THE POTENTIAL TO REDUCE TBI PATHOLOGY
The potential of nutrient and/or dietary interventions to reduce TBI pathology has been demonstrated to affect at least four primary objectives to moderate cognitive and emotional distress following TBI:
-
to reduce energy crisis and promote membrane repair,
-
to elevate the potential for synaptic plasticity,
TABLE C-6 Main Nutrients Examined in Animal Models of TBI
|
Nutrient |
Type of Model |
|
Omega-3 fatty acids (DHA) |
Concussion model of TBI (Mills, 2010; Wu et al., 2006, 2007) |
|
|
Models of spinal cord injury (Huang, 2007) |
|
Curcumin (turmeric) |
Concussion models of TBI (Sharma, 2010; Wu et al., 2006) |
|
Flavonoids |
Reduction of stroke in humans (Keli, 1996) Plaque formation in Alzehimer’s Disease animal models (Joseph, 2003) |
|
Vitamin E |
Models of concussion (Wu et al., 2006, 2010) |
|
Ketogenic diet |
Models of head injury (Hu, 2009; Prins, 2005) |
|
Dietary restriction or fasting |
Models of head injury (Davis, 2008) |
|
Caffeine |
Models of head injury (Li et al., 2008) |
|
SOURCE: Gomez-Pinilla and Ying, 2010 (workshop presentation). |
|
-
to support neuronal signaling, and
-
to stimulate mechanisms that can provide broad protection.
Several dietary components that can provide some modification to these processes have been identified in the following sections (Table C-6). Some of the effective dosage for the use of select nutrients in the brain have been identified in animal models of neurological disorders. Although dosage is an important issue, it is less of a factor for the use of nutrients based on their safety profile.
n-3 Fatty Acids
The study of n-3 fatty acids have provided some of the strongest evidence for the profound effects that dietary factors can have on the brain. The n-3 is a large family of fatty acids in which the docosahexaenoic acid (DHA) is one of the most relevant forms for brain function. It is important to consider, however, that DHA may act together with other n-3 fatty acids such as eicosapentaenoic acid (EPA), which also has demonstrated neuroprotective abilities. DHA acid is a key component of neuronal membranes at sites of signal transduction at the synapse, such that its action is vital for the maintenance of neuronal structure and function (Gomez-Pinilla, 2008). Because of the inefficiency of mammals to produce DHA from precursors, supplementation of DHA in the diet is important for insuring proper function of neurons during homeostatic conditions and after injury. Evidence suggests that DHA serves to improve neuronal function by supporting synaptic membrane fluidity (Suzuki et al., 1998) and regulating gene expression and cell signaling (Salem et al., 2001). This implies that insufficient DHA can result in neuronal dysfunction affecting a broad array of functional modalities.
The literature shows positive results for the effects of DHA in the injured central nervous system. It has been found that dietary DHA when provided for a few weeks before the onset of the injury can promote resistance against the effects of brain trauma (Wu et al., 2004a, 2007). In particular, animals exposed to experimental brain trauma that had been supplemented with DHA showed nearby normal performance in the Morris water maze and nearby normal levels of BDNF-related synaptic markers in the hippocampus. DHA also has
shown to overcome the effects of the injury when supplemented in the diet of animals after the injury onset, acting on the brain (Wu et al., 2004a, 2007) and spinal cord (Huang et al., 2007; Mills et al., 2010). These studies have shown that DHA can influence the injured brain by maintaining normal levels of BDNF-associated synaptic plasticity and reducing injury-related oxidative stress (Wu et al., 2007, 2008), and these DHA effects were accompanied by nearby normal cognitive abilities. As discussed in a later section, the concurrent application of exercise to animals fed on DHA dietary supplementation has been shown to have additional beneficial effects on synaptic plasticity and cognition (Wu et al., 2008).
Dietary Polyphenols
Polyphenols are found in plants and are characterized by the presence of one or more phenol groups. Curcuminoids and flavonoids are the main polyphenol subtypes with recognized actions on the brain. An inverse correlation between dietary flavonoids consumption and the incidence of stroke was found in a cohort of 552 men aged 50–69 years who were followed for 15 years (Keli et al., 1996). It has been shown that dietary supplementation of blueberry extracts for eight weeks can reverse cognitive deficits in spatial learning ability in aged rats (Andres-Lacueva et al., 2005). Blueberry extracts in the diet also have shown to reduce plaques in an Alzheimer’s disease (AD) animal model (Joseph et al., 2003), apparently acting on signaling pathways important for memory formation. Although polyphenols are known for their powerful antioxidant capacity, their antioxidant effects on the brain seem to follow multiple mechanisms related to the chemistry of each flavonoid. The flavanol epicatechin found in cocoa has been shown to cross the blood-brain barrier (BBB) after ingestion in food or drink, and its consumption improved retention of spatial memory and angiogenesis in the water maze (van Praag, 2009). The property to penetrate the BBB seems to extend to other polyphenols as the flavonoid epigallocatechin gallate (Suganuma et al., 1998), and the citrus flavonoids naringenin and hesperitin (Youdim et al., 2003) have been reported to enter the brain after a gastric administration.
Curcumin
Curcumin is a major chemical component of the turmeric plant (Curcuma longa) and has been widely used as a spice and food preservative in India. Curcumin has shown excellent efficacy in counteracting neuronal dysfunction in several models of neurodegenerative diseases such as AD and focal cerebral ischemia (see Gomez-Pinilla, 2008 for review). Recent findings show that curcumin stimulates proliferation of embryonic progenitor cells and neurogenesis in the adult hippocampus (Kim et al., 2008). Curcumin also has been shown to protect the hippocampus and to counteract learning impairment resulting from experimental TBI, in a process involving the action of the BDNF system (Wu et al., 2006). There is substantial evidence from in vitro studies indicating that curcumin has strong antioxidant capacity exerted by increasing free radical scavengers and reducing lipid peroxidation (Wei et al., 2006). As discussed earlier, a disruption in energy metabolism is a major sequela in the acute pathology of TBI, thereby compromising synaptic function and the capacity of the brain to respond to challenges. Interestingly, recent studies have shown the potential of curcumin to restore molecular events important for energy homeostasis following TBI (Figure C-9). Study results (Sharma et al., 2010) showed that four weeks of curcumin dietary supplementation before a fluid percussion injury counteracted a decrease in the levels of AMP-activated protein kinase (AMPK), ubiquitous mitochondrial creatine kinase (uMtCK), and cytochrome c oxidase II (COX-II). These molecular systems play a crucial role in main-
FIGURE C-9 Possible events by which TBI can affect brain function by disrupting plasma membrane homeostasis. TBI increases free radical formation in the mitochondria, thereby promoting oxidation of membrane lipids, as detected by the lipid peroxidation marker 4-HNE. These events may lead to membrane dysfunction as evidenced by reduced levels of iPLA2 with subsequent effects on membrane DHA. These events may lower membrane function as evidenced by reduced levels of STX-3 and GAP-43. The loss of membrane flexibility may compromise function of membrane embedded receptors such as the subunit NR2B of the NMDA receptor. These alterations can result in abnormal neuronal signaling, which can reduce cognitive capacity. Interventions such as exercise and curcumin with demonstrated antioxidant abilities may be able to restore membrane homeostasis and brain function after TBI.
SOURCE: Modified from Sharma et al., 2010.
taining energy homeostasis; therefore, results seem to portray the capacity of curcumin to attenuate the period of metabolic dysfunction and to foster functional recovery.
Cerebral edema is a major sequela in the TBI pathology that largely contributes to reducing neuronal function and neural repair events. It has been known for a few years already that aquaporins water membrane channels play a role in edema formation after TBI (Neal et al., 2007). In agreement with its strong anti-inflammatory capacity (Menon and Sudheer, 2007), it has recently been shown that a single injection of curcumin 15 minutes prior to or 30 minutes after a cortical impact injury can reduce brain water contents and improve neurological outcome. These effects of curcumin seem associated with the blocking of aquaporin-4 expression (Laird et al., 2010). In addition, the protective effect of curcumin was associated with a significant attenuation in the acute pericontusional expression of interleukin-1b, a pro-inflammatory cytokine.
Green Tea
Green tea is rich in flavonoids (Graham, 1992) out of which the catechins epigallocatechingallate (EGCG), epigallocatechin (EGC), epicatechin (EC), and epicatechin-3-gallate (ECG) are the most abundant. The intake of the compound catechin has been associated with a variety of positive effects in rodents inflicted with ischemia-induced degeneration, including antioxidative, anti-inflammatory, and anti-apoptotic (Sutherland et al., 2006). In particular, a four-year follow-up study that included 5,910 individuals showed that the incidence of stroke was two-fold higher in those individuals who consumed less than 5 cups per day (Sato et al., 1989). Green tea consumption also has been related to the prevention of various structural and biochemical and behavioral characteristics of aging. For example, long-term exposure of rodents to green tea has been shown to reverse some of the degenerative effects of aging in the hippocampus (Assuncao et al., 2009; Li et al., 2009) and has shown to prevent memory regression and DNA oxidative damage in aged mice.
Resveratrol
Resveratrol is a non-flavonoid polyphenolic found in grapes, red wine, and berries. There are two isomeric forms of resveratrol, and the transresveratrol (trans-3,4,5-trihydroxystilbene) is the biologically inactive form. A number of studies have demonstrated the antioxidant, anti-inflammatory, antimutagenic, and anticarcinogenic effects of resveratrol (de la Lastra and Villegas, 2005; Jang et al., 1997; Soleas et al., 1997). Interestingly, several epidemiological studies indicate an inverse correlation of wine consumption and the incidence of AD (Lindsay et al., 2002; Orgogozo et al., 1997; Truelsen et al., 2002). It is well-known that reducing food intake or caloric restriction extends lifespan in a wide range of species. Recently, it has been found that resveratrol can mimic certain aspects of calorie restriction such as increasing production of sirtuin proteins (Baur et al., 2006). The sirtuin is a phylogenetically conserved family of enzymes that catalyze NAD-dependent protein deacetylation, and is essential for lifespan extension elicited by caloric restriction and other stressors (Anderson et al., 2003). Interestingly, resveratrol’s neuroprotective aptitude appears to be the result of its effective capability to promote caloric homeostasis acting on the mitochondria resulting in low oxidative stress and more efficient neuronal function.
Ketogenic Diet
The ketogenic diet, which is high in fat (mainly medium-chain triglycerides), moderate in protein, and low in carbohydrates, has been used for the treatment of seizure disorders for more than 50 years and has lately been regarded as protective for other types of neurological disorders. The ketogenic diet has been shown to reduce the extent of neuronal degeneration in developmental animals affected by TBI (Hu et al., 2009; Prins et al., 2005). It has recently been shown in vitro (Samoilova et al., 2010) that treatment with the ketone body D-β-hydroxybutyrate counteracts the consequences of chronic hypoglycemia, oxygen-glucose deprivation, and excitotoxicity, and that these effects are independent of seizure control. The hypothesis behind these results is that ketosis may serve as an alternative energy source during the TBI metabolic depression phase, or may help reduce metabolic stress. These ideas seem indirectly supported by the results of studies showing that reduction of calories by fasting, which is neuroprotective in animal models of TBI, elicits a stage of brain ketosis (Davis et al., 2008).
As discussed earlier, restoration of energy balance is a major goal for overcoming the
pathobiology of TBI. In these terms, dietary agents that promote metabolic homeostasis or the calorie content of the diet can play crucial roles in restoring neuronal plasticity and brain function. Accordingly, strategies such as reducing the caloric content of the diet or the frequency of feeding seem to play a protective role on the brain. For example, fasting every other day has been shown to protect neurons in the hippocampus against excitotoxicity-induced death in rodent studies (Bruce-Keller et al., 1999). It can be noted that restriction of calories can increase levels of BDNF, which may contribute to support neuronal function. The effects of caloric restriction are also extended to the spinal cord. For example, it has been shown that reducing the frequency of food consumption can improve functional recovery after a partial injury to the spinal cord (Graham, 1992; Plunet et al., 2008).
Vitamin E
Vitamin E, which is naturally found in certain oils, nuts, and spinach, has shown promise in protecting the brain against the effects of TBI (Wu et al., 2010) by reducing free radical contents in the brain, which would otherwise impede optimal function of neurons. Vitamin E also has shown positive effects on reducing memory decay in aging individuals (Perkins et al., 1999). A different study in aging mice revealed the benefits of vitamin E by showing a correlation between the amount of ingested vitamin E and improved neurological performance, survival, and brain mitochondrial function (Navarro et al., 2005).
What Diets to Avoid
Although certain foods seem to contribute positively to enhance neuronal health, diets that are rich in saturated fats and sugar can do the opposite. Molteni and colleagues have shown that rats fed a diet high in saturated fats and refined sugars (similar in content to “junk food”) for a period of one to two months, performed significantly worse on the spatial learning Water maze test (Molteni et al., 2002). The increased levels of oxidative stress induced by this diet result in decreased levels of hippocampal BDNF, thereby reducing cognitive abilities; however, all of these effects were counteracted by antioxidant treatment with vitamin E (Wu et al., 2004b). Even more alarming is the fact that consumption of this high-fat diet for a period of three weeks made the effects of experimental TBI worse, in terms of reducing levels of BDNF-related synaptic plasticity and protracted learning and memory ability (Wu et al., 2003). Interestingly, the application of voluntary exercise concurrent to the consumption of the diet reduced its deleterious effects on cognition and synaptic plasticity (Molteni et al., 2004).
DIET AND EXERCISE COLLABORATE TO PRESERVE MEMBRANE STRUCTURE, BRAIN METABOLISM, AND SYNAPTIC PLASTICITY
Much like a healthful diet, physical activity is thought to benefit neuronal function. According to recent studies, the combination of diet and exercise can deliver more beneficial effects than either intervention alone. Studies show that exercise applied after experimental traumatic brain injury have beneficial effects, but these effects seem to depend on the length of the postinjury resting period and the severity of the injury (Griesbach et al., 2007). Because of the potential of certain diets to restore energy homeostasis, dietary management can be regarded as an intervention that can increase the efficacy of exercise after TBI.
The complementary influence of diet and exercise on the brain can be exerted at various levels of interactions, which circumvent around mechanisms implicated with the control of
energy homeostasis and synaptic plasticity (Gomez-Pinilla et al., 2008). Probably the best demonstrated interaction between exercise and diet involves the consumption of the n-3 polyunsaturated fatty acid DHA. Dietary supplementation with DHA and exercise influences hippocampal plasticity and cognitive function activating similar molecular systems, and the DHA effects are enhanced by the concurrent application of exercise (Chytrova et al., 2010; Wu et al., 2008). According to these studies, exercise seems to act on mechanisms that preserve DHA on the plasma membrane with important implications for neuronal signaling. In addition, the concurrent effects of the DHA diet and exercise involve BDNF-mediated synaptic plasticity such as the Akt signaling system (Wu et al., 2008). The results of these studies are significant and suggest the inherent capacity of the brain to benefit from the effects of DHA dietary supplementation and exercise. More in-depth studies (Gomez-Pinilla and Ying, 2010) have shown that the DHA diet and exercise exerted differential effects on molecular systems controlling important aspects of brain homeostasis, associated with food intake (obesity receptor for leptin, growth hormone receptor), energy metabolism (AMPK, SIRT1), and stress (glucocorticoid receptor, 11β-Hydroxysteroid dehydrogenase type 1), and all of them with the capacity to influence cognition. In agreement with the involvement of the hypothalamus and the hippocampus on energy homeostasis and behavior, these two regions showed distinct susceptibilities to the actions of diet and exercise. On another front, the combination of a flavonoid-enriched diet and exercise has been shown to increase the expression of genes that have a positive effect on neuronal plasticity while decreasing genes involved with deleterious processes, such as inflammation and cell death (van Praag, 2009). Exercise also has proven to be effective in reducing the deleterious effects of unhealthful diets, i.e., a diet high in saturated fat and sucrose. The concurrent exposure to exercise compensated for the effects of this diet on reducing the levels of BDNF-related synaptic plasticity and cognitive function (Molteni et al., 2004).
Control of cell energy metabolism seems to be a common denominator for the effects of foods and exercise on the brain, as several of the processes outlined above subside on the management of cellular energy. Now we know that there is a direct association between pathways associated with metabolism and synaptic plasticity, and this association can determine important aspects of behavioral plasticity such as learning and memory (Vaynman et al., 2006). Proteomic studies have revealed that voluntary exercise affects the expression pattern and post-translational modification of protein classes in the hippocampus associated with energy metabolism and synaptic plasticity (Ding et al., 2006). In particular, exercise modulates molecular systems in the brain associated with energy balance and energy transduction, which have the capacity to affect learning and memory, i.e., AMPK, ghrelin, ubiquitous mitochondrial creatine kinase (uMtCK), uncoupling protein 2 (UCP2), and insulin-like growth factor-I (IGF-I) (Gomez-Pinilla et al., 2008). The overall evidence supports the idea that select diets and exercise can influence synaptic plasticity, neuronal signaling, and cognitive function by acting on critical mediators of energy metabolism. The results of these investigations have direct implications for the therapeutic use of diet and exercise for the treatment of TBI as the pathobiology of TBI involves a period of metabolic dysfunction. Therefore, an important aspect of dietary therapy is to restore energy homeostasis, thereby increasing the capacity of the brain for plasticity.
CONCLUSIONS
The diffuse nature of TBI can compromise fundamental and broad aspects of neuronal signaling that are required for mental operation. In turn, the broad mode of action of dietary factors makes of them a unique tool that can be implemented to counteract various aspects
|
BOX C-1 Implications of Research on Neuroprotective Properties of Various Nutrients
|
involved in the TBI pathobiology (Box C-1). Specific diets and exercise routines have been shown in animal studies to influence select molecular systems that can make the brain more resistant to damage, facilitate synaptic transmission, and improve cognitive abilities. New evidence shows that dietary supplementations of DHA and curcumin have important actions on the mechanisms that maintain membrane physiology and neuronal signaling, and have a great potential to circumvent TBI pathobiology. Emerging studies indicate that exercise is capable of boosting the healthful effects of certain diets such as n-3 fatty acids. It also has been observed that exercise can counteract some of the deleterious effects of a saturated-fat diet on synaptic plasticity and cognitive function of rats. Therapy based on DHA, curcumin, and exercise can benefit TBI and have long-term consequences on molecular systems responsible for maintaining synaptic function, underlying higher order operations such as learning and memory, and emotions. The overall evidence indicates that the broad neuroprotection provided by diet and exercise could be implemented to counteract the diffuse nature of TBI and other neurological disorders. Although there is still much to be understood in the scientific front, there is sufficient information that can be applied to solve practical problems in the military and the community. A practical and feasible strategy in the short-term would be to use current information to implement dietary policies in the military or other institutions, and the community as a whole.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health awards NS50465, NS56413, and NS068473.
REFERENCES
Anderson, R. M., M. Latorre-Esteves, A. R. Neves, S. Lavu, O. Medvedik, C. Taylor, K. T. Howitz, H. Santos, and D. A. Sinclair. 2003. Yeast life-span extension by calorie restriction is independent of NAD fluctuation. Science 302(5653):2124-2126.
Andres-Lacueva, C., B. Shukitt-Hale, R. L. Galli, O. Jauregui, R. M. Lamuela-Raventos, and J. A. Joseph. 2005. Anthocyanins in aged blueberry-fed rats are found centrally and may enhance memory. Nutritional Neuroscience 8(2):111-120.
Assuncao, M., M. J. Santos-Marques, F. Carvalho, N. V. Lukoyanov, and J. P. Andrade. 2009. Chronic green tea consumption prevents age-related changes in rat hippocampal formation. Neurobiology of Aging 32(4):707–717.
Baur, J. A., K. J. Pearson, N. L. Price, H. A. Jamieson, C. Lerin, A. Kalra, V. V. Prabhu, J. S. Allard, G. Lopez-Lluch, K. Lewis, P. J. Pistell, S. Poosala, K. G. Becker, O. Boss, D. Gwinn, M. Wang, S. Ramaswamy, K. W. Fishbein, R. G. Spencer, E. G. Lakatta, D. Le Couteur, R. J. Shaw, P. Navas, P. Puigserver, D. K. Ingram, R. De Cabo, and D. A. Sinclair. 2006. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444(7117):337–342.
Bruce-Keller, A. J., G. Umberger, R. McFall, and M. P. Mattson. 1999. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Annals of Neurology 45(1):8–15.
Chytrova, G., Z. Ying, and F. Gomez-Pinilla. 2010. Exercise contributes to the effects of DHA dietary supplementation by acting on membrane-related synaptic systems. Brain Research 1341:32–40.
Colle, L. M., L. J. Holmes, and H. M. Pappius. 1986. Correlation between behavioral status and cerebral glucose utilization in rats following freezing lesion. Brain Research 397(1):27–36.
Davis, L. M., J. R. Pauly, R. D. Readnower, J. M. Rho, and P. G. Sullivan. 2008. Fasting is neuroprotective following traumatic brain injury. Journal of Neuroscience Research 86(8):1812–1822.
de la Lastra, C. A., and I. Villegas. 2005. Resveratrol as an anti-inflammatory and anti-aging agent: Mechanisms and clinical implications. Molecular Nutrition and Food Research 49(5):405–430.
Dietrich, W. D., O. Alonso, R. Busto, and M. D. Ginsberg. 1994. Widespread metabolic depression and reduced somatosensory circuit activation following traumatic brain injury in rats. Journal of Neurotrauma 11(6):629–640.
Ding, Q., S. Vaynman, P. Souda, J. P. Whitelegge, and F. Gomez-Pinilla. 2006. Exercise affects energy metabolism and neural plasticity-related proteins in the hippocampus as revealed by proteomic analysis. European Journal of Neuroscience 24(5):1265–1276.
Egan, M. F., M. Kojima, J. H. Callicott, T. E. Goldberg, B. S. Kolachana, A. Bertolino, E. Zaitsev, B. Gold, D. Goldman, M. Dean, B. Lu, and D. R. Weinberger. 2003. The BDNF Val66Met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112(2):257–269.
Ginsberg, M. D., W. Zhao, O. F. Alonso, J. Y. Loor-Estades, W. D. Dietrich, and R. Busto. 1997. Uncoupling of local cerebral glucose metabolism and blood flow after acute fluid-percussion injury in rats. American Journal of Physiology 272(6 Pt 2):H2859–2868.
Gomez-Pinilla, F. 2008. Brain foods: The effects of nutrients on brain function. Nature Reviews. Neuroscience 9(7):568–578.
Gomez-Pinilla, F., and Z. Ying. 2010. Differential effects of exercise and dietary docosahexaenoic acid on molecular systems associated with control of allostasis in the hypothalamus and hippocampus. Neuroscience 168(1):130–137.
Gomez-Pinilla, F., S. Vaynman, and Z. Ying. 2008. Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. European Journal of Neuroscience 28(11):2278–2287.
Graham, H. N. 1992. Green tea composition, consumption, and polyphenol chemistry. Preventive Medicine 21(3):334–350.
Griesbach, G. S., F. Gomez-Pinilla, and D. A. Hovda. 2007. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. Journal of Neurotrauma 24(7):1161–1171.
Hariri, A. R., T. E. Goldberg, V. S. Mattay, B. S. Kolachana, J. H. Callicott, M. F. Egan, and D. R. Weinberger. 2003. Brain-derived neurotrophic factor Val66Met polymorphism affects human memory-related hippocampal activity and predicts memory performance. Journal of Neuroscience 23(17):6690–6694.
Hovda, D. A., R. L. Sutton, and D. M. Feeney. 1987. Recovery of tactile placing after visual cortex ablation in cat: A behavioral and metabolic study of diaschisis. Experimental Neurology 97(2):391–402.
Hovda, D. A., A. Yoshino, T. Kawamata, Y. Katayama, and D. P. Becker. 1991. Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: A cytochrome oxidase histochemistry study. Brain Research 567(1):1–10.
Hu, Z. G., H. D. Wang, L. Qiao, W. Yan, Q. F. Tan, and H. X. Yin. 2009. The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Injury 23(5):459–465.
Huang, W. L., V. R. King, O. E. Curran, S. C. Dyall, R. E. Ward, N. Lal, J. V. Priestley, and A. T. Michael-Titus. 2007. A combination of intravenous and dietary docosahexaenoic acid significantly improves outcome after spinal cord injury. Brain 130(Pt 11):3004–3019.
Jang, M., L. Cai, G. O. Udeani, K. V. Slowing, C. F. Thomas, C. W. Beecher, H. H. Fong, N. R. Farnsworth, A. D. Kinghorn, R. G. Mehta, R. C. Moon, and J. M. Pezzuto. 1997. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275(5297):218–220.
Joseph, J. A., N. A. Denisova, G. Arendash, M. Gordon, D. Diamond, B. Shukitt-Hale, and D. Morgan. 2003. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutritional Neuroscience 6(3):153–162.
Keli, S. O., M. G. Hertog, E. J. Feskens, and D. Kromhout. 1996. Dietary flavonoids, antioxidant vitamins, and incidence of stroke: The Zutphen Study. Archives of Internal Medicine 156(6):637–642.
Kim, S. J., T. G. Son, H. R. Park, M. Park, M. S. Kim, H. S. Kim, H. Y. Chung, M. P. Mattson, and J. Lee. 2008. Curcumin stimulates proliferation of embryonic neural progenitor cells and neurogenesis in the adult hippocampus. Journal of Biological Chemistry 283(21):14497–14505.
Laird, M. D., S. Sukumari-Ramesh, A. E. Swift, S. E. Meiler, J. R. Vender, and K. M. Dhandapani. 2010. Curcumin attenuates cerebral edema following traumatic brain injury in mice: A possible role for aquaporin-4? Journal of Neurochemistry 113(3):637–648.
Li, Q., H. F. Zhao, Z. F. Zhang, Z. G. Liu, X. R. Pei, J. B. Wang, and Y. Li. 2009. Long-term green tea catechin administration prevents spatial learning and memory impairment in senescence-accelerated mouse prone-8 mice by decreasing A[beta]1-42 oligomers and upregulating synaptic plasticity-related proteins in the hippocampus. Neuroscience 163(3):741–749.
Li, W., S. Dai, J. An, P. Li, X. Chen, R. Xiong, P. Liu, H. Wang, Y. Zhao, M. Zhu, X. Liu, P. Zhu, J. F. Chen, and Y. Zhou. 2008. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience 151(4):1198–1207.
Lindsay, J., D. Laurin, R. Verreault, R. Hebert, B. Helliwell, G. B. Hill, and I. McDowell. 2002. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology 156(5):445–453.
Lyons, W. E., L. A. Mamounas, G. A. Ricaurte, V. Coppola, S. W. Reid, S. H. Bora, C. Wihler, V. E. Koliatsos, and L. Tessarollo. 1999. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proceedings of the National Academy of Sciences of the United States of America 96(26):15239–15244.
Menon, V. P., and A. R. Sudheer. 2007. Antioxidant and anti-inflammatory properties of curcumin. Advances in Experimental Medicine and Biology 595:105–125.
Mills, J. D., J. E. Bailes, C. L. Sedney, H. Hutchins, and B. Sears. 2010. Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. Journal of Neurosurgery 114(1):77–84.
Molteni, R., R. J. Barnard, Z. Ying, C. K. Roberts, and F. Gomez-Pinilla. 2002. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 112(4):803–814.
Molteni, R., A. Wu, S. Vaynman, Z. Ying, R. J. Barnard, and F. Gomez-Pinilla. 2004. Exercise reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience 123(2):429–440.
Moore, T. H., T. L. Osteen, T. F. Chatziioannou, D. A. Hovda, and T. R. Cherry. 2000. Quantitative assessment of longitudinal metabolic changes in vivo after traumatic brain injury in the adult rat using FDG-microPET. Journal of Cerebral Blood Flow and Metabolism 20(10):1492–1501.
Navarro, A., C. Gomez, M. J. Sanchez-Pino, H. Gonzalez, M. J. Bandez, A. D. Boveris, and A. Boveris. 2005. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. American Journal of Phsyiology. Regulatory, Integrative, and Comparative Physiology 289(5):R1392–1399.
Neal, C. J., E. Y. Lee, A. Gyorgy, J. M. Ecklund, D. V. Agoston, and G. S. Ling. 2007. Effect of penetrating brain injury on aquaporin-4 expression using a rat model. Journal of Neurotrauma 24(10):1609–1617.
Orgogozo, J. M., J. F. Dartigues, S. Lafont, L. Letenneur, D. Commenges, R. Salamon, S. Renaud, and M. B. Breteler. 1997. Wine consumption and dementia in the elderly: A prospective community study in the Bordeaux area. Revue Neurologique (Paris) 153(3):185–192.
Perkins, A. J., H. C. Hendrie, C. M. Callahan, S. Gao, F. W. Unverzagt, Y. Xu, K. S. Hall, and S. L. Hui. 1999. Association of antioxidants with memory in a multiethnic elderly sample using the Third National Health and Nutrition Examination Survey. American Journal of Epidemiology 150(1):37–44.
Plunet, W. T., F. Streijger, C. K. Lam, J. H. Lee, J. Liu, and W. Tetzlaff. 2008. Dietary restriction started after spinal cord injury improves functional recovery. Experimental Neurology 213(1):28–35.
Prins, M. L., L. S. Fujima, and D. A. Hovda. 2005. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. Journal of Neuroscience Research 82(3):413–420.
Salem, N., Jr., B. Litman, H. Y. Kim, and K. Gawrisch. 2001. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 36(9):945–959.
Samoilova, M., M. Weisspapir, P. Abdelmalik, A. A. Velumian, and P. L. Carlen. 2010. Chronic in vitro ketosis is neuroprotective but not anti-convulsant. Journal of Neurochemistry 113(4):826–835.
Sato, Y., H. Nakatsuka, T. Watanabe, S. Hisamichi, H. Shimizu, S. Fujisaku, Y. Ichinowatari, Y. Ida, S. Suda, K. Kato, et al. 1989. Possible contribution of green tea drinking habits to the prevention of stroke. Tohoku Journal of Experimental Medicine 157(4):337–343.
Sharma, S., Z. Ying, and F. Gomez-Pinilla. 2010. A pyrazole curcumin derivative restores membrane homeostasis disrupted after brain trauma. Experimental Neurology 226(1):191–199.
Soleas, G. J., E. P. Diamandis, and D. M. Goldberg. 1997. Resveratrol: A molecule whose time has come? And gone? Clinical Biochemistry 30(2):91–113.
Suganuma, M., S. Okabe, M. Oniyama, Y. Tada, H. Ito, and H. Fujiki. 1998. Wide distribution of [3H](–)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 19(10):1771–1776.
Sutherland, B. A., R. M. Rahman, and I. Appleton. 2006. Mechanisms of action of green tea catechins, with a focus on ischemia-induced neurodegeneration. Journal of Nutritional Biochemistry 17(5):291–306.
Suzuki, H., S. J. Park, M. Tamura, and S. Ando. 1998. Effect of the long-term feeding of dietary lipids on the learning ability, fatty acid composition of brain stem phospholipids and synaptic membrane fluidity in adult mice: A comparison of sardine oil diet with palm oil diet. Mechanisms of Ageing and Development 101(1–2):119–128.
Truelsen, T., D. Thudium, and M. Gronbaek. 2002. Amount and type of alcohol and risk of dementia: The Copenhagen City Heart Study. Neurology 59(9):1313–1319.
Tsuchida, A., T. Nonomura, T. Nakagawa, Y. Itakura, M. Ono-Kishino, M. Yamanaka, E. Sugaru, M. Taiji, and H. Noguchi. 2002. Brain-derived neurotrophic factor ameliorates lipid metabolism in diabetic mice. Diabetes, Obesity & Metabolism 4(4):262–269.
Vagnozzi, R., S. Signoretti, B. Tavazzi, R. Floris, A. Ludovici, S. Marziali, G. Tarascio, A. M. Amorini, V. Di Pietro, R. Delfini, and G. Lazzarino. 2008. Temporal window of metabolic brain vulnerability to concussion: A pilot 1H-magnetic resonance spectroscopic study in concussed athletes—Part III. Neurosurgery 62(6):1286–1295; discussion 1295–1286.
van Praag, H. 2009. Exercise and the brain: Something to chew on. Trends in Neurosciences 32(5):283–290.
Vaynman, S., Z. Ying, A. Wu, and F. Gomez-Pinilla. 2006. Coupling energy metabolism with a mechanism to support brain-derived neurotrophic factor-mediated synaptic plasticity. Neuroscience 139(4):1221–1234.
Wei, Q. Y., W. F. Chen, B. Zhou, L. Yang, and Z. L. Liu. 2006. Inhibition of lipid peroxidation and protein oxidation in rat liver mitochondria by curcumin and its analogues. Biochimica et Biophysica Acta 1760(1):70–77.
Wu, A., Molteni R, Ying Z, and G.-P. F. 2003. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience 119(2):365–375.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2004a. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. Journal of Neurotrauma 21(10):1457–1467.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2004b. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. European Journal of Neuroscience 19(7):1699–1707.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2006. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Experimental Neurology 197(2):309–317.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2007. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. Journal of Neurotrauma 24(10):1587–1595.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2008. Docosahexaenoic acid dietary supplementation enhances the effects of exercise on synaptic plasticity and cognition. Neuroscience 155(3):751–759.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2010. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabilitation and Neural Repair 24(3):290–298.
Yoshino, A., D. A. Hovda, T. Kawamata, Y. Katayama, and D. P. Becker. 1991. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Research 561(1):106–119.
Youdim, K. A., M. S. Dobbie, G. Kuhnle, A. R. Proteggente, N. J. Abbott, and C. Rice-Evans. 2003. Interaction between flavonoids and the blood-brain barrier: In vitro studies. Journal of Neurochemistry 85(1):180–192.
Resolvins and Protectins: Specialized Pro-Resolving Mediators in Inflammation and Organ Protection: Metabolomics of Catabasis
Charles N. Serhan21
INTRODUCTION
A highly regulated inflammatory response and its timely resolution is the ideal outcome of an acute inflammatory response essential for ongoing health. Hence the cellular and molecular mechanisms that govern natural resolution are vital. Using an unbiased metabolomic-based systems approach to profile self-limited inflammatory exudates, namely, by studying acute inflammatory responses that resolve on their own the author and colleagues identified novel potent chemical mediators. These new chemical mediators constitute a genus of specialized pro-resolving lipid mediators (SPMs) comprised of three new families coined the resolvins, protectins, and most recently the maresins biosynthesized from n-3 fatty acids. These novel local chemical signals join the lipoxin and aspirin-triggered lipoxins as potent anti-inflammatory and pro-resolving lipid mediators formed from the n-6 essential fatty acid arachidonic acid (Serhan, 2007). SPMs are each stereoselective in their actions and as a defining bioaction for this genus; each family member controls both the duration and magnitude of the inflammatory response as well as reduces leukocyte, mediated injury from within and pain signals. Mapping these endogenous resolution circuits had already provided new avenues to appreciate the molecular basis of many widely occurring diseases that are associated with uncontrolled inflammation. The focus of this review is to overview our current understanding and recent advances on the biosynthesis and actions of these novel anti-inflammatory, pro-resolving and protective lipid mediators biosynthesized from dietary essential fatty acids including eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Also, emphasis herein is on the neuroprotective actions of these local mediators that may be relevant in trauma and brain injury and means to enhance their local biosynthesis from the recent literature.
Currently the most widely used anti-inflammatory therapies are directed at inhibiting specific enzymes and/or antagonizing specific receptors for pro-inflammatory mediators (Brennan and McInnes, 2008). Both selective cyclooxygenase inhibitors and anti-tumor necrosis factor α (TNF-α) are examples of this clinical approach. The goal of this approach is to block the production of pro-inflammatory chemical mediators that reduce both the signs and symptoms of inflammation and local tissue damage (Brennan and McInnes, 2008; Flower, 2003). Research efforts within the author’s laboratory focus on profiling self-limited inflammation and uncovering novel mechanisms that terminate the local acute inflammatory response and stimulate resolution with the return of the tissue to homeostasis in murine systems in vivo—a process known as catabasis or the return from disease or the battlefront. Identification of these biochemical and cellular processes demonstrated for the first time that the process of resolution, once considered a passive process, is actually an active, programmed process at the level of the tissue (for recent reviews see Serhan, 2007; Serhan et al., 2008). The resolution as a passive event was thought to simply burn out with the dilution of chemotactic gradients that recruit leukocytes to an infection or invading organism.
Natural resolution of self-limited challenges as well as limited second organ injury proved to be an active process in that we found evidence for temporal activation of bio-
chemical pathways and mediators that instruct the tissue and inflammatory white cells to resolve. Research in the author’s laboratory focused on the potential use of these endogenous agonists to stimulate natural resolution of inflammation rather than targeting inhibition or antagonism of inflammation. This approach also has opened a new understanding of the mechanisms underlying chronic inflammatory diseases as well as a new area in molecular pharmacology, namely resolution pharmacology. The essential n-3 fatty acids, in particular EPA and DHA, are precursors to a several families of mediators in this new genus of potent lipid mediators (LM) that are both pro-resolving and anti-inflammatory (SPM).
AN IDEAL OUTCOME
An acute inflammatory response initiated by microbial invasion or tissue damage is characterized by the cardinal signs of inflammation, i.e., heat, redness, swelling and pain (Majno, 1975). These signs and symptoms are accompanied by a well-known set of cellular microscopic events, including edema and the accumulation of leukocytes, specifically polymorphonuclear leukocytes (PMNs), followed by monocytes that differentiate locally to macrophages (Cotran et al., 1999) (Figure C-10). The local acute inflammatory response
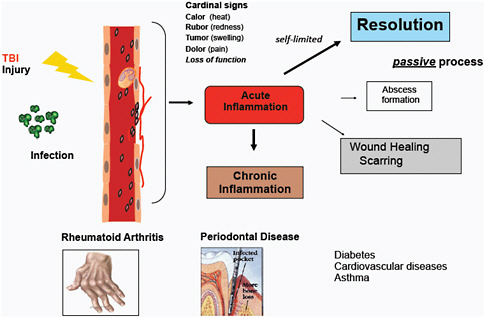
FIGURE C-10 Decision paths in acute inflammation. Several outcomes of acute inflammation caused by infection or injury are possible, including progression to chronic inflammation, tissue fibrosis and wound healing/scarring or, in the ideal scenario, complete resolution (Majno, 1975). The cardinal signs of inflammation—calor (heat), rubor (redness), tumor (swelling), dolor (pain), and loss of function—have been known as the visual signs of inflammation apparent to ancient civilizations. Chronic inflammation was viewed as the persistence of an acute inflammatory response. The chronic inflammatory diseases widely observed in the West are rheumatoid arthritis and periodontal disease. There is now considerable interest in inflammation because it is considered to be the pathophysiologic basis of many diseases that were traditionally not considered to be inflammatory in their pathobiology. These include diabetes, cardiovascular diseases, and asthma, to name a few.
is protective for the host and serves to maintain tissue homeostasis. If uncontrolled, this vital response can become deleterious for the host and the process can progress to chronic inflammation, scarring and fibrosis rather than terminate or resolve. In many cases, the fundamental cause of tissue damage is excessive leukocyte accumulation (Cassatella, 2003). In traumatic brain injury (TBI), for example excessive neutrophil infiltration amplifies local tissue damage (Cotran et al., 1999; Majno and Joris, 2004).
The Resolution Program
In self-limited resolving inflammatory reactions, leukocyte recruitment is coupled with release of local factors that prevent further or excessive trafficking of leukocytes, allowing for resolution (Serhan et al., 2000, 2002). We found that, early in the initiation phase of an inflammatory response, pro-inflammatory mediators such as prostaglandins and leukotrienes play an important role (Samuelsson et al., 1987). Monkeys fed a diet lacking essential fatty acids were found to be defective at mounting an efficient inflammatory response (Palmblad et al., 1988). This highlights the importance of arachidonate-derived eicosanoids or the n-6 essential fatty acid contribution to controlled acute inflammation. In this report from Palmblad et al. (1988), both neutrophil (PMN) chemotaxis and superoxide generation in response to the chemotactic peptide formyl-methiony-leucylphenylalanine (fMLF) were markedly abrogated when the cells were isolated from monkeys fed n-3 essential fatty acids. This is important to our appreciation of the action of dietary n-3 in non-human primates because the PMNs that normally defends the body can release reactive oxygen species ideally intended to kill invading organisms phagocytized by white cells. Instead, when they are summoned into surrounding tissues that are already damaged these cells can inadvertently spill noxious reactive oxygen species that can further destroy tissue. The finding that dietary n-3 reduces this potentially deleterious response of white cells ex vivo provides evidence from primates that leukocyte-mediated tissue damage can be reduced with dietary essential fatty acid. The molecular mechanism for this reduction in leukocyte mediated events was not known at the time of the Palmblad et al. (1988) report but appears to be related to the now known actions of the SPM (see below).
Progression from acute to chronic inflammation as in many widely occurring human diseases of concern in public health such as periodontal disease, arthritis (Koopman and Moreland, 2005), and cardiovascular disease (Libby, 2008), is widely viewed as an excess of pro-inflammatory mediators (Van Dyke and Serhan, 2006). Complete termination of an acute inflammatory insult also is pertinent for restoration of tissue homeostasis and is necessary for ongoing health. Key to this process is the complete removal of leukocytes from inflammatory sites without leaving remnants of the host’s combat between leukocytes, invading microbes, and/or other initiators of inflammation that can seed or amplify the local inflammatory response. Evidence from the author’s laboratory and many others (e.g., Morris et al., 2009) now indicates that the resolving phase of inflammation is not merely a passive process as once believed, but actively takes place as a programmed response at the tissue level (reviewed in Serhan, 2007; Serhan et al., 2008), which can be viewed as analogous to the cellular level or programmed cell death (Cohen et al., 1992). Although mononuclear cells can sometimes contribute to pro-inflammatory responses (Cotran et al., 1999), they also are critical in wound healing, tissue repair and remodeling in a non-inflammatory, non-phlogistic (not fever-causing) manner (Serhan and Savill, 2005). This important homeostatic role of monocytes and macrophages is termed efferocytosis (Tabas, 2010).
SPECIALIZED PRO-RESOLVING LIPID MEDIATORS AND NUTRITION: NOT JUST N-6 VS. N-3 IN THE CONTROL OF INFLAMMATION—A TEMPORAL PROGRESSION TO CATABASIS
The new genus of essential fatty acid-derived autacoids were coined specialized proresolving mediators (SPMs) because they possess families of potent anti-inflammatory, proresolving, and protective mediators in experimental animal models of disease (reviewed in Serhan, 2007). These include the lipoxins from n-6 arachidonic acid (Levy et al., 2001) as well as the n-3-derived resolvins, protectins, and the newly identified maresins, which are not reviewed herein (see Serhan et al., 2009). These novel families of endogenous lipid-derived mediators were originally isolated from self-limited inflammatory murine exudates captured during the natural resolution phase (Figures C-11 and C-12). Each of these local chemical mediators is actively biosynthesized via distinct cellular and in some cases transcellular, enzymatic pathways that are stereocontrolled to produce molecules that are stereoselective in their actions. Hence SPMs are potent agonists that control the duration and magnitude of inflammation by acting on specific receptors (e.g., G protein-coupled receptor [GPCR]) on separate cell populations to stimulate the overall resolution of inflammation (Serhan and Chiang, 2008).
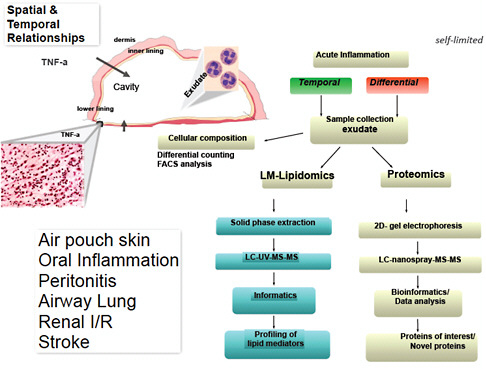
FIGURE C-11 Systems approach to metabolomics of resolution. Illustration of the systems approach taken for the differential analysis of inflammatory exudates. For these initial studies, the murine air pouch was used because it provided a convenient means to assess tissue-level responses by studying the histology as well as the temporal and spatial relationships between infiltrating leukocytes and inflammatory mediators that are initiated by pro-inflammatory stimuli such as bacteria, microbial products, or cytokines such as TNF-α. This differential temporal analysis of inflammatory exudates comparing cellular composition, lipid mediator lipidomics, and proteomics also has been carried out in oral inflammation, peritonitis, airway and lung inflammation, renal ischemia-reperfusion injury, and stroke (see text for further details).
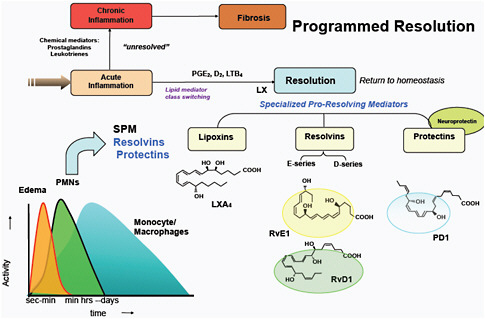
FIGURE C-12 Specialized pro-resolving mediators are generated during inflammatory resolution: ideal outcome of inflammation. Using a rigorous definition of acute inflammation and its resolution, (Cotran et al., 1999) illustrated lower left, we found that acute inflammation initiates a lipid mediator class switch from prostaglandins and leukotrienes in the initial phases to lipoxins and pro-resolving mediators to resolution and the return to homeostasis. Alternatively, an unresolved acute inflammatory response can persist and become chronic inflammation, and in this case lipid mediators such as prostaglandins and leukotrienes are elevated and lead from chronic inflammation to tissue fibrosis. Resolution is an active process switched on by specialized pro-resolving mediators (SPMs), including E-series resolvins derived from n-3 polyunsaturaed fatty acid (PUFA) eicosapentaenoic acid (Serhan et al., 2000), D-series resolvins biosynthesized from dDHA (Serhan et al., 2002; Sun et al., 2007), as well as the protectins (Serhan et al., 2002, 2006). From these results we can now consider the tissue-level events of acute inflammation as programmed resolution (see text for further details).
A systems metabolomics approach was taken using mass spectrometry to analyze the endogenous mediators and mechanisms temporally biosynthesized in vivo by exudates to actively resolve inflammation (Figure C-11). For this, the murine dorsal air pouch system proved to be very useful because self-limited inflammatory exudates could easily be obtained (Serhan et al., 2000, 2002). This system permitted direct exudate analysis in terms of lipidomics and lipid mediator metabolomic profiling (e.g., bioactive autacoids, as well as their inactive precursors and further metabolites), proteomics, and cellular composition by monitoring leukocyte trafficking. Utilizing this approach made it possible to determine when and where different families of local mediators were biosynthesized during resolution (Bannenberg et al., 2005; Serhan, 2007).
Lipoxins were the first anti-inflammatory and pro-resolving lipid mediators recognized, signaling the resolution of acute contained inflammation (Serhan, 2005). Lipoxins are lipoxygenase-derived eicosanoids derived enzymatically from arachidonic acid, an n-6 essential fatty acid that is released from phospholipid stores during inflammation (Samuelsson et al., 1987). In humans they are biosynthesized via transcellular metabolic processes engaged during leukocyte interactions with mucosal cells, i.e., epithelia of the gastrointestinal tract or bronchial tissue, and within the vasculature during platelet–leukocyte interactions
(Serhan, 2007). The murine dorsal air pouch was used to determine the formation and roles of endogenous lipoxin A4 (LXA4) in the resolution of acute inflammation (Levy et al., 2001). Upon initiation of inflammation with TNF-α, there was a typical acute-phase response denoted by rapid PMN infiltration preceded by generation of local prostaglandins and leukotrienes. Unexpectedly, the eicosanoids then underwent a temporal progression in local mediators we termed a “class switch”. As exudates evolve, the eicosanoid profiles switch and the lipid mediators produced within the milieu of the exudate changes (Levy et al., 2001). Within the inflammatory exudate, arachidonate-derived eicosanoids changed from initial production of prostaglandins and leukotrienes to lipoxins, which halted further recruitment of neutrophils. This class switch was driven in part by COX-derived prostaglandins E2 and D2 that regulate the transcription of enzymes involved in lipoxin biosynthesis (Levy et al., 2001). Hence, the concept that “alpha signals omega,” the beginning signals the end in inflammation was introduced (Serhan and Savill, 2005). This became evident because the appearance of lipoxins within inflammatory exudates was concomitant with the loss of PMN and resolution of inflammation (Levy et al., 2001).
In the inflammatory milieu, neutrophils undergo either apoptosis or necrotic cell death at the site of battle and must be removed. As part of the resolution circuit, lipoxins (LX) are biosynthesized (Levy et al., 2001) and signal macrophages to engulf/take up the apoptotic PMN (Godson et al., 2000). LX also proved to be potent chemoattractants for mononuclear cells, but in a non-phlogistic manner. LX activates monocyte infiltration without stimulating release of pro-inflammatory chemokines or activation of pro-inflammatory gene pathways in these cells. Thus, LX actively reduced the entry of PMN to the site of inflammation while accelerating uptake of apoptotic PMN (Serhan, 2007). LX are potent anti-inflammatory mediators that are formed and act in picogram to nanogram amounts within human tissues and in animal disease models (Serhan, 2005). They have the specific pro-resolution actions of limiting PMN recruitment, chemotaxis, and adhesion to the site of inflammation, acting essentially as a braking signal for PMN-mediated tissue injury (Morris et al., 2009; Serhan, 2005). Additionally, LX stable analogs activate endogenous anti-microbial defense mechanisms (Canny et al., 2002), and thus, unlike many other anti-inflammatory treatments, LX are not immunosuppressive.
SPMS ARE BIOSYNTHESIZED FROM THE OMEGA-3 ESSENTIAL FATTY ACIDS, EPA AND DHA
Dietary n-3 PUFA are known to carry protective effects in the cardiovascular system, many inflammatory disorders, and neural function (Leon et al., 2008; Salem et al., 2001; Simopoulos, 2008). Importantly, Hibbeln and colleagues have shown that reduced serum levels of DHA can lead to severe physiological outcomes including depression and increased suicide (1998). This reduction in circulating levels of n-3 correlated with low consumption in humans. Hence Hibbeln introduced the concept of nutritional armor to help emphasize the important role of dietary DHA and to underscore the need to balance this potential for DHA and other essential fatty acid deficits in the diet, in particular those of the military diet given the stress and potential for tissue trauma. The molecular mechanisms by which n-3 PUFAs exert their biological effects in general were incomplete and needed to be elucidated at the cellular and molecular levels. The prevailing theory on the actions of n-3 fatty acids (DHA, EPA) is that they compete for arachidonic acid in phospholipid stores, blocking the biosynthesis of eicosanoids that are pro-inflammatory such as leukotrienes and prostaglandins as well as vasoactive thromboxane (Lands, 2005). This competition between n-3 and n-6 pathways is evident with results from many studies and is demonstrable in vitro with isolated cells and enzymes with high concentrations of essential fatty acids (e.g., micro- to
millimolar range) particularly DHA and EPA. New results indicate that many of the n-6 eicosanoids play important protective roles in resolution and host defense. Thus, these new findings raise critical questions regarding reducing n-6 essential fatty acid levels as relevant in human physiology. To this end, the author has addressed the question “Are the essential n-3 PUFAs such as EPA and DHA enzymatically converted locally in vivo to novel bioactive mediators? Do these local mediators serve as effectors or agonists in vivo?” Accordingly, using an unbiased metabolomic approach, mice given n-3 PUFA biosynthesized novel lipid mediators during the resolution phase of acute inflammation. These mediators are potent agonists of many responses relevant in the immune system and organ protection, particularly neuroprotection. Given the pathophysiology that accompanies TBI it is highly likely that the SPMs will have a beneficial role in TBI by limiting further tissue damage and protecting neurons from uncontrolled inflammatory cells summoned to the TBI-damaged tissue site (vide infra) (Serhan et al., 2000; 2002).
RESOLVINS: RESOLUTION-PHASE INTERACTION PRODUCTS FROM N-3 ESSENTIAL FATTY ACID
Resolvins are a family of new local mediators enzymatically produced within resolving inflammatory exudates. They were initially identified using a systems approach with LC-MS-MS-based lipidomics and informatics, and subsequently complete structural elucidation of these bioactive mediators and related compounds was achieved (Arita et al., 2005a; Hong et al., 2003; Serhan et al., 2000, 2002, 2006). The term resolvins or resolution-phase interaction products refers to endogenous compounds biosynthesized from the major n-3 fatty acids EPA and DHA, denoted E series (RvE) and D series (RvD) resolvins, respectively (Serhan et al., 2002) (Figures C-13 and C-14). Similar to lipoxins, resolvins also can be produced
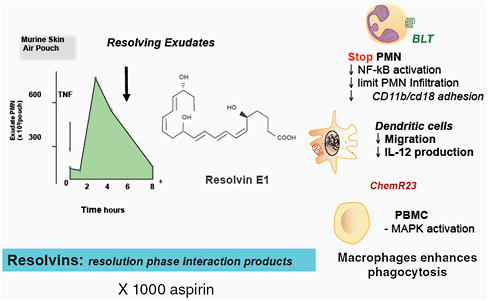
FIGURE C-13 Resolvin E1: Structure and actions. Addition of TNF to murine air pouch leads to rapid infiltration of PMN into the inflammatory exudate. As the PMN levels declined, increased production of resolvin E1 derived from EPA was first identified (Serhan et al., 2000). The complete stereochemistry and the mechanism of action for RvE1 has been established (Arita et al., 2005a, 2007). Resolvins are defined as resolution phase interaction products and proved to be, on a molar basis, log orders more potent than traditional nonsteroidal anti-inflammatory drugs including aspirin.
FIGURE C-14 Biosynthetic scheme of resolvins. The complete stereochemistry of RvD1 and RvD2 has been established.
SOURCE: Spite et al., 2009a; Sun et al., 2007.
by a COX-2-initiated pathway in the presence of aspirin to give “aspirin-triggered” (AT) forms. Accruing evidence indicates that resolvins possess potent anti-inflammatory and immunoregulatory actions that include blocking the production of pro-inflammatory mediators and regulating leukocyte trafficking to inflammatory sites (reviewed in Serhan et al., 2008) as well as clearance of neutrophils from mucosal surfaces (Campbell et al., 2007). Specifically, resolvins limit PMN transendothelial migration in vitro and infiltration in vivo (Serhan et al., 2002; Sun et al., 2007). The potency of these compounds is notable, with concentrations as low as 10 nM producing approximately 50 percent reduction in PMN transmigration. A more detailed description of the specific mechanistic actions of resolvins is discussed in Serhan (2007 and references cited within). Recent results established the stereochemistry and the stereoselective bioactions of RvD1, AT-RvD1, RvE1, and PD1/NPD1 (vide infra) as well as their further enzymatic inactivation because at the end of resolution these signal are “turned off” (Arita et al., 2005a; Serhan et al., 2006; Sun et al., 2007).
It is important to note that the actions of these endogenous SPM are mediated through specific receptors. RvE1 acts as an agonist on at least two GPCRs, namely ChemR23 and as a partial agonist on the LTB4 receptor (BLT1), thus competing with LTB4 for binding (Arita et al., 2005a, 2006). Recent research has revealed that RvE1 stimulates phosphorylation of Akt in a time- and dose-dependent manner via direct activation of ChemR23 (Ohira et al., 2009). This agonist of resolution therefore displays a distinct mechanism of action compared to LXA4 that inhibits downstream tyrosine phosphorylation in eosinophils (Starosta et al., 2008). Additionally, a recent study identified two separate GPCRs that RvD1 specifically binds on human leukocytes, namely the LXA4 receptor (ALX) and GPR32, an orphan receptor, which were validated using a GPCR β-arrestin coupled system (Krishnamoorthy et al., 2010). Identification of receptors for other n-3-derived SPM is in progress and is likely to be
high-affinity GPCRs based on the potency of these newly discovered agonists of resolution. Thus at least two GPCRs for each SPM, RvE1, RvD1, and LXA4, are shared by AT-LXA4. Hence the concept that one ligand can act on a repertoire of receptors is not surprising in view of recent results on neuronal responses with the formyl peptide receptors including FPR2, peptides, and LXA4, which unexpectedly evokes chemosensory actions (Riviere et al., 2009).
PROTECTINS AND NEUROPROTECTIN D1: ORGAN-PROTECTIVE MEDIATORS
In addition to D-series resolvins, DHA is also a precursor for the family of protectins. These mediators are biosynthesized via a separate pathway (Figure C-15). Protectins are distinguished by their conjugated triene-containing structure (Serhan et al., 2006). The name “protectins” was coined from the observed anti-inflammatory and protective actions in neural tissues and systems (Hong et al., 2003). The prefix neuroprotectin gives the tissue location of its generation and local actions, such as neuroprotectin D1 (NPD1) (Mukherjee et al., 2004; Serhan et al., 2006). Like the resolvins, protectins stop PMN infiltration and enhance macrophage phagocytosis of apoptotic (dead cells) PMN (Hong et al., 2003; Serhan et al., 2006). They are biosynthesized by and act on glial cells and reduce cytokine expression (Hong et al., 2003).
FIGURE C-15 Protectins: Structure and actions. The complete stereochemistry of (NPD1) or protectin D1 (PD1) when generated in inflammatory exudates has been established (Serhan et al., 2006). NPD1 stops further neutrophilic infiltration in a number of in vivo animal systems as well as reduces microglial cell cytokine expression. These initial actions defined the biological functions and roles of PD1. In addition, in collaboration with Nicolas Bazan and colleagues, we have found that NPD1 reduces stroke damage and retinal injury (see Bazan et al., 2010).
Of special interest for this report, NPD1 reduces both retinal and corneal injury and stroke damage (Marcheselli et al., 2003), and improves corneal wound healing in mouse models (Gronert et al., 2005) (see Serhan, 2007; Serhan et al., 2008, and references cited within). DHA is well known for its role in neuronal systems and, along with arachidonic acid (AA), is a major PUFA found in the retina (Bazan et al., 2010). The available results provide the biological basis for the actions of NPD1 derived from DHA. In recent reviews we considered in detail the results obtained from several independent lines of investigation required to address the structure of the potent bioactive NPD1/PD1 (see Bazan et al., 2010). As with other bioactive mediators, such as the eicosanoids (Samuelsson et al., 1987), it is important to establish the stereochemistry of the compound and/or mediator. Notably, subtle structural changes in these and related products can be less active, inactive, or, even in some cases display opposing biologic actions as a result of subtle changes in stereochemistry that are recognized in biologic systems. To confirm the proposed basic structure and establish the complete stereochemistry, these studies on the 10,17S-docosatriene termed NPD1/PD1 included results from (a) biosynthesis studies; (b) matching of materials prepared by total organic synthesis with defined stereochemistry; and (c) the actions of these and related compounds in biological systems (Ariel et al., 2005; Hong et al., 2003; Marcheselli et al., 2010; Mukherjee et al., 2004; Serhan et al., 2002, 2006). We also consider these findings as they appeared in the literature with the goal of providing a clear and rigorous account of the evidence that supports the structure and bioactions of NPD1/PD1. Investigations along these lines were required to establish the complete structure and potent actions of NPD1/PD1 and related endogenous products biosynthesized from DHA because the small amounts of NPD1 attainable from biological systems precluded direct stereochemical analyses of the products identified in retinal pigmented epithelial (RPE) cells. For a recent review, see Bazan et al. (2010).
Briefly, the novel 10,17S-docosatriene was coined NPD1/PD1 given its potent actions in vivo noted above, which were identified first in Hong et al. (2003) and Serhan et al. (2002). Its basic structure was established and displayed potent anti-inflammatory actions, i.e., reducing PMN numbers in vivo and reducing the production of inflammatory cytokines by glial cells. Moreover, during the resolution phase of peritonitis, unesterified DHA levels increase in resolving exudates, where it appears to promote catabasis or the return to homeostasis following tissue insult, via conversion to D-series resolvins and 10,17S-docosatrienes (Bannenberg et al., 2005) by shortening the resolution interval of an inflammatory response in vivo (Schwab et al., 2007).
In collaboration with Dr. Bazan and colleagues at Louisiana State University, we found next that the DHA-derived 10,17S-docosatriene was generated in vivo in the brain during experimental stroke in the ipsilateral cerebral hemisphere following focal ischemia (Figures C-15, C-16, and C-17). We also demonstrated its potent bioactions in stroke, where NPD1/PD1 limited the entry of leukocytes, downregulated both COX-2 expression and NFκB activation, and decreased infarct volume (Figure 8 from Marcheselli et al., 2003). Of interest, 10,17S-docosatriene is also formed in the human retinal pigment epithelial cell line ARPE-19 and introduced the term neuroprotectin D1 based on its neuroprotective bioactivity in the stroke model and RPE cells (Mukherjee et al., 2004). Also NPD1 proved to be a potent signal to inactivate pro-apoptotic and pro-inflammatory signaling. The capital D in NPD1 refers to its being the first identified neuroprotective mediator derived from DHA (Mukherjee et al., 2004).
In parallel to these investigations, biosynthesis and function studies were carried out with human TH2-skewed peripheral blood mononuclear cells (PBMC) (Ariel et al., 2005). When produced by human PBMC, PD1 promotes T-cell apoptosis via the formation of lipid
FIGURE C-16 NPD1/PD1 biosynthesis and actions. NPD1 was identified initially in murine exudates and next in murine brain cells. Fish such as trout also produce NPD1/PD1, indicating that this is a primordial structure and pathway. In the immune system, T cells skew to a Th2 cell in the presence of interleukin-4, increase their 15-LO type 1, and convert DHA to PD1. The actions of PD1 are denoted in the inset boxes. The biosynthesis of PD1 and its complete stereochemistry have been established. The conversion of DHA to PD1 enzymatically proceeds via the enzymatic epoxidation and formation of a key 16(17)-epoxy-DHA intermediate that is enzymatically converted to the complete stereochemistry required for full biological action in PD1 (see text for further details).
raft-encoded signaling complexes and reduced T-cell traffic in vivo. Matching materials prepared by total organic synthesis determined the complete stereochemistry of the PBMC DHA-derived product. NPD1/PD1 generated by human PBMC carried the complete stereochemistry of (10R,17S)-dihydroxydocosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid and was matched to the most potent bioactive product (Figure C-16) using several dihydroxytriene-containing DHA-derived products isolated from human PBMC, human PMN, and murine exudates (Ariel et al., 2005; Serhan et al., 2006).
With the stereochemistry of NPD1/PD1 established, its identification in human material was sought and found in exhaled breath condensates from human asthmatics (Levy et al., 2007) as well as in human brain tissue. NPD1 is present in both control brain tissues and at reduced levels in brain tissues from patients with Alzheimer’s disease (Lukiw et al., 2005, and Table 1). In addition, PD1 is a major product in bone marrow of female rats fed EPA and DHA (Poulsen et al., 2008). PD1 is also generated in vivo during ischemia-reperfusion of renal tissues, where it has profound actions, reducing the deleterious consequences of ischemia-reperfusion in renal tissues (Duffield et al., 2006) in agreement with our earlier findings in brain tissues (Marcheselli et al., 2003).
With the complete stereochemistry and synthetic compound in hand, it was possible to demonstrate for the first time that PD1 activates resolution programs in vivo and shortens the resolution time of experimental inflammation in animal models (Schwab et al., 2007).
FIGURE C-17 Neuroprotective action of NPD1/PD1. NPD1 reduces stroke volume and leukocyte-mediated tissue damage in a murine model of focal ischemia in stroke. Figure is from Marcheselli et al., J. Biol. Chem. 2003; 278:43807-43817. See Bazan et al. (2010) and text for further details.
Also, with the total organic synthesis route of NPD1/PD1, it was possible to radiolabel and isolate 3H-NPD1/PD1 made from the synthetic intermediate. With this radiolabel, we define for the specific binding sites present with ARPE-19 cells (Kd ~ 31 pM/mg cell protein) for 3H-NPD1/PD1 as well as specific binding to human neutrophils (Kd of ~25 nM) (Marcheselli et al., 2010).
Recent results using an LC-MS-MS profiling approach demonstrated that PD1 is made during the resolution of Lyme disease infections in mice (Blaho et al., 2009). NPD1 inhibits retinal ganglion cell death (Qin et al., 2008), is renal protective (Hassan and Gronert, 2009), and regulates adiponectin (González-Périz et al., 2009). Of interest, the double dioxygenation product 10S,17S-diHDHA, isomer of NPD1/PD1, was recently shown to have actions on platelets, reducing platelet aggregation at 0.3 μM, 1 μM, and higher concentrations (Chen et al., 2009). In peritonitis, this isomer also showed biological activity but was less potent than NPD1/PD1 (Serhan et al., 2006). It is noteworthy that NPD1/PD1 and the resolvins are produced by trout tissues including trout brain from endogenous DHA, suggesting that these structures are highly conserved from fish to humans (Hong et al., 2005) and that we still have much to learn regarding the bioactions and functions of NPD1/PD1, the D-series and E-series resolvins, and related products in human physiology and pathophysiology as well as in biological systems such as fish or in bone marrow, where the actions of NPD1/PD1 remain to be fully appreciated.
OMEGA-3 ESSENTIAL FATTY ACID AND SUBSTRATE AVAILABILITY IN RESOLUTION OF INFLAMMATION
To elucidate the role of EPA, DHA, and AA during inflammation in vivo, we studied disease models in wild-type mice that overexpress the C. elegans fat-1 gene to compare the effects of n-3 and -6 PUFAs. This gene converts n-6 PUFA into n-3, resulting in elevated tis-
sue levels of n-3 PUFA within the fat-1 overexpressing mice. In a model of oxygen-induced retinopathy, a protective effect against pathological angiogenesis was found in the retina when there was a lower ratio of n-6:n-3 PUFA. Wild-type mice lacking the fat-1 transgene had more extensive vaso-obliteration and more severe retinal neovascularization compared with fat-1 mice (Connor et al., 2007). In mice fed n-3 PUFA, biosynthetic markers of NPD1 and RvE1 were detected in the retinal tissues. In mice without n-3 PUFA supplementation, administration of RvD1, RvE1, or NPD1 gave protection from vaso-obliteration and neovascularization (Connor et al., 2007), as well as suture-induced ocular inflammation (Jin et al., 2009). These fat-1 mice also have increased levels of resolvins and protectins in the colon and are protected from colitis (Hudert et al., 2006). In evolving exudates, rapid appearance of unesterified n-3 PUFA parallels the initial increase in edema (Kasuga et al., 2008). Thus, n-3 precursors for resolving and protectin biosynthesis in exudates become available directly from the peripheral circulation. This contrasts with the pool or storage of AA and other essential fatty acid that requires phospholipase A2 for liberation for example in inflammatory cells (Dennis, 2000; Lands, 2005) and in retina RPE cells (Bazan et al., 2010).
SPM IN INFLAMMATORY EXPERIMENTAL DISEASE SYSTEMS
Inflammatory bowel disorders, such as colitis, are characterized by a relapsing inflammatory process as a result of local mucosal damage and abnormal mucosal responses. In a well-appreciated experimental colitis in mice, mice are challenged with either an intrarectal antigenic hapten, 2,4,6-trinitrobenzene sulfonic acid (TNBS) or to induce colitis, RvE1, which was protective against bowel inflammation (Arita et al., 2005b). With treatment of as little as 1 μg of RvE1 per mouse, there was dramatic reduction in mortality, and weight loss, and less severe histologic display of colitis, namely reduction in the associated inflammatory cells, such as PMN and lymphocytes, compared with mice with TNBS-induced colitis. RvE1 is also protective in Porphyromonas gingivalis-induced periodontal disease in rabbits, where it appears to stimulate tissue regeneration of the periodontium (Hasturk et al., 2007). Another pro-resolving action of RvE1 was evident in a murine model of fatty liver disease, where RvE1 administration to obese mice significantly alleviated hepatic steatosis and restored the loss of insulin sensitivity (Gonzalez-Periz et al., 2009).
The complete stereochemical assignment for RvD2 (Figure C-11) was recently established as 7S,16R,17S-trihydroxy-4Z,8E,10Z,12E,14E,19Z-docosahexaenoic acid (Spite et al., 2009a). RvD2 proved very potent suggesting it may have a broad role in vivo and potential application in disease. Sepsis remains a clinical challenge with increasing prevalence and mortality rates. In these cases, infection progresses rapidly unless contained and cleared by phagocytes, and epithelial and endothelial barrier dysfunction results in immune suppression, multiple-organ failure, and death. Administration of n-3 PUFA in some of these cases of sepsis has shown favorable outcomes (Singer et al., 2008), although the mechanism of action is still being studied. In an established mouse model of sepsis, initiated by “mid-grade” cecal ligation and puncture (CLP) surgery, RvD2-methyl ester (RvD2-ME; 100ng) was protective (Spite et al., 2009a). After 12 hours, mice that underwent CLP surgery had severe bacterial burden both locally within the peritoneum and systemically, which was accompanied by a significant leukocyte infiltrate to the peritoneal cavity. RvD2-ME treatment immediately following CLP significantly reduced both blood and peritoneal bacterial levels and dramatically limited local PMN influx. Septic mice were hypothermic 12 hours after CLP and displayed a drastic decrease in activity levels, whereas RvD2-treated mice remained active within their cages and their body temperatures were similar to sham-operated control
mice. The proportion of mice that survived seven days following “mid-grade” CLP was approximately 36 percent. RvD2-ME was able to double the survival rates (Figure C-14). Interestingly, whereas all of the mice that did not survive in the vehicle-treated group died by 36 hours, 25 percent of the mice that died in the RvD2-treated group lived until 48 hours post-CLP, suggesting that RvD2 may increase the “therapeutic window” that could afford additional time for further interventions such as antibiotics. Further analysis of peritoneal exudates showed a “cytokine-storm” of both pro-inflammatory cytokines and other mediators associated with detrimental outcomes in sepsis, as well as elevated pro-inflammatory lipid mediators LTB4 and PGE2. RvD2-ME significantly blunted overzealous cytokine production and pro-inflammatory lipid mediators measured 12 hours post-CLP. Dissection of mouse inguinal lymph nodes revealed that bacteria were disseminated throughout this organ in vehicle-CLP mice, whereas RvD2 enhanced phagocyte-dependent bacterial containment and clearance. Corroboratory results were obtained in vitro with human PMN exposed to RvD2 (1 and 10 nM) that showed increased phagocytosis and killing of E. coli (Spite et al., 2009a). These novel findings highlight that RvD2 is protective in mucosal barrier breakage and leakage leading to sepsis.
A recent study found that RvE1 is also anti-infective, enhancing clearance of bacteria from mouse lungs in a model of pneumonia, leading to increased survival (Seki et al., 2010). Thus, pro-resolving mediators display anti-inflammatory, anti-fibrotic, and recently demonstrated anti-infective actions in several widely used laboratory models of inflammation (see Table C-7). Together, these results indicate SPM, i.e., resolvins, are not immunosuppressive but rather enhance the innate anti-microbial systems in phagocytes and mucosal epithelial cells (Canny et al., 2002).
STEM CELLS AND RECENT RESULTS OF INTEREST FOR TRAUMATIC BRAIN INJURY
The identification and biosynthesis of resolvins and protectins from DHA in brain (Serhan et al., 2002) and microglial cells (Hong et al., 2003), and in view of their anti-inflammatory and pro-resolving actions in acute inflammation using in vivo models, suggested that these compounds also may be active in neural tissues. Indeed, neuroprotectin D1 limited neutrophilic infiltration in ischemic brain as well as reduced pro-inflammatory gene expression, suggesting that this compound had potent neuroprotective actions in local ischemia-reperfusion in the brain (Marcheselli et al., 2003), which also was observed in renal protection (Duffield et al., 2006; Hassan and Gronert, 2009) (Figure C-15). Given the importance of neural stem cells in organ regeneration, we examined the actions of LT B4 and LX A4 and found that murine neural stem cells produced LT B4 and possess leukotriene receptors. LX A4 regulated proliferation and differentiation of these cells. Of interest, the 15-epimer isomer of LXA4 (which is more stable in vivo as it is not inactivated as rapidly as LXA4) reduced neural stem cell growth at concentrations as low as 1 nanomolar. Gene chip analysis demonstrated that LT B4 and LXA4 gave opposing gene expression profiles with neural stem cells. LTB4 induced proliferation and differentiation of neural stem cells into neurons, and the regulatory actions of LXA4 suggested that these new pathways may be relevant in restoring stem cells (Wada et al., 2006). Using a metabolomic approach with stem cells and evaluation of redox status, Yanes et al. recently found that NPD1 is produced, which in turn showed potent actions at the 10-nanomolar range in promoting neural differentiation as well as cardiac stem cell differentiation (Yanes et al., 2010). Hence, the local molecular response to injury and local inflammation may generate these local mediators,
LX A4 and NPD1, to regulate organ stem cells to initiate tissue repair and regulate the local inflammatory response.
The neuroprotection effects of LX A4 also have been confirmed and extended by others. For example, LX A4 is neuroprotective in experimental stroke (Sobrado et al., 2009), and LX A4 methyl ester reduces neutrophilic infiltration and protects rat brain from focal cerebral ischemia-reperfusion (Ye et al., 2010). In addition to the actions on leukocytes, LX A4 also regulates cytokine production from human astrocytoma cells (Decker et al., 2009). Taken together, these findings in murine systems suggest that the arachidonic acid-derived SPMs, namely lipoxins, are neuroprotective by downregulating local inflammatory response and limiting further inflammation via tissue destruction by limiting neutrophil-mediated tissue damage. Hence, arachidonic acid as substrate and an essential fatty acid plays a role in neuroprotection and particularly one that is regulated by dietary intake of essential fatty acids. Along these lines, Huang et al. reported that intravenous and dietary DHA significantly improves spinal cord injury in rats. Improvements and neuroprotection were obtained when DHA was injected intraveneously with sustained dietary supplementation (Huang et al., 2007).
GAPS IN CURRENT KNOWLEDGE: TOO MUCH OF A GOOD THING?
Recently, using a rat model of TBI, Bailes and Mills (2010) found that dietary supplementation with DHA reduced acceleration of injury. These are encouraging findings suggesting that DHA delivery can have a neuroprotective effect, likely via the local biosynthesis of pro-resolving and anti-inflammatory mediators such as NPD1 and D-series resolvins produced from the exogenous DHA. However, there is a potential negative side for excess delivery of DHA above “optimal dietary levels,” the limits of which remain unknown at present. When excess DHA is available to undergo nonenzymatic autooxidation, neurotoxic products can be produced, such as trans-4-hydroxy-2-hexenal (Long et al., 2008). Likewise, in isolated neuroblastoma cells, we recently found that resolvins and protectins are not produced from DHA by these tumor cells in vitro but rather exogenous DHA is converted to hydroperoxy acid intermediates that are cytotoxic (Gleissman et al., 2010). For a cancer cell type, this may be a beneficial approach to killing tumor cells with exogenous DHA. The point taken from these experiments is that excess DHA can lead to oxidation products that can further amplify local inflammation and tissue damage at the cellular level. Along these lines, the enzymatically produced resolvin D1 reduces inflammation initiated by lipid oxidation products (Spite et al., 2009b). Taken together, these results suggest that optimal dietary levels of DHA could enhance production of neuroprotective local mediators. Thus, a clear starting point in humans is to establish the daily dose required for DHA and the levels that ought to be considered in excess for healthy individuals. In this regard, potential gender and age differences need to be taken into consideration for future studies. In considering TBI on the battlefield, the currently available animal results suggest that intravenous delivery of DHA in addition to dietary supplementation can have neuroprotective actions. These neuroprotective actions may be mediated by enhanced local production of resolvins and protectins. Indeed, if local conversion of dietary DHA to resolvins and protectins can be established in humans with TBI and stress, then dietary DHA could speed recovery time for military personnel. Because DHA is a natural product and has been shown in many studies to be safe in humans (Lands, 2005), DHA supplementation can be recommended for military personnel. This also can open the opportunity to gain direct evidence for DHA effectiveness in clinical studies with military personnel (see below).
TABLE C-7 SPM Actions in Animal Disease Models Relevant to TBI and DHAa
|
Disease Model |
Species |
Action(s) |
References |
|
Resolvin E1 |
|||
|
Periodontitis |
Rabbit |
Reduces neutrophil infiltration; prevents connective tissue and bone loss; promotes healing of diseased tissues; regenerates lost soft tissue and bone |
(Hasturk et al., 2006, 2007) |
|
Peritonitis |
Mouse |
Stops neutrophil recruitment; regulates chemokine/cytokine production Promotes lymphatic removal of phagocytes |
(Arita et al., 2005a; Bannenberg et al., 2005; Schwab et al., 2007) |
|
Dorsal air pouch |
Mouse |
Stops neutrophil recruitment |
(Serhan et al., 2000) |
|
Retinopathy |
Mouse |
Protects against neovascularization |
(Connor et al., 2007; Jin et al., 2009) |
|
Colitis |
Mouse |
Decreases neutrophil recruitment and pro-inflammatory gene expression; improves survival; reduces weight loss |
(Arita et al., 2005b) |
|
Pneumonia |
Mouse |
Improves survival; decreases neutrophil infiltration; enhances bacterial clearance; reduces pro-inflammatory cytokines and chemokines |
(Seki et al., 2010) |
|
Resolvin D1 |
|||
|
Peritonitis |
Mouse |
Stops neutrophil recruitment |
(Hong et al., 2003; Spite et al., 2009b; Sun et al., 2007) |
|
Dorsal skin air pouch |
Mouse |
Stops neutrophil recruitment |
(Hong et al., 2003; Serhan et al., 2002) |
|
Kidney ischemia-reperfusion |
Mouse |
Protects from ischemia-reperfusion-induced kidney damage and loss of function; regulates macrophages and protects from fibrosis |
(Duffield et al., 2006) |
|
Retinopathy |
Mouse |
Protects against neovascularization |
(Connor et al., 2007; Jin et al., 2009) |
|
Resolvin D2 |
|||
|
Peritonitis |
Mouse |
Stops neutrophil recruitment |
(Spite et al., 2009a) |
|
Sepsis |
Mouse |
Improves survival; reduces pro-inflammatory cytokines; regulates neutrophil recruitment; enhances bacterial containment |
(Spite et al., 2009a) |
|
Protectin D1 |
|||
|
Peritonitis |
Mouse |
Stops neutrophil recruitment; regulates chemokine/cytokine production; promotes lymphatic removal of phagocytes; regulates T-cell migration |
(Ariel et al., 2005; Arita et al., 2005a; Bannenberg et al., 2005; Schwab et al., 2007) |
SUMMARY
In summation, acute inflammation initiated by neutrophils in response to injury or microbial invasion is, ideally, a self-limited response that is protective for the host. Excessive uncontrolled inflammatory responses can lead to chronic disorders as well as further tissue injury, as in the case of TBI. Neutrophil-derived pro-inflammatory mediators, including leukotrienes and prostaglandins, can amplify this process. Within contained inflammatory exudates, we found that neutrophils change phenotype to initiate resolution and begin to biosynthesize pro-resolving and protective local mediators from essential fatty acids including AA, DHA, and EPA. There is an active catabasis to return tissues to a homeostatic healthy state from the battle of host defense to microbes during inflammatory episodes
(Bannenberg et al., 2005). SPM such as LX, protectins, and resolvins biosynthesized locally from essential fatty acids accelerate this process and the return to tissue homeostasis (Schwab et al., 2007). Each of these SPMs is temporally and spatially biosynthesized to actively regulate resolution by acting on specific receptors (Serhan et al., 2008), initiating anti-inflammatory and pro-resolving signals to terminate the inflammatory response and limit further tissue injury. Hence, dietary essential fatty acids in experimental animal models reviewed herein directly regulate the magnitude and duration of local acute inflammation via the production of potent local chemical mediators that are pro-resolving. These experimental findings in animal models that demonstrate pathophysiologic events relevant to TBI (Table C-7), taken together, suggest that increasing the n-3 essential fatty acids in military personnel to boost their n-3 status may have a positive impact on traumatic tissue injury to limit the magnitude of the response. Helping to optimize the host’s response to injury and local inflammation with nutritional substrates also may represent a cost-effective means to reduce recovery times in the field. Our recent findings indicate that resolvins also are potent modulators reducing pain in murine systems (Bang et al., 2010; Sommer and Birklein, 2010; Xu et al., 2010).
The answer to the question “Is there enough evidence from TBI animal models to conclude that military personnel should boost their n-3 status to protect against the effects of TBI?” is yes, but additional information from both human and animal studies is needed to directly support the cause-and-effect relationships between increasing DHA status and reducing TBI and other indications in military personnel. Because the identification of DHA metabolome in resolution of acute inflammation with the production of potent local protective and anti-inflammatory mediators in murine systems is relatively recent (see Serhan, 2007), additional results are needed to establish cause-and-effect relationships in TBI and, for example, increased resolvins and neuroprotectins or SPM and reduced tissue inflammation and local damage. The results reviewed herein suggest that the protective actions of DHA in rat TBI (Bailes and Mills, 2010) are mediated locally by resolvins and protectins, given their potent actions in in vivo models of inflammation in animals (Table C-7).
With these metabolomic tools and structures now available in synthetic form (Serhan, 2010), these suggestions can be directly tested rigorously in the field to assess their validity for military implementation. For example, human studies are needed to establish:
-
Is DHA a “green pro-drug”? Is it converted locally with trauma to D-series resolvins and protectins, which in turn are bioactive mediators that limit tissue inflammation and further tissue damage?
-
What is the relationship between serum/plasma DHA levels and reduced TBI and/or ear injury in military personnel? Is there a gender selectivity for DHA conversion, actions, and overall impact?
-
Do resolvins and/or protectins from DHA mediate cellular protection and reduce TBI damage and recovery time?
-
Can excess serum DHA levels lead to enhanced autooxidation pathways in stressed military personnel or protection and enhanced SPM biosynthesis?
-
What are the pathway biomarkers for resolvins and protectins that can be monitored in peripheral blood of military personnel? For example, do 17-HDHA, 14-HDHA, 22-hydroxy-DHA, RvD5, 7,17-diHDHA, NPD1, RvD1, or RvD2 correlate with DHA levels and/or the degree of protection in TBI and/or ear trauma? Do they reduce recovery times for military personnel?
In addition to these human studies, mechanism of action studies can be carried out with animal experiments. These also can include direct comparisons between administration of NPD1, RvD1, RvD2 versus DHA in animal models of TBI and ear injury. The role of substrate (e.g., DHA) levelscan be assessed in transgenic mice with elevated tissue DHA such as the fat-1 mouse (Hudert et al., 2006), which show protection from inflammatory disorders as well as elevated levels of resolvins and protectins. Murine models with transgenic over-expressing proresolving receptors (Devchand et al., 2003; Krishnamoorthy et al., 2010) can be used to determine the role of resolvin receptors in protection from TBI. These animal and human studies can be implemented in parallel to increase the DHA status in military personnel in order to gain mechanistic insight and fill gaps in current knowledge of the role of essential fatty acids and n-3 status in reducing trauma and shortening the time required for recovery of military personnel.
ACKNOWLEDGMENTS
The authors thank Mary H. Small for expert assistance with manuscript preparation and the members of the laboratory and collaborators for their expertise and efforts in the reports referenced herein. Research in the author’s laboratory reviewed here was supported by National Institutes of Health grants DK-074448 and GM-38765 (C.N.S).
Conflict of Interest Statement
The author is inventor on patents assigned to Brigham and Women’s Hospital and Partners HealthCare on the composition of matter, uses, and clinical development of anti-inflammatory and pro-resolving lipid mediators. These are licensed for clinical development. The author retains founder stock in Resolvyx Pharmaceuticals.
ABBREVIATIONS
AA arachidonic acid
AT-RvD1 aspirin-triggered-resolvin D1, 7S,8R,17R-trihydroxy-docosa-4Z,9E,11E,13Z,15E,19Z-hexaenoic acid
DHA docosahexaenoic acid
EPA eicosapentaenoic acid
LX lipoxin
LXA4 5S, 6R,15S-trihydroxy-7,9,13-trans-11-cis-eicosatetraenoic acid
PD1/NPD1 protectin D1/neuroprotectin D1, 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid
PMN polymorphonuclear leukocytes
Protectins biosynthesized from DHA, containing conjugated triene structures and are protective mediators
PUFA polyunsaturated fatty acid
Resolvins structurally unique and potent bioactive local mediators; E series from EPA and D series from DHA
RvD1 resolvin D1, 7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid
RvD2 resolvin D2, 7S,16R,17S-trihydroxy-4Z,8E,10Z,12E,14E,19Z-docosahexaenoic acid
RvE1 resolvin E1, 5S,12R,18R-trihydroxy-6Z,8E,10E,14Z,16E-eicosapentaenoic acid
REFERENCES
Ariel, A., P.-L. Li, W. Wang, W.-X. Tang, G. Fredman, S. Hong, K. H. Gotlinger, and C. N. Serhan. 2005. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. The Journal of Biomedical Chemistry 280:43079–43086.
Arita, M., F. Bianchini, J. Aliberti, A. Sher, N. Chiang, S. Hong, R. Yang, N. A. Petasis, and C. N. Serhan. 2005a. Stereochemical assignment, anti-inflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. The Journal of Experimental Medicine 201:713–722.
Arita, M., M. Yoshida, S. Hong, E. Tjonahen, J. N. Glickman, N. A. Petasis, R. S. Blumberg, and C. N. Serhan. 2005b. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proceedings of the National Academies of Sciences of the United States of America 102:7671–7676.
Arita, M., S. Oh, T. Chonan, S. Hong, S. Elangovan, Y.-P. Sun, J. Uddin, N. A. Petasis, and C. N. Serhan. 2006. Metabolic inactivation of resolvin E1 and stabilization of its anti-inflammatory actions. The Journal of Biomedical Chemistry 281:22847–22854.
Arita, M., T. Ohira, Y. P. Sun, S. Elangovan, N. Chiang, and C. N. Serhan. 2007. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and chemR23 to regulate inflammation. Journal of Immunology 178:3912–3917.
Bailes, J. E., and J. D. Mills. 2010. Docosahexaenoic acid (DHA) reduces traumatic axonal injury in a rodent head injury model. Journal of Neurotrauma 27(9):1617–1624.
Bang, S., S. Yoo, T. J. Yang, H. Cho, Y. G. Kim, and S. W. Hwang. 2010. Resolvin D1 attenuates activation of sensory transient receptor potential channels leading to multiple anti-nociception. British Journal of Pharmacology 161:707–720.
Bannenberg, G. L., N. Chiang, A. Ariel, M. Arita, E. Tjonahen, K. H. Gotlinger, S. Hong, and C. N. Serhan. 2005. Molecular circuits of resolution: Formation and actions of resolvins and protectins. Journal of Immunology 174:4345–4355.
Bazan, N. G., J. M. Calandria, and C. N. Serhan. 2010. Rescue and repair during photoreceptor cell renewal mediated by docosahexaenoic acid-derived neuroprotectin D1. Journal of Lipid Research 51:2018–2031.
Blaho, V. A., M. W. Buczynski, C. R. Brown, and E. A. Dennis. 2009. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. The Journal of Biomedical Chemistry 284:21599–21612.
Brennan, F. M., and I. B. McInnes. 2008. Evidence that cytokines play a role in rheumatoid arthritis. Journal of Clinical Investigation 118(11):3537–3545.
Campbell, E. L., N. A. Louis, S. E. Tomassetti, G. O. Canny, M. Arita, C. N. Serhan, and S. P. Colgan. 2007. Resolvin E1 promotes mucosal surface clearance of neutrophils: A new paradigm for inflammatory resolution. The Journal of the Federation of American Societies for Experimental Biology 21:3162–3170.
Canny, G., O. Levy, G. T. Furuta, S. Narravula-Alipati, R. B. Sisson, C. N. Serhan, and S. P. Colgan. 2002. Lipid mediator-induced expression of bactericidal/permeability-increasing protein (bpi) in human mucosal epithelia. Proceedings of the National Academies of Sciences of the United States of America 99:3902–3907.
Cassatella, M. A., ed. 2003. The neutrophil. Vol. 83, Chemical Immunology and Allergy. Basel: Karger.
Chen, P., B. Fenet, S. Michaud, N. Tomczyk, E. Véricel, M. Lagarde, and M. Guichardant. 2009. Full characterization of PDX, a neuroprotectin/protectin D1 isomer, which inhibits blood platelet aggregation. FEBS Letters 583:3478–3484.
Cohen, J. J., R. C. Duke, V. A. Fadok, and K. S. Sellins. 1992. Apoptosis and programmed cell death in immunity. Annual Review of Immunology 10:267–293.
Connor, K. M., J. P. SanGiovanni, C. Lofqvist, C. M. Aderman, J. Chen, A. Higuchi, S. Hong, E. A. Pravda, S. Majchrzak, D. Carper, A. Hellstrom, J. X. Kang, E. Y. Chew, N. N. Salem, Jr., C. N. Serhan, and L. E. H. Smith. 2007. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nature Medicine 13:868–873.
Cotran, R. S., V. Kumar, and T. Collins, eds. 1999. Robbins pathologic basis of disease. 6th ed. Philadelphia: W.B. Saunders Co.
Decker, Y., G. McBean, and C. Godson. 2009. Lipoxin A4 inhibits IL-1beta-induced IL-8 and ICAM-1 expression in 1321N1 human astrocytoma cells. American Journal of Physiology. Cell Physiology 296:1420–1427.
Dennis, E. A. 2000. Phospholipase A2 in eicosanoid generation. American Journal of Respiratory and Critical Care Medicine 161:S32–S35.
Devchand, P. R., M. Arita, S. Hong, G. Bannenberg, R.-L. Moussignac, K. Gronert, and C. N. Serhan. 2003. Human ALX receptor regulates neutrophil recruitment in transgenic mice: Roles in inflammation and host-defense. The Journal of the Federation of American Societies for Experimental Biology 17:652–659.
Duffield, J. S., S. Hong, V. Vaidya, Y. Lu, G. Fredman, C. N. Serhan, and J. V. Bonventre. 2006. Resolvin D series and protectin D1 mitigate acute kidney injury. Journal of Immunology 177:5902–5911.
Flower, R. J. 2003. The development of COX2 inhibitors. Nature Reviews Drug Discovery 2(3):179–191.
Gleissman, H., R. Yang, K. Martinod, M. Lindskog, C. N. Serhan, J. I. Johnsen, and P. Kogner. 2010. Docosa-hexaenoic acid metabolome in neural tumors: Identification of cytotoxic intermediates. The Journal of the Federation of American Societies for Experimental Biology 24:906–915.
Godson, C., S. Mitchell, K. Harvey, N. A. Petasis, N. Hogg, and H. R. Brady. 2000. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. Journal of Immunology 164:1663–1667.
González-Périz, A., R. Horrillo, N. Ferre, K. Gronert, B. Dong, E. Moran-Salvador, E. Titos, M. Martinez-Clemente, M. Lopez-Parra, V. Arroyo, and J. Claria. 2009. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: A role for resolvins and protectins. The Journal of the Federation of American Societies for Experimental Biology 23(6):1946–1957.
Gronert, K., N. Maheshwari, N. Khan, I. R. Hassan, M. Dunn, and M. L. Schwartzman. 2005. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. Journal of Biological Chemistry 280:15267–15278.
Hassan, I. R., and K. Gronert. 2009. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. Journal of Immunology 182:3223–3232.
Hasturk, H., A. Kantarci, T. Ohira, M. Arita, N. Ebrahimi, N. Chiang, N. A. Petasis, B. D. Levy, C. N. Serhan, and T. E. Van Dyke. 2006. RvE1 protects from local inflammation and osteoclast mediated bone destruction in periodontitis. The Journal of the Federation of American Societies for Experimental Biology 20:401–403.
Hasturk, H., A. Kantarci, E. Goguet-Surmenian, A. Blackwood, C. Andry, C. N. Serhan, and T. E. Van Dyke. 2007. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. Journal of Immunology 179:7021–7029.
Hibbeln, J. R. 1998. Fish consumption and major depression. Lancet 351:1213.
Hong, S., K. Gronert, P. Devchand, R.-L. Moussignac, and C. N. Serhan. 2003. Novel docosatrienes and 17s-resolvins generated from docosahexaenoic acid in murine brain, human blood and glial cells: Autacoids in anti-inflammation. Journal of Biological Chemistry 278:14677–14687.
Hong, S., E. Tjonahen, E. L. Morgan, L. Yu, C. N. Serhan, and A. F. Rowley. 2005. Rainbow trout (Oncorhynchus mykiss) brain cells biosynthesize novel docosahexaenoic acid-derived resolvins and protectins—mediator lipidomic analysis. Prostaglandins & Other Lipid Mediators 78:107–116.
Huang, W. L., V. R. King, O. E. Curran, S. C. Dyall, R. E. Ward, N. Lal, J. V. Priestley, and A. T. Michael-Titus. 2007. A combination of intravenous and dietary docosahexaenoic acid significantly improves outcome after spinal cord injury. Brain Research 130:3004–3019.
Hudert, C. A., K. H. Weylandt, J. Wang, Y. Lu, S. Hong, A. Dignass, C. N. Serhan, and J. X. Kang. 2006. Transgenic mice rich in endogenous n-3 fatty acids are protected from colitis. Proceedings of the National Academies of Sciences of the United States of America 103:11276–11281.
Jin, Y., M. Arita, Q. Zhang, D. R. Saban, S. K. Chauhan, N. Chiang, C. N. Serhan, and R. Dana. 2009. Anti-angiogenesis effect of the novel anti-inflammatory and pro-resolving lipid mediators. Investigative Ophthalmology and Visual Science 50(10):4743–4752.
Kasuga, K., R. Yang, T. F. Porter, N. Agrawal, N. A. Petasis, D. Irimia, M. Toner, and C. N. Serhan. 2008. Rapid appearance of resolvin precursors in inflammatory exudates: Novel mechanisms in resolution. Journal of Immunology 181(12):8677–8687.
Koopman, W. J., and L. W. Moreland. 2005. Arthritis and allied conditions: A textbook of rheumatology. 15th ed. Philadelphia, Pa.; London: Lippincott Williams & Wilkins.
Krishnamoorthy, S., A. Recchiuti, N. Chiang, S. Yacoubian, C.-H. Lee, R. Yang, N. A. Petasis, and C. N. Serhan. 2010. Resolvin D1 binds human phagocytes with evidence for pro-resolving receptors. Proceedings of the National Academies of Sciences of the United States of America 107:1660–1665.
Lands, W. E. M. 2005. Fish, omega-3 and human health. 2nd ed. Champaign, IL: AOCS Press.
Leon, H., M. C. Shibata, S. Sivakumaran, M. Dorgan, T. Chatterley, and R. T. Tsuyuki. 2008. Effect of fish oil on arrhythmias and mortality: Systematic review. British Medical Journal 337:a2931.
Levy, B. D., C. B. Clish, B. Schmidt, K. Gronert, and C. N. Serhan. 2001. Lipid mediator class switching during acute inflammation: Signals in resolution. Nature Immunology 2:612–619.
Levy, B. D., P. Kohli, K. Gotlinger, O. Haworth, S. Hong, S. Kazani, E. Israel, K. J. Haley, and C. N. Serhan. 2007. Protectin D1 is generated in asthma and dampens airway inflammation and hyper-responsiveness. Journal of Immunology 178:496–502.
Libby, P. 2008. Role of inflammation in atherosclerosis associated with rheumatoid arthritis. American Journal of Medicine 121(10 Suppl. 1):S21–31.
Long, E. K., T. C. Murphy, L. J. Leiphon, J. Watt, J. D. Morrow, G. L. Milne, J. R. H. Howard, and M. J. Picklo, Sr. 2008. Trans-4-hydroxy-2-hexenal is a neurotoxic product of docosahexaenoic (22:6; n-3) acid oxidation. Journal of Neurochemistry 105:714–724.
Lukiw, W. J., J. G. Cui, V. L. Marcheselli, M. Bodker, A. Botkjaer, K. Gotlinger, C. N. Serhan, and N. G. Bazan. 2005. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. Journal of Clinical Investigations 115:2774–2783.
Majno, G. 1975. The healing hand: Man and wound in the ancient world. Cambridge, MA: Harvard University Press.
Majno, G., and I. Joris. 2004. Cells, tissues, and disease: Principles of general pathology. 2nd ed. New York: Oxford University Press.
Marcheselli, V. L., S. Hong, W. J. Lukiw, X. Hua Tian, K. Gronert, A. Musto, M. Hardy, J. M. Gimenez, N. Chiang, C. N. Serhan, and N. G. Bazan. 2003. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. Journal of Biological Chemistry 278:43807–43817.
Marcheselli, V. L., P. K. Mukherjee, M. Arita, S. Hong, R. Antony, K. Sheets, N. Petasis, C. N. Serhan, and N. G. Bazan. 2010. Neuroprotectin D1/protectin D1 stereoselective and specific binding with human retinal pigment epithelial cells and neutrophils. Prostaglandins Leukotrienes and Essential Fatty Acids 82:27–34.
Morris, T., M. Stables, A. Hobbs, P. de Souza, P. Colville-Nash, T. Warner, J. Newson, G. Bellingan, and D. W. Gilroy. 2009. Effects of low-dose aspirin on acute inflammatory responses in humans. Journal of Immunology 183:2089–2096.
Mukherjee, P. K., V. L. Marcheselli, C. N. Serhan, and N. G. Bazan. 2004. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proceedings of the National Academies of Sciences of the United States of America 101:8491–8496.
Ohira, T., M. Arita, K. Omori, A. Recchiuti, T. E. Van Dyke, and C. N. Serhan. 2009. Resolvin e1 receptor activation signals phosphorylation and phagocytosis. Journal of Biological Chemistry 285:3451–3461.
Palmblad, J., R. W. Wannemacher, N. Salem, Jr., D. B. Kuhns, and D. G. Wright. 1988. Essential fatty acid deficiency and neutrophil function: Studies of lipid-free total parenteral nutrition in monkeys. The Journal of Laboratory and Clinical Medicine 111(6):634–644.
Poulsen, R. C., K. H. Gotlinger, C. N. Serhan, and M. C. Kruger. 2008. Identification of inflammatory and proresolving lipid mediators in bone marrow and their profile alteration with ovariectomy and omega-3 intake. American Journal of Hematology 83:437–445.
Qin, Q., K. A. Patil, K. Gronert, and S. C. Sharma. 2008. Neuroprotectin D1 inhibits retinal ganglion cell death following axotomy. Prostaglandins Leukotrienes and Essential Fatty Acids 79:201–207.
Riviere, S., L. Challet, D. Fluegge, M. Spehr, and I. Rodriguez. 2009. Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature 459(7246):574–577.
Salem, N., Jr., B. Litman, H. Y. Kim, and K. Gawrisch. 2001. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 36(9):945–959.
Samuelsson, B., S. E. Dahlen, J. A. Lindgren, C. A. Rouzer, and C. N. Serhan. 1987. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 237(4819):1171–1176.
Schwab, J. M., N. Chiang, M. Arita, and C. N. Serhan. 2007. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447:869–874.
Seki, H., K. Fukunaga, M. Arita, H. Arai, H. Nakanishi, R. Taguchi, T. Miyasho, R. Takamiya, K. Asano, A. Ishizaka, J. Takeda, and B. D. Levy. 2010. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. Journal of Immunology 184(2):836–843.
Serhan, C. N., guest ed. 2005. Special issue on lipoxins and aspirin-triggered lipoxins. Prostaglandins Leukotrienes and Essential Fatty Acids 73(3–4):139–321.
Serhan, C. N. 2007. Resolution phases of inflammation: Novel endogenous anti-inflammatory and pro-resolving lipid mediators and pathways. Annual Reviews in Immunology 25:101–137.
Serhan, C. N. 2010. Novel resolution mechanisms in acute inflammation: To resolve or not? American Journal of Pathology 177(4):1576–1591.
Serhan, C. N., and N. Chiang. 2008. Endogenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. British Journal of Pharmacology 153(Suppl. 1):S200–215.
Serhan, C. N., and J. Savill. 2005. Resolution of inflammation: The beginning programs the end. Nature Immunology 6:1191–1197.
Serhan, C. N., C. B. Clish, J. Brannon, S. P. Colgan, N. Chiang, and K. Gronert. 2000. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. Journal of Experimental Medicine 192:1197–1204.
Serhan, C. N., S. Hong, K. Gronert, S. P. Colgan, P. R. Devchand, G. Mirick, and R.-L. Moussignac. 2002. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro-inflammation signals. Journal of Experimental Medicine 196:1025–1037.
Serhan, C. N., K. Gotlinger, S. Hong, Y. Lu, J. Siegelman, T. Baer, R. Yang, S. P. Colgan, and N. A. Petasis. 2006. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: Assignments of dihydroxy-containing docosatrienes. Journal of Immunology 176:1848–1859.
Serhan, C. N., N. Chiang, and T. E. Van Dyke. 2008. Resolving inflammation: Dual anti-inflammatory and proresolution lipid mediators. Nature Reviews. Immunology 8:249–261.
Serhan, C. N., R. Yang, K. Martinod, K. Kasuga, P. S. Pillai, T. F. Porter, S. F. Oh, and M. Spite. 2009. Maresins: Novel macrophage mediators with potent anti-inflammatory and pro-resolving actions. Journal of Experimental Medicine 206:15–23.
Simopoulos, A. P. 2008. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Experimental Biology and Medicine (Maywood, NJ) 233(6):674–688.
Singer, P., H. Shapiro, M. Theilla, R. Anbar, J. Singer, and J. Cohen. 2008. Anti-inflammatory properties of omega-3 fatty acids in critical illness: Novel mechanisms and an integrative perspective. Intensive Care Medicine 34(9):1580–1592.
Sobrado, M., M. P. Pereira, I. Ballesteros, O. Hurtado, D. Fernández-López, J. M. Pradillo, J. R. Caso, J. Vivancos, F. Nombela, J. Serena, I. Lizasoain, and M. A. Moro. 2009. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARgamma-dependent, neuroprotective effects of rosiglitazone in experimental stroke. Journal of Neuroscience 29(12):3875–3884.
Sommer, C., and F. Birklein. 2010. Fighting off pain with resolvins. Nature. Medicine 16:518–520.
Spite, M., L. V. Norling, L. Summers, R. Yang, D. Cooper, N. A. Petasis, R. J. Flower, M. Perretti, and C. N. Serhan. 2009a. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461:1287–1291.
Spite, M., L. Summers, T. F. Porter, S. Srivastava, A. Bhatnagar, and C. N. Serhan. 2009b. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. British Journal of Pharmacology 158:1062–1073.
Starosta, V., K. Pazdrak, I. Boldogh, T. Svider, and A. Kurosky. 2008. Lipoxin A4 counterregulates GM-CSF signaling in eosinophilic granulocytes. Journal of Immunology 181(12):8688–8699.
Sun, Y.-P., S. F. Oh, J. Uddin, R. Yang, K. Gotlinger, E. Campbell, S. P. Colgan, N. A. Petasis, and C. N. Serhan. 2007. Resolvin D1 and its aspirin-triggered 17R epimer: Stereochemical assignments, anti-inflammatory properties and enzymatic inactivation. Journal of Biological Chemistry 282:9323–9334.
Tabas, I.2010. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Reviews in Immunology 10:36–46.
Van Dyke, T. E., and C. N. Serhan. 2006. A novel approach to resolving inflammation. Scientific American presents Oral and Whole Body Health: 42–45.
Wada, K., M. Arita, A. Nakajima, K. Katayama, C. Kudo, Y. Kamisaki, and C. N. Serhan. 2006. Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. The Journal of the Federation of American Societies for Experimental Biology 20:1785–1792.
Xu, Z.-Z., L. Zhang, T. Liu, J.-Y. Park, T. Berta, R. Yang, C. N. Serhan, and R.-R. Ji. 2010. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nature Medicine 16:592–597.
Yanes, O., J. Clark, D. M. Wong, G. G. Patti, A. Sánchez-Ruiz, H. P. Benton, S. A. Trauger, C. Desponts, S. Ding, and G. Siuzdak. 2010. Metabolic oxidation regulates embryonic stem cell differentiation. Nature Chemical Biology 6(6):411–417.
Ye, X.-H., Y. Wu, P.-P. Guo, J. Wang, S.-Y. Yuan, Y. Shang, and S.-L. Yao. 2010. Lipoxin a4 analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Research 1323:174–183.
Potential Efficacy and Mechanism of Action of the Flavanol (–)-Epicatechin in Acute Brain Trauma22
Sylvain Doré23
OVERVIEW
One objective of this research effort over the years has been to provide an integrated mechanism for research into the potential benefits, mechanism(s), and optimal doses of plant-derived flavonoids for the prevention or mitigation of neurologically based illness.
Although many of the polyphenols and flavonoids have been suggested to provide neuroprotection via direct antioxidant properties, we propose that some of these compounds, instead, activate an endogenous cellular pathway that builds resistance to free radical and inflammatory brain damage. More specifically, we have recently reported that the flavanol, (–)-epicatechin, which is naturally enriched in some standardized dark chocolate products, may protect the brain after a stroke by increasing cellular signals already known to protect nerve cells from damage. Ninety minutes after feeding mice a single modest dose of epicatechin, we induced an ischemic stroke by almost entirely cutting off blood supply to the animals’ brains. In the animals that had ingested the epicatechin, we found they had suffered significantly less brain damage than those that had not received the compound. Although most treatments against stroke in humans must be given within a 2- to 3-hour time window to be effective, epicatechin appeared to limit further neuronal damage when given to mice 3.5 hours after a stroke, but not at 6 hours after a stroke. We further showed that intracellular Nrf2/heme oxygenase 1 pathways appear to be of importance because eliminating one or the other abolished most protective epicatechin effects. Thus, by using preclinical models, we can begin to address the efficacy and mechanisms of action, necessary first steps in planning translational clinical tests in humans and helping health-care providers and the public understand how such bioactive nutrients may have both preventive and therapeutic beneficial effects. This work provides an understanding of the pathways by which natural bioactive compounds such as epicatechin can shield nerve cells in the brain from damage provoked by stroke, brain trauma, or age-related neurodegenerative disorders.
BACKGROUND
Polyphenols are found in most plant-derived foods and beverages, and add to the sensory and nutritional qualities of foods. More than 8,000 polyphenolic structures have been identified, but edible plants contain only several hundred of those known (Bravo, 1998; Manach et al., 2004; Ross and Kasum, 2002). They are often involved in the plant’s defensive response against different types of stress, such as ultraviolet radiation, pathogens, and physical damage (Bravo, 1998; Manach et al., 2004). Because plants usually produce these polyphenols as a defensive mechanism, environmental conditions, such as soil type, sun exposure, and rainfall, along with other characteristics such as genetic factors, germination, degree of ripeness, processing and storage, and species variety, can affect the polyphenol concentration (Bravo, 1998; Manach et al., 2004; Ross and Kasum, 2002). Even pieces of fruit from the same tree can have substantial differences in their polyphenol concentrations, owing to diverse exposures to sunlight or other environmental factors. Because of the large variability, the content of polyphenols in foods is usually poorly characterized (Manach et al., 2004).
All polyphenols contain an aromatic ring with one or more hydroxyl group. Most also have at least one sugar residue (a glycoside) attached to the hydroxyl groups. They are classified into different groups depending on the number of phenol rings and chemical groups bound to the rings (Bravo, 1998; Manach et al., 2004; Ross and Kasum, 2002). Polyphenols are found in a wide range of molecular sizes. For example, phenolic acids are simple compounds, whereas the tannins are highly polymerized molecules (Bravo, 1998). Flavonoids make up most of the polyphenols and form the most biologically active group in mammals (Bravo, 1998). Table C-8 summarizes the main classes of polyphenols, some representative phenolics in the groups, and their dietary sources.
Polyphenols are usually recognized for their purported antioxidant capabilities. In test tubes, polyphenols (often at high pharmacological levels) can react with radicals to form
TABLE C-8 Major Subclasses of Polyphenols, Compounds, and Food Sources
|
Polyphenol Group |
Representative compounds |
Common sources |
|
Flavonoid |
||
|
Flavonols |
Quercetin, myricetin, kaempferol |
Onions, apples, kale, red wine, green and black tea, broccoli, berries |
|
Flavanones |
Hesperetin, naringenin, eriodictyol, tangeretin |
Citrus fruit, tomatoes, mint |
|
Flavanols-Flavan-3-ol (proanthocyanidins and catechins) |
Epicatechin, epigallocatechin, epicatechin-3-gallate |
Apricots, green and black tea, red wine, peanut skins, chocolate |
|
Anthocyanins |
Cyanidin, pelargonidin, malvidin, dephinidin |
Berries, grapes, wine, tea, eggplant, cabbage, beans, onions, radishes |
|
Isoflavones |
Genistein, diadzein, glycitein (phytoestrogens) |
Lentils, chickpeas, alfalfa, clover, flaxseed, soybeans |
|
Flavones |
Apigenin, luteolin, diosmetin, tangeretin, nobiletin, sinensetin, wogonin |
Parsley, thyme, celery, citrus fruit rind |
|
Phenolic acid |
||
|
Benzoic acid derivative |
Gallic acid, vanillic, syringic, hydroxybenzoic |
Tea, strawberries, raspberries, blackberries, black radish, onions |
|
Cinnamic acid derivative |
p-coumaric, caffeic, ferulic, sinapic acids, vanillin, syringaldehyde, p-hydroxybenzaldehyde |
Blueberries, kiwis, plums, cherries, apples, cereal grains, coffee |
|
Lignan |
Secoisolariciresinol |
Linseed, lentils, cereals, garlic, asparagus, carrots, pears, prunes |
|
Stilbene |
Resveratrol |
Grapes, red wine |
|
Saponin |
Ginsenoside |
Ginseng root |
|
Other |
Curcumin |
Turmeric |
polyphenol radicals, which are more stable and less reactive because of the ability of the phenol group to absorb extra electrons. Most polyphenols are conjugated by methylation, sulfation, or glucuronidation during metabolism. The antioxidant capability can be determined by the type of conjugate and its location on the polyphenol structure (Bravo, 1998; Esposito et al., 2002; Manach et al., 2004; Nijveldt et al., 2001; Williams et al., 2004). In addition to their antioxidant ability, at high concentrations the polyphenols can inhibit the activities of several enzymes, including lipoxygenase, cyclooxygenase, xanthine oxidase, phospholipase A2, ATPases, aldole reductase, phoshodiesterases, topoisomerase I and II, protein kinase C, phosphoinositide 3-kinase, protein kinase B (Akt/PKB), and mitogen-activated protein kinases (Esposito et al., 2002; Korkina and Afanas’ev, 1997; Nijveldt et al., 2001; Skibola and Smith, 2000; Williams et al., 2004). Some polyphenols have weak estrogenic properties, and others can inhibit the enzymes involved in estrogen metabolism, aromatase, and 17β-hydroxysteroid oxidoductase (Skibola and Smith, 2000).
The reduction of several diseases has been linked to polyphenols. Cardioprotection and a reduction in the incidence of certain types of cancer have been correlated with consumption of phenolic antioxidants (Bravo, 1998; Ross and Kasum, 2002; Skibola and Smith, 2000). Evidence also indicates that polyphenols have antiallergenic, antiviral, antibiotic, an-
tidiarrheal, antiulcer, and anti-inflammatory properties. Polyphenols have been used to treat hypertension, vascular fragility, allergies, and hypercholesterolemia (Bravo, 1998; Korkina and Afanas’ev, 1997; Nijveldt et al., 2001), and have been implicated in the prevention of neurodegenerative diseases by their ability to protect neurons against oxidative stress. Even ascorbate, at concentrations 10-fold higher than those of the polyphenols, did not offer as much neuroprotection (Esposito et al., 2002; Williams et al., 2004). Polyphenols attenuate ischemia–reperfusion injury, suggesting that they might interfere with nitric oxide synthase activity, thereby inhibiting lipid peroxidation, decreasing the number of immobilized leukocytes during reperfusion, and reducing complement activation, resulting in a diminished inflammatory response (Nijveldt et al., 2001). In addition to their antioxidant actions, they also influence neuroprotective and neurorestorative signal transduction mechanisms (Williams et al., 2004).
Epidemiological studies show an inverse relationship between stroke and polyphenol consumption (Ross and Kasum, 2002). The dietary intake of polyphenols varies greatly among different societies. Isoflavone intake from soy consumption ranges from 20 to 240 mg/day for Asians and from 1 to 9 mg/day in the United States and Western populations (Manach et al., 2004; Skibola and Smith, 2000). Agricultural practices also can affect dietary intake of polyphenols. A region where a particular plant is grown will probably have the greatest consumption (Manach et al., 2004). The total consumption of flavonols, flavanones, flavanols, and isoflavones in Western cultures is estimated to be 100 to 150 mg/day. An accurate estimate of dietary intake of all polyphenols ingested is difficult to attain because of poor characterization of polyphenols in foods and the great variability of polyphenol concentration and “quality” within foods (Manach et al., 2004).
DISEASE STATES THAT ARE POTENTIALLY PREVENTABLE OR TREATABLE WITH FLAVONOIDS
Inflammatory Pain
Chronic pain is a leading cause of disability and high health-care costs in North America, Europe, and Australia. Although it is likely to be a significant problem in the developing countries as well, epidemiological data are not easily accessible. A recent systematic review of 13 studies reported the prevalence of chronic pain in developed nations to range from 10 percent to 55 percent (Harstall and Ospina, 2003). These data indicate a higher prevalence of chronic pain among females than males. It is estimated that the annual cost of medical care and lost productivity in the United States as a result of chronic pain is estimated at $120 billion (Harstall and Ospina, 2003). With the aging of the “baby boomers,” the incidence and cost of pain management is likely to increase. Pain practitioners have recognized that chronic pain is not merely a symptom, but is a disease with far-reaching consequences on several aspects of life: mood, concentration, motor performance, sleep, and social relations.
Arthritis ranks among the most common chronic pain conditions in the elderly, with studies suggesting that about half of Americans over 65 years suffer from the disease (CDC, 2003; Feinglass et al., 2003). Although the elderly have the highest risk, approximately two-thirds of those afflicted with arthritis are below 65 years old. A third of the 66 million Americans with arthritis must restrict their daily activities because of pain (Verbrugge and Juarez, 2006). A variety of disease states, such as rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, lupus, and inflammatory bowel disease (e.g., Crohn’s disease and ulcerative colitis), are associated with inflammation of joints, resulting in pain, swelling, and loss of function in the affected limbs. Felson (2004) recently reviewed factors associated with
pain in those with osteoarthritis. Those with pain were more likely to have effusions, bone marrow edema, synovitis and synovial hypertrophy, and tendinitis and bursitis around the joint. Policy reports from the U.S. Department of Health and Human Services in 2000 called for more research on this common disabling condition.
In terms of overall burden on the population, arthritis ranks high in America and worldwide for medical costs, lost income, and lost years of disability-free life (Murry and Lopez, 1996; Reginster, 2002). Analysis of data from the National Health Interview Survey Disability Supplement Phase Two revealed that arthritis-disabled individuals get out less often than other disabled individuals, and have more disabilities related to personal care, household management, physical tasks, transportation, and work (Verbrugge and Juarez, 2006). The disabilities result in fatigue, long task time, and pain. At the beginning of this century, it was estimated that arthritis treatment in America would exceed 2 percent of the gross domestic product, with estimated medical care costs for people with arthritis of $15 billion annually and total costs (medical care and lost productivity) of almost $65 billion annually (Felson et al., 2000).
Ischemia and Hemorrhagic Stroke
Stroke
Stroke is a major cause of morbidity and mortality across all industrialized countries. It is the third leading cause of death in America and the number one cause of adult disability. A stroke occurs when a blood clot blocks an artery or when a blood vessel breaks, interrupting blood flow to an area of the brain. When either of these things happens, brain cells begin to die and brain damage occurs. As brain cells die, speech, movement, and memory can be lost. How a stroke patient is affected depends on the location of the stroke in the brain and the extent of brain damaged. For example, someone who has a small stroke may experience only minor problems, such as weakness of an arm or leg, whereas larger strokes may cause unilateral paralysis or loss of speech. Some people recover completely from strokes, but more than two-thirds of survivors will have some type of disability.
Types of Stroke
Ischemic Stroke Although blood clotting is normal and essential for stopping blood loss from wounds, in the case of stroke, blood clots are dangerous because they can block arteries and cut off blood flow, a process called ischemia. Ischemia can be caused by embolic and thrombotic strokes. In an embolic stroke, a blood clot forms somewhere in the body (usually the heart) and then travels through the bloodstream to the brain, where it eventually travels to a blood vessel small enough to block its passage. The clot lodges there, blocking the blood vessel and causing a stroke. The medical term for this type of blood clot is embolus. In thrombotic stroke, blood flow is impaired because of blockage to one or more of the arteries that supply blood to the brain. The process leading to this blockage is known as thrombosis. Ischemic strokes also can occur as the result of an unhealthy blood vessel clogged with a buildup of fatty deposits and cholesterol. The body regards these buildups as multiple tiny, repeated injuries, and reacts as it would to bleeding from a wound; it responds by forming clots. Initial treatment for ischemic stroke involves removing the blockage and restoring blood flow. Tissue plasminogen activator (t-PA) is a medication that can break up blood clots and restore blood flow when administered within three hours of the event.
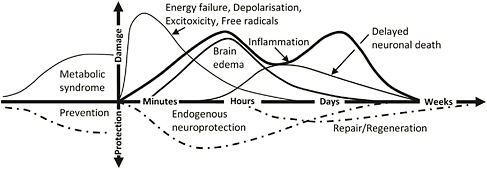
FIGURE C-18 Simplified pathobiology of stroke-induced damage, endogenous repair, and regeneration.
SOURCE: Adapted from Dirnagl et al., 1999.
Hemorrhagic Stroke Strokes caused by the breakage or rupture of a blood vessel in the brain are called hemorrhagic strokes. Hemorrhages can be caused by a number of disorders that affect the blood vessels, including long-standing high blood pressure and cerebral aneurysms (weak or thin spots in the vessel wall). Aneurysms, usually present at birth, develop over a number of years, and usually do not cause detectable problems until they break. There are two types of hemorrhagic stroke: intracerebral and subarachnoid. In an intracerebral hemorrhage, primarily caused by hypertension, bleeding occurs from vessels within the brain itself. In a subarachnoid hemorrhage, an aneurysm bursts in a large artery on or near the thin, delicate membrane surrounding the brain. Blood spills into the area around the brain, thereby contaminating the cerebral spinal fluid that normally surrounds the brain. Treatment for hemorrhagic stroke usually requires surgery to relieve intracranial pressure caused by bleeding. Most of the damage caused by this type of stroke results from the physical disruption of brain tissue. Surgical treatment for hemorrhagic stroke caused by an aneurysm or defective blood vessel can prevent additional strokes. Surgery may be performed to seal off the defective blood vessel and redirect blood flow to other vessels that supply blood to the same region of the brain. Endovascular treatment involves inserting a long, thin, flexible tube (catheter) into a major artery, usually in the thigh, guiding it to the aneurysm or the defective blood vessel, and inserting tiny platinum coils, called stents, into the blood vessel through the catheter. Stents support the blood vessel to prevent further damage and additional strokes.
It is likely that the most effective therapeutic intervention should begin as soon as possible after the onset of stroke (Figure C-18). Considering that most stroke patients seek treatment after several hours, an intervention that could limit inflammation and delayed neuronal cell death (apoptosis-like) would be highly desirable. Flavonoids could fill this need by inducing the endogenous system, such as heme oxygenase, which we and others have shown to have antioxidant, anti-inflammatory, and anti-delayed-cell-death activity.
PUTATIVE MECHANISMS OF FLAVONOIDS
It is estimated that 80 percent of strokes are preventable, but there are no simple solutions. Some measures include not smoking, exercising regularly, maintaining an appropriate body weight, and limiting dietary intake of salt, alcohol, and saturated fat. People with hypertension, diabetes, or high cholesterol can reduce their risk for stroke through proper medication and appropriate lifestyle modifications.
A vast amount of recent literature proposes that it is the polyphenols in numerous natural extracts that provide the beneficial effects, and that the benefits are mainly attributable to their potential to act as antioxidants. Among the polyphenols, the most abundant, and those with the greatest bioactivity, are the flavonoids, which are found in natural extracts, plants, and fruits. The antioxidant properties of natural extracts could be associated with the presence of flavonoids. We propose that the antioxidant properties likely result from the initiation of a cascade of intracellular events that lead to activation of endogenous antioxidant pathways. We believe that this mechanism is the most likely, as flavonoids do not reach sufficient plasma levels to neutralize free radicals directly. Consequently, the flavonoids are likely to stimulate an intracellular signaling pathway that leads to cytoprotection (Figure C-19).
Several genes and proteins have been shown to be potential targets of flavonoids. Heme oxygenase (HO) is a strong candidate. We have shown that several polyphenols are potent inducers of HO protein levels, activity, and cytoprotection (Zhuang et al., 2003). The main function of HO is to cleave heme (iron-protoporphyrin-IX). That reaction liberates iron and generates carbon monoxide and biliverdin, which are rapidly converted to bilirubin. Modulation of HO activity and levels most likely provides cytoprotection against free radical damage through degradation of heme (a prooxidant) into biliverdin/bilirubin (antioxidants). Using in vitro and in vivo models, we have shown that HO can be neuroprotective (Doré, 2002). Heme oxygenase and its metabolites also have been associated with antiapoptotic and anti-inflammatory actions, and are known to have a vasodilatory effect.
Several other enzymatic systems have been suggested to be either directly or indirectly
FIGURE C-19 As a working hypothesis, we have proposed that flavonoids can be protective by enhancing the levels of heme oxygenase and, consequently, its multiple beneficial functions.
NOTE: ARE = antioxidant response element, BV = biliverdin, BR = bilirubin, CO = carbon monoxide, Fe2+ = ferrous iron, Keap 1 = Kelch-like ECH-associated protein 1, Nrf2 = NF-E2-related factor 2.
modified by different members of the flavonoid family. For example, inflammation and carcinogenesis cause upregulation of cyclooxygenase-2 (COX-2) and tumor necrosis factor alpha (TNFα), which consequently lead to increased production and release of pro-inflammatory prostaglandins and cytokines. The inflammatory response can then activate and sensitize nearby nerve fibers, and result in persistent pain and hyperalgesia (Abbadie, 2005; Mantyh et al., 2002; Sorkin et al., 1997; Watkins et al., 1994; Woolf et al., 1997). Hence, prophylactic or therapeutic interventions aimed at inhibiting COX activity can lead to cytoprotection. A number of flavonoids have shown promising anti-inflammatory properties. For example, using animal models of cancer and inflammation, investigators demonstrated that flavonoids in tea suppress arachidonic acid metabolism, leukotriene production, and COX activity (Huang et al., 2006; Metz et al., 2000). Cyanidin-3-glucoside from blackberries was efficacious in inhibiting COX-2 and TNF expression, an effect that led to inhibition of carcinogenesis (Cooke et al., 2006; Ding et al., 2006). Our published and preliminary studies indicate that flavonoids, such as (–)-epicatechin and cyanidin-3-glucoside, have significant COX-1 and COX-2 inhibitory activities (Seeram et al., 2001). Because COX-1 inhibition can lead to deleterious effects such as gastric bleeding, and COX-2 inhibition has antiinflammatory effects, it is important to clearly determine the effect of flavonoids on COX enzyme activity from both a therapeutic and safety standpoint.
Here, we propose that the protective properties of flavonoids primarily stem from HO-1 induction, through which they provide the brain with resistance to a variety of neurological stresses (Figure C-20). Flavonoids have been shown to affect cerebral blood flow, cell death, and inflammatory processes, all of which are important therapeutic targets, because they are known to be factors in the development of acute and/or chronic neurodegenerative diseases. These vascular properties become especially important when considering that reduction in cerebral blood flow and lack of oxygen, followed by a reperfusion phase are likely to affect specific neurons and/or the cell types that are particularly vulnerable to free radical damage. Consequently, preventing cell death is likely to have a beneficial effect on the rate of neuroinflammation and its consequences. Considering their actions and favorable effects, one can make a valid hypothesis that polyphenol flavonoids could precondition neurons against induced stress damage.
RATIONALE
One of the most popular explanations given for the benefits of bioactive nutrients is that they have antioxidant properties, but recent conflicting results reported in the literature question the validity of such general statements. Therefore, we have decided to concentrate on better understanding the purported properties of the functional foods by using various methodologies and models. Also, in numerous natural extracts, polyphenols are often cited as the main biologically active (or bioactive) components. Considering the wide variety of compounds included in the polyphenol class, we have decided to focus on the flavonoids. We already have demonstrated the neuroprotective efficacy of some flavonoids, and have developed a working hypothesis to be tested and developed in cellular and in vivo models. The research design of this endeavor will allow us to determine the efficacy of preventive and therapeutic protocols, and optimize the time course and doses with the preclinical models. That information then can be put to the challenge in a clinical trial.
Our research thus far has focused on the effect of flavonoids in ischemic stroke, though such work could be extended to other brain trauma. Again, taking into consideration the purported anti-inflammatory and antioxidant abilities of flavonoids, their actions are likely to have significant influence on the outcome of ischemia in the brain, an organ especially vul-
nerable to inflammatory and oxidative stress injury. One of the richest flavonoid-containing foods is cocoa. It has been suggested for centuries to have medicinal properties. It appears to possess nearly twice the antioxidant level of red wine and up to three times that of green tea. The flavonoids in cocoa are principally flavanols, such as epicatechin and catechin. Based on reports that flavonoids have cardioprotective effects through blood flow regulation, their consumption has been suggested for use as potential preventive medicine and for treatment of neurological disorders. Brain cell death in acute and chronic neurodegenerative conditions is suggested to be mediated by free radical damage. Flavonoid structures normally lead one to predict their antioxidant properties, but the underlying cellular mechanisms have not been found. Knowing that the HO enzyme plays various roles in oxidative stress, blood flow, and cell death, we tested the hypothesis that HO activity could participate in flavonoid neuroprotective function. Preliminary results reveal that cultured neurons treated with flavonoids induce HO-1 expression. Pretreatment of neurons is sufficient to provide protection, demonstrating that co-treatment during induced oxidative stress is not necessary. This finding suggests that flavonoids may not be the direct cause of the protection, but that they might induce a neuroprotective pathway. The protective effect of the flavonoids is abolished by an HO inhibitor or a protein synthesis inhibitor. Therefore, specific HO-1 induction may be a novel mechanism by which flavonoids are protective. Our results show that cocoa flavanols affect stroke outcomes. In parallel, we have been investigating mechanisms of action in cultured neurons derived from wild-type control and knockout mice. This was one of the first studies to show a specific pathway by which cocoa-derived flavanol compounds can protect neurons in culture and in the brain. The study also revealed a mechanism by which those compounds might provide the brain resistance in acute and/or chronic neurological disorders.
DISCUSSION
Although the general public is targeted daily with claims that a “natural” compound will reduce inflammation, lessen cognitive decline, boost memory, prevent neurological diseases, etc., individuals and practitioners have the right to know what is effective and what is the proper dose. Moreover, although most claims of efficacy are based on potential antioxidant and anti-inflammatory properties, the most recent critical reviews suggest that such statements are probably inaccurate. There is also the misconception that because these extracts are natural, they do not have side effects, and that the more one consumes, the more benefits one would receive. When these extracts are used in clinical trials, it is important to know the appropriate dose range and maximal dose to be tested. Researchers should take advantage of well-established preclinical models to apply rigorous scientific testing to address these issues. Our goals over the years have been to examine alternative hypotheses regarding the direct antioxidant actions of phytochemicals. We have been investigating an indirect mechanism by which bioactive compounds could build intracellular or organ resistance to free radical damage. Stimulation of an endogenous defense pathway would then attenuate inflammatory and cell death cascades.
Our emphasis here has been based on several factors:
-
Elucidating the underlying mechanisms of action of nutraceutical therapies will facilitate their integration into conventional medical care. Thus, our goal has been to focus on addressing and testing mechanisms of action based on the most recent research. Accumulation of laboratory data has allowed us to develop hypotheses
-
to be tested, notably on the role of the transcriptional factor Nrf2 and HO as an antioxidant-protective enzyme. We believe that identifying the potential mechanisms of action can contribute to better acceptance and integration of nutraceutical therapies into conventional medicine. The information will be of benefit to both patients and practitioners.
-
Mechanistic studies of nutraceutical therapies will improve the identification of key study endpoints and, thus, will strengthen the design of nutraceutical clinical trials. Preventive medicine is a strategy by which natural or synthetic chemicals or biological agents are used to prevent health or medical problems before they arise. Despite advances in the treatment of numerous neurological disorders, morbidity and mortality remain high. Because most neurological diseases have no cure, prevention strategies can save many lives. Epidemiological data have suggested that numerous natural, biologically based practices may prevent diseases. Studies in our research laboratories have provided evidence that natural extracts and isolated compounds can be beneficial. Because preventive clinical trials typically require thousands of patients and decades of follow-up, it is important to consider the development of suitable surrogate markers that can be used to monitor the effect of bioactive treatment on a short-term and long-term basis. Efforts from various teams have been devoted to identifying key endpoints and testing potential surrogate markers. Such tools would help the research community to design future clinical trials of nutraceuticals.
-
It has been postulated that the use of adequately defined products and optimal dosage schedules in studies decreases the risk of failures that might discourage further research into otherwise promising modalities. Therefore, for optimal study design, it is important to: (a) determine active ingredients, pharmacology, bioavailability, and optimal dosing; (b) identify surrogate markers; and (c) assess study feasibility. The goal is that outcomes of such studies will form the basis for designing larger trials with an enhanced ability to detect a meaningful positive effect, if any, of the therapy under study.
Thus, to investigate the mechanisms of action, we have been focusing on one family of active ingredients present in numerous bioactive nutrients-the flavanols and their metabolites. By examining specific compounds, we can test specific hypotheses and potential actions. In so doing, we can better address pharmacology, bioavailability, and optimal dosing of the compounds. Appropriate support will allow various teams of investigators, including our own, to continue designing optimal protocols to use a given compound, will potentially help us to identify surrogate markers, and will certainly allow us to improve feasibility and determine the therapeutic and preventive efficacy of given members of the flavonoid family. We also understand that by testing an active ingredient to better address mechanisms of action, it is also important, in most cases, to test naturally enriched standardized extracts.
In the literature, recent results from various clinical trials of natural biologically active approaches have yielded negative data. These negative results have raised concerns, especially regarding appropriate dosages. Negative results are informative; however, carefully conducted dose-response studies that use preclinical models are needed. In response to the goals of this specific Institute of Medicine study, it is important to use and further develop preclinical models to begin resolving some of these issues that are often associated with bioactive nutraceutical research.
The general public and funding agencies are paying close attention to biologically based practices, including foods and food components, dietary supplements, and functional foods. One of the purported active ingredients often cited in food supplements is a group of com
pounds called polyphenols. To limit the complexity of this wide family of compounds, we will first focus our efforts on a subset of the polyphenols that is postulated to provide the main bioactivity: the flavonoids. We already have reported substantial evidence regarding the neuroprotective effects of flavonoids in both in vitro and in vivo models, though we recognize that additional research is most likely required. Once mechanisms of action have been better explored and delineated, the plan will be to test these natural compounds and/or enriched extracts. We believe that results from this research endeavor could be applied to various acute neurodegenerative disorders.
CONCLUSIONS
Bioactive nutrients often are used because of their purported beneficial properties, such as free radical scavenging and/or inhibition of enzymes involved in the inflammatory cascade. Although these beneficial effects have been shown in test tubes or in cultured cells, we and others believe that the mechanism responsible for these effects is not through direct neutralization of free radicals. It is unlikely that any of the bioactive compounds or their metabolites reach sufficient levels in the bloodstream to directly fend off free radical-induced cellular damage or inhibit enzymes. Thus, the specific mechanisms of action of most of these compounds are still unknown. Based on our ongoing research, we have been proposing that these compounds, even at very low levels in the organism, would be sufficient to initiate an intracellular defense against environmental or pathological stress. Our central working hypothesis is that flavanols, instead of acting as direct antioxidants, stimulate the Nrf2 pathway and induce an antioxidant/protective enzyme, such as HO-1, thus providing cellular and tissue resistance to damage generated by oxidative stress. We already have found that HO-1 is inducible by the flavanols. HO has been characterized as an antioxidant enzyme that catalyzes the degradation of heme, a prooxidant, into the antioxidants, biliverdin and bilirubin.
By addressing optimal doses, limitations, side effects, and mechanisms of action, such research could help both the general public and health-care providers make informed decisions on whether or not the given biologically active modalities should be accepted for prevention or as adjunct treatment for specific medical conditions. Multidisciplinary approaches, including disciplines such as neurobiology, pharmacology, imaging, biomarkers, and cellular/molecular biology, should be encouraged. We believe that the synergistic projects proposed have potentially high impact and present innovative concepts. The goal is that such efforts would result in synergistic discoveries and optimal plans for the most rigorously executed, randomized clinical trials.
REFERENCES
Abbadie, C. 2005. Chemokines, chemokine receptors and pain. Trends in Immunology 26(10):529–534.
Bravo, L. 1998. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutrition Reviews 56(11):317–333.
CDC (Centers for Disease Control and Prevention). 2003. Public health and aging: Projected prevalence of self-reported arthritis or chronic joint symptoms among persons aged ≥ 65 years—United States, 2005–2030. Morbidity and Mortality Weekly Report 52(21):489–491.
Cooke, D., M. Schwarz, D. Boocock, P. Winterhalter, W. P. Steward, A. J. Gescher, and T. H. Marczylo. 2006. Effect of cyanidin-3-glucoside and an anthocyanin mixture from bilberry on adenoma development in the ApcMin mouse model of intestinal carcinogenesis—relationship with tissue anthocyanin levels. International Journal of Cancer 119(9):2213–2220.
Ding, M., R. Feng, S. Y. Wang, L. Bowman, Y. Lu, Y. Qian, V. Castranova, B. H. Jiang, and X. Shi. 2006. Cyanidin-3-glucoside, a natural product derived from blackberry, exhibits chemopreventive and chemotherapeutic activity. The Journal of Biological Chemistry 281(25):17359–17368.
Dirnagl, U., C. Iadecola, and M. A. Moskowitz. 1999. Pathobiology of ischaemic stroke: An integrated view. Trends in Neurosciences 22(9):391–397.
Doré, S. 2002. Decreased activity of the antioxidant heme oxygenase enzyme: Implications in ischemia and in Alzheimer’s disease. Free Radical Biology & Medicine 32(12):1276–1282.
Doré, S. 2005. Unique properties of polyphenol stilbenes in the brain: More than direct antioxidant actions; gene/protein regulatory activity. Neuro-Signals 14(1–2):61–70.
Esposito, E., D. Rotilio, V. Di Matteo, C. Di Giulio, M. Cacchio, and S. Algeri. 2002. A review of specific dietary antioxidants and the effects on biochemical mechanisms related to neurodegenerative processes. Neurobiology of Aging 23(5):719–735.
Feinglass, J., C. Nelson, T. Lawther, and R. W. Chang. 2003. Chronic joint symptoms and prior arthritis diagnosis in community surveys: Implications for arthritis prevalence estimates. Public Health Reports 118(3):230–239.
Felson, D. T. 2004. Risk factors for osteoarthritis: Understanding joint vulnerability. Clinical Orthopaedics and Related Research 427 (Suppl.):S16–S21.
Felson, D. T., R. C. Lawrence, P. A. Dieppe, R. Hirsch, C. G. Helmick, J. M. Jordan, R. S. Kington, N. E. Lane, M. C. Nevitt, Y. Zhang, M. Sowers, T. McAlindon, T. D. Spector, A. R. Poole, S. Z. Yanovski, G. Ateshian, L. Sharma, J. A. Buckwalter, K. D. Brandt, and J. F. Fries. 2000. Osteoarthritis: New insights. Part 1: The disease and its risk factors. Annals of Internal Medicine 133(8):635–646.
Harstall, C., and M. Ospina. 2003. How prevalent is chronic pain? Pain Clinic Updates 11(2).
Huang, M. T., Y. Liu, D. Ramji, C. Y. Lo, G. Ghai, S. Dushenkov, and C. T. Ho. 2006. Inhibitory effects of black tea theaflavin derivatives on 12-o-tetradecanoylphorbol-13-acetate-induced inflammation and arachidonic acid metabolism in mouse ears. Molecular Nutrition and Food Research 50(2):115–122.
Korkina, L. G., and I. B. Afanas’ev. 1997. Antioxidant and chelating properties of flavonoids. Advances in Pharmacology 38:151–163.
Manach, C., A. Scalbert, C. Morand, C. Remesy, and L. Jimenez. 2004. Polyphenols: Food sources and bioavailability. American Journal of Clinical Nutrition 79(5):727–747.
Mantyh, P. W., D. R. Clohisy, M. Koltzenburg, and S. P. Hunt. 2002. Molecular mechanisms of cancer pain. Nature Reviews. Cancer 2(3):201–209.
Metz, N., A. Lobstein, Y. Schneider, F. Gosse, R. Schleiffer, R. Anton, and F. Raul. 2000. Suppression of azoxy-methane-induced preneoplastic lesions and inhibition of cyclooxygenase-2 activity in the colonic mucosa of rats drinking a crude green tea extract. Nutrition and Cancer 38(1):60–64.
Murry, D. J., and A. D. E. Lopez. 1996. The global burden of disease: A comprehensive assessment of mortality and disability from disease, injuries, and risk factors in 1990 and projected to 2020. In Global Burden of Disease and Injury Series, Vol. I. Cambridge, MA: Harvard School of Public Health.
Nijveldt, R. J., E. van Nood, D. E. van Hoorn, P. G. Boelens, K. van Norren, and P. A. van Leeuwen. 2001. Flavonoids: A review of probable mechanisms of action and potential applications. American Journal of Clinical Nutrition 74(4):418–425.
Reginster, J. Y. 2002. The prevalence and burden of arthritis. Rheumatology (Oxford) 41(Suppl. 1):3–6.
Ross, J. A., and C. M. Kasum. 2002. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annual Review of Nutrition 22:19–34.
Seeram, N. P., R. A. Momin, M. G. Nair, and L. D. Bourquin. 2001. Cyclooxygenase inhibitory and antioxidant cyanidin glycosides in cherries and berries. Phytomedicine 8(5):362–369.
Skibola, C. F., and M. T. Smith. 2000. Potential health impacts of excessive flavonoid intake. Free Radical Biology & Medicine 29(3–4):375–383.
Sorkin, L. S., W. H. Xiao, R. Wagner, and R. R. Myers. 1997. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience 81(1):255–262.
Verbrugge, L. M., and L. Juarez. 2006. Profile of arthritis disability: II. Arthritis and Rheumatism 55(1):102–113.
Watkins, L. R., E. P. Wiertelak, L. E. Goehler, K. P. Smith, D. Martin, and S. F. Maier. 1994. Characterization of cytokine-induced hyperalgesia. Brain Research 654(1):15–26.
Williams, R. J., J. P. Spencer, and C. Rice-Evans. 2004. Flavonoids: Antioxidants or signalling molecules? Free Radical Biology & Medicine 36(7):838–849.
Woolf, C. J., A. Allchorne, B. Safieh-Garabedian, and S. Poole. 1997. Cytokines, nerve growth factor and inflammatory hyperalgesia: The contribution of tumour necrosis factor alpha. British Journal of Pharmacology 121(3):417–424.
Zhuang, H., Y. S. Kim, R. C. Koehler, and S. Doré. 2003. Potential mechanism by which resveratrol, a red wine constituent, protects neurons. Annals of the New York Academy of Sciences 993:276–286; discussion 287–278.
Overview of Therapeutics for Traumatic Brain Injury
Edward D. Hall24
INTRODUCTION
The pathophysiology of traumatic brain injury (TBI) is divisible into two components. The first component is the “primary” mechanical injury to the brain tissue involving the shearing, stretching, or twisting of neuronal axons and dendrites and vascular elements. The only way to minimize this primary mechanical insult involves avoidance of the injury or the use of protective head gear to minimize any blunt impact or penetrating trauma to the skull and brain contained within. The only therapeutic approach to compensating for the brain damage done by the primary mechanical event involves finding a means to (1) stimulate the process of neurogenesis to form new neurons to replace those that have been lost or angiogenesis to form new blood vessels to restore adequate tissue perfusion and oxygen delivery, (2) to enhance the regeneration of the new axons from injured, but surviving, neuronal cell bodies, or (3) to somehow increase the plasticity of the axons and nerve terminals of surviving (i.e., uninjured) neurons so that they can branch and form new synaptic connections to functionally compensate for the degenerated cells and their lost postsynaptic connections. Although a number of pharmacological and gene therapeutic approaches for achieving each of these options are under investigation, the translation of these to clinical use is still many years away.
The second component of the TBI-induced brain damage involves a complex pathophysiological cascade of events that is set in motion by the primary injury, develops over the first seconds, minutes, hours, and days, and leads to progressive “secondary” microvascular, neuronal, and glial cell degeneration. This cascade has been shown to be extraordinarily complex, and the individual molecular events are linked through a number of feed-forward and feed-back pathways. However, based upon our knowledge of much of the secondary injury process, a number of therapeutic targets have been identified by which posttraumatic brain damage can be attenuated either pharmacologically, targeting individual secondary injury mechanisms, or by induction of brain cooling (i.e., moderate local or systemic hypothermia; decrease in brain temperature from ~37 to 33°C), which has been shown to simultaneously interfere with multiple secondary injury molecular targets.
Although TBI can victimize active individuals at any age, most injuries occur in young adults in the second and third decades of life. This includes a large number of blast-induced TBIs to our war fighters in the Iraq and Afghanistan theaters in the War on Terror mainly caused by improvised explosive devices (IEDs). Moreover, the majority of civilian and military TBI patients now survive their neurological insults owing to improvements in emergency, neurological intensive care and surgical treatments. Nevertheless, the need for intensive rehabilitation and the reality of prolonged disability exacts a significant toll on the individual, his or her family, and society. Effective ways of maintaining or recovering function could markedly improve the outlook for those with TBI enabling higher levels of independence and productivity. However, at present, there are no clinically proven and Food and Drug Administration-approved pharmacological therapies for acute treatment of TBI patients aimed at mitigating the damaging neurological effects of their injuries. Moreover, the efficacy and optimal application of moderate systemic hypothermia remains to be established. Nevertheless, the possibility of an effective “neuroprotective” treatment
FIGURE C-21 Schematic of posttraumatic secondary injury showing the cascade of pathophysiological and pathochemical players and their inter-relationships. Reprinted with permission from Hall.
NOTE: AA = arachidonic acid; AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; COX1 = cyclooxygenase 1; COX2 = cyclooxygenase 2; LTs = leukotrienes; NMDA = N-methyl-D-aspartic acid; NOS = nitric oxide synthase; ONOO– = peroxynitrite; PGF2α = prostaglandin 2α; TBI = traumatic brain injury; TXA2 = thromboxane A2; 5-LO = 5-lipoxygenase
SOURCE: Hall, 2007.
is derived from the fact that although some of the neurodegeneration is due to the primary mechanical trauma, the majority of posttraumatic damage is the result of secondary events that exacerbate the effects of the primary injury. Figure C-21 displays a schematic diagram that includes many of the key molecular players in secondary injury that have been experimentally targeted by pharmacological neuroprotective drug candidates and in the case of glutamate receptor-medicated excitotoxicity, intracellular calcium (Ca2+) overload, and reactive oxygen-induced oxidative damage have been the subject of clinical trials during the past 2 decades.
However, as shown in Table C-9, several compounds that have targeted these mechanisms in phase II and III clinical trials in moderately and/or severely injured TBI patients have failed to produce a significant improvement in survival and neurological outcomes.
TABLE C-9 Compounds That Have Failed to Produce an Overall Improvement in Phase III (or large Phase II) TBI Trials
|
Compound and Pharmaceutical Sponsor |
Mechanism(s) of Action |
|
Nimodipine (Nimotop; Bayer) |
L-type Ca2+ channel blocker |
|
Selfotel (CGS19755; Ciba-Geigy) |
competitive NMDA antagonist |
|
D-CPP-ene (Sandoz) |
competitive NMDA antagonist |
|
Aptiganel (CNS1102; Cambridge Neuroscience) |
non-competitive NMDA antagonist |
|
CP 101,606 (Traxoprodil; Pfizer) |
non-competitive NR2B subtype selective NMDA antagonist |
|
PEG-SOD (PEG-orgatine; Sterling-Winthrop) |
free radical scavenger |
|
Tirilazad (U-74006F, Freedox; Upjohn) |
lipid peroxidation inhibitor |
|
Dexanabinol (HU211; Pharmos) |
non-competitive NMDA antagonist; antioxidant; TNF-α inhibitor |
MAJOR FINDINGS FROM THE 2008 NIH COMBINATION THERAPIES FOR TBI WORKSHOP
The failure of these clinical trials to demonstrate the clinical effectiveness of these various neuroprotective agents has been attributed to a number of shortcomings in discovery, development, and clinical testing including (1) an insufficient understanding of the secondary injury mechanisms, time courses, and inter-relationships, (2) inadequate preclinical testing in regards to dose-response, definition of therapeutic blood levels, optimal duration of treatment and the therapeutic post-TBI time window in which it is possible to interrupt the targeted injury mechanism, and (3) poorly conceived clinical trial design, insensitive endpoints, and lack of recognition that not all TBIs have the same pathophysiological characteristics (i.e., there are subgroups of TBI patients that might selectively respond to particular types of drugs) (Narayan et al., 2002). In regard to the possibility that particular drugs may be effective in some types of TBIs, the antioxidant/lipid peroxidation inhibitor tirilazad, listed in Table C-9, has been reported to significantly improve survival in male TBI patients who displayed traumatic subarachnoid hemorrhage (tSAH) despite the fact that this subgroup effect was statistically obscured in the overall group of moderate and severe TBI patients enrolled in the trial (Marshall et al., 1998). Similarly, there is evidence that the L-type Ca2+ channel blocker nimodipine may also be selectively effective in tSAH patients (Harders et al., 1996).
Recommendations from the 2008 National Institutes of Health (NIH) Combination Therapies for TBI Workshop
In addition to the reasons for the disappointing failures of past TBI therapeutic trials already mentioned, it has been suggested that the multi-factorial post-TBI secondary injury cascade (Figure C-21) may require treatment with a combination of therapeutic agents that simultaneously interrupt the cascade at multiple points in order to achieve a clinically significant effect in human TBI. This possibility was considered in detail at an NIH workshop held in 2008 from which several recommendations were elaborated (Margulies and Hicks, 2009). The objectives of the workshop were to:
-
identify promising therapies (mainly pharmacological) that would logically be expected to produce an additive neuroprotective effect when combined,
-
consider the challenges involved in preclinical and clinical testing of combination treatments, and
-
optimize strategies for developing combination therapies or single agents that may be capable of affecting multiple secondary injury mechanisms simultaneously.
Table C-10 lists some of the leading therapeutic approaches that were identified by the workshop attendees.
The combination therapy workshop also detailed several recommendations related to the future preclinical and clinical testing of combination therapies for TBI. These include
-
selecting combinations that target different and complimentary mechanisms of action,
-
validating surrogate markers to monitor treatment effects in animal models and humans,
-
developing in vitro, animal, and clinical platforms for coordinated studies across laboratories,
-
using efficient designs for preclinical and clinical trials and data analysis,
-
becoming informed of FDA regulations that need to be considered in moving toward TBI clinical trials with combinations of single agents that may only be effective when used together (similar to many cancer chemotherapeutic strategies),
-
adopting a uniform standard of care for clinical trials, and mimicking those standards in preclinical studies, and
-
establishing a shared database of positive and negative clinical and preclinical data.
Evidence that a combination of two mechanistically distinct neuroprotective drugs might achieve a significantly better degree of neuroprotective efficacy than each monotherapy has been observed in at least one instance. A phase III clinical trial in aneurysmal SAH showed that the combined use of the Ca2+ channel blocker nimodipine and the lipid peroxidation inhibitor tirilazad resulted in significantly greater decrease in mortality and an improvement in the distribution of Glasgow Outcome Scale scores compared to that observed in patients treated with nimodipine alone (Kassell et al., 1996).
INTRINSIC DIFFERENCES IN POSTTRAUMATIC BRAIN AND SPINAL CORD NEUROPATHOLOGY AND IMPLICATIONS FOR NUTRITIONAL INTERVENTIONS FOLLOWING INJURY
The major structural difference between the brain and spinal cord is that the former has a higher ratio of gray matter (containing neuronal cell bodies, dendrites, and synaptic connections) to white matter (composed of myelinated axonal fiber tracks) while the spinal cord has a higher white to gray matter ratio. In other words, compared to the brain, which is an exceedingly complicated structure, the spinal cord is predominantly a cable that carries sensory information from the periphery up to the brain and brain-initiated motor commands down to the muscles of the body. However, despite this difference in structural and functional complexity, an extensive body of research accumulated over the past three decades has demonstrated that there is a great deal of overlap in the molecular pathophysiology of TBI and spinal cord injury (SCI). All of the leading molecular mechanisms involved in secondary injury following acute TBI also are implicated in the secondary injury after SCI. These include glutamate-mediated excitotoxicity, intracellular Ca2+ overload, and free radical-induced oxidative damage. Indeed, the TBI secondary injury cascade schematic shown in Figure C-21 is essentially accurate for SCI.
Recent research in my own laboratory has compared the time course of reactive oxygen species oxidative damage in rodent models of TBI and SCI and finds that this secondary in-
jury mechanism is initiated during the first posttraumatic hour in both instances. In the case of the injured brain, oxidative damage products tend to peak during the first 3 hours and return to the preinjury baseline by 24–48 hours (Deng et al., 2007). In contrast, oxidative damage products do not peak in the injured spinal cord until 24 hours, but they persist for at least a week (Carrico et al., 2009). Similarly, the peak of brain mitochondrial oxidative damage and bioenergetic dysfunction occurs at 12 hours after injury (Singh et al., 2006), but not until 24 hours in the case of mitochondrial isolated from the injured spinal cord (Sullivan et al., 2007). Despite these time course differences, acute treatment with the reactive oxygen scavenger tempol is able to attenuate oxidative damage and preserve mitochondrial function equally well in both TBI and SCI (Deng-Bryant et al., 2008; Xiong and Hall, 2009). On the other hand, additional studies have shown that spinal cord mitochondria produce more reactive oxygen species compared to brain mitochondria (Sullivan et al., 2004), and spinal cord mitochondria are more sensitive to bioenergetic impairment by oxidative damage products than are brain mitochondria (Vaishnav et al., 2010). What these differences mean from a translational perspective in regard to trying to achieve neuroprotective effects with either acute postinjury pharmacological antioxidant administration or the prophylactic protective effects of preinjury dietary antioxidant (e.g., alpha tocopherol, ascorbic acid, CoQ10) intake is that while these strategies should be effective in either TBI or SCI, it may take larger doses and a longer duration of treatment to achieve protection after SCI.
LEADING ANTIOXIDATION, ANTI-INFLAMMATORY THERAPEUTICS FOR TBI AND THEIR APPLICABILITY TO BLAST INJURIES
The study of blast-induced TBI is a relatively new aspect of neurotrauma research that has taken off during the past few years largely because of its prevalence in the War on Terror. However, the available pathophysiological data obtained from animal models of blast injuries strongly suggest that the pathophysiological secondary injury cascade after blast TBI includes most, if not all, of the molecular mechanisms that have been documented to occur in mechanical blunt impact or penetrating types of TBI. For instance, studies in blast TBI models thus far have confirmed that the general features of mechanical TBI including diffuse axonal injury, glutamate-mediated excitotoxicity, free radical generation and oxidative damage, disruption of Ca2+ homeostasis, calcium-activated proteolysis, blood-brain barrier breakdown, tissue hemorrhage, cerebral vasospasm, inflammation, and altered brain metabolism also are seen in blast-induced TBI (Margulies and Hicks, 2009). Thus, even though preclinical studies focused on blast injuries have been mainly directed at model development and description of neuropathology and pathophysiology, rather than experimental therapeutics, it is reasonable to predict that as the blast TBI field moves forward, single or combination therapies (see Table C-10) that are demonstrated to be neuroprotective or neurorestorative in models of mechanical trauma and in humans, also might be useful for the treatment of blast TBI.
Findings on Creatine and TBI and Applicability to Blast Injuries
Creatine is an amino acid that is endogenously produced in the liver, kidney, and pancreas from the precursor amino acids glycine, methionine, or arginine by the action of the enzyme creatine kinase. Creatine monohydrate has been used extensively by athletes as a performance-enhancing supplement and been shown to increase creatine levels in brain as well as muscle tissue (Andres et al., 2008). The creatine kinase/phosphocreatine system plays a major role in neuronal energy metabolism, and creatine supplementation has been shown
TABLE C-10 Currently Promising TBI Therapies—Interventions with Some Reasonable Amount of Preliminary Preclinical or Clinical Data Supporting Their Neuroprotective or Neurorestorative Efficacy
|
Intervention |
|
|
Citicoline |
membrane repair |
|
Erythropoietin |
multiple neuroprotective and neurorestorative actions |
|
Hypothermia |
inhibits many secondary injury mechanisms |
|
Progesterone |
multiple neuroprotective actions |
|
Cyclosporine A |
mitochondrial protection |
|
Statins |
increased nitric oxide produce → improved cerebral blood flow (CBF) |
|
Hypertonic saline |
improved CBF, reduce edema, etc. |
to increase the process of neurogenesis (Andres et al., 2008). Moreover, creatine supplementation has been shown to be neuroprotective in animal models of hypoxic brain damage (Balestrino et al., 1999; Holtzman et al., 1998), Parkinson’s disease (Matthews et al., 1998), and amyotrophic lateral sclerosis (Klivenyi et al., 1999).
Based upon these findings, there has been interest in exploring changes in brain creatine levels and the potential neuroprotective effects of creatine supplementation in the rat controlled cortical impact TBI model, which is characterized by a contusion injury. It has been reported that creatine and phosphocreatine levels are decreased in the peri-contusional cortical tissue by 35 percent (Schuhmann et al., 2003). A single study using the same rat TBI model has examined the neuroprotective effects of creatine supplementation 3 mg/g body weight for 1, 3, or 5 days before injury, and observed that creatine supplementation for 3 days reduced cortical tissue damage by 21 percent whereas 5 days caused a 36 percent decrease. These results show that the protective effects are related to the duration of supplementation, but are apparent within a few days. However, additional experiments in the same study showed that 4 weeks of dietary creatine supplementation (1 percent in the diet) could achieve a 50 percent reduction in cerebral cortical damage (Sullivan et al., 2000). Furthermore, cortical synaptic mitochondria isolated at 4 weeks from creatine-supplemented TBI animals showed significantly less free radical formation and improved mitochondrial membrane potentials, Ca2+ homeostasis, and adenosine triphospate (ATP) levels compared to non-supplemented brain-injured rats. A subsequent investigation by the same laboratory found that creatine-supplemented TBI rats showed significantly lower levels of brain lactate during the first 6 post-injury hours providing further evidence of neuronal mitochondrial functional preservation as a result of chronic creatine supplementation. While these results tend to support the potential brain-protective value of dietary creatine supplementation in either athletes engaged in contact sports or war fighters, additional preclinical research is needed to accurately define the creatine dose-response characteristics, the needed duration of treatment, efficacy in different types of TBI, and its safety during chronic use.
NUTRIENTS OR DIETS THAT HAVE SHOWN PROMISE AND WARRANT FURTHER RESEARCH REGARDING ACUTE NEUROPROTECTION OF IMPROVED RECOVERY FROM TBI
Beyond the potential neuroprotective benefits of dietary creatine supplementation, perhaps the most convincing body of data regarding the beneficial effects of nutrients or diets has to do with the protective effects of various compounds that possess antioxidant (i.e., free radical scavenging) properties. The phenolic compounds curcumin (Sharma et al., 2009;
FIGURE C-22 Chemical structures of various nutraceuticals possessing antioxidant properties that are protective in preclinical neurotrauma models. Each of them has the ability to scavenge (i.e., donate an electron) to a lipid peroxyl radical (LOO•) to interrupt the process of lipid peroxidation. In the case of resveratrol, curcumin and vitamin E, the electron donation comes from the single or multiple phenolic hydroxyl groups. Moreover, certain fruits, vegetables, dark chocolate and green tea which are all high in the content of polyphenolic antioxidants have also been reported to be neuroprotective.
Wu et al., 2006), vitamin E (Badhe et al., 2007; Clifton et al., 1989; Conte et al., 2004; Flanary and Streit, 2006; Hall et al., 1992a, 1992b; Wu et al., 2010), and resveratrol (Ates et al., 2007; Sonmez et al., 2007); the indoleamine melatonin (Beni et al., 2004; Cirak et al., 1999; Mesenge et al., 1998; Ozdemir et al., 2005a, 2005b); and alpha lipoic acid (Toklu et al., 2009) have all been shown to exert a protective effect in animal models of TBI (Figure C-22). Additionally, melatonin (Gul et al., 2005; Liu et al., 2004; Samantaray et al., 2008) and vitamin E (Hall and Wolf, 1986; Hall et al., 1992b) have been reported to exert vascular and/or neuronal protective in models of SCI. In each of these cases, the protective effects were demonstrated to be due to their antioxidant properties (i.e. a reduction in oxidative damage). However, it should be noted that daily high dose supplementation with vitamin E can take as much as eight weeks of before a significant increase in central nervous system parenchymal tissue levels occurs (Machlin and Gabriel, 1982). Therefore, the full extent of the neuroprotective effects of chronic vitamin E supplementation is probably not fully developed until that time. Consistent with that, it has been shown that a chronic two-fold dietary supplementation with vitamin E required 16 weeks before cerebrovascular protection was observed in an animal model of subarachnoid hemorrhage (SAH) (Travis and Hall, 1987).
In addition to chemically scavenging free radicals, there is increasing interest in the possibility of nutraceutically or pharmacologically inducing endogenous enzymatic antioxidant defense mechanisms. In that regard, experimental attention has recently been increasingly focused on the transcription factor Nuclear factor E2-related factor (Nrf2) which interacts with the antioxidant response element (ARE) of various cytoprotective and free radical
detoxifying genes. Evidence is rapidly accumulating which shows that the induction of the Nrf2/ARE pathway is a potential therapeutic target for acute neurological injury and chronic neurodegenerative diseases (Calkins et al., 2009). An increasing number of naturally occurring or synthetic compounds have been shown to be able to induce Nrf2 activation and to exert tissue protective effects in brain and other organ systems. Nrf2 is normally repressed in the cytoplasm by binding to the Kelch-like ECH-associated protein 1 (Keap1), where it is rapidly degraded by the ubiquitin-proteasome system. However, one of the prototype Nrf2/ARE pathway activators is sulforaphane, a compound found in high concentrations in broccoli that has been demonstrated to inhibit the proteasomal degradation of Nrf2 resulting in its nuclear accumulation and hence an increased activation of Nrf2-regulated genes. In a rat TBI model, Nrf2 protein, but not mRNA levels, have been shown to increase in injured and surrounding brain areas after TBI along with an increase in mRNA levels of the antioxidant enzymes heme oxygenase-1 (HO-1) and NADPH quinone oxidoreductase-1 (NQO1). This appears to represent an endogenous neuroprotective oxidative stress response (Yan et al., 2009; Yan et al., 2008). However, this response can be pharmacologically enhanced. For instance, sulforaphane administration has been reported to protect the blood brain barrier (Zhao et al., 2007), reduce brain edema (Zhao et al., 2005) and improve cognitive recovery (Dash et al., 2009) in rodent TBI models. Curcumin, one of the phenolic radical scavengers mentioned above, has also been shown to upregulate Nrf2 levels and HO-1 expression and to protect rat brains against focal ischemia (Yang et al., 2009).
SUMMARY
Much of the damage and neurological dysfunction after TBI or SCI is not due to the effects of the primary mechanical trauma, but rather to a molecular cascade of secondary events. In animal models, these have been shown to be potentially modifiable by promptly administered pharmacological treatments that target individual secondary injury factors such as glutamate-mediated excitotoxicity, free radical production and oxidative damage, intracellular Ca2+ overload, mitochondrial dysfunction, etc. However, phase II and III clinical trials of compounds that target specific factors and are neuroprotective in animal models have failed to produce a significant improvement in outcome in populations of moderately or severely injured TBI patients. The only exception of note has been the observation that certain TBI patient subgroups may be selectively benefitted by certain types of compounds (e.g. reduction in mortality in patients with traumatic SAH by the antioxidant tirilazad or the L-type Ca2+ channel blocker nimodipine).
One of the possible reasons for the failure of various compounds to produce a convincing effect in clinical trials may be that the secondary injury cascade is too complex to be adequately inhibited by interference with a single pathomechanism. Thus, contemporary neuroprotective research includes the testing of combination pharmacotherapies that are made up of two agents that simultaneously act at complimentary target sites based upon the idea that a more robust and/or a less variable effect may be achievable. Another option along these lines is to utilize what are variously called “multi-potential,” “multi-mechanistic,” or “pleiotropic” compounds that possess the ability to protect the injured brain by multiple mechanisms. Two such compounds that are being examined in clinical TBI trials are the hormone progesterone and the immunosuppressive agent cyclosporine A, each of which have shown impressive neuroprotective properties in rodent TBI models.
Neuroprotective strategies that are shown to work in civilian TBIs can also reasonably be expected to be beneficial in military blast-induced TBIs and SCI in which the secondary injuries cascades are similar. In addition to monotherapies or combination therapies that
might be shown to be acute postinjury treatments for civilian and military injuries in the years to come, there is also evidence that certain dietary supplements (e.g. creatine, various antioxidants, Nrf2/ARE pathway activators) may be observed to be both safe and effective for prophylactic neuroprotection in both civilians and military personnel.
REFERENCES
Andres, R. H., A. D. Ducray, U. Schlattner, T. Wallimann, and H. R. Widmer. 2008. Functions and effects of creatine in the central nervous system. Brain Research Bulletin 76(4):329–343.
Ates, O., S. Cayli, E. Altinoz, I. Gurses, N. Yucel, M. Sener, A. Kocak, and S. Yologlu. 2007. Neuroprotection by resveratrol against traumatic brain injury in rats. Molecular and Cellular Biochemistry 294(1–2):137–144.
Badhe, P., J. Thorat, B. D. Diyora, R. Mamidanna, P. Sayal, S. Badhe, and A. K. Sharma. 2007. Neuroprotective effects of vitamin E in cold induced cerebral injury in guinea pigs. Indian Journal of Experimental Biology 45(2):180–184.
Balestrino, M., R. Rebaudo, and G. Lunardi. 1999. Exogenous creatine delays anoxic depolarization and protects from hypoxic damage: Dose-effect relationship. Brain Research 816(1):124–130.
Beni, S. M., R. Kohen, R. J. Reiter, D. X. Tan, and E. Shohami. 2004. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-kappaB and AP-1. The Federal American Societies for Experimental Biology Journal 18(1):149–151.
Calkins, M. J., D. A. Johnson, J. A. Townsend, M. R. Vargas, J. A. Dowell, T. P. Williamson, A. D. Kraft, J. M. Lee, J. Li, and J. A. Johnson. 2009. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxidants and Redox Signaling 11(3):497–508.
Carrico, K. M., R. Vaishnav, and E. D. Hall. 2009. Temporal and spatial dynamics of peroxynitrite-induced oxidative damage after spinal cord contusion injury. Journal of Neurotrauma 26(8):1369–1378.
Cirak, B., N. Rousan, A. Kocak, O. Palaoglu, S. Palaoglu, and K. Kilic. 1999. Melatonin as a free radical scavenger in experimental head trauma. Pediatric Neurosurgery 31(6):298–301.
Clifton, G. L., B. G. Lyeth, L. W. Jenkins, W. C. Taft, R. J. DeLorenzo, and R. L. Hayes. 1989. Effect of D, alphatocopheryl succinate and polyethylene glycol on performance tests after fluid percussion brain injury. Journal of Neurotrauma 6(2):71–81.
Conte, V., K. Uryu, S. Fujimoto, Y. Yao, J. Rokach, L. Longhi, J. Q. Trojanowski, V. M. Lee, T. K. McIntosh, and D. Pratico. 2004. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. Journal of Neurochemistry 90(3):758–764.
Dash, P. K., J. Zhao, S. A. Orsi, M. Zhang, and A. N. Moore. 2009. Sulforaphane improves cognitive function administered following traumatic brain injury. Neuroscience Letters 460(2):103–107.
Deng-Bryant, Y., I. N. Singh, K. M. Carrico, and E. D. Hall. 2008. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. Journal of Cerebral Blood Flow and Metabolism 28(6):1114–1126.
Deng, Y., B. M. Thompson, X. Gao, and E. D. Hall. 2007. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Experimental Neurology 205(1):154–165.
Flanary, B. E., and W. J. Streit. 2006. Alpha-tocopherol (vitamin E) induces rapid, nonsustained proliferation in cultured rat microglia. Glia 53(6):669–674.
Gul, S., S. E. Celik, M. Kalayci, M. Tasyurekli, N. Cokar, and T. Bilge. 2005. Dose-dependent neuroprotective effects of melatonin on experimental spinal cord injury in rats. Surgical Neurology 64(4):355–361.
Hall, E. D. 2007. Stroke/traumatic brain and spinal cord injuries. In Comprehensive medicinal chemistry II. 2nd ed. 8 vols. Vol. 6, edited by M. Williams. Oxford, UK: Elsevier. Pp. 253–277.
Hall, E. D., J. M. Braughler, and J. M. McCall. 1992a. Antioxidant effects in brain and spinal cord injury. Journal of Neurotrauma 9(Suppl 1):S165–S172.
Hall, E. D., and D. L. Wolf. 1986. A pharmacological analysis of the pathophysiological mechanisms of posttraumatic spinal cord ischemia. Journal of Neurosurgery 64(6):951–961.
Hall, E. D., P. A. Yonkers, P. K. Andrus, J. W. Cox, and D. K. Anderson. 1992b. Biochemistry and pharmacology of lipid antioxidants in acute brain and spinal cord injury. Journal of Neurotrauma 9(Suppl 2):S425–S442.
Harders, A., A. Kakarieka, and R. Braakman. 1996. Traumatic subarachnoid hemorrhage and its treatment with nimodipine. German TSAH Study Group. Journal of Neurosurgery 85(1):82–89.
Holtzman, D., A. Togliatti, I. Khait, and F. Jensen. 1998. Creatine increases survival and suppresses seizures in the hypoxic immature rat. Pediatric Research 44(3):410–414.
Kassell, N. F., E. C. Haley, J r., C. Apperson-Hansen, and W. M. Alves. 1996. Randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: A cooperative study in Europe, Australia, and New Zealand. Journal of Neurosurgery 84(2):221–228.
Klivenyi, P., R. J. Ferrante, R. T. Matthews, M. B. Bogdanov, A. M. Klein, O. A. Andreassen, G. Mueller, M. Wermer, R. Kaddurah-Daouk, and M. F. Beal. 1999. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nature Medicine 5(3):347–350.
Liu, J. B., T. S. Tang, H. L. Yang, and D. S. Xiao. 2004. Antioxidation of melatonin against spinal cord injury in rats. Chinese Medical Journal (English) 117(4):571–575.
Machlin, L., and E. Gabriel. 1982. Kinetics of tissue alpha tocopherol uptake and depletion following administration of high levels of vitamin E. Annals of the New York Academy of Sciences 393:48–59.
Margulies, S., and R. Hicks. 2009. Combination therapies for traumatic brain injury: Prospective considerations. Journal of Neurotrauma 26(6):925–939.
Marshall, L. F., A. I. Maas, S. B. Marshall, A. Bricolo, M. Fearnside, F. Iannotti, M. R. Klauber, J. Lagarrigue, R. Lobato, L. Persson, J. D. Pickard, J. Piek, F. Servadei, G. N. Wellis, G. F. Morris, E. D. Means, and B. Musch. 1998. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. Journal of Neurosurgery 89(4):519–525.
Matthews, R. T., L. Yang, B. G. Jenkins, R. J. Ferrante, B. R. Rosen, R. Kaddurah-Daouk, and M. F. Beal. 1998. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. The Journal of Neuroscience 18(1):156–163.
Mesenge, C., I. Margaill, C. Verrecchia, M. Allix, R. G. Boulu, and M. Plotkine. 1998. Protective effect of melatonin in a model of traumatic brain injury in mice. Journal of Pineal Research 25(1):41–46.
Narayan, R. K., M. E. Michel, B. Ansell, A. Baethmann, A. Biegon, M. B. Bracken, M. R. Bullock, S. C. Choi, G. L. Clifton, C. F. Contant, W. M. Coplin, W. D. Dietrich, J. Ghajar, S. M. Grady, R. G. Grossman, E. D. Hall, W. Heetderks, D. A. Hovda, J. Jallo, R. L. Katz, N. Knoller, P. M. Kochanek, A. I. Maas, J. Majde, D. W. Marion, A. Marmarou, L. F. Marshall, T. K. McIntosh, E. Miller, N. Mohberg, J. P. Muizelaar, L. H. Pitts, P. Quinn, G. Riesenfeld, C. S. Robertson, K. I. Strauss, G. Teasdale, N. Temkin, R. Tuma, C. Wade, M. D. Walker, M. Weinrich, J. Whyte, J. Wilberger, A. B. Young, and L. Yurkewicz. 2002. Clinical trials in head injury. Journal of Neurotrauma 19(5):503–557.
Ozdemir, D., K. Tugyan, N. Uysal, U. Sonmez, A. Sonmez, O. Acikgoz, N. Ozdemir, M. Duman, and H. Ozkan. 2005a. Protective effect of melatonin against head trauma-induced hippocampal damage and spatial memory deficits in immature rats. Neuroscience Letters 385(3):234–239.
Ozdemir, D., N. Uysal, S. Gonenc, O. Acikgoz, A. Sonmez, A. Topcu, N. Ozdemir, M. Duman, I. Semin, and H. Ozkan. 2005b. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiological Research 54(6):631–637.
Samantaray, S., E. A. Sribnick, A. Das, V. H. Knaryan, D. D. Matzelle, A. V. Yallapragada, R. J. Reiter, S. K. Ray, and N. L. Banik. 2008. Melatonin attenuates calpain upregulation, axonal damage and neuronal death in spinal cord injury in rats. Journal of Pineal Research 44(4):348–357.
Schuhmann, M. U., D. Stiller, M. Skardelly, J. Bernarding, P. M. Klinge, A. Samii, M. Samii, and T. Brinker. 2003. Metabolic changes in the vicinity of brain contusions: A proton magnetic resonance spectroscopy and histology study. Journal of Neurotrauma 20(8):725–743.
Sharma, S., Y. Zhuang, Z. Ying, A. Wu, and F. Gomez-Pinilla. 2009. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience 161(4):1037–1044.
Singh, I. N., P. G. Sullivan, Y. Deng, L. H. Mbye, and E. D. Hall. 2006. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: Implications for neuroprotective therapy. Journal of Cerebral Blood Flow and Metabolism 26:1407–1418.
Sonmez, U., A. Sonmez, G. Erbil, I. Tekmen, and B. Baykara. 2007. Neuroprotective effects of resveratrol against traumatic brain injury in immature rats. Neuroscience Letters 420(2):133–137.
Sullivan, P. G., J. D. Geiger, M. P. Mattson, and S. W. Scheff. 2000. Dietary supplement creatine protects against traumatic brain injury. Annals of Neurology 48(5):723–729.
Sullivan, P. G., S. Krishnamurthy, S. P. Patel, J. D. Pandya, and A. G. Rabchevsky. 2007. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. Journal of Neurotrauma 24(6):991–999.
Sullivan, P. G., A. G. Rabchevsky, J. N. Keller, M. Lovell, A. Sodhi, R. P. Hart, and S. W. Scheff. 2004. Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. Journal of Comparative Neurology 474(4):524–534.
Toklu, H. Z., T. Hakan, N. Biber, S. Solakoglu, A. V. Ogunc, and G. Sener. 2009. The protective effect of alpha lipoic acid against traumatic brain injury in rats. Free Radical Research 43(7):658–667.
Travis, M. A., and E. D. Hall. 1987. The effects of chronic two-fold dietary vitamin e supplementation on subarachnoid hemorrhage-induced brain hypoperfusion. Brain Research 418(2):366–370.
Vaishnav, R. A., I. N. Singh, D. M. Miller, and E. D. Hall. 2010. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. Journal of Neurotrauma 27(7):1311–1320.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2006. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Experimental Neurology 197(2):309–317.
Wu, A., Z. Ying, and F. Gomez-Pinilla. 2010. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabilitation and Neural Repair 24(3):290–298.
Xiong, Y., and E. D. Hall. 2009. Pharmacological evidence for a role of peroxynitrite in the pathophysiology of spinal cord injury. Experimental Neurology 216(1):105–114.
Yan, W., H. D. Wang, X. M. Feng, Y. S. Ding, W. Jin, and K. Tang. 2009. The expression of NF-E2-related factor 2 in the rat brain after traumatic brain injury. The Journal of Trauma 66(5):1431–1435.
Yan, W., H. D. Wang, Z. G. Hu, Q. F. Wang, and H. X. Yin. 2008. Activation of Nrf2-are pathway in brain after traumatic brain injury. Neuroscience Letters 431(2):150–154.
Yang, C., X. Zhang, H. Fan, and Y. Liu. 2009. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Research 1282:133–141.
Zhao, J., A. N. Moore, G. L. Clifton, and P. K. Dash. 2005. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. Journal of Neuroscience Research 82(4):499–506.
Zhao, J., A. N. Moore, J. B. Redell, and P. K. Dash. 2007. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. Journal of Neuroscience 27(38):10240–10248.
Glycemic Control in the Critically Ill and in Brain Injury Patients
Stanley A. Nasraway, Jr.25
GLYCEMIC CONTROL IN THE CRITICALLY ILL: OVERVIEW
The publication in 2001 of the now famous Leuven I trial (Van Den Berghe et al., 2001) showed that tight glycemic control in a population of surgical critically ill patients could improve survival and could reduce multiple organ failure and nosocomial infection. This past decade as witnessed a great deal research dedicated to confirming these initial findings.
Leuven I (Van Den Berghe et al., 2001) unleashed a torrent of skepticism, excitement and investigation into tight glycemic control. Google searches of “tight glycemic control” and “intensive insulin” produce 80,900 and 334,000 results, respectively. After entering a new decade, where are we? There is a great deal that we do not know, in part, because this field of discovery has been disadvantaged by inconsistencies in research methodology. Among differences in the studies are casetype selection, targeted ranges of blood glucose, inconsistency in the frequency of blood glucose monitoring, variability in the accuracy of glucometer devices used, in the methods used to define euglycemia, whether insulin dosing was driven by paper protocol or software algorithm and nonstandardization in caloric intake. Starting with Leuven I, all of the prospective studies conducted to date are vulnerable to significant methodologic criticisms (Nasraway Jr. and Rattan, 2010). We also really have no conclusive understanding on the biologic plausibility to explain how intensive insulin would decrease death or organ failure or nosocomial infection. Is it through anti-inflammatory pathways, because insulin is a vasodilator that may increase microperfusion, or by other unrealized mechanisms of action? In some ways, the scientific evolution of this field resembles that of Sepsis research from 1985–2005, in which the study of anti-inflammatory compounds was severely hindered by lack of standardization in the total treatment for patients with severe sepsis, with too many confounding and uncontrolled variables (Nasraway Jr., 1999).
After all of these studies, what do we actually know? What are the consistent threads? This is what we know with certainty:
-
Hyperglycemia is bad. Falciglia (2009) convincingly showed in an analysis of 259,040 intensive care unit (ICU) patients that hyperglycemia (glucose > 110 mg/dL) was associated with mortality independent of illness severity, type of ICU or length of stay. Consistent with the findings of others, the two-thirds of patients who are nondiabetic benefit more from insulin than do diabetics.
-
Hypoglycemia is bad. An incidental and constant observation from many studies is that severe hypoglycemia (glucose < 40 mg/dL) in a population of patients by logistic regression is associated with a 6-fold increase in death (Griesdale et al., 2009). It would not be surprising to find with additional study that even mild hypoglycemia has long lasting but subtle neurologic consequences that are not clinically evident or measured. Hypoglycemia is particularly detrimental to the brain, which neither produces nor stores glucose, but is entirely dependent upon cerebral glucose delivery.
-
Critically ill patients typically sustain large swings in blood glucose, even with insulin administration (Finney et al., 2003). Sustaining the blood glucose within a target range in a hypermetabolic patient with changing gluconeogenic drivers in a 24-hour day is enormously challenging, frequently outstripping the crude tools used at the bedside to measure blood glucose and respond to its variation in concentration.
-
Software-driven insulin dosing is better than paper-driven insulin protocols. Software integrates all of the glucose measurements and all of the previous insulin adjustments to determine the next best insulin dose. Software appears to reduce glucose variability and sustain glucose within the target range for prolonged periods of time (Juneja et al., 2009). There are now many software programs tested and/or available.
-
Handheld blood glucometers, originally intended for use by Type I diabetic outpatients in the 1980s, are not accurate enough in the ICU environment (Kanji et al., 2005), and are very laborious to use. In the United States, the Food and Drug Administration in March of 2010 hosted a public inquiry into glucose meters, after which it is redefining what it will accept in the way of accuracy by blood glucose measurement devices in the hospital setting going forward. It has asked the international standards body to reset its limits for accuracy for glucometers. Current generation handheld devices now in use will not make the cut.
-
The more frequent the blood glucose measurement, even with handheld glucometers, the less hypoglycemia experienced by patients and the tighter the glycemic control (Cook et al., 2008b). Frequency is crucial, however laborious it may be.
What Can We Expect Going Forward?
We can expect that the world will continue to use intensive insulin, but that the range that defines “tight” will be narrowed as it becomes more achievable. We can expect that there will be more emphasis on defining hypoglycemia, and in avoiding it with greater rigor. We can expect a movement towards insulin-dosing software, as the development of many programs appears to be simple, and competition will force down the cost of purchase and use. Software-insulin dosing has hidden advantages: it forces more blood glucose monitoring and also provides an instant database for analysis. We will someday be using glucometers that are engineered to be more accurate, especially in the hypoglycemic range, avoiding pitfalls in today’s instruments due to chemical interferences and specific disease conditions. Importantly, these devices will be continuous or near continuous, and by their nature will be
less arduous. At the same time, manufacturers will need to make these devices affordable, or their uptake will be slowed. The frequency of blood glucose measurements by these devices will dramatically make safer the continuous administration of insulin.
Improving the accuracy of blood glucose measurements and standardizing the determination of insulin dosing with better methods will produce better quality research, synergizing global convergence on tight glycemic control, reduced glucose variability and better patient outcomes.
GLYCEMIC CONTROL IN ACUTE BRAIN INJURY
Research into blood glucose management for patients with acute brain injury has been a representative microcosm of the larger field of glycemic control in the critically ill. Numerous studies have demonstrated that hyperglycemia in patients after stroke or other forms of acute brain injury is deleterious and worsens outcome (Bilotta et al., 2009; Cook et al., 2008a). Van den Berghe and colleagues (2005) retrospectively analyzed 63 patients from their original study; these patients had sustained isolated brain injury. Patients who had been randomized to intensive insulin therapy sustained decreases in the mean and maximal intracranial pressure. This, in turn, was associated with improved long-term recovery in comparison with those patients who had received conventional glycemic management.
However, a very important study examined the effects of tight glycemic control on cerebral glucose metabolism after severe brain injury. Tight glycemic control, even without systemic hypoglycemia, was associated with decreased brain glucose and increases in brain energy crises (Oddo et al., 2008). The latter was associated with an increase in death. The study has raised questions about the value of very tight glycemic control.
There have since been four prospective randomized controlled trials examining the benefits of intensive insulin in patients with subarachnoid hemorrhage (Bilotta et al., 2007), traumatic brain injury (Bilotta et al., 2008; Coester et al., 2010), or in a heterogeneous group of mechanically ventilated critically ill neurologic patients (Green et al., 2010). Overall, there were no differences in the rates of infection, neurologic recovery, or mortality rate. The results of these studies have frustrated advocates for very tight glycemic control.
It is clear that severe hyperglycemia in patients with acute brain injury is deleterious. However blood glucose concentrations which are normal, but tightly regulated, may also be deleterious with a reduction in brain glucose availability. As a result the best overall recommendation has been to achieve a broader range of glycemic control while avoiding hypoglycemia in this especially sensitive population. Bilotta et al. (2009) have suggested a blood glucose concentration target range of 80–155 mg/dL.
REFERENCES
Bilotta, F., R. Caramia, I. Cernak, F. P. Paoloni, A. Doronzio, V. Cuzzone, A. Santoro, and G. Rosa. 2008. Intensive insulin therapy after severe traumatic brain injury: A randomized clinical trial. Neurocritical Care 9(2):159–166.
Bilotta, F., F. Giovannini, R. Caramia, and G. Rosa. 2009. Glycemia management in neurocritical care patients: A review. Journal of Neurosurgical Anesthesiology 21(1):2–9.
Bilotta, F., A. Spinelli, F. Giovannini, A. Doronzio, R. Delfini, and G. Rosa. 2007. The effect of intensive insulin therapy on infection rate, vasospasm, neurologic outcome, and mortality in neurointensive care unit after intracranial aneurysm clipping in patients with acute subarachnoid hemorrhage: A randomized prospective pilot trial. Journal of Neurosurgical Anesthesiology 19(3):156–160.
Coester, A., C. R. Neumann, and M. I. Schmidt. 2010. Intensive insulin therapy in severe traumatic brain injury: A randomized trial. Journal of Trauma—Injury, Infection and Critical Care 68(4):904–911.
Cook, A. M., A. Peppard, and B. Magnuson. 2008a. Nutrition considerations in traumatic brain injury. Nutrition in Clinical Practice 23(6):608–620.
Cook, C. B., V. Abad, G. Kongable, Y. Hanseon, and D. McMahon. 2008b. The status of glucose control in U.S. intensive care units. Critical Care Medicine 36(12):A68.
Falciglia, M., R. W. Freyberg, P. L. Almenoff, D. A. D’Alessio, and M. L. Render. 2009. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Critical Care Medicine 37(12):3001–3009.
Food and Drug Administration (FDA). 2010. FDA/CDRH public meeting: Blood glucose meters March 16–17, 2010. http://www.fda.gov/MedicalDevices/NewsEvents/WorkshopsConferences/ucm187406.htm#transcripts (accessed November 7, 2010).
Finney, S. J., C. Zekveld, A. Elia, and T. W. Evans. 2003. Glucose control and mortality in critically ill patients. Journal of the American Medical Association 290(15):2041–2047.
Green, D. M., K. H. O’Phelan, S. L. Bassin, C. W. J. Chang, T. S. Stern, and S. M. Asai. 2010. Intensive versus conventional insulin therapy in critically ill neurologic patients. Neurocritical Care 13(3):299–306.
Griesdale, D. E. G., R. J. De Souza Rd, R. M. Van Dam, D. K. Heyland, D. J. Cook, A. Malhotra, R. Dhaliwal, W. R. Henderson, D. R. Chittock, S. Finfer, and D. Talmor. 2009. Intensive insulin therapy and mortality among critically ill patients: A meta-analysis including nice-sugar study data. Canadian Medical Association Journal 180(8):821–827.
Juneja, R., C. P. Roudebush, S. A. Nasraway, A. A. Golas, J. Jacobi, J. Carroll, D. Nelson, V. J. Abad, and S. J. Flanders. 2009. Computerized intensive insulin dosing can mitigate hypoglycemia and achieve tight glycemic control when glucose measurement is performed frequently and on time. Critical Care (London, England) 13(5).
Kanji, S., J. Buffie, B. Hutton, P. S. Bunting, A. Singh, K. McDonald, D. Fergusson, L. A. McIntyre, and P. C. Hebert. 2005. Reliability of point-of-care testing for glucose measurement in critically ill adults. Critical Care Medicine 33(12):2778–2785.
Nasraway Jr., S. A. 1999. Sepsis research: We must change course. Critical Care Medicine 27(2):427–430.
Nasraway Jr., S. A., and R. Rattan. 2010. Tight glycemic control: What do we really know, and what should we expect? Critical Care (London, England) 14(5):198.
Oddo, M., J. M. Schmidt, E. Carrera, N. Badjatia, E. S. Connolly, M. Presciutti, N. D. Ostapkovich, J. M. Levine, P. L. Roux, and S. A. Mayer. 2008. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: A microdialysis study. Critical Care Medicine 36(12):3233–3238.
Van Den Berghe, G., K. Schoonheydt, P. Becx, F. Bruyninckx, and P. J. Wouters. 2005. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology 64(8):1348–1353.
Van Den Berghe, G., P. Wouters, F. Weekers, C. Verwaest, F. Bruyninckx, M. Schetz, D. Vlasselaers, P. Ferdinande, P. Lauwers, and R. Bouillon. 2001. Intensive insulin therapy in critically ill patients. The New England Journal of Medicine 345(19):1359–1367.
Mitochondrial Dysfunction Following Traumatic Brain Injury (TBI): Potential of Creatine as a Neuroprotective Strategy
Patrick G. Sullivan26
INTRODUCTION
Although TBI is a major healthcare problem in the United States, there are currently no pharmacological interventions approved for clinical treatment of this condition. TBI affects about 7 million individuals each year in North America. However, athletes—particularly in full-contact sports such as boxing, football, hockey and soccer—are exposed to single and repeated concussions at a much higher incidence than the general population, which can result in long-term neurological dysfunction and even death (Clark, 1998). Regardless
of rule changes, improved protective equipment, and conditioning, approximately 300,000 people still experience sport-related TBI annually (Cantu, 1997; Thurman et al., 1998). Furthermore, accumulating clinical evidence, as well as experience in contemporary military operations, suggests that substantial short-term and long-term neurologic deficits can be caused without a direct contact to the head (Cernak et al., 1999; DePalma et al., 2005; Elder and Cristian, 2009; Ling et al., 2009; Trudeau et al., 1998). With an estimated 15 percent of troops serving in Iraq sustaining some level of neurological impairment following blast exposure, TBI is the signature injury of this war and makes troops another high incident population for TBI (Hoge et al., 2008).
Although the neuropathology of TBI is not completely elucidated, several lines of evidence have demonstrated that mitochondrial dysfunction is a major feature of TBI. Mitochondria have also been found to play a pivotal role in neuronal cell survival and death following injury. Mitochondria serve as the powerhouse of the cell by maintaining ratios of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) that thermodynamically favor the hydrolysis of ATP to ADP + Pi. Proton pumping by components of the electron transport system (ETS) generates a membrane potential (∆Ψ) that can then be used to phosphorylate ADP to ATP or used to sequester Ca2+ into the mitochondrial matrix. This allows mitochondria to act as Ca2+ sinks for the cell as well as to stay in tune with changes in cytosolic Ca2+ levels. However, excessive mitochondrial Ca2+ uptake following TBI can result in formation of the mitochondrial permeability transition pore (mPTP) (Sullivan et al., 2005). A consequence of mPTP formation is a loss of membrane potential, which causes the uncoupling of electron transport from ATP production. The release of pro-apoptotic molecules (i.e., cytochrome C, Smac/Diablo, and apoptosis-inducing factor) from the mitochondria is, in part, orchestrated by mPTP and leads to the activation of cellular death pathways. An additional consequence of mPTP formation is the production of reactive oxygen species (ROS), which contribute to cellular damage by oxidizing cellular proteins and lipids (Mazzeo et al., 2009). Thus, the fine line between cell survival and cell death relies on mitochondrial integrity and, ultimately, the state of mitochondria following TBI.
Creatine is a molecule that is produced both endogenously and acquired exogenously through diet where it plays a prominent role in buffering cellular energy stores by increasing levels of phosphocreatine. Thus, the creatine/phosphocreatine system can increase overall cellular bioenergetics following injury/insult by acting as an energy storehouse. Additionally, increases in creatine can stabilize creatine kinase which has been demonstrated to interact with components of the mPTP and inhibit permeability transition (Beutner et al., 1996, 1998; O’Gorman et al., 1997). Inhibition of the mPTP has been demonstrated to reduce damage following TBI (Sullivan et al., 2005). Together, these data may point to creatine as a viable prophylactic treatment for certain populations engaged in activities that increase their chance for sustaining a TBI. However, limited preclinical data is available concerning the use of creatine following TBI, making this an untapped resource that should be further explored.
TRAUMATIC BRAIN INJURY
Although treatment options designed to improve survival of their injuries are limited to minimizing acute brain edema, decreasing intracranial pressure, and the prevention of peripheral complications, there is no current treatment aimed at the loss of neural tissue that occurs following TBI. Perhaps the most insidious aspect of TBI is that it can occur without any obvious signs of injury to the patient’s body. Medical reports dating back to World War I have recorded medical incidences of mysterious neurological disorders. Physicians in the British armed forces began to label the bulk of these phenomenon with the term “shell
shock” (SS) (Jones et al., 2007). Although some cases were attributed to psychosis, SS was responsible for 14 percent of all discharges from the British armed forces, and accounted for over one-third of all discharges of nonwounded soldiers by 1917. The controversial definition of the disorder, its method of treatment, public controversy, and stigma over diagnosis delayed the development of treatment protocols and eventually caused the British army to ban the use of the term “shell shock” from reports. However, with the start of World War II, it became readily apparent that disavowing the existence of this disorder did not prevent another epidemic.
In response to the army regulations, alternative terminology arose in its place, such as postconcussional syndrome (PCS) or posttrauma concussion state. Physicians began realizing many of the soldiers that suffered from this concussed state had been in a close proximity to an explosion, and thus, leading them to speculate that some force was affecting neural tissue without affecting the rest of the body. It was also realized that patients with a severe head injury would present with immediate neurologic symptoms that would trend toward recovery; whereas PCS would have delayed onset of neurologic symptoms with a trend toward worsening symptoms (Jones et al., 2007). Since soldiers and civilians can often suffer immense psychiatric morbidity without realizing the need for medical treatment that normally stems from a physical injury, this delayed development of symptoms in mild to moderate TBI patients is perhaps the most unfortunate aspect of this condition. A recent online poll indicated that 42 percent of respondents who suffered a TBI failed to seek medical care (Setnik and Bazarian, 2007); a rate that is considerably higher than the Centers for Disease Control and Prevention estimate of 25 percent. It has been observed clinically that even mild or moderate TBI can require neurosurgical intervention, and any delay in treatment could prove to be costly in terms of cognitive and functional recovery (Setnik and Bazarian, 2007).
Of the more than 1.5 million military personnel deployed since 2001 to the Middle East, approximately 25 percent of the injured service members have reported brain injury. Given the statistic from the poll above, this is probably an extreme underestimate with regards to military peronnel. Unpublished data from the Department of Defense indicates that blast injuries are the leading cause of TBI in war zones; consequently, TBI has been labeled as a signature injury of the current Middle Eastern conflicts (Hoge et al., 2008). In addition to cognitive deficits, this injury population also has an increased predisposition to the development of post traumatic stress disorder (PTSD).
Within the civilian population of the United States, about 2 percent of the population (5.3 million) is currently living with disabilities that are the direct result of TBI (Langlois et al., 2006). TBI has a bimodal age distribution of incidence such that the peaks are found in young (< 25) and elderly (> 75) populations (Langlois et al., 2006; Rutland-Brown et al., 2006). Due to the high incidence and the development of chronic symptoms associated with TBI, the medical costs within the United States alone have been estimated at over $50 billion dollars per year. These dismal figures do not factor in the cost to social and family dynamics that occur following TBI. Despite being obvious that TBI is a devastating military and civilian health care problem in the United States, there are currently no pharmacological treatments approved for clinical treatment of this condition. Several lines of evidence have indicated that mitochondrial dysfunction is a prominent feature of TBI, and mitochondria are known to play a pivotal role in neuronal cell survival and death following injury. As such, there is a clear need for the development of mitochondrial-targeted neuroprotective therapies for the treatment of TBI.
MITOCHONDRIA AND TBI
Several studies in recent years have shown that mitochondria play a pivotal role in neuronal cell survival in addition to mitochondrial dysfunction being considered an early, prominent event in central nervous system (CNS) injury that can cause neuronal cell death (Fiskum, 2000; Sullivan et al., 2004, 2005). Experimental data also indicates that excitotoxicity may be the initial upstream mechanism that leads to TBI-induced neuronal cell death (Choi et al., 1990; Faden et al., 1989). In order to discuss mitochondrial dysfunction, however, we must first address normal mitochondrial function. Mitochondria are double-membraned organelles that orchestrate oxidative phosphorylation. Specifically, mitochondria act as the “powerhouses” of cells by taking products from the Krebs cycle (citric acid cycle), fatty acid oxidation, and amino acid oxidation and producing most of the cell’s supply of ATP—the energy source used to power virtually all cellular functions. In fact, in the cells of evolutionarily “higher animals,” greater than 95 percent of all ATP is produced by oxidative phosphorylation within mitochondria. Mitochondrial function is dependent upon the generation and maintenance of the mitochondrial ∆Ψ, which is used to drive ATP production. ∆Ψ is generated by the translocation of protons across the inner mitochondrial membrane via the electron transport system (ETS), culminating in the reduction of O2 to H2O. This store of potential energy (the electrochemical gradient) can then be coupled to ATP production as protons flow back through the ATP synthase and complete the proton circuit. The potential can also be used to drive Ca2+ into the mitochondrial matrix via the electrogenic uniporter when cytosolic levels increase (Gunter et al., 1994). When cytosolic levels decrease, mitochondria pump Ca2+ out to precisely regulate cytosolic Ca2+ homeostasis.
During excitotoxic insults, such as the result of TBI, Ca2+ uptake into mitochondria has been shown to increase ROS production, inhibit ATP synthesis, and induce mitochondrial permeability transitions. It is important to note that inhibition of mitochondrial Ca2+ uptake by reducing ∆Ψ (chemical uncoupling) following excitotoxic insults is neuroprotective, emphasizing the pivotal role of mitochondrial Ca2+ uptake in TBI-induced neuronal cell death (Pandya et al., 2007; Sullivan et al., 2004). Studies from our group have demonstrated that changes in mitochondrial Ca2+ levels/cycling are coupled with increases in oxidative damage and significant mitochondrial dysfunction, which occurs acutely and is progressive for up to 48 hours postinjury (Maragos and Korde, 2004; Mbye et al., 2008; Pandya et al., 2009; Sullivan et al., 2004, 2005). The opening of the mitochondrial permeability transition pore (mPTP) is suggested to be a key mediator in this process.
While several studies have demonstrated mitochondrial failure in rodent TBI models over the past 15 years, only recently have careful time course studies been carried out to better understand the temporal profile of bioenergetic failure. In the mouse controlled cortical impact (CCI) model of TBI, we have shown that mitochondrial failure is significant by 3 hours within the cortical tissue surrounding the injury site and follows a progressive failure that peaks at 12 hours (Singh et al., 2006). The onset of mitochondrial dysfunction has been demonstrated to be even more rapid in the tissue considered to be the injury core and penumbra following CCI. In these studies, it is apparent that a significant loss of mitochondrial bioenergetics begins as early as 1 hour post-injury and continues for up to 48 hours postinjury (Gilmer et al., 2009; Pandya et al., 2007, 2009). Furthermore, mitochondrial Ca2+ overload, which directly initiates mPTP formation, was found to coincide with the loss of mitochondrial bioenergetics. However, both mitochondrial bioenergetics and Ca2+ loading were most amendable to treatment with a mitochondrial uncoupler administered within a 6 hour post-injury window (Pandya et al., 2009). Thus, these data sets show that a critical time for intervention occurs at t < 6 hours post-injury. In fact, in order for any
mitochondrial–targeted compound to maximally rescue mitochondria at the epicenter of the injury, administration within a three hour post-injury window would be needed; while administration within the first six hours would prevent mitochondrial failure in the cortical tissue surrounding the epicenter. Given these findings, a prophylactic approach with a safe compound is very logical for persons at an increased risk for TBI, such as athletes and military personal in active war zones.
CREATINE
Creatine (N-(aminoiminomethy)-N-methyl glycine) is an amino acid endogenously produced from glycine, methionine, and arginine in the liver, kidney, and pancreas and is also supplied in diets containing meat products. While as much as 95 percent of the total pool of creatine is contained in muscle, high levels of creatine have also been demonstrated in the brain (Mujika and Padilla, 1997). For athletes, creatine is used to increase levels of phospho-creatine (which serves as a phosphate donor to generate ATP) and thereby decrease muscle fatigue during—and improve recovery after—repeated bouts of high intensity exercise (Mujika and Padilla, 1997). Importantly, creatine is also the main shuttle to transport energy from the mitochondria to locales in the cytosol via phosphocreatine. This is accomplished by generation of phosphocreatine from mitochondrial ATP in the intermembrane space via phosphocreatine kinase. Phosphocreatine can then be shuttled to various sites within the cell and used to regenerate ATP from ADP. This allows phosphocreatine to serve as a spatial/temporal buffer for ATP produced by oxidative phosphorylation in mitochondria. Higher levels of phosphocreatine therefore result in a higher reserve of ATP that is available for cells following injury and may account for the neuroprotection afforded by creatine supplementation. In fact, creatine supplementation has been placed into several human clinical trials for various CNS disorders including amyotrophic lateral sclerosis, Charcot-Marie-Tooth disease, Huntington’s disease, and Parkinson’s disease with mixed results (see Gualano et al., 2010, for review). In an effort to boost neuronal ATP and bioenergetics, all these trials used started creatine treatment after the disease state had been reached (Adhihetty and Beal, 2008).
It is also apparent that many of the neuroprotective functions that creatine has been shown to afford cannot be attributed to changes in cellular bioenergetics. One of the most striking examples is the anti-apoptotic effect, which has been attributed to the prevention or delay of the mPTP, that elevated creatine levels have been reported to produce (Adhihetty and Beal, 2008; Andres et al., 2008). Additionally, creatine kinase is now recognized as a component of the mPTP, and its activation inhibits the induction of the mPTP (Beutner et al., 1996; Beutner et al., 1998; O’Gorman et al., 1997). Given that bioenergetic failure and mPTP activation have been documented as key players in TBI-induced neuropathology, creatine supplementation would be expected to offer neuroprotection following experimental TBI (Sullivan et al., 2005).
CREATINE SUPPLEMENTATION AND TBI
Creatine supplementation has been shown to be neurprotective following TBI in both mice and rats. Our laboratory demonstrated in 2000 that chronic administration of creatine ameliorated cortical tissue damage by 36 percent in mice and 50 perecent in rats depending upon the regimen and dosage used during the pretreatment (Sullivan et al.). In mice, pretreatment with 3g/kg (intraperitoneal injections) for a minimum of three days prior to injury was required to demonstrate significant neuroprotection. In rats, animals that were
fed a dietary supplementation of 1 percent creatine for four weeks demonstrated significant neuroprotection that was linked to improved mitochondrial bioenergetics, increased ATP levels, and an increased threshold for activation of the mPTP. Further experiments have reported that two weeks of dietary supplementation of creatine (0.5 and 1 percent) prior to injury was sufficient to significantly reduce lactate and free fatty acid levels following TBI (Scheff and Dhillon, 2004). Additionally, all animals feed a creatine supplemented diet had significantly less cortical tissue damage compared to non-supplemented controls. To date these are the only studies assessing the use of creatine for the treatment of TBI.
CLOSING REMARKS
Creatine may be a viable prophylactic treatment for TBI based on its proposed target mechanisms including stabilization of cellular bioenergetics and inhibition of mPTP activation. Yet, a Medline search using the terms “traumatic brain injury” and “creatine supplementation” yields only six hits, of which only one is relevant. This may seem surprising considering the robust neuroprotective effects demonstrated by creatine pretreatment. However, the need to preload the system with creatine (or with any other compound) has historically reduced enthusiasm for funding this line of research as it relates to the treatment of TBI. This, of course, has left many unanswered questions:
-
What is the optimal dosage of creatine (i.e., dose-response)?
-
What is the minimum amount of pretreatment needed, or the therapeutic window of opportunity?
-
Is postinjury treatment beneficial in combination with pretreatment?
-
What is the optimal route of administration?
-
Can having creatine onboard enhance or hinder other neuroprotective treatments (i.e., does prophylactic creatine alter the TBI patient profile)?
Based on the safety profile of creatine and current experimental data, it is obvious that the potential for using creatine following TBI has not been explored sufficiently; however, creatine supplementation may offer a much needed therapeutic approach for targeting TBI in specific populations.
REFERENCES
Adhihetty, P. J., and M. F. Beal. 2008. Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromolecular Medicine 10(4):275–290.
Andres, R. H., A. D. Ducray, U. Schlattner, T. Wallimann, and H. R. Widmer. 2008. Functions and effects of creatine in the central nervous system. Brain Research Bulletin 76(4):329–343.
Beutner, G., A. Ruck, B. Riede, and D. Brdiczka. 1998. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochimica et Biophysica Acta 1368(1):7–18.
Beutner, G., A. Ruck, B. Riede, W. Welte, and D. Brdiczka. 1996. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Letters 396(2–3):189–195.
Cantu, R. C. 1997. Athletic head injuries. Clinics in Sports Medicine 16(3):531–542.
Cernak, I., J. Savic, D. Ignjatovic, and M. Jevtic. 1999. Blast injury from explosive munitions. Journal of Trauma 47(1):96–103; discussion 103–104.
Choi, D. W., H. Monyer, R. G. Giffard, M. P. Goldberg, and C. W. Christine. 1990. Acute brain injury, nmda receptors, and hydrogen ions: Observations in cortical cell cultures. Advances in Experimental Medicine and Biology 268:501–504.
Clark, K. 1998. Epidemiology of athletic head injury. Clinics in Sports Medicine 17(1):1–12.
DePalma, R. G., D. G. Burris, H. R. Champion, and M. J. Hodgson. 2005. Blast injuries. The New England Journal of Medicine 352(13):1335–1342.
Elder, G. A., and A. Cristian. 2009. Blast-related mild traumatic brain injury: Mechanisms of injury and impact on clinical care. Mount Sinai Journal of Medicine 76(2):111–118.
Faden, A. I., P. Demediuk, S. S. Panter, and R. Vink. 1989. The role of excitatory amino acids and nmda receptors in traumatic brain injury. Science 244(4906):798–800.
Fiskum, G. 2000. Mitochondrial participation in ischemic and traumatic neural cell death. Journal of Neurotrauma 17(10):843–855.
Gilmer, L. K., K. N. Roberts, K. Joy, P. G. Sullivan, and S. W. Scheff. 2009. Early mitochondrial dysfunction after cortical contusion injury. Journal of Neurotrauma 26(8):1271–1280.
Gualano, B., G. G. Artioli, J. R. Poortmans, and A. H. Lancha Junior. 2010. Exploring the therapeutic role of creatine supplementation. Amino Acids 38(1):31–44.
Gunter, T. E., K. K. Gunter, S. Sheu, and C. E. Gavin. 1994. Mitochondrial calcium transport: Physiological and pathological relevance. The American Journal of Physiology 267:C313–C339.
Hoge, C. W., D. McGurk, J. L. Thomas, A. L. Cox, C. C. Engel, and C. A. Castro. 2008. Mild traumatic brain injury in U.S. soldiers returning from Iraq. The New England Journal of Medicine 358(5):453–463.
Jones, E., N. T. Fear, and S. Wessely. 2007. Shell shock and mild traumatic brain injury: A historical review. American Journal of Psychiatry 164(11):1641–1645.
Langlois, J. A., W. Rutland-Brown, and M. M. Wald. 2006. The epidemiology and impact of traumatic brain injury: A brief overview. Journal of Head Trauma and Rehabilitation 21(5):375–378.
Ling, G., F. Bandak, R. Armonda, G. Grant, and J. Ecklund. 2009. Explosive blast neurotrauma. Journal of Neurotrauma 26(6):815–825.
Maragos, W. F., and A. S. Korde. 2004. Mitochondrial uncoupling as a potential therapeutic target in acute central nervous system injury. Journal of Neurochemistry 91(2):257–262.
Mazzeo, A. T., A. Beat, A. Singh, and M. R. Bullock. 2009. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Experimental Neurology 218(2):363–370.
Mbye, L. H., I. N. Singh, P. G. Sullivan, J. E. Springer, and E. D. Hall. 2008. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Experimental Neurology 209(1):243–253.
Mujika, I., and S. Padilla. 1997. Creatine supplementation as an ergogenic acid for sports performance in highly trained athletes: A critical review. International Journal of Sports Medicine 18(7):491–496.
O’Gorman, E., G. Beutner, M. Dolder, A. P. Koretsky, D. Brdiczka, and T. Wallimann. 1997. The role of creatine kinase in inhibition of mitochondrial permeability transition. FEBS Letters 414(2):253–257.
Pandya, J. D., J. R. Pauly, V. N. Nukala, A. H. Sebastian, K. M. Day, A. S. Korde, W. F. Maragos, E. D. Hall, and P. G. Sullivan. 2007. Post-injury administration of mitochondrial uncouplers increases tissue sparing and improves behavioral outcome following traumatic brain injury in rodents. Journal of Neurotrauma 24(5):798–811.
Pandya, J. D., J. R. Pauly, and P. G. Sullivan. 2009. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Experimental Neurology 218(2):381–389.
Rutland-Brown, W., J. A. Langlois, K. E. Thomas, and Y. L. Xi. 2006. Incidence of traumatic brain injury in the United States, 2003. Journal of Head Trauma Rehabilitation 21(6):544–548.
Scheff, S. W., and H. S. Dhillon. 2004. Creatine-enhanced diet alters levels of lactate and free fatty acids after experimental brain injury. Neurochemical Research 29(2):469–479.
Setnik, L., and J. J. Bazarian. 2007. The characteristics of patients who do not seek medical treatment for traumatic brain injury. Brain Injury 21(1):1–9.
Singh, I. N., P. G. Sullivan, Y. Deng, L. H. Mbye, and E. D. Hall. 2006. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: Implications for neuroprotective therapy. Journal of Cerebral Blood Flow and Metabolism 26(11):1407–1418.
Sullivan, P. G., J. D. Geiger, M. P. Mattson, and S. W. Scheff. 2000. Dietary supplement creatine protects against traumatic brain injury. Annals of Neurology 48(5):723–729.
Sullivan, P. G., A. G. Rabchevsky, P. C. Waldmeier, and J. E. Springer. 2005. Mitochondrial permeability transition in cns trauma: Cause or effect of neuronal cell death? Journal of Neuroscience Research 79(1–2):231–239.
Sullivan, P. G., J. E. Springer, E. D. Hall, and S. W. Scheff. 2004. Mitochondrial uncoupling as a therapeutic target following neuronal injury. Journal of Bioenergetics and Biomembranes 36(4):353–356.
Thurman, D. J., C. M. Branche, and J. E. Sniezek. 1998. The epidemiology of sports-related traumatic brain injuries in the United States: Recent developments. Journal of Head Trauma Rehabilitation 13(2):1–8.
Trudeau, D. L., J. Anderson, L. M. Hansen, D. N. Shagalov, J. Schmoller, S. Nugent, and S. Barton. 1998. Findings of mild traumatic brain injury in combat veterans with PTSD and a history of blast concussion. Journal of Neuropsychiatry and Clinical Neurosciences 10(3):308–313.
Nutritional Care of Active Duty Patients with TBI
Stephanie Sands27
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of death and disability in the United States. Among military personnel serving in Operation Enduring Freedom/Operation Iraqi Freedom (OEF/OIF), the likelihood of traumatic brain and other polytrauma injuries is significantly elevated. As defined by the U.S. Veterans Health Administration, polytrauma is “two or more injuries to physical regions or organ systems, one of which may be life threatening, resulting in physical, cognitive, psychological, or psychosocial impairments and functional disability” (United States Department of Veterans Affairs, 2009). Such injuries are often a result of rocket-propelled grenades, improvised explosive devises, gunshot wounds, and landmines. In 2005, the United States Congress established four Polytrauma Rehabilitation Centers, one of which is the James A. Haley Veterans’ Hospital (JAHVH) in Tampa, Florida (Scott et al., 2006). This report is largely based on nutritional management and monitoring of complex variables of patients who have suffered TBI and polytrauma in the sub-acute and long-term setting at JAHVH.
SUB-ACUTE NUTRITIONAL MANAGEMENT
Following severe trauma and acute TBI, striking metabolic changes involving an accelerated catabolic rate and extensive nitrogen losses proportional to the severity of injury are common (Cook and Hatton, 2007). The hypermetabolic response is related to increases in energy expenditure, oxygen consumption, carbon dioxide production as well as primary mediators such as catecholamines, corticosteroids, and inflammatory cytokines (Berry, 2009; Esper, 2004). Because the brain functions as a regulator for metabolic activity, disruptions caused by TBI result in a cascade of hormonal modifications, irregular cellular metabolism, and dynamic cerebral and systemic inflammatory response as an effort to circulate substrate required at the cellular level. The end result of these alterations involves systemic catabolism causing an increase in basal metabolism, oxygen consumption, glycogenolysis, hyperglycemia, proteolysis, muscle wasting, and energy requirements (Cook et al., 2008). Optimal timing of nutrition, fluid, and electrolyte management may improve the overall clinical course in TBI patients. The fundamental goal of nutritional intervention is to provide adequate calories and protein sufficient to meet the demands of hypermetabolism and increased protein breakdown as a means of preserving lean body mass while maintaining skin integrity, immune function, gastrointestinal mucosal integrity, wound healing, and nitrogen balance during rehabilitation.
While providing nutritional care of polytrauma patients during the months following injury, one of the most challenging decisions is the accurate assessment and provision of essential calories as the complications related to under- or overfeeding can compromise rehabilitation prognosis. Nutrition support should be aimed toward current physiologic
reactions and should not exacerbate complications of the current phase (stress, catabolic, anabolic). Underfeeding can result in decreased respiratory muscle strength, decreased ventilator drive, failure to wean from mechanical ventilation, impaired organ function, immunosuppression, poor wound healing, increased risk of nosocomial infection, and low transport protein levels. This cachexia (i.e. wasting syndrome or loss of weight, muscle atrophy, fatigue, weakness, and significant loss of appetite in someone who is not actively trying to lose weight) can impact mobility, functional rehabilitation and overall length of stay as well as the development of complications such as decubitus ulcers, pneumonia, urinary tract infections, and venous thromboembolism. Among the complications of overfeeding include the risk of refeeding syndrome,28 hyperglycemia, azotemia, hypertriglyceridemia, electrolyte imbalance, immunosuppression, alterations in hydration status, hepatic steatosis, and failure to wean from mechanical ventilation (Cook et al., 2008; Esper, 2004).
Calorie Provision
Energy expenditure has been investigated extensively and has been shown to be elevated following acute TBI (Cook and Hatton, 2007; Rajpal and Johnston, 2009). Two methods for determining energy requirements involve measurement of resting metabolic rate using indirect calorimetry (IC), or estimating energy needs with the use of predictive equations and clinical judgment. Although IC is considered the “gold standard” for determining energy expenditure in TBI, many clinicians do not have access to resources necessary to measure metabolic rate. Additionally, calorie requirements may vary day to day in this population secondary to symptoms such as sympathetic storming, fevers, or muscle contractions. More commonly, clinicians use one of more than 200 predictive equations that have been developed for estimating energy expenditure (McCarthy et al., 2008). For example, the Brain Trauma Foundation (BTF) recommends use of the Harris Benedict Equation multiplied by a stress factor of 1.4 with an observed variance of 1.2–2.5 (Bratton et al., 2007), and the American Society of Enteral and Parenteral Nutrition recommends 25–30 kcal/kg in critically ill patients. However, it should be noted that there is a limited amount of literature available following the critical care setting or for patients with further polytrauma injuries in addition to TBI. Because utilizing the above methods to determine energy expenditure can be imprecise considering the complexity of this patient population, the following variables have been observed or proven to alter metabolic rate (Berry, 2009; Cook et al., 2008; Dickerson and Roth-Yousey, 2005; Esper, 2004; Frankenfield, 2006; Rajpal and Johnston, 2009):
-
Severity of trauma and additional injuries, burns, or wounds
-
Time since injury, depending on the ongoing stress response and degree of healing
-
Physiologic effects-blood pressure, heart rate, respiratory rate, sympathetic storming such as seen in Paroxysmal Autonomic Instability with Dystonia (Blackman et al., 2004), body temperature (diaphoresis, hyperthermia, and medically induced hypothermia)
-
Physical activity (restlessness, agitation) or muscular dysfunction (posturing, dystonia)
-
Level of consciousness (Glasgow Coma Score)
-
Cognitive Functioning (Rancho Los Amigos Scale)
-
Neuroendocrine disruption
-
Sepsis and inflammatory response
-
Medications: Central nervous system agents-sedatives, anticonvulsants, analgesics, narcotics, hypnotics, barbiturates; autonomic neuromuscular blocking agents; cardiovascular agents-beta-adrenergic blockers; steroids, inotropic agents.
-
Ventilator support
-
Thermic effect of food generated by caloric intake
-
Preinjury nutritional status and/or malnutrition
Protein Requirements
Protein requirements following TBI are grossly elevated. Protein catabolism peaks 8 to 14 days after injury with documented nitrogen losses up to 30 grams per day. This extreme protein breakdown can cause a 10 percent loss of lean body mass during the first seven days of injury (Cook and Hatton, 2007; Gleghorn et al., 2005; Rajpal and Johnston, 2009). Urinary urea nitrogen (UUN) values can be monitored to determine a state of nitrogen balance. Although it is often unrealistic to obtain nitrogen balance during the first week after injury regardless of nutritional provision, nitrogen balance is an important way to measure the adequacy of caloric intake and metabolism in the weeks that follow (Esper, 2004; Rajpal and Johnston, 2009). The BTF recommends protein provision of 1.5–2.0 g/kg of body weight in TBI patients (Cook and Hatton, 2007; Cook et al., 2008). Although hepatic production of transport proteins (albumin, prealbumin, transferring) is reduced during states of inflammation regardless of nutrition, monitoring their trends can be helpful to determine recovery from the inflammatory process along with overall clinical improvement such as wound healing, infection resolution, and weaning from ventilator support. Some facilities also incorporate specific amino acids into their nutritional programs such as glutamine, arginine, or branched chain amino acids (Esper, 2004; Rajpal and Johnston, 2009).
Method of Feeding
Another area of consideration during nutritional care of TBI patients is the method of feeding. Initially, it has to be determined whether to use parenteral (PN) or enteral (EN) nutrition. It is generally accepted that EN is preferable over PN, with the exception of cases such as barbiturate coma, multiple vasopressors (risk of bowel necrosis), or prolonged periods of being supine. When it is determined that EN is desirable, many clinicians debate whether to obtain small bowel or gastric access given that there is limited consensus that postpyloric feedings have demonstrated improved outcomes. TBI patients often exhibit gastrointestinal dysfunction with increased incidences of aspiration pneumonia, diarrhea, vomiting, abdominal distention, and increased gastric residuals. Impaired gastric emptying is often present secondary to decreased lower esophageal sphincter tone, vagus nerve damage, elevated levels of endogenous opioids/endorphins, elevated intracranial pressure, or medication side effects (Cook et al., 2008; Esper, 2004; Ott et al., 1991; Rajpal and Johnston, 2009). Often nasogastric or nasoenteral tubes are placed until it is determined that longer term EN access is needed. The placement of longer term EN access via percutaneous endoscopic gastrostomy (PEG) tubes often proves successful in establishing well-tolerated feeding access (Cook and Hatton, 2007). Despite the potential feeding difficulties in many TBI patients, the majority of these patients are able to receive safe and adequate nutrition through EN. Approaches to improve EN tolerance include head of bed elevation (30–45 degrees), continuous tube feeding at low infusion rates advanced per tolerance, the use of pro-motility agents, using concentrated enteral formulas to decrease total volume, and consideration of small bowel versus gastric feeds (Cook et al., 2008).
TABLE C-11 Sample of Drug-Nutrition Interactions Common in TBI Medical Management
|
Medication |
Examples of Nutrition Implications |
|
Antipsychotics (Ziprasidone, Olanzapine, Risperidone) |
Linked to weight gain |
|
Barbiturates |
May lower metabolic rate or cause constipation |
|
Bisacodyl |
Risk of hypocalcemia and decreased fat absorption |
|
Bromocriptine |
Potential of nausea/vomiting/constipation, elevated residuals |
|
Carbamazepine |
Increased risk of hyponatremia, can cause formation of orange rubbery precipitate when combined with water/dilatants |
|
Corticosteroids |
Risk of hyperglycemia; osteoporosis and gastric ulcer risk with chronic use |
|
Mannitol |
Monitor for hypokalemia, hypomagnesemia, hypovolemia |
|
Metoclopramide |
Possible for changes in mental status/cognition |
|
Mirtazapine |
Related to increased appetite and weight gain |
|
Narcotic analgesics |
Delayed emptying/constipation (especially in opioid usage) |
|
Oxandrolone |
Linked to elevated liver enzymes and/or lipid panela |
|
Phenytoin |
Absorption may be impaired with provided with enteral nutrition, possible decline in folate, vitamin D |
|
Propofol |
Provides lipid calories (pro-inflammatory fat source) |
|
Stimulants (Methylphenidate, Dextroamphetamine amphetamine) |
May cause decreased appetite and weight loss |
|
Vasopressors |
Decreases gut perfusion |
|
Zolpidem |
May cause appetite changes, binge eating, nocturnal eatinga |
|
aAnecdotal observations noted at JAHVH; UpToDate Online 18.3. SOURCE: Cook and Hatton, 2007. |
|
Medication Interactions
Drug-nutrient interactions require consideration when providing medical nutrition therapy to TBI patients. Registered dietitians review patient medications as a part of nutritional assessment to identify nutritional implications. For example, enteral nutrition is typically held one to two hours before and after the administration of phenytoin to prevent absorptive changes and chelation. Other medications may lower electrolyte and micronutrient levels, or increase the risk of weight gain or loss. Table C-11 provides examples of some interactions encountered in the clinical setting.
Sample of 12 Polytrauma Patients
As discussed previously, there is a limited amount of research evaluating nutritional needs of TBI patients following the acute critical period or with multiple polytrauma injuries in addition to TBI. Figures C-23 through C-28 represent a snapshot of 12 patients in acute rehabilitation at JAHVH. The categories illustrated include patient age, time since injury, method of feeding, percent of usual body weight lost and caloric provision required to facilitate weight maintenance of weight gain of one to two pounds per week. The illustrations depict at typical distribution of patients which changes from day to day.
FIGURE C-23 Distribution of 12 polytrauma patients according to age (captured 9/15/2010 at JAHVH).
FIGURE C-24 Distribution of 12 polytrauma patients according to the number of months since injury (captured 9/15/2010 at JAHVH).
FIGURE C-25 Distribution of 12 polytrauma patients according to the method of feeding (captured 9/15/2010 at JAHVH).
FIGURE C-26 Distribution of 12 polytrauma patients according to the percent of pre-injury weight loss since injury (captured 9/15/2010 at JAHVH).
FIGURE C-27 Distribution of 12 polytrauma patients according to number of calories per kilogram to attain weight maintenance or gain (1–2 lbs per week) (captured 9/15/2010 at JAHVH).
FIGURE C-28 Distribution of 12 polytrauma patients according to percent of estimated resting metabolic rate using the Harris Benedict Equation to attain weight maintenance or gain (1–2 lbs per week) (captured 9/15/2010 at JAHVH)
LONG TERM NUTRITIONAL CONSIDERATIONS
While the concerns of severe weight loss following TBI in active duty service members are significant, the opposing issue regarding unintentional weight gain has scarcely been discussed in the literature. Following TBI, changes such as alterations in the brain’s regulation of hunger and satiety, neuroendocrine dysfunction, brain injury-induced hyperphagia, medication related side effects, cognitive impairments, or emotional coping may impact the ability to maintain a healthy weight. In the post-acute rehabilitation setting at JAHVH, a number of polytrauma patients experience detrimental weight gain and dyslipidemia.
Under normal conditions, the brain functions to regulate energy homeostasis by constant transmission of signals that influence energy intake and ultimately body weight (Woods and D’Alessio, 2008). Satiation signals such as cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), peptide tyrosine-tyrosine (PYY) and apolipoprotein A-IV (apo A-IV) are secreted in response to specific macronutrient stimuli. These peptides, many of which are synthesized in the brain in addition to the gastrointestinal tract, are released in response to food ingestion and act to reduce meal size. Adiposity signals, insulin and leptin, are secreted relative to the amount of body fat and are transported across the blood-brain barrier to interact with neuronal receptors predominately in the hypothalamus. Because TBI may impair the transmission of these signals, many patients experience an altered sensation of hunger and satiety.
TABLE C-12 Nutritional Implications of Post Traumatic Hypopituitarism
|
Hormone Insufficiency/Deficiency |
Nutritional Implications |
|
Adrenocorticotrophic hormone (ACTH) (0–19 percent of TBI patients with deficiencies) |
Nausea/vomiting, abdominal pain, anorexia, weight loss, hypotension, tachycardia, hyponatremia, hypoglycemia, normocytic anemia |
|
Growth hormone (GH) (6–33 percent of TBI patients with deficiencies) |
Osteoporosis, dyslipidemia, atherosclerosis, visceral obesity, reduced lean body mass |
|
Hypothyroidism-Thyroid stimulating hormone (TSH) (1–10 percent of TBI patients with deficiencies) |
Weight gain, hypotension, myopathy, dyspnea, periorbital edema, bradycardia, normocytic anemia, mild hyponatremia |
|
Luteinizing hormone (LH)/Follicle-stimulating hormone (FSH) (2–20 percent of TBI patients with deficiencies) |
Reduced muscle mass and exercise tolerance (men), decreased breast tissue and bone mineral density (women) |
|
SOURCE: Compiled from Schneider et al., 2007. |
|
Disturbances of the neuroendocrine system following TBI have nutritional implications that should be considered. Schneider et al (2007) identified 19 clinical studies that report the prevalence of endocrine dysfunction ranges from 15–68 percent in TBI patients. Most researchers agree upon the association of neuroendocrine changes and the severity of brain injury, however variables such as secondary brain damage and medical complications make analysis and prediction more complicated (Rothman et al., 2007). Klose and associates (2007) reported that TBI patients with posttraumatic hypopituitarism display symptoms such as adverse lipid profiles, unfavorable body composition, and worsened perceived health-related quality of life (lowered energy, sleep, increased social isolation) compared to those with preserved pituitary function. Table C-12 reflects sample nutritional implications of post-traumatic hypopituitarism.
Although there is a paucity of information regarding brain injury-induced hyperphagia, clinicians working in TBI are likely to encounter patients with an abnormally increased appetite for and consumption of food. This can be especially problematic in a significantly impaired patient with limited self-awareness. Rao and Lyketsos (2000) describe a complex syndrome they refer to as Behavioral Dyscontrol Disorder, Major Variant. This syndrome has mood, cognitive, and behavioral manifestations in both acute and chronic stages of 5–70 percent of TBI. The behaviors reported in this report include impulsivity, aggression, hyperactivity, hyperphagia, and pica. One such scenario at JAHVH resulted in the removal of a patient-accessible family refrigerator secondary to a patient with an insatiable appetite and weight gain. This patient experienced weight gain at a rate of 15 pounds per month, with a total gain of 110 pounds over seven months. He required constant nursing supervision to prevent further instances of excessive eating and consumption of non-food substances like coffee grounds. Instances of rapid and nearly uncontrollable weight gain as described are not uncommon among this population. A multidisciplinary approach should be utilized to treat such conditions by developing environmental modification strategies, behavioral therapy, psychotherapy, and family therapy.
Another contributing factor to the weight gain experienced in the postacute setting following TBI is medication side effects. Common medications prescribed following brain injury such as antidepressants, anxiolytics, anticonvulsants, and antipsychotics may promote increased appetite and unintentional weight gain. Healthcare providers should be aware of these side effects and consider weight-neutral alternatives.
Cognitive impairments among service members who have suffered TBI greatly impact the successfulness of nutrition counseling to a degree that cannot be overstated. For example, consider how executive dysfunction may inhibit the ability to make healthful choices secondary to difficulties in planning, problem solving, organizing, sequencing, self-regulation and monitoring, judgment, set-shifting, impulse control, initiation, and motivation (Rao and Lyketsos, 2000). Tasks such as grocery shopping, reading a recipe, preparing a meal, or understanding and applying healthful eating guidelines can be nearly impossible for some patients. Furthermore, among patients with varying levels of memory impairment, the ability to remember nutritional recommendations may be compromised in addition to recalling and identifying problematic eating behaviors. The combination of impaired hunger and satiety cues as well as short-term memory loss often results in patients who cannot remember having eaten just minutes after mealtime and are likely to overeat as well.
Lastly, the emotional strain of suffering from TBI can play a significant role in the ability to make smart choices and maintain a healthy weight. Coping with symptoms such as depression, boredom, demoralization, anxiety, irritability, anger, the feeling of loss, discouragement, and posttraumatic stress disorder can lead to uncontrolled emotional eating. Additionally, families of those who have experienced a TBI are susceptible to encourage the use of food as a coping and comforting mechanism. These patients greatly benefit from a team approach to identify coping strategies and alternatives to eating as well as nutrition education to encourage healthy choices.
COMPLEMENTARY AND ALTERNATIVE MEDICINE
Incorporation of complementary and alternative medicine (CAM) into treatment regimens has become more prevalent among both acute and chronically ill patients. Many researchers are investigating the role of CAM in providing resilience to brain injury or as a treatment modality following an injury. A large portion of posttraumatic neurodegeneration is a result of secondary damage from a pathochemical and pathophysiological cascade during the first minutes, hours, and days following an injury (Hall et al., 2010). Many investigators seek to discover the optimal timing of neuroprotective substances to prevent exacerbation of damage caused by the primary injury. However, there are many challenges that come with both performing and interpreting research relating to CAM. As described by Mullin (2009):
-
The majority of CAM providers are non-physician based, utilizing techniques and tools that are more experiential than evidence-based.
-
CAM often focuses on treatment of symptoms, which can be subjective, rather than the underlying diagnosis of Western-based medicine.
-
Blinding is often compromised and many publications labeled as randomized control trials are actually not blinded.
-
Publication biases are created when investigators, reviewers, and editors submit or accept manuscripts based on the strength or direction of the findings.
-
Access to CAM literature is incomplete; one such example includes mainstream databases such as MEDLINE, which indexes only 10 percent of CAM journal worldwide.
-
Negative CAM findings are more likely to be published in mainstream medical journals, whereas most studies published in leading CAM journals have positive results.
-
-
-
Studies outside of the United States are more likely to be positive than those in the United States.
-
For example, while completing a PubMed literature review on supplements and TBI over the past five years, Table C-13 depicts the number of articles referenced with various supplements. It is important to note that the majority of these references utilize animal studies to answer their research question. While understanding the possibility of publication bias, one may wonder how to identify the CAM articles being published and indexed in other locations.
Clinicians attempting to provide evidence-based guidelines to patients and families are likely to encounter difficulties interpreting the literature and making recommendations. Reasons for this include the publication bias as described above, the lack of clear data, and the number of supplements being promoted. There is a copious amount of marketing which targets TBI patients anywhere along the spectrum of severity and time since injury. Patients and their families are likely to encounter or trial a number of supplements which are promoted to treat side effects of TBI or improve brain health and functioning. Table C-14 reflects a list of supplements being taken among different levels of TBI severity as disclosed by patients and families over one month at JAHVH. An especially susceptible population includes the caregivers of emerging conscious patients. Some programs seek to discover a beneficial cocktail of nutraceuticals which may assist in promoting consciousness. Incidences of patients receiving 45 different nutraceuticals, some of which are administered two or three times a day, as an effort to cause the patient to emerge have been encountered. While assisting patients in making informed decisions regarding supplements, factors should be evaluated such as the risk of causing harm if taken in excessive doses, decreasing medication side effects or modifying the action, lowering seizure threshold, instigating other deficiencies, or mislabeling or adulterating supplements.
TABLE C-13 Depiction of PubMed Literature Review of TBI and Various Supplements Over the Past 5 Years (2005–2010)
TABLE C-14 Illustration of Supplements Encountered Among TBI patients, Ranging from Mild to Severe Injuries (JAHVH from 8/20/2010–9/20/2010)
|
Antioxidant vitamins |
Cinnamon |
Ginseng |
Omega-6 fatty acids |
|
Apigenin |
Citric Acid |
Green tea |
Resveratrol |
|
B Vitamins (mega-doses) |
CoEnzyme Q10 |
Huperzine |
RNA |
|
Baicalein Butcher’s Broom |
Cognitex (a-glyceryl phosphoryl choline, ginger, rosemary, phosphatidylserine, pregnolone, vinpocentine, leucoselect phytosome, wild blueberry, sensoril ashwagandha, perluxan) |
Individual amino acids or blends (most commonly: branched chain, tyrosine, glutamine, arginine) |
Rutin |
|
Caffeine |
Corella |
Lipoic acid |
Spirulina |
|
Capsaicin |
Creatine |
Luteolin |
St. John’s wart |
|
Carnitine (L-configuration) |
Curcumin |
Magnesium |
Tocopherol |
|
Carnosine (L-configuration) |
D-Ribose |
Milk thistle |
Valarian root |
|
Catechin |
Feverfew |
Mycelia extract |
Vinpoceti |
|
Choline |
Ginkgo biloba |
n-3 fatty acids |
Zinc |
CONCLUSION
Nutritional assessment, monitoring, and evaluation should be a priority throughout the course of TBI and polytrauma injuries among active-duty service members. Registered dietitians have the educational background to coordinate acute nutritional support and subacute nutritional management based on the variety of nutritional conditions prevalent following TBI. Furthermore, a multidisciplinary team approach is critical to discuss progress, treatment plans, and goals for overall best outcomes.
REFERENCES
Berry, A. 2009. Neurocritical Care 101: Learning the lingo for effective nutrition management. Support Line 31(6):3–11.
Blackman, J. A., P. D. Patrick, M. L. Buck, and R. S. Rust Jr. 2004. Paroxysmal autonomic instability with dystonia after brain injury. Archives of Neurology 61(3):321–328.
Bratton, S., D. Chestnut, J. Ghajar, F. Hammond, O. Harris, R. Hartl, J. Schouten, L. Shutter, S. Timmons, J. Ullman, W. Videtta, J. Wilberger, and D. Wright. 2007. Nutrition. Journal of Neurotrauma 24(1 Suppl.):S77–S82.
Cook, A., and J. Hatton. 2007. Neurological impairment. In The A.S.P.E.N. Nutrition support core curriculum: A case-based approach-the adult patient. 2nd ed., edited by M. Gottschlich, M. DeLegge, T. Mattox, C. Mueller and P. Worthingon. Silver Spring, MD: American Society for Parenteral and Enteral Nutrition. Pp. 424–439.
Cook, A. M., A. Peppard, and B. Magnuson. 2008. Nutrition considerations in traumatic brain injury. Nutrition in Clinical Practice 23(6):608–620.
Dickerson, R. N., and L. Roth-Yousey. 2005. Medication effects on metabolic rate: A systematic review (part 1). Journal of the American Dietetic Association 105(5):835–843.
Esper, D. 2004. Metabolic response and nutrition management in patients with severe head injury. Support Line 26(2):9–13.
Frankenfield, D. 2006. Energy expenditure and protein requirements after traumatic injury. Nutrition in Clinical Practice 21(5):430–437.
Gleghorn, E., K. Amorde-Spalding, and M. DeLegge. 2005. Neurologic diseases. In A.S.P.E.N. Nutrition support manual. 2nd ed., edited by R. Merritt. Silver Spring, MD: American Society for Parenteral and Enteral Nutrition. Pp. 246–250.
Hall, E. D., R. A. Vaishnav, and A. G. Mustafa. 2010. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7(1):51–61.
Klose, M. C., T. Watt, J. Brennum, and U. Feldt-Rasmussen. 2007. Posttraumatic hypopituitarism is associated with an unfavorable body composition and lipid profile, and decreased quality of life 12 months after injury. Journal of Clinical Endocrinology and Metabolism 92(10):3861–3868.
McCarthy, M. S., J. Fabling, R. Martindale, and S. A. Meyer. 2008. Nutrition support of the traumatically injured warfighter. Critical Care Nursing Clinics of North America 20(1):59–65.
Mullin, G. E. 2009. Issues in complementary and alternative nutrition treatments. Nutrition in Clinical Practice 24(5):543–548.
Ott, L., B. Young, R. Phillips, C. McClain, L. Adams, R. Dempsey, P. Tibbs, and U. Yun Ryo. 1991. Altered gastric emptying in the head-injured patient: Relationship to feeding intolerance. Journal of Neurosurgery 74(5):738–742.
Rajpal, V., and J. Johnston. 2009. Nutrition management of traumatic brain injury patients. Support Line 31(1):10–19.
Rao, V., and C. Lyketsos. 2000. Neuropsychiatric sequelae of traumatic brain injury. Psychosomatics 41(2):95–103.
Rothman, M. S., D. B. Arciniegas, C. M. Filley, and M. E. Wierman. 2007. The neuroendocrine effects of traumatic brain injury. Journal of Neuropsychiatry and Clinical Neurosciences 19(4):363–372.
Schneider, H. J., I. Kreitschmann-Andermahr, E. Ghigo, G. K. Stalla, and A. Agha. 2007. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. Journal of the American Medical Association 298(12):1429–1438.
Scott, S. G., H. G. Belanger, R. D. Vanderploeg, J. Massengale, and J. Scholten. 2006. Mechanism-of-injury approach to evaluating patients with blast-related polytrauma. Journal of the American Osteopathic Association 106(5):265–270.
United States Department of Veterans Affairs. 2009. VA polytrauma systems of care. http://www.polytrauma.va.gov/definitions.asp#polytrauma (accessed 10/15/10).
Woods, S. C., and D. A. D’Alessio. 2008. Central control of body weight and appetite. Journal of Clinical Endocrinology and Metabolism 93(11 Suppl. 1):S37–S50.































































































































