
5
Alternative Approaches to Animal Testing for Biodefense Countermeasures
This chapter examines alternative approaches to the use of animals in the development of medical countermeasures against biothreats. It presents the concept of the Three Rs as a basis to safeguard good science while improving laboratory animal welfare. It briefly describes a number of in vitro and in vivo methods that support the Three Rs and humane endpoints. The Committee on Animal Models for Assessing Countermeasures to Bioterrorism Agents concludes that absolute replacement of animal models in countermeasure development is not possible at this time and that in vitro and in silico methods are not advanced enough yet (in part due to absence of human data) to reliably replace animals in biodefense research. Recognizing that the premise of the Animal Rule is the use of animals and that the Animal Welfare Act (AWA; 7 USC § 2131-2159) requires the consideration of alternatives, the Committee recommends to embrace and further develop alternative options to (1) take advantage of new (clinical and epidemiological) data; (2) correlate those new findings with outcomes from established animal models; (3) improve the welfare of animals used in countermeasure development and testing; and (4) strive, where possible, to replace nonhuman primates as the animal of choice in biodefense research. The Committee reiterates the fundamental need for data from human populations (versus laboratory animal species) as the crucial driver for the development of in vitro and in silico pathways. Further, the Committee concludes that changing the standard practice of animal experimentation to approximate the clinical course of treatment that humans may receive could provide a more reasonable expectation of the usefulness of certain countermeasures during development.
GENERAL PRINCIPLES OF ALTERNATIVE APPROACHES
In 1959, Russell and Burch formulated three principles to reduce the numbers of animals used in experimentation. These are known as the Three Rs: refinement, reduction, and replacement (absolute or relative).1 The validation process of regulatory testing has over the years incorporated many methods that support one or more of these principles for two main reasons: (1) only regulated and standardized animal tests that are repeatedly carried out over a long period of time (typical timeframe 12 years) and tests that have enormous costs warrant a formal validation process to be accepted as such; and (2) in the area of safety assessments, it is especially difficult to abandon an established test because safety standards could be lowered. In other areas of research and development, especially in the agent discovery phase, replacement of older with more advanced methods is more common due to constant pressure for more predictive and less costly tests.
In the case of developing countermeasures for bioterrorism agents where absolute dependence on animal models for efficacy testing serves to replace human clinical trials, the Three Rs provide a good framework to reduce animal use and minimize the animals’pain and distress.2 Because adoption of alternatives was driven by both animal welfare considerations and scientific advances in our understanding of biological phenomena, their utilization is often in the best interest of the study. Although alternatives do not compensate for the lack of clinical efficacy trials with human participants, they can enable technologies from which more information is gained than that gained by animal tests alone. In addition, they reduce the use of precious and expensive resources and reduce animal pain and distress, often resulting in improved quality of research outcomes (NCR 2008, 2009, 2011; Wolfer et al. 2004).
Applying the Principle of Refinement
The best possible treatment of animals starts with attention to husbandry (AWA; 7 USC § 2131-2159). Social housing and enriched environment within the cages, especially for the highly social nonhuman primates often used in these studies, while taking into consideration the scientific needs of the study, represent key strategies to avoid or reduce distress3 (NRC 2008). The use of analgesics and anesthetics is mandatory, not only for the alleviation of pain and distress, but also because it represents a more realistic approximation to the treatment of human patients.4,5
The development and use of protocol-appropriate humane endpoints, especially the early termination of studies at time points that indicate that animals are unlikely to recover, minimizes pain
_________________________
1 The utility and applicability of the Three Rs has been described in various documents. For additional information, see the Guide for the Care and Use of Laboratory Animals, Eighth Edition and references therein (NRC 2011), and the website for the National Centre for the Replacement, Refinement and Reduction of Animals in Research (http://www.nc3rs.org.uk/).
2 The usefulness of the Three Rs and the employment of alternatives in regulatory safety testing are not discussed here as safety testing does not fall under the auspices of the Animal Rule.
3 The ability to provide enriched environment or social housing in biocontainment facilities may be difficult, extremely limited, or impossible. Although it is a necessary husbandry and animal welfare provision, methods to enrich the environment or house social animals in a biocontainment facility have not been studied.
4 It should be noted that the quality of care for human patients is not universally identical and that analgesics and anesthetics may not be available in natural outbreak settings. Therefore, different animal research protocols may be needed for the development of treatments under these conditions.
5 Non-administration of analgesics should be scientifically justifiable and accepted by an Institutional Animal Care and Use Committee (IACUC; Animal Welfare Act; 7 USC § 2131-2159) and employed as rarely as possible.
and distress without compromising the result of the study6 (Chapter 5 of Recognition and Alleviation of Pain in Laboratory Animals (NRC 2008); Nemzek et al. 2004; NRC 2011; Olfert and Godson 2000). Conversely, insistence on death or even moribundity as an endpoint is questionable, as signs of irreversible decline are well established for all common laboratory animal species.7 It is important that early termination studies include complete necropsies and histopathological examination accompanied by appropriate agent isolation from tissues to determine if the killed animal was, in fact, unlikely to survive. Further, early endpoints ought to be verified before embarking on larger studies to ensure that studies are not needlessly repeated. At a minimum, if natural history or descriptive studies require an understanding of events proximate to death or a time-to-death estimate, then these data should be used where possible as a historical benchmark to estimate fatal outcome without needing to actually follow a full disease course in a moribund animal.
Looking at the Numbers: Reduction
Most measures to reduce the number of animals used are often justified in terms of avoiding pain or distress and of saving resources, especially in studies conducted in biocontainment facilities. Well-chosen statistical methods, such as appropriate power calculations of group numbers, should always be part of the experimental design, and knowledge of historical data about the variance of the anticipated results can help to select the appropriate sample size. Tiered testing strategies (e.g., treating individual animals or small groups sequentially and not in parallel) and use of qualified pilot studies allow for studies to be terminated early if no effect is observed in the first few animals.8 As discussed in the previous section, it may be possible to avoid the use of untreated contemporary control groups if historical data can be employed. Similarly, sharing control groups among multiple experiments performed at the same time may also lower the number of animals required. Careful selection of dosages and omission of unrealistic treatment groups further reduces animal use.
The broader use of inbred murids has helped to reduce the variability of experimental results and thus the size and number of groups required. Consequently, inbred animals are often selected for this reason, although their use does not reflect the variability of the human population (also stated in Chapter 2, Table 2-9; for an approach that embraces genetically diverse murine models, see page 69). The use of early, informative endpoints derived from complementary in vitro methods (e.g., estimation of effective or maximal tolerated doses by effective concentrations in vitro and in vitro metabolism studies with hepatocytes or microsomes to exclude the use of species that do not reflect human metabolism) can further reduce animal numbers and refine the experiments by minimizing pain and distress. Other noninvasive methods, such as imaging technologies and telemetry, that allow
_________________________
6 Biomarkers and signatures of toxicity that are often derived from nonanimal models are useful tools that make animal studies more sensitive or facilitate earlier termination (with humane endpoints) without loss of information.
7 The development of (early termination) endpoints and guidelines for when animals should be euthanized is a highly desirable animal welfare practice, and as such discussed in laboratory animal care and use regulations. Under the AWA, such provisions are part of the research protocol and subject to IACUC approval. The Committee recognizes that the above can be at odds with the 3rd criterion of the Animal Rule that “the animal study endpoint is clearly related to the desired benefit in humans, which is generally the enhancement of survival or prevention of major morbidity” (FDA 2002, p 37989). That is why pilot studies and determination of biomarkers or endpoints through small sample sizes are important. Further, careful and focused clinical observation of animals can identify entrance into an irreversible state of decline followed by immediate euthanasia.
8 Such approaches may require larger group sizes for statistical power.
monitoring of the same animal over the entire course of an experiment, instead of sacrificing more animals at different time-points, also reduce group-sizes while preserving statistical power.9
Focusing on Replacement
While opportunities for in vitro replacements are available to a greater extent in toxicology and safety assessments (e.g., NICEATM and ICCVAM test method evaluations; http://iccvam.niehs.nih.gov/methods/methods.htm), non-sentient test systems in infectious disease research generally and in studies under the Animal Rule particularly are limited. Some opportunities lie in ex vivo approaches where animals or human volunteers are treated with the product in development and only tissues or blood are subsequently exposed to pathogens or used for measurements. Tissue engineering methods, including artificial organs and organotypic cultures, can sometimes reproduce the physiological environment to study aspects of the course of infection, but extrapolation to the in vivo conditions and the systemic multifactorial components of host defense are limited. For further exploration of this topic as it pertains to studies under the Animal Rule, see section In vitro tools and replacement strategies (below).
ANIMAL EFFICACY STUDIES ARE CLINICAL TRIALS
Clinical trial designs for efficacy mandate that human subjects must be protected from undue risk when participating in clinical research activities (45 CFR 46 [2009]; National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research 1979). This protection includes the provision of clinical standard of care in addition to the product being evaluated (unless the standard of care is contraindicated). However, efficacy testing of a new drug or vaccine in animals routinely involves the administration of only the test product. Although this has been the standard practice in animal research protocols, such reliance solely on the test article to demonstrate efficacy may produce a misleading model of what the human counterpart may actually experience and lead to false or incomplete data on the effectiveness of the test product. Patients with dyspnea and acid and base imbalances due to pulmonary insufficiency, for example, would have to be provided with oxygen and intravenous electrolytes while enrolled in a trial to test the efficacy of a novel bronchodilator. Similarly, patients with congestive heart failure participating in an efficacy trial for a drug intended to increase the strength of myocardial contractions, would concomitantly be given diuretics (presuming sufficient kidney function) to minimize pulmonary congestion. In either example, although the test product could be efficacious for neutralizing or reversing the initial insult, the patient could still succumb from underlying or preexisting complications that were not treated by the test product (Miller and Silverman 2004).
The same clinical standard of supportive care certainly applies to persons exposed to a bioterrorism agent, regardless of whether they are lightly exposed and asymptomatic or suffering from organ failure. Severe dehydration and hypotension resulting from a highly infectious pathogen (e.g., acute diarrhea, septic shock, and hemorrhagic fever) would be treated with blood volume and blood pressure restoratives even though the origin of disease was microbial and the test product being evaluated was an antibiotic or antiviral drug.
The incorporation of supportive veterinary care in animal efficacy testing of countermeasures against biothreats is recognized by regulators as both reasonable and informative. In the 2009 Draft
_________________________
9 The use for in vivo imaging strategies in product development for biothreats remains largely unrealized. For these techniques to be used in the preclinical arena, validation of imaging as a correlate of bacterial or viral burden is necessary.
Guidance for Industry concept paper, the FDA described important components to be included in preclinical protocols for demonstrating efficacy under the Animal Rule. In that document, it is advised that “studies should be designed to mimic the clinical scenario and achieve meaningful outcomes comparable to the endpoints desired in humans. In some instances, supportive care should be administered to the animals as part of the study design” (FDA 2009a; see Appendix B). Not providing similar medical interventions to an animal subject when assessing preclinical efficacy may result in false (negative) conclusions about corresponding efficacy in humans. A test product could be sufficiently effective when combined with reasonable supportive care, but it could “fail” if evaluated alone. The acute need for additional biodefense medical countermeasures is not served when candidate drugs and vaccines are abandoned that may, indeed, prove efficacious when tested in a comprehensive medical fashion.
Employing an expanded spectrum of clinical care rather than relying on a single test product for efficacy testing has other advantages beyond not prematurely discarding promising drugs and vaccines. By more closely mimicking the broader scope of clinical care provided to patients, one may identify which specific ancillary care regimens, if any, contribute most to the efficacy of the test product. This information might lead to a critical component for the final label of the approved drug. In addition, longer survival of animal subjects due to an expanded repertoire of clinical support could result in better predictive models. If animals die too quickly,10 the pharmacokinetics and drug metabolism of the test product or the absence of effects of increased time on an effective immune response may not replicate or approximate the expected timelines in patients, resulting in misleading findings.11 Furthermore, subtle yet important differences in test products or dosages may become evident over a longer time frame of therapy due to prolonged animal survival. Objections to including an expanded array of clinical care for animal subjects in efficacy testing protocols usually involve one of two themes. First, anything administered to the animal besides the test product could interfere with the “true” efficacy properties of the product in question, possibly leading to (false) positive conclusions when the test product is not otherwise strong enough as a therapeutic or preventative agent. A counterargument to that objection is that efficacy should address only the specific effects of that pathogen or toxin; if nonspecific complications represent the actual disease, then one should focus on efficacy testing specific to those sequelae.12
The second objection is that supportive-care components are not compatible with 21 CFR Part 58 (Good Laboratory Practice for Nonclinical Laboratory Studies; GLP) because the components may introduce high levels of variation that cannot withstand a quality-assurance audit of that study, or they may create many expensive complexities. If, however, quantitative thresholds are established for anticipated clinical signs (e.g., fever) and standardized supportive interventions in advance, such an
_________________________
10 Lethality in animals may be due to secondary causes, such as severe dehydration or hypothermia as a consequence of being too sick to eat, drink, or move around rather than specific or primary effects of the disease or the product tested.
11 Metabolism and thus pharmacokinetics can differ between humans and animals (Martignoni et al. 2006). The difference is relevant not only for extrapolations to human kinetics but also for its impact on drug efficacy studies, i.e., where, when, and for how long are effective tissue concentrations reached. Furthermore, drug pharmacokinetics may change under disease conditions and may be affected by the severity of disease.
12 As discussed in chapter 3, the application for licensure of a human monoclonal antibody against inhalational anthrax by Human Genome Sciences is on hold pending additional studies. One of the concluding remarks of the FDA-appointed committee of experts was that “there was no study with the antibiotic as the control arm” (FDA 2009b). While standard clinical care against inhalational anthrax is primarily the administration of antibiotics, the implication is that it should be a necessary component of the animal efficacy trial, both as a separate arm of the trial and as a combination treatment with the test product. The same trial design should apply when the standard of care in humans is supportive therapy alone.
expanded study design can be carried out in keeping with GLP principles. An example of this approach would be to combine body-temperature monitoring via a standard operating procedure in anticipation of fever as a clinical sign and initiation of antipyretic therapy in accordance with that standard operating procedure when the body temperature rises above a predetermined threshold as documented in the veterinary literature.
The Committee recognizes that the above is a different approach to using animals and generating data from animal trials. It is meant to be more comprehensive and apply considerations from human clinical trials (or treatment in the field) to animal studies in order to make more meaningful contributions to the interpretation of data as might be applied to humans. This strategy, i.e., provision of supportive care to animals subjected to severe disease, is not only more humane but may allow fewer animal numbers to be used in accordance with the principles of the Three Rs, as these are among the common causes of reduced validity of animal studies presented in Chapter 2, Table 2-9. Even though these actions can be performed in multiple grades of moderation without converting the laboratory into an intensive care unit, the practical difficulties of establishing this methodology in biocontainment facilities indicate the need for careful deliberation and study of the basic principles of such an approach and the creation of guidelines for the care and use of animals in research done under biocontainment conditions. The safety requirements of working in a biocontainment environment and the potential increased costs of implementing such types of animal trials are of considerable importance, especially for long-term studies.
IN VITRO TOOLS AND REPLACEMENT STRATEGIES
In the United States, a discussion of the future of the field of toxicology was prompted by the National Research Council report Toxicity Testing in the 21st Century: A Vision and A Strategy (NRC 2007). In 2008, several U.S. agencies, including the FDA, announced a coalition to facilitate this reports’ implementation: “We propose a shift from primarily in vivo animal studies to in vitro assays, in vivo assays with lower organisms, and computational modeling for toxicity assessments” (Collins et al. 2008, p 906).
In Vitro and In Silico Methods
Biothreat agents are prime candidates for accelerated development and regulatory clearance of countermeasures by using animal models as alternatives to human clinical trials. This acceleration suggests the need to develop new and innovative strategies for collecting data and observations about how humans respond to these pathogens. Without this information the relevance of the animal models cannot be adequately ascertained. The same need exists for information obtained from the animal models to help develop and interpret new in vitro and computational in silico (IV/IS) methods. Advances in molecular characterization and in computational power have made it possible to consider approaches that do not even require the use of living systems, or at a minimum, accelerate the capacity of these artificial systems.
Many of the elements for a fully integrated IV/IS product development and approval strategy exist today, especially for anti-infectives against known agents that can be cultured in vitro. Standard techniques include high-throughput screening for drug discovery, in vitro testing of antimicrobial efficacy and drug resistance in many bacterial and viral pathogens, testing of some aspects of toxicity and pharmacokinetics and pharmacodynamics and computer modeling of structure-activity relationships. While in vitro assays for preclinical toxicity testing have been used extensively for several decades (reviewed in Judson et al. 2010), reliable assays for systemic toxicities, although improving,
remain a challenge (Adler et al. 2011; Hartung et al. 2011). Similarly, advances in computational capacity allow for increasingly complex modeling, but the animal models remain a critical bridge to test and confirm biosignatures (biomarkers) and other effects identified through studies of the natural history of the disease, relevant human clinical trials, or other animal model work. These biosignatures and pathways so identified and tested could then be a bridge to IV/IS models. However, despite technological advances the absence of suitable high-quality comparative data impedes the realization of this process (see previous discussion in Chapters 2 and 4 on the importance of human data).
The use of biomarkers in drug discovery and development is a related nascent area with great promise (pharmacogenomics; Hamburg and Collins 2010).13 Suitable biomarkers, such as gene polymorphisms or gene expression profiles, can be determined in vivo (in animal models, natural infections, or clinical trials), and be used to predict whether a new candidate therapeutic is likely to be effective or, based on markers associated with adverse events, which individuals might be at increased risk for adverse reactions. This approach of moving from in vivo identification to in vitro testing of biomarkers has been used in the last few years for several biodefense-related agents, including monkeypox and anthrax. Time course studies of biomarker expression (i.e., gene expression arrays) following experimental infection of animals suggest that it may be possible (at least in some situations) to determine how long the patient has been infected, and whether optimal treatment varies depending on time after infection (Alkhalil et al. 2010; Das et al. 2008).
The immune system is a prime example of complex (and still incompletely understood) interactions of multiple cell types not yet amenable to IV/IS testing or modeling. An “artificial” functional in vitro immune system could facilitate the identification of candidate vaccines and therapeutics for immunosuppression or immune enhancement and could also help eliminate candidate therapeutics with undesirable immunologic properties. Although this work is still in the nascent stages, progress has been reported (Gaucher et al. 2008; Schanen and Drake 2008). In theory, this system, which depends on cell migration and maturation, will mimic what occurs in vivo and its output will more reliably reflect anticipated outcomes. Once created, this engineered tissue system can be manipulated and dynamic endpoints determined. For example, the Modular Immune In Vitro Constructs (MIMIC®) System, a simulated human immune system, enables testing of the adaptive immune response to vaccine antigens directly on microtiter plates and can provide multiple replicates of immune system activation and response from a single individual to different antigens ex vivo (Higbee et al. 2009). In recent years, a number of cell assemblages have been “engineered” in vitro to functionally mimic corresponding organs in the body (Fernandez and Khademhosseini 2010; Ingber et al. 2006; Mammoto and Ingber 2010). For instance, the development of micropatterned cocultures of human hepatocytes and supportive stromal cells permitted the growth of Hepatitis C virus (HCV) in vitro for the first time and, serving as a high-throughput platform, could allow rapid in vitro screening of candidate anti-HCV therapeutics for both efficacy and toxicity (Ploss et al. 2010). However, for this technology to assist in the development of countermeasures, the system must demonstrate that it accurately reflects human infections by using pathogens for which large amounts of (preferably) human in vivo data exist to test its reliability. Until such data are available, the system may be used to explore differences between multiple species (including humans) to further refine animal models or point to more accurate in silico representations of the human system.
_________________________
13 The term “pharmacoepigenetics” is sometimes used when gene expression, rather than DNA gene sequence variants, is the biomarker (Baer-Dubowska et al. 2011).
Computational Modeling
At present, computational approaches are most useful in the basic science phase of drug and vaccine development, specifically in identifying targets and biomarkers. These approaches are often helpful in the selection of the appropriate animal model because they allow the pathology and response to an agent to be defined in great detail. In this context, the computational approaches reduce animal testing by focusing future studies on the most promising leads and potentially by identifying biomarkers to develop humane endpoints for follow-up studies.
In their current form, IV/IS tools and strategies cannot serve as complete replacements for animal models. For complete replacements to be possible, it will be necessary to further define the functional and regulatory networks within the mammalian host and develop modeling approaches that allow prediction of how those networks (and ultimately the host’s physiology) will behave when perturbed by infection or toxin. For infectious diseases, interactions of significance comprise pathogen and host responses, including the role of specific and nonspecific immune responses. The development of the first protease inhibitor against HIV-1 was a dramatic advance about two decades ago, and is probably the best known example of a new therapeutic developed by computational methods from structural information alone, but the hope is for many more examples in the future as both structural biology and computational expertise advance (Miller et al. 1989; for a general review on computer-aided drug design see Talele et al. 2010).
More recently, computational modeling has played an important role in the development of vaccines against influenza. For instance, the identification of highly conserved epitope sequences of the influenza virus elicited broadly reactive neutralizing antibodies that are currently pursued as potential “universal” influenza vaccines (Ekiert et al. 2010; Fleishman et al. 2011; Kang et al. 2011; Toussaint et al. 2011; Wang et al. 2010). Further, the identification of preexisting, cross-reacting epitopes against H1N1 viruses on human T-cells were used to test candidate vaccines against not only influenza viruses but other pathogens as well (Schanen et al. 2011).
In Vivo Tools to Improve Efficacy Testing
Surrogate animal models: smallpox
Orthopoxviruses are large DNA viruses that can infect a variety of vertebrate animals. Interestingly, a strong tropism effect is observed among members of the family Orthopoxviridae; thus, in most cases, a given orthopoxvirus infects only one host. Smallpox, caused by the variola virus, is an extremely virulent respiratory infection observed only in humans. Monkeypoxvirus infects a number of animal species, one of which is nonhuman primates. One of the primary virulence strategies observed during both of these infections is the generation of a large variety of viral immunomodulator proteins that prevent the host from mounting a protective immune response (Smith 1999).
Today smallpox is eradicated and no new infections of humans occur anywhere in the world (WHO 2011). Although a large amount of clinical information, including autopsy data, is available from past epidemics, the available scientific methods of the times did not allow for evaluation of the host response; thus comparison of mechanistic data with information obtained from current animal models is limited. Because of the stringent tropism effects, it is very difficult to infect animals with variola virus. One alternative method to overcome this hurdle is to create a surrogate animal model in which to establish, through the use of a different orthopox virus, similar pathophysiology and clinical disease to that observed in humans with smallpox. Jahrling and colleagues (2004) developed a non-human primate model of variola through the introduction of high doses (108 plaque-forming units; PFU) of virus intravenously and effectively bypassing the initial oropharyngeal site of virus replication. Due to the very limited and tightly controlled nature of variola virus research permitted by the World Health
Organization, further refinements of this model are difficult and expensive to achieve, although efforts are still ongoing to improve that model. To that effect, Hooper and colleagues (2004) proposed that a nonhuman primate model of monkeypox given infective doses that closely represent the typical exposure to this virus, would be more relevant to and accurately present smallpox than the use of nonphysiological doses and routes of infection developed with variola virus. One reason for the potential effectiveness of this strategy is the ability of the orthopoxviruses to produce strong, cross-reactive immune responses in animals of different species. An additional benefit of this strategy may be the relevance to “newer” types of human orthopoxvirus diseases, such as human monkeypox, which have emerged as serious public health burdens in places where endemic smallpox was observed in the past (Rimoin et al. 2010). Animals of several species can serve as natural reservoirs for the monkeypox virus (Khodakevich et al. 1986, 1987a, 1987b); in this case, using surrogate animal models susceptible to monkeypox virus would be a more natural approach than the persistent use of variola virus on organisms with no natural affinity for this agent. Moreover, because the tropism effect is likely to occur with other pathogens, the monkeypox strategy may become a paradigm with future use as well (McFadden 2005).
Systems approaches to infectious diseases
Virtually all human diseases are a manifestation of interactions among many inherited polymorphic genes and environmental factors (Churchill et al. 2004; Cookson et al. 2009; Kotb 2010; Kotb et al. 2008; Thompson 1995; Villar et al. 2004; Voit et al. 2008; Williams 2006). Traditional reductionist approaches to develop disease models based on gene-by-gene comparisons or extrapolations have been universally applicable. Broader systems approaches may be useful in this regard because they can reveal how disease variables influence one another within a whole organism; provide a roadmap to expedite the discovery of networks of pathways that modulate disease susceptibility and outcomes; and reveal those networks likely to be good candidates for the development of more targeted rapid diagnostics and effective therapeutics.
Although animals with limited genetic diversity have several advantages (see above), translating findings from these animals to humans is not always useful. Whereas nonhuman primates offer sufficient genetic variation for the implementation of a systems perspective (Sasaki et al. 2009; Wolfe et al. 1998), the number of replicate studies needed to generate these data is limited by ethical considerations, inadequate stocks, and prohibitive cost. Inbred rodents, although useful for generating the quantity of data needed for systems evaluation, are characterized by little genetic heterogeneity. To address these challenges, novel animal models have been developed from which discoveries, made with a systems genetics or biological approach, are likely to translate to humans more readily. For example, recombinant inbred mice (Advanced, or the next generation Collaborative Cross strains) are generated and bred to maximize the number of recombinations in each of their chromosomes thereby diversifying their genetic context and exposing a wider spectrum of disease phenotypes (Durrant et al. 2011; Kotb 2010; Williams et al. 2001).
When infected, these strains exhibit a wide spectrum of disease phenotypes because, as is the case in humans, random assortment of many polymorphic loci can accentuate resistance or susceptibility to a particular disease. Accordingly, findings in these genetically diverse populations can significantly enhance the translation of experimental research findings to the clinical setting to prevent or improve the management of complex infectious diseases. Network-based systems approaches and pathway-to-pathway comparisons between species are now more likely to expedite the discovery of targets and networks and the translation of research across species than gene-by-gene comparisons (for other comparative biological approaches see discussion on compartmentalization, Chapter 4, p 56).
PROMISES AND CHALLENGES FOR THE FUTURE
FDA Commissioner Margaret Hamburg recently wrote “We must bring 21st century approaches to 21st century products and problems” (Hamburg 2011). This is a time of rapid and unprecedented development of enabling biotechnologies that hold great promise for the future, but it also presents several serious challenges. Despite the diversity of currently available approaches and promising technologies, no approaches can at this time fully address the shortfalls of using animal models as complete surrogates for humans. The Committee notes the following concerns:
- As stated in Chapters 2 and 4, there is a need to develop new and innovative strategies for collecting data about how humans will respond to pathogens of concern. Without this information, there can be little useful comparison to animal models (or qualification thereof, see Chapter 4), the effectiveness and predictability of biomarkers is curtailed, and the animal data to be used for the development and interpretation of meaningful IV/IS methods will not be accurate. Further, original data (positive or negative; human and animal) may not be systematically shared with the wider research community (as also discussed in Chapter 3, p 44). The lack of sharing causes the fragmentation of knowledge and prevents the comparison of inputs and outcomes,14 which may be particularly important in the event of an “unknown-unknown” emergency. Therefore, this information should be collected systematically, consistently, and accurately and be made available to the research community to enable progress toward standardization of methods and qualification of models, and to address ethical concerns regarding the potential nonproductive or duplicative use of animals or the unnecessary duplication of studies and waste of resources.
- The provision of supportive veterinary care during animal efficacy trials for countermeasures is a means to improve data gathering from animal models to enhance the efficiency and productivity of this research field. In the Draft Guidance for Industry the FDA states that “studies should be designed to mimic the clinical scenario and achieve meaningful outcomes comparable to the endpoints desired in humans. In some instances, supportive care should be administered to the animals as part of the study design” (FDA 2009). The Animal Rule does not require that a test product exhibit added benefit over conventional therapy (“…the drug product is reasonably likely to produce clinical benefit in humans.”; FDA 2002, p 37995), but if conventional therapy is beneficial for human patients then it is a reasonable measure to include in the study design. Furthermore, studies that include provision of the standard of care as one arm were suggested at the public meeting to evaluate the licensure application of raxibacumab under the Animal Rule (FDA 2009b). Since for most countermeasures in development there is no other standard of care than supportive therapy, it is appropriate to include it when evaluating the test products. Experience with such study designs and experimental protocols may be helpful in the event of an efficacy trial for a countermeasure against an “unknown-unknown”. Due to the nature of biocontainment, defining the basic principles of such an approach —including guidelines for the care and use of animals in research done in biocontainment facilities— is recommended.
_________________________
14 In addition to data sharing being one of the principle tenets of responsible conduct of research (see the Office of Research Integrity’s Introduction to the Responsible Conduct of Research, http://ori.hhs.gov/education/products/RCRintro/), it also is a fundamental tool of “the economy of knowledge production” (Nat Genet 2011).
- The Public Health Emergency Medical Countermeasures Enterprise (PHEMCE) Review emphasized the primary role for regulatory science15 in biodefense research (DHHS 2010). As shown in Figure 5-1, a window of opportunity may exist in which regulatory science can help to overcome the limited use of advanced in vitro and in vivo technologies in the development of medical countermeasures. It is desirable to develop criteria for choosing the most suitable methods, and essential to do this in a way that will allow effective utilization of IV/IS technologies while not inhibiting advances. Steps in the product development process have a clear potential use for IV/IS as an adjunct method but the use of the whole animal will not be replaced in the process. A research strategy to address these gaps would be useful as well as improve areas in which in vitro assays are already showing promise. A place to begin would be an analysis of the discovery, development, and approval process for medical countermeasures to identify (1) where the most important scientific gaps exist in terms of utilizing alternative methods to animal models and how to address them; (2) the specific areas where the use of in vitro and in silico methods could be sufficient or as an adjunct to the use of animals; and (3) the criteria for choosing and utilizing the most suitable technologies to replace animal use in biodefense research.
- Regulations that require humane treatment of animals in research (such as the AWA as discussed above) do not impose principled limits on the use of animals, i.e., pain and distress caused by the research protocols are to be minimized only when and to the extent consistent with the needs of science (Walker and King 2011). However, the needs of science in this research field should be weighed against the potential advances in knowledge and benefits to the warfighters as well as against the duration and severity of animal pain and distress. In previous sections, the report outlines the need for the development of humane endpoints and biomarkers, for the administration of supportive clinical care, and for the alleviation of pain and distress. Medical countermeasures research and development for biodefense currently depends on the continued use of nonhuman primates, as discussed in chapter 2, and will probably remain so until such time that robust alternatives (either absolute or relative) to their use are available.16 However, the report’s conclusions and recommendations could help reduce a key tension in animal research, namely that the animals that most resemble humans are simultaneously viewed as most necessary for research that is impermissible in humans and as having greater moral value because they resemble humans. The recommended comprehensive strategy of implementing the Three Rs, utilizing compartmentalization and systems biology, and enhancing collection and analysis of human data reduces dependency on nonhuman primates by maximizing the value of data derived from all research.17The Committee recommends that, where possible, the TMT should encourage efforts to replace nonhuman primates as the animal of choice in biodefense research. In addition, unhindered access to data (as discussed above) and publishing of all results —including negative ones— are critical steps to ensure that this data is indeed useful, animals are used judiciously, and unnecessary duplication of work is avoided (Bateson 2011).
_________________________
15 “The development and use of new tools, standards, and approaches to more efficiently develop products and to more effectively evaluate product safety, efficacy, and quality” (FDA 2010).
16 The authors of the recent Review of Research Using Non-Human Primates “agreed that in many cases the use of NHPs was justifiable even in the context of current understanding of animal welfare and advances in knowledge that might now render some work on living animals unnecessary” (Bateson 2011, p 1).
17 To cite from the Review of Research Using Non-Human Primates, “it is an ethical imperative that maximum benefit be derived from studies employing NHPs” (Bateson 2011, p 3).
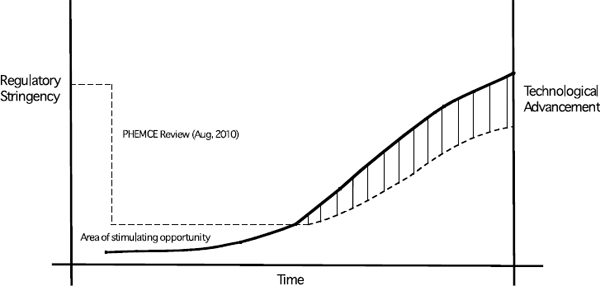
FIGURE 5-1 Regulatory science proceeds as a function of regulatory stringency and technological advancement. Whereas stringency is necessary to safeguard the safety and efficacy of products, it can be better achieved as newer technologies and reliable models provide a better approximation of the human system (or a relevant component of the human system). Greater innovation or investment in many of the suggested approaches above may be achieved by adjusting the real or perceived stringency of the current regulatory framework. As technologies, models or approaches are discovered that provide better fidelity with a human system (or the relevant component of the human system), then standardization may be achieved and stringency increased based on demonstration of the model’s reliability.
Adler S, Basketter D, Creton S, Pelkonen O, van Benthem J, Zuang V, Andersen KE, Angers-Loustau A, Aptula A, Bal-Price A., Benfenati E, Bernauer U, Bessems Jos, Bois FY, Boobis A, Brandon E, Bremer S, Broschard T, Casati S, Coecke S, Corvi R, Cronin M, Daston G, Dekant W, Felter S, Grignard E, Gundert-Remy U, Heinonen T, Kimber I, Kleinjans J, Komulainen H, Kreiling R, Kreysa J, Batista Leite S, Loizou G, Maxwell G, Mazzatorta P, Munn S, Pfuhler S, Phrakonkham P, Piersma A, Poth A, Prieto P, Repetto G, Rogiers V, Schoeters G, Schwarz M, Serafimova R, Tahti H, Testai E, van Delft J, van Loveren H, Vinken M, Worth A, Zaldivar JM. 2011. Alternative (non-animal) methods for cosmetics testing: current status and future prospects - 2010. Arch Toxicol 85(5):367–485.
Alkhalil A, Hammamieh R, Hardick J, Ichou MA, Jett M, Ibrahim S. 2010. Gene expression profiling of monkeypox virus-infected cells reveals novel interfaces for host-virus interactions. Virol J 7:173.
Baer-DubowskaW, Majchrzak-Celińska A, Cichocki M. 2011. Pharmacoepigenetics: A new approach to predicting individual drug responses and targeting new drugs. Pharmacol Rep 63(2):293-304.
Bateson P. 2011. Review of Research Using Non-Human Primates. Available online (http://www.mrc.ac.uk/Utilities/Documentrecord/index.htm?d=MRC008083), accessed September 2011.
Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, Bleich A, Bogue M, Broman KW, Buck KJ, Buckler E, Burmeister M, Chesler EJ, Cheverud JM, Clapcote S, Cook MN, Cox RD, Crabbe JC, Crusio WE, Darvasi A, Deschepper CF, Doerge RW, Farber CR, Forejt J, Gaile D, Garlow SJ, Geiger H, Gershenfeld H, Gordon T, Gu J, Gu W, de Haan G, Hayes NL, Heller C, Himmelbauer H, Hitzemann R, Hunter K, Hsu HC, Iraqi FA, Ivandic B, Jacob HJ, Jansen RC, Jepsen KJ, Johnson DK, Johnson TE, Kempermann G, Kendziorski C, Kotb M, Kooy RF, Liamas B, Lammert F, Lassalle JM, Lowenstein PR, Lu L, Lusis A, Manly KF, Marcucio R, Matthews D, Medrano JF, Miller DR, Mittleman G, Mock BA, Mogil JS, Montagutelli X, Morahan G, Morris DG, Mott R, Nadeau JH, Nagase H, Nowakowski RS, O’Hara BF, Osadchuk AV, Page GP, Paigen B, Paigen K, Palmer AA, Pan HJ, Peltonen-Palotie L, Peirce J, Pomp D, Pravenec M, Prows DR, Qi Z, Reeves RH, Roder J, Rosen GD, Schadt EE, Schalkwyk LC, Seltzer Z, Shimomura K, Shou S, Sillanpää MJ, Siracusa LD, Snoeck HW, Spearow JL,
Svenson K, Tarantino LM, Threadgill D, Toth LA, Valdar W, de Villena FP, Warden C, Whatley S, Williams RW, Wiltshire T, Yi N, Zhang D, Zhang M, Zou F; Complex Trait Consortium. 2004. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36(11):1133-1137.
Collins FS, Gray GM, Bucher JR. 2008. Toxicology: Transforming environmental health protection. Science 319(5865):906-907.
Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. 2009. Mapping complex disease traits with global gene expression. Nat Rev Genet 10(3):184-194.
Das R, Hammamieh R, Neill R, Ludwig GV, Eker S, Lincoln P, Ramamoorthy P, Dhokalia A, Mani S, Mendis C, Cummings C, Kearney B, Royaee A, Huang XZ, Paranavitana C, Smith L, Peel S, Kanesa-Thasan N, Hoover D, Lindler LE, Yang D, Henchal E, Jett M. 2008. Early indicators of exposure to biological threat agents using host gene profiles in peripheral blood mononuclear cells. BMC Infect Dis 8:104.
DHHS [U.S. Department of Health and Human Services]. 2010. The Public Health Emergency Medical Countermeasures Enterprise Review: Transforming the Enterprise to Meet Long-Range National Needs. Assistant Secretary for Preparedness and Response, U.S. Department of Health and Human Services. Available online (https://www.medicalcountermeasures.gov/documents/MCMReviewFinalcover508.pdf), accessed May 2011.
Durant C, Tayem H, Yalcin B, Cleak J, Goodstadt L, Pardo-Manuel de Villena P, Mott R, Iraqi FA. 2011. Collaborative Cross mice and the power to map host susceptibility to Aspergillus fumigatus infection. Genome Res 21(8):1239-1248.
Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. 2011. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333(6044):843-850.
FDA [Food and Drug Administration]. 2002. New Drug and Biological Drug Products; Evidence Needed to Demonstrate Effectiveness of New Drugs When Human Efficacy Studies Are Not Ethical or Feasible. Fed Regist 67(105):37988-37998.
FDA. 2009a. Guidance for Industry. Animal Models – Essential Elements to Address Efficacy Under the Animal Rule. Available online (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078923.pdf), accessed May 2011.
FDA. 2009b. Summary Minutes of the Anti-infective Drugs Advisory Committee Meeting, October 27, 2009. Center for Drug Evaluation and Research, Food and Drug Administration. Available online (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AntiInfectiveDrugsAdvisoryCommittee/UCM196436.pdf), accessed September 23, 2011.
FDA. 2010. NIH and FDA Announce Collaborative Initiative to Fast-Track Innovations to the Public. FDA-NIH News Release: February 24, 2010. Available online (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm201706.htm), accessed June 2011.
Fernandez JG, Khademhosseini A. 2010. Micro-masonry: Construction of 3D structures by microscale self-assembly. Adv Mater 22(23):2538-2541.
Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. 2011. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332(6031):816-821.
Gaucher D., Therrien R, Kettaf N, Angermann BR, Boucher G, Filali-Mouhim A, Moser JM, Mehta RS, Drake DR, III, Castro E, Akondy R, Rinfret A, Yassine-Diab B, Said EA, Chouikh Y, Cameron MJ, Clum R, Kelvin D, Somogyi R, Greller LD, Balderas RS, Wilkinson P, Pantaleo G, Tartaglia J, Haddad EK, Sékaly RP. 2008. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med 205(13):3119-3131.
Hamburg MA. 2011. Advancing regulatory science. Science 331(6020):987.
Hamburg MA, Collins FS. 2010. The path to personalized medicine. N Engl J Med 363:301-304. [Erratum, N Engl J Med 2010; 363:1092.]
Hartung T, Blaauboer BJ, Bosgra S, Carney E, Coenen J, Conolly RB, Corsini E, Green S, Faustman EM, Gaspari A, Hayashi M, Wallace Hayes A, Hengstler JG, Knudsen LE, Knudsen TB, McKim JM, Pfaller W and Roggen EL. 2011. An expert consortium review of the EC-commissioned report “Alternative (Non-Animal) Methods for Cosmetics Testing: Current Status and Future Prospects – 2010”. ALTEX 28(3):183-209.
Higbee RG, Byers AM, Dhir V, Drake D, Fahlenkamp HG, Gangur J, Kachurin A, Kachurina O, Leistritz D, Ma Y, Mehta R, Mishkin E, Moser J, Mosquera L, Nguyen M, Parkhill R, Pawar S, Poisson L, Sanchez-Schmitz G, Schanen B, Singh I, Song H, Tapia T, Warren W, Wittman V. 2009. An immunologic model for rapid vaccine assessment –a clinical trial in a test tube. Altern Lab Anim 37(suppl 1):19-27.
Hooper JW, Thompson E, Wilhelmsen C, Zimmerman M, Ichou MA, Steffen SE, Schmaljohn CS, Schmaljohn AL, Jahrling PB. 2004. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J Virol 78(9):4433-4443.
Ingber DE, Mow VC, Butler D, Niklason L, Huard J, Mao J, Yannas I, Kaplan D, Vunjak-Novakovic G. 2006. Tissue engineering and developmental biology: Going biomimetic. Tissue Eng 12(12):3265-3268.
Jahrling PB, Hensley LE, Martinez MJ, LeDuc JW, Rubins KH, Relman DA, Huggins JW. 2004. Exploring the potential of variola virus infection of cynomologus macaques as a model for human smallpox. Proc Natl Acad Sci USA 101(42):15196-15200.
Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, Reif DM, Rotroff DM, Shah I, Richard AM, Dix DJ. 2010. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast project. Environ Health Perspect 118(4):485-492.
Kang SM, Song JM, Compans RW. 2011. Novel vaccines against influenza viruses. Virus Res. Oct 1. [Epub ahead of print] doi:10.1016/j.virusres.2011.09.037.
Khodakevich L, Jezek Z, Kinzanzka K. 1986. Isolation of monkeypox virus from wild squirrel infected in nature. Lancet 1(8472):98–99.
Khodakevich L, Szczeniowski M, Manbu-ma-Disu, Jezek Z, Marennikova S, Nakano J, Messinger D. 1987a. The role of squirrels in sustaining monkeypox virus transmission. Trop Geogr Med 39(2):115–122.
Khodakevich L, Szczeniowski M, Manbu-ma-Disu, Jezek Z, Marennikova S, Nakano J, Meier F. 1987b. Monkeypox virus in relation to the ecological features surrounding human settlements in Bumba zone, Zaire. Trop Geogr Med 39(1):56–63.
Kotb M. 2010. Septomics: Application of systems approaches to sepsis - potential for personalized management of septic patients. Curr Pharmacogenomics Person Med 8(2):97-107.
Kotb M, Fathey N, Aziz R, Rowe S, Williams RW, Lu L. 2008. Unbiased forward genetics and systems biology approaches to understanding how gene-environment interactions work to predict susceptibility and outcomes of infections. Novartis Found Symp 293:156-65; discussion 165-167, 181-183.
Mammoto T, Ingber DE. 2010. Mechanical control of tissue and organ development. Development 137(9):1407-1420.
Martignioni M, Groothuis GM, de Kanter R. 2006. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2(6):875-894.
McFadden G. 2005. Poxvirus tropism. Nat Rev Microbiol 3(3): 201-213.
Miller FG, Silverman HJ. 2004. The ethical relevance of the standard of care in the design of clinical trials. Am J Respir Crit Care Med 169(5):562-564.
Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB, Wlodawer A. 1989. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution. Science 246(4934):1149-1152.
National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. 1979. The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research. Available online (http://ohsr.od.nih.gov/guidelines/belmont.html), accessed May 2011.
Nature Genetics. 2011. No second thoughts about data access. 43(5):389.
Nemzek JA, Xiao H-Y, Minard AE, Bolgos GL, Remick DG. 2004. Humane endpoints in shock research. Shock 21(1):17-25.
NRC [National Research Council]. 2007. Toxicity Testing in the 21st Century: A Vision and A Strategy. Washington, DC: National Academies Press.
NRC. 2008. Recognition and Alleviation of Distress in Laboratory Animals. Washington, DC: National Academies Press.
NRC. 2009. Recognition and Alleviation of Pain in Laboratory Animals. Washington, DC: National Academies Press.
NRC. 2011. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academies Press.
Olfert ED, Godson DL. 2000. Humane endpoints for infectious disease animal models. ILAR J 41(2):99-104.
Ploss A, Khetani SR, Jones CT, Syder AJ, Trehan K, Gaysinskaya VA, Mu K, Ritola K, Rice CM, Bhatia SN. 2010. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc Natl Acad Sci USA 107(7):3141-3145.
Rimoin AW, Mulembakani PM, Johnston SC, Lloyd Smith JO, Kisalu NK, Kinkela TL, Blumberg S, Thomassen HA, Pike BL, Fair JN, Wolfe ND, Shongo RL, Graham BS, Formenty P, Okitokolonda E, Hensley LE, Meyer H, Wright LL, Muyembe J-J. 2010. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. Proc Nat Acad Sci USA 107(37):16262-16267.
Russell WMS, Burch RL. 1959. The Principles of Humane Experimental Technique. London: Methuen. Available online (http://altweb.jhsph.edu/pubs/books/humane_exp/het-toc), accessed March 2011.
Sasaki E, Suemizu H, Shimada A, Hanazawa K, Oiwa R, Kamioka M, Tomioka I, Sotomaru Y, Hirakawa R, Eto T, Shiozawa S, Maeda T, Ito M, Ito R, Kito C, Yagihashi C, Kawai K, Miyoshi H, Tanioka Y, Tamaoki N, Habu S, Okano H, Nomura T. 2009. Generation of transgenic non-human primates with germline transmission. Nature. 459 (7246):523-527.
Schanen BC, De Groot AS, Moise L, Ardito M, McClaine E, Martin W, Wittman V, Warren WL, Drake DR, III. 2011. Coupling sensitive in vitro and in silico techniques to assess cross-reactive CD4(+) T cells against the swine-origin H1N1 influenza virus. Vaccine 29(17):3299-3309.
Schanen BC, Drake DR, III. 2008. A novel approach for the generation of human dendritic cells from blood monocytes in the absence of exogenous factors. J Immunol Methods 335(1):53-64.
Smith GL. 1999. Vaccinia virus immune evasion. Immunol Lett 65(1-2):55-62.
Talele TT, Khedkar SA, Rigby AC. 2010. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem 10(1):127-141.
Thomson G. 1995. Analysis of complex human genetic traits: An ordered-notation method and new tests for mode of inheritance. Am J Hum Genet 57(2):474-486.
Toussaint NC, Mama Y, Kohlbacher O, Louzoun Y. 2011. Universal peptide vaccines – Optimal peptide vaccine design based on viral sequence conservation. Vaccine 29(47):8745-8753.
Villar J Maca-Meyer N, Pérez-Méndez I, Flores C. 2004. Bench-to-bedside review: Understanding genetic predisposition to sepsis. Crit Care 8(3):180-189.
Voit EO, Qi Z, Miller GW. 2008. Steps of modeling complex biological systems. Pharmacopsychiatry 41(suppl 1):S78-S84.
Walker RL, King NMP. 2011. Biodefense research and the U.S. regulatory structure. Whither nonhuman primate moral standing? Kennedy Inst Ethics J 2193):277-310.
Wang TT, Tan GS, Hai R, Pica N, Ngai L, Ekiert DC, Wilson IA, García-Sastre A, Moran TM, Palese P. 2010. Vaccination with a synthetic peptide from the influenza virus hemagglutinin provides protection against distinct viral subtypes. Proc Natl Acad Sci 107(44):18979-18984.
WHO [World Health Organization]. 2011. Smallpox: Fact sheet. Available online (http://www.who.int/mediacentre/factsheets/smallpox/en/), accessed March 2011.
Williams RW. 2006. Expression genetics and the phenotype revolution. Mamm Genome 17(6):496-502.
Williams RW Gu J, Lu L. 2001. The genetic structure of recombinant inbred mice: High-resolution consensus maps for complex trait analysis. Genome Biol 2(11):46.
Wolfe ND, Escalante AA, Karesh WB, Kilbourn A, Spielman A, Lai AA. 1998. Wild primate populations in emerging infectious disease research: The missing link? Emerg Infect Dis 4 (2):149-158.
Wolfer DP, Litvin O, Morf S, Nitsch RM, Lipp H-P, Würbel H. 2004. Laboratory animal welfare: Cage enrichment and mouse behavior. Nature 432(7019):821–822.















