
The Evolution of Drug Resistance and
the Curious Orthodoxy of Aggressive
Chemotherapy
![]()
ANDREW F. READ,*†§TROY DAY,‡AND SILVIE HUIJBEN*
The evolution of drug-resistant pathogens is a major challenge for 21st century medicine. Drug use practices vigorously advocated as resistance management tools by professional bodies, public health agencies, and medical schools represent some of humankind’s largest attempts to manage evolution. It is our contention that these practices have poor theoretical and empirical justification for a broad spectrum of diseases. For instance, rapid elimination of pathogens can reduce the probability that de novo resistance mutations occur. This idea often motivates the medical orthodoxy that patients should complete drug courses even when they no longer feel sick. Yet “radical pathogen cure” maximizes the evolutionary advantage of any resistant pathogens that are present. It could promote the very evolution it is intended to retard. The guiding principle should be to impose no more selection than is absolutely necessary. We illustrate these arguments in the context of malaria; they likely apply to a wide range of infections as well as cancer and public health insecticides. Intuition is unreliable even in simple evolutionary contexts; in a social milieu where in-host competition can radically alter the fitness costs and benefits of resistance, expert opinion will be insufficient. An evidence-based approach to resistance management is required.
___________________
*Center for Infectious Disease Dynamics, Departments of Biology and Entomology, Pennsylvania State University, University Park, PA 16802; †Fogarty International Center, National Institutes of Health, Bethesda, MD 20892; and ‡Departments of Mathematics, Statistics, and Biology, Queen’s University, Kingston, ON, Canada K7L 3N6. §To whom correspondence should be addressed. E-mail: a.read@psu.edu.
The evolution of drug-resistant pathogens significantly affects human well-being and health budgets. Consequently, existing and new antimicrobials should be viewed as precious resources in need of careful stewardship (Owens, 2008; Spellberg et al., 2008). An important aspiration is to maximize the therapeutically useful life span of a compound, the time a given antimicrobial yields clinical benefits before drug efficacy is undermined by resistance evolution. Attempting to do so is essentially an exercise in evolutionary management.
Various practices are widely thought to be effective resistance management strategies (American Academy of Microbiology, 2009; World Health Organization, 2010a; zur Wiesch et al., 2011). For instance, there is near-universal agreement that combination drug therapy, the coad-ministration of drugs with unrelated modes of action, prolongs the useful life of the component compounds for diseases as diverse as leprosy, HIV, malaria, and tuberculosis (TB). Another practice is the restriction of treatment to those patients who need it on clinical grounds, so as to reduce unnecessary selection for resistance. This philosophy underpins restrictions on the use of antibiotics in hospitals and in the community at large, and it has led to calls for reductions in drug use in animal feed.
A third practice thought to be an effective resistance management strategy is the use of drugs to clear all target pathogens from a patient as fast as possible. We hereafter refer to this practice as “radical pathogen cure.” For a wide variety of infectious diseases, recommended drug doses, interdose intervals, and treatment durations (which together constitute “patient treatment regimens”) are designed to achieve complete pathogen elimination as fast as possible. This is often the basis for physicians exhorting their patients to finish a drug course long after they feel better (long-course chemotherapy). Our claim is that aggressive chemotherapy cannot be assumed to be an effective resistance management strategy a priori. This is because radical pathogen cure necessarily confers the strongest possible evolutionary advantage on the very pathogens that cause drugs to fail.
At one level, our argument is simple. Elementary population genetics shows that, all else being equal, the stronger the strength of selection, the more rapid is the spread of a favored allele (Maynard Smith, 1989a). For drug use, the strength of selection is determined by how many people are being treated and, among the treated people, the treatment regimen. The more aggressive the regimen, the greater is the selection pressure in favor of resistance. Because overwhelming chemical force necessarily confers the strongest possible selective advantage on any pathogen capable of resisting it, radical pathogen cure can very effectively drive resistant pathogens through a population. As we will argue, this
problem is especially important when there is genetic diversity among pathogens within an infected individual.
AIMS OF PATIENT TREATMENT
Ignoring economic considerations, patient treatment should seek to achieve the following:
(i) Make the patient healthy.
(ii) Prevent the patient from infecting others.
(iii) Prevent the spread of resistant pathogens to others.
The first aim concerns the health of the patient being treated. The second and third aims concern the effects of patient treatment on the health of others.
A single strategy cannot simultaneously best achieve all three aims. In the limit, zero treatment will usually be the best resistance management strategy. It is important to identify and justify compromises because this makes explicit problems in need of solution and is a prerequisite for evidence-based resistance management. There may come a time when resistance management strategies are required that put overall public health ahead of patient health (Foster and Grundmann, 2006). We do not think the problems of resistant pathogens are yet so dire as to require this. In our view, the current scientific challenge is to identify, among patient treatment regimens that are similarly effective at restoring health and preventing transmission, those regimens that best effect resistance management.
The aim of resistance management is to prevent clinical failures caused by high-level resistance. Resistance is often a continuous trait, and there can be varying degrees of intermediate resistance. Sometimes referred to as “tolerance,” intermediate resistance confers the ability to survive concentrations of drug below those considered therapeutic (Fig. 11.1). We define high-level resistance as that which undermines patient health by causing therapeutic failure. It is the rate of spread of high-level resistance that needs to be managed because this determines the therapeutically useful life span of a drug.
The useful life span of a drug is determined by two processes. The first is the rate at which genetic events conferring high-level resistance on an individual pathogen actually occur. For simplicity, we refer to these events as de novo mutations, but we use this to include any heritable change that confers de novo high-level resistance on a pathogen individual. For example, in bacteria, this event can be the acquisition by lateral transfer of genetic material from another species. The second process affecting the
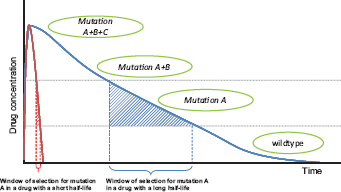
FIGURE 11.1 Hypothetical path to drug resistance. Solid curves show drug concentration in a treated patient for two drugs with very different half-lives; concentrations wane when treatment ceases. In this schematic, wild-type parasites can survive very low concentrations, with mutations A, B, and C conferring the ability to survive (“tolerate”) successively higher drug concentrations. High-level resistance (full clinical resistance) is where treatment has a negligible direct impact on pathogens with all three mutations. The windows of selection for mutation A are shown. In those windows, parasites with mutation A have a selective advantage over wild-type parasites. Note that the duration of the window depends critically on the drug half-life, which for antimalarial drugs can vary from hours (e.g., artemisinin), to weeks (e.g., SP), to months (e.g., mefloquine).
rate of evolution is the strength of selection acting on this genetic change. Because both mutational and selection processes together determine the useful life span of a drug, resistance evolution can be retarded by managing mutations, selection, or, ideally, both. Our view is that conventional wisdom focuses too much on managing mutational events (genetic origins), often with the consequence that the selection pressures are ignored.
A REAL-WORLD CONTEXT
Our logic likely applies to a very wide range of pathogens, but, as we discuss further below, there will not be simple generalities. To make things more concrete, we base our discussion on malaria, a disease that typifies the clinical and financial problems posed by drug resistance.
Resistance has evolved to all classes of frontline antimalarial drugs (Hyde, 2005), and several have had to be withdrawn from use in many countries. The eventual failure of drugs in the face of parasite evolution is now accepted as inevitable by the World Health Organization (WHO) (Roll Back Malaria, 2008) and others (American Academy of Microbiology,
2009). A key component of the Global Malaria Action Plan is an explicit plan for a discovery pipeline to deliver replacement drugs continuously (Roll Back Malaria, 2008). This pipeline will cost more than U.S. $2.5 billion in research and development for the coming decade and, once the currently inadequate drug arsenal is rebuilt, U.S. $1.5 billion thereafter for every decade until malaria is eradicated (Roll Back Malaria, 2008). Even if we assume that an unlimited supply of drug classes can be discovered, more than money is at stake. Drugs can fail more rapidly than the time it takes to get them through modern regulatory processes, and the cost in terms of human suffering is high. National authorities switch their choice of first-line drug only when forced to by declining patient cure rates; thus, disease burdens are considerable. WHO currently recommends that a drug be withdrawn once treatment failure rates attributable to resistance reach 10% (World Health Organization, 2010a, p. 8). In practice, governments of poor countries do not have this luxury and often wait longer before drug withdrawal is implemented (World Health Organization, 2006, p. 15).
Severe (life-threatening) malaria involves the dysfunction of vital organs; for patients in this state, the sole aim of treatment is to prevent death. Uncomplicated malaria constitutes the bulk of treated cases and those that can drive transmission chains, and hence resistance evolution. The WHO Guidelines for the Treatment of Malaria (World Health Organization, 2010a, p. 6) state: “The objective of treating uncomplicated malaria is to cure the infection as rapidly as possible,” with cure being defined as “the elimination from the body of the parasites that caused the illness.” Patient treatment regimens recommended in the WHO guidelines are those designed to achieve rapid and full elimination.
It is clear that radical pathogen cure can, in the absence of resistance, achieve the first two aims of patient treatment (restore health and prevent disease transmission). The consensus view is that it can also achieve the third aim: “Resistance can be prevented, or its onset slowed considerably” by “ensuring very high cure rates through full adherence to correct dosing regimens” (World Health Organization, 2010a, p. 6). This is the orthodoxy that concerns us.
The strength of selection on resistance is primarily determined by the fate of resistant parasites in treated and untreated hosts. Resistant strains gain an advantage in treated hosts but often pay a cost in untreated hosts. In both types of host, the social milieu of strains within individual infections plays a very important role in mediating these costs and benefits. To explain why, we need to summarize some within-host ecology.
Genetic Diversity of Infections
Human malaria infections normally consist of more than one asexually proliferating parasite lineage (“clone”). Thus, the majority of Plasmodium falciparum clones in the world share their human hosts with at least one other lineage (Read and Taylor, 2001). Mixed infections arise from inoculations of genetically diverse parasites by a single mosquito or contemporaneous bites by multiple mosquitoes infected with different parasites. Consequently, the coexistence of drug-sensitive and drug-resistant parasites is common, and indeed may even be the rule (Day et al., 1992; Arnot, 1998; Babiker et al., 1999; Smith et al., 1999; Bruce et al., 2000; Jafari et al., 2004; Juliano et al., 2007, 2010; McCollum et al., 2008; Zhong et al., 2008; Owusu-Agyei et al., 2009).
A substantial body of epidemiological evidence is consistent with crowding effects within infections, whereby the population densities of individual genotypes are suppressed when other genotypes are present (Daubersies et al., 1996; Mercereau-Puijalon, 1996; Smith et al., 1999; Bruce et al., 2000; Hastings, 2003; Talisuna et al., 2003, 2006; Färnert, 2008; Harrington et al., 2009; Orjuela-Sánchez et al., 2009; Baliraine et al., 2010). For example, parasite densities are unrelated to the number of clones per host, and high turnover rates are observed in mixed-genotype infections.
Direct experimental evidence of crowding cannot be ethically obtained from human infections because formally demonstrating competition requires deliberate infection and/or the withholding of treatment (Read and Taylor, 2001). However, in a rodent malaria model, P. chabaudi in laboratory mice, we and others have experimentally demonstrated that densities of individual clones within an infection are severely suppressed when coinfecting clones are present (Jarra and Brown, 1985; Taylor et al., 1997a,b; Taylor and Read, 1998; de Roode et al., 2003, 2004a,b, 2005a,b; Raberg et al., 2006; Wargo et al., 2007; Huijben et al., 2010; Pollitt et al., 2011). This competitive suppression substantially reduces the density of transmission stages (Wargo et al., 2007; Huijben et al., 2010), and hence transmission of individual clones to mosquitoes (Taylor et al., 1997a; Taylor and Read, 1998; de Roode et al., 2004a). To date, there is no evidence of direct interference competition analogous to bacteriocin-mediated competition in bacteria (Riley and Gordon, 1999). Instead, the competition between coinfecting malaria parasites probably arises from competition for resources. Most likely, this competition is for access to red blood cells (Hellriegel, 1992; Yap and Stevenson, 1994; Hetzel and Anderson, 1996; Haydon et al., 2003; Antia et al., 2008; Mideo et al., 2008; Kochin et al., 2010; Miller et al., 2010; Pollitt et al., 2011), although other resources, such as glucose, may also be involved (de Roode et al., 2003). Immune-mediated apparent competition, wherein the immune
response provoked by one strain suppresses the population densities of a coinfecting strain (Read and Taylor, 2001), likely also plays a major role (Mota et al., 2001; Raberg et al., 2006).
This in-host competition has profound effects on the evolution of drug resistance because it affects the fitness costs and benefits of resistance. We take these in turn.
Costs of Resistance
It is generally assumed that resistant pathogens are less fit than their wild-type ancestors in the absence of drug treatment and that this is the main force slowing the evolution of resistance. In malaria, there is good evidence of this (Hastings and Donnelly, 2005; Walliker et al., 2005; Babiker et al., 2009; World Health Organization, 2010a). One consequence of the social ecology within a host is that it acts as a serious multiplier of these costs of resistance. Costs of resistance arise from metabolic inefficiencies associated with efflux or detoxification mechanisms, which can include negative pleiotropic effects on other cellular and biochemical processes or reduced biochemical efficiencies associated with target site mutations (Hastings and Donnelly, 2005). These reductions in performance can be quite small (e.g., a few percent), but small differences can be greatly magnified by competition between clones. For example, in mice, the social context of the infection can translate modest differences in performance into differences well in excess of 90% (Fig. 11.2). The social context within which resistant strains are circulating is thus a potent determinant of the fitness costs of resistance, the main brake on the spread of drug resistance.
Benefits of Resistance
The flip side of this ecology is that the fitness advantages resistant parasites experience in treated hosts are greatly magnified in mixed-clone infections. Consider the consequences of radical pathogen cure where competition is occurring. Aggressive chemotherapy will kill all sensitive or tolerant parasites. This will result in competitive release and enhanced transmission of any highly resistant strains that are present. In rodent models, this is precisely what happens (de Roode et al., 2004a; Wargo et al., 2007; Huijben et al., 2010) (Fig. 11.3). Thus, radical parasitological cure enhances the transmission of the resistant strains. The impact of this competitive release on the rate of spread of resistance can be very substantial, as was first recognized by Hastings and colleagues (Hastings, 1997; Mackinnon and Hastings, 1998; Hastings and D’Alessandro, 2000). Where multiclone infections dominate, this within-host ecology
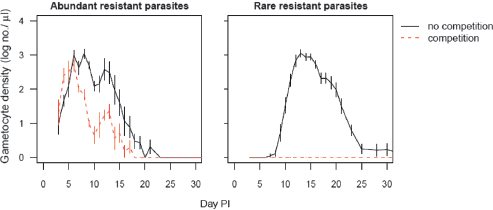
FIGURE 11.2 Costs of resistance are greatly affected by competition. Transmission stage densities of the resistant P. chabaudi clone in laboratory mice in the absence of drug treatment are shown. Infections were initiated with 106 (Left) or 101 (Right) resistant parasites and either no sensitive parasites (no competition, solid lines) or 106 sensitive parasites (competition, dashed lines). Performance of the resistant clone alone includes any physiological costs to resistance. When the resistant clone shares a host with a sensitive clone, performance is greatly reduced, and is effectively zero when rare in the inoculum (Right). Thus, the costs of resistance depend critically on whether competitors are present and the frequency of resistant parasites in an infection. PI, post-infection. Plotted points are the mean (±SEM) densities in peripheral blood from 5 to 10 mice per group, estimated by quantitative PCR using protocols described elsewhere (Huijben et al., 2010).
can be the primary determinant of the speed at which resistance spreads, and a far more important selective force than the simple survival advantage conferred by resistance (Hastings, 1997, 2003, 2006; Mackinnon and Hastings, 1998; Hastings and D’Alessandro, 2000; Mackinnon, 2005; Talisuna et al., 2006).
For instance, in an infection composed of two equally represented clones, aggressive treatment can effectively double the absolute fitness of the resistant strain if that strain can fully exploit the “infection-space” created by the removal of its competitor. If the resistant clone was rare before treatment, the effect can be substantially greater (Fig. 11.3). In nature, there is wide variation in the number of clones per person. Next-generation sequencing techniques are already discovering patients with more than 15 P. falciparum clones (Juliano et al., 2010), some of which are represented at frequencies significantly less than 1%. Were those rare clones drug-resistant, aggressive chemotherapy could increase transmission success of resistant parasites >100-fold.
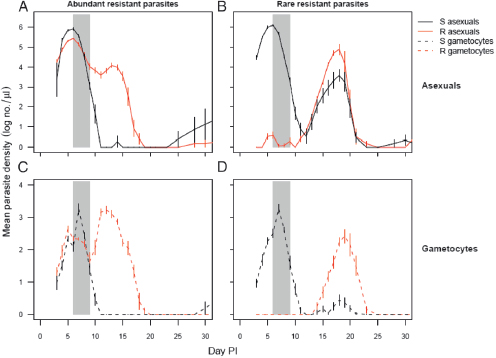
FIGURE 11.3 Competitive release of drug resistance. Infections of P. chabaudi were initiated in laboratory mice with 106 sensitive parasites (dark lines) and either 106 (A and C) or 101 (B and D) resistant parasites (gray lines). Panels A and B, densities of asexual parasites (within-host replicative stages). Panels C and D, densities of gametocytes (transmission stages). Gray bars indicate period of drug treatment (four daily doses of 8 mg/kg of pyrimethamine). R, resistant; S, sensitive; PI, post-infection. Drug treatment rapidly suppresses sensitive parasites, allowing resistant parasites to dominate post-treatment populations; the expansion following competitive release is especially marked when the resistant clone is rare. In untreated mice, resistant parasite densities are markedly lower than sensitive parasite densities throughout the infections, particularly when they were rare initially (compare with Fig. 11.2, which details the transmission stage densities of resistant parasites in the untreated mice in the same experiment). Plotted points are the mean (±SEM) densities in peripheral blood from 5 to 10 mice per group, estimated by quantitative PCR using protocols described elsewhere (Huijben et al., 2010).
Putting this slightly more formally, highly resistant parasites have a relative fitness advantage in treated hosts simply because drug treatment reduces the fitness of susceptible parasites. This advantage plays out even if all infections in a population consist of just a single clone. When hosts are infected with multiple lineages, however, the removal of competitors by drug treatment also leads to absolute fitness gains if
resistant clones are able to capitalize on the newly emptied niche space in the host. These absolute fitness gains can be very, very large when resistant parasites are otherwise kept at very low numbers by competitive suppression.
Whence Conventional Wisdom?
Thus, radical parasite cure, by rapidly eliminating sensitive competitor strains, confers very strong selection in favor of resistance. Despite this, radical parasite cure is frequently advocated as a resistance management strategy. This conventional wisdom is based on two arguments. Both have to do with managing the initial mutational inputs into the system, essentially trying to prolong the time until high-level resistance appears in the first place. The first argument is that aggressive chemotherapy maximally reduces parasite numbers, and thus the probability that resistance mutations will occur in a treated patient [e.g., White (2004) and World Health Organization (2010a, p. 129)]. This clearly has to be true.
The second argument is essentially a subtle variation of the first. The idea is that when multiple independent mutations are required to confer high-level resistance, it is essential to try to minimize positive selection in favor of any partially resistant mutant because these partially resistant mutants can be important mutational stepping stones toward full (high-level) resistance (Hastings and Watkins, 2006). Partially resistant parasites only have an evolutionary advantage at lower drug concentrations; thus, from a resistance management perspective, it is important to minimize the probability that such parasites encounter those lower concentrations. Low drug concentrations in a patient can arise in several ways, not least after a course of chemotherapy has finished and the drug is being metabolized or excreted from the body (Fig. 11.1). During some of that time, there is a period [the “selection window” (Stepniewska and White, 2008)] when parasites that are able to survive low drug doses have a selective advantage. The aim of aggressive chemotherapy is to ensure that no parasites from the treated infection remain alive during the selection window, thus reducing the number of parasites in the overall population experiencing that source of selection for low-level resistance.
DOUBLE-EDGED SWORD
Thus, aggressive chemotherapy is a double-edged sword for resistance management. It can reduce the chances of high-level resistance arising de novo in an infection. But when an infection does contain resistant parasites, either from de novo mutation or acquired by transmission from other hosts, it gives those parasites the greatest possible evolution-
ary advantage both within individual hosts and in the population as a whole. How do the opposing evolutionary pressures generated by radical cure combine in different circumstances to determine the useful life span of a drug? There will be circumstances when overwhelming chemical force retards evolution and other times when it drives things very rapidly. We contend that for no infectious disease do we have sufficient theory and empiricism to determine which outcome is more important. It seems unlikely that any general rule will apply even for a single disease, let alone across disease systems.
Consider again the case of malaria. There will be many cases where the resistance management gains of radical pathogen cure (reduced mutational inputs) will not outweigh its costs (maximal selection for high-level resistance). For instance, where high-level resistance is conferred by a single point mutation [e.g., atovaquone (White, 2004)], the mutational stepping stone argument is clearly irrelevant. Moreover, there are about 1012 parasites in an infection at the time radical cure commences (White, 2004), so that every point mutation in the genome can potentially occur in a single infection. There are at least one-quarter of a billion symptomatic cases of malaria each year (World Health Organization, 2010b), so that at least 1020 parasites could see a new drug each year. Among these 1020 parasites, it is quite plausible that there already exists at least a single parasite completely resistant to most yet-to-be invented drugs. Aggressive chemotherapy can reduce the chances of de novo resistance mutations occurring in treated patients, but it can make no impact on the probability that such mutations occurred before treatment. Aggressive use of a new drug will very effectively find these resistant “needles in the haystack.”
Even when we can be confident that mutational inputs in patients receiving treatment do limit the rate of evolutionary change (something that is extremely hard to know, especially for new drugs), there is an important quantitative argument to be had about the advantage of managing mutational inputs by aggressive chemotherapy. This is because aggressive treatment regimens increase the probability that any high-level resistance that has arisen de novo will avoid stochastic loss and reach transmissible frequencies. It is extremely challenging for a very rare resistant mutant to replicate to transmissible densities in a host [e.g., Mackinnon (2005), Pongtavornpinyo et al. (2009), and Hastings (2011a)], not least because it will likely compete with the ancestral strain from which it arose. The performance of the mutant can be especially poor if de novo resistance is associated with large fitness costs. Large costs can erode as compensatory mutations accumulate (Levin et al., 2000; zur Wiesch et al., 2011), but this requires persistence and large population sizes, both of which are countered by competition. Thus, even when
aggressive chemotherapy reduces the probability that de novo mutations occur, it can, by eliminating competitors, increase the population-wide probability that de novo mutations survive to transmit from hosts, and hence escape stochastic loss.
Moreover, there are ways to manage mutational inputs that do not have the unfortunate consequence of simultaneously maximizing selection for the very mutations they are trying to prevent. Combination therapy is an example. As WHO puts it (World Health Organization, 2010a), if resistance to one drug has a per parasite probability of 10-12 of spontaneously arising, the probability of resistance to two drugs with independent modes of action arising spontaneously in the same parasite is 10-24, a van-ishingly small probability. The duration of the selection window (Fig. 11.1) depends critically on the half-life of the particular drug. The window can be weeks long in some cases [sulfadoxine-pyrimethamine (SP)] or just a few hours in others (artemisinin and its derivatives). Judicious choice of a drug or drug combination can thus affect the likelihood of stepping stones to high-level resistance.
EVIDENCE-BASED RESISTANCE MANAGEMENT
The foregoing suggests to us that radical parasite cure is not a priori the best way to manage resistance and that it could even promote the very evolution it is intended to retard. The scientific challenge is to determine how the contrasting evolutionary consequences of aggressive chemotherapy determine the rate of resistance evolution and whether, among the vast array of possible regimens, there are other ways of treating patients that would better delay resistance.
It might be, of course, that the other aims of patient treatment (restore health and prevent infectiousness) can be achieved only by radical parasite cure (Hastings, 2011b). If radical parasite cure is indeed critical for clinical management, an empirical question, we might be stuck with evolutionary mismanagement as an unavoidable side effect. If so, it is important to recognize this. Claims that resistance evolution is retarded by aggressive treatment regimens might be obscuring a serious evolutionary problem in need of solution.
Rational development of treatment regimens that deliver effective resistance management requires a sound knowledge base (Read and Huijben, 2009; Goncalves and Paul, 2011; zur Wiesch et al., 2011), and there is considerable scope for investigating the evolutionary consequences of different treatment regimens for a wide range of diseases. Ideally, these would involve quantitative comparisons of how contrasting regimens affect each of the aims of patient treatment: health, infectiousness, and resistance management. In principle, such studies can be done
on animal models [e.g., de Roode et al. (2004a), Wargo et al. (2007), and Huijben et al. (2010)] and, in a more limited way, on humans [e.g., Harrington et al. (2009)]. It is possible to measure the evolutionary consequences of competing resistance management strategies in hospitals (Brown and Nathwani, 2005; Martínez et al., 2006; R. L. Smith et al., 2008), and it might even be possible in human communities. Penilla et al. (2007) randomly allocated 24 villages in Mexico to one of four different methods of applying public health insecticides and compared the rate of rise of resistant mosquitoes over several years. None of the putative resistance management strategies slowed the spread of phenotypic resistance. Empirical assessments of evolutionary outcomes are problematic for a drug against which resistance has yet to arise, but once high-level resistance has arisen, there is an ethical imperative to do such studies.
Mathematical models have much to offer, but the challenges are formidable even in silico. Consider malaria. As we argued above, the strength and direction of selection are critically affected by the interactions between competing pathogen lineages within a patient and how drug treatment affects this ecology. Treatment determines what is transmitted, and changes in the force of infection will, in turn, affect the genetic diversity within an infection, and hence the ecology. Such feedbacks defy standard population genetics approaches, which track gene frequencies without explicit population dynamics (Mackinnon, 2005). Evolutionary-epidemiological models [e.g., Gandon and Day (2009)] are computationally intensive, and we are unaware of any real-world context in which resistance evolution is adequately modeled. Unfortunately, the complexity of the situation does not make it go away. Quantitative predictions of the impact of different treatment regimens on the useful life of a drug have to involve this social ecology. Such modeling efforts would also evaluate the resistance management consequences of reductions in disease transmission by other measures, such as mass drug administration or transmission-blocking interventions [e.g., World Health Organization (2011)]. These too will reduce force of infection, and hence alter the in-host ecology. Reductions in force of infection might reduce the benefits of resistance by reducing the multiplicity of infection, and hence the levels of competitive release; however, as argued above, the costs of resistance will also be lower if there is less competition.
The difficulty of adequately capturing the relevant features in a mathematical model points to an important bottom line: Intuition (expert opinion), a very poor guide to evolutionary trajectories at the best of times, is really going to struggle in this context.
HOW TO TREAT PATIENTS?
A corollary of our observation that radical pathogen cure can very seriously promote the evolution of resistance is that less aggressive drug treatment could prolong the useful life span of a drug. Because even small changes in relative fitness can alter the useful therapeutical life span of a drug by decades (Hastings and Donnelly, 2005), there is a strong case for investigating the clinical consequences of lighter touch chemotherapy.
Drug treatment is often continued after patient health is restored; this is a major reason why patients fail to complete prescribed drug courses. Could there be room to harness the in-host ecology to reduce the fitness advantages of resistance, in effect retaining some drug-sensitive pathogens to suppress resistance (Wargo et al., 2007; Read and Huijben, 2009; Huijben et al., 2010)? Critically, patient health does not necessarily require immediate parasite elimination by drugs. To affect clinical recovery, the immune system often just needs to battle fewer parasites or have a longer time period over which to ramp up. It may be, for instance, that only minimal intervention with drugs is required before immunity controls and clears disease-causing pathogens. This could involve a very short course of treatment with a rapidly clearing drug (or drug combination), perhaps repeated at well-spaced intervals. Given a bit of help, immunity can deal very effectively with resistant parasites without imposing any selection for resistance (Cravo et al., 2001; Rice, 2008a,b; Taubes, 2008). Some currently heretical rules, such as “stop taking drugs when you feel better, and take them again if you get sick,” bear examination in such contexts. Critical questions are how best to combine dose and duration, how much it is necessary to have an impact on pathogen densities at first treatment, and how far apart pulses of treatment should be.
A general principle that should guide the rational development of patient treatment guidelines is to impose no more selection for resistance than is absolutely necessary. There might be cases where rules like “hit hard and hit early” (Ehrlich, 1913) or “ensure very high cure rates” (World Health Organization, 2010a) are consistent with this, but we doubt that they apply across a wide swath of diseases. For instance, de novo resistance mutants are a major threat to the health of patients infected with highly mutable pathogens like HIV. In such a case, it probably is wise to use aggressive chemotherapy to reduce pathogen biomass, and hence the probability of de novo mutations. For many diseases, however, patients are at far higher risk of acquiring resistance from other patients. In TB, for example, up to 99% of cases of drug-resistant infections are acquired from the community (Luciani et al., 2009). In these circumstances, the merits of managing de novo mutations with aggressive chemotherapy are less clear. Chloroquine became ineffective against malaria because the highly resistant progeny of a single parasite in Asia spread across
the entire African continent (Wootton et al., 2002; Talisuna et al., 2004). SP, another inexpensive and initially highly effective antimalarial, was similarly undermined by vast epidemics derived from very few genetic events (Roper et al., 2004). Those resistant parasites enjoyed maximum evolutionary advantage in patients who adhered to regimens effecting radical cure of susceptible parasites.
More broadly, resistance management strategies will probably have to be tailored to particular drug-bug combinations and epidemiological circumstances. For instance, where single-clone infections dominate (acute childhood diseases or malaria where force of infection is low), the relative fitness of resistant and sensitive strains will be quite different from situations where most infections have a high multiplicity of infection. Where there is lateral transfer of resistance genes from the environment (many bacteria), persistent subpopulations [e.g., Escherichia coli (Levin and Rozen, 2006)], or infection sites that are difficult to treat [e.g., TB (Dye, 2009)], or where treated stages are diploid [e.g., helminths (Prichard and Tait, 2001)], things could again be different. Where the social interactions between coinfecting strains differ from those we have described for malaria [e.g., West et al. (2006)], things could be different again.
It might also be that patient treatment regimens need to be modified as resistance evolution proceeds. Perhaps, for instance, aggressive chemotherapy can reduce the probability that mutations to high-level resistance will occur. If so, it could be worth moving to less aggressive regimens as soon as high-level resistance is detected in a region. Regimens involving lower doses or shorter treatments will impose weaker selection on that new resistance. Such a switch may be difficult in practice. Health messaging may require constancy, or it may be that by the time unambiguous evidence of high-level resistance has been obtained and policy changed, it is already too late.
CODA
Arguments somewhat analogous to ours have also been made for bacterial diseases (Lipsitch and Samore, 2002; Rice, 2008a,b). Aggressive chemotherapy could be particularly problematic in the case of many bacterial infections, where exhortations for patients to adhere to long-course regimens probably generate sustained selection on gut commensals to harbor resistance genes. These can be readily passed to any disease-causing bacteria that subsequently invade. Our discussion also has strong parallels with the management of Clostridium difficile in hospitals, where aggressive use of broad-spectrum antibiotics is responsible for the competitive release of the more virulent C. difficile (Vonberg et al., 2008).
An analogous situation also occurs in cancer therapy, where cell lineages within a tumor compete for access to space and nutrients. There, the argument has recently been made that less aggressive chemotherapy might sustain life better than overwhelming drug treatment, which simply removes the competitively more able susceptible cell lineages, allowing drug-resistant lineages to kill the host (Gatenby, 2009; Gatenby et al., 2009). Mouse experiments support this: Conventionally treated mice died of drug-resistant tumors, but less aggressively treated mice survived (Gatenby et al., 2009). Elsewhere, we and others have also argued that by concentrating on malaria control rather than vector control, selection for insecticide-resistant mosquitoes can be managed and even eliminated, obviating the need for an insecticide discovery pipeline (Koella et al., 2009; Read et al., 2009; Gourley et al., 2011). In all this, the key issue is to impose only the selection needed to achieve health gains and no more.
There is widespread agreement that stewardship of antimicrobials means restricting their use to only those patients who need them. We suggest that a similar default philosophy of sparing use should apply at the within-host level to patient treatment regimens. Overwhelming chemical force may at times be required, but we need to be very clear about when and why that is. Aggressive chemotherapy will, under a wide range of circumstances, spread resistance.
ACKNOWLEDGMENTS
Our arguments benefited from discussion with V. Barclay, C. Bergstrom, S. Bonhoeffer, N. Colegrave, M. Ferdig, A. Griffin, J. Hansen, J. Juliano, M. Laufer, B. Levin, J. Lloyd Smith, M. Mackinnon, S. Meshnick, N. Mideo, S. Nee, R. Paul, J. de Roode, P. Schneider, F. Taddei, and A. Wargo, not all of whom agree with our conclusions. We thank J. Antonovics, A. Bell, K. Foster, M. Greischar, S. Reece, and an anonymous referee for comments on the manuscript and members of the Research and Policy in Infectious Disease Dynamics Program of the Science and Technology Directorate, Department of Homeland Security, and the Fogarty International Center, National Institutes of Health, for stimulating discussion. Award R01GM089932 from the National Institute of General Medical Sciences supported the empirical work reported here; work under Awards R01AI089819 and U19AI089676 from the National Institute of Allergy and Infectious Diseases contributed to conceptual development. This work greatly benefited from a Fellowship to A.F.R. at the Wissenschaftskolleg zu Berlin.
















