
Important Points Highlighted by Individual Speakers
• There needs to be agreement upon standards for both analytical and clinical validation.
• Clinical data will need to be linked to genomic databases in order to further understanding of the phenotypic effects of genetic variants.
• Genomic data should be put into meaningful formats in order to be most useful to health care providers.
• Clinical actions need to be determined through collaborative efforts involving physicians, patients, their families, and laboratories.
Technologies such as whole-genome sequencing generate a tremendous amount of data, and reducing those data down to clinically applicable information will require a robust analysis process. As Debra Leonard from Weill Cornell Medical Center introduced the speakers, she stated some of the key questions and challenges for analysis of genomic data: What standards will be applied to the analysis of genomic data? How will new knowledge be incorporated into previously analyzed data? Who will be responsible for re-contacting physicians and patients as new data are gathered and existing data are re-analyzed?
Federico Monzon from the Methodist Hospital Research Institute described how cancer screenings have evolved over time. For example, physicians who tested for breast cancer were originally limited to studying the morphology of the breast. Later, the science progressed to evaluating single biomarkers, such as analyzing estrogen receptors in tissue. Since that time, testing for breast cancer has advanced through biomarker panels, expression profiles, targeted sequencing of specific genes, exome sequencing, and, finally, to whole-genome sequencing. While this progression of tests has led to a greater understanding of disease morphology, it has also required laboratories and physicians to complete more complex analyses of the test results. As Monzon said, “This is an evolution of testing which is driven by our better understanding of disease, and with that better understanding of disease comes better clinical testing that we are doing in our laboratories.”
Based on his experience with the genomic testing of cancer tissues, Monzon offered several challenges that laboratories can expect to face when performing their analyses. Chief among them is demonstrating the analytical and clinical validity of the genetic test, no matter whether the test is for a single gene or for the whole genome. A laboratory is responsible for determining whether a test is able to detect all described variants—whether in sequences, transcripts, or some other biological indicator—as well as for validating that the data are correct when the test finds something new. Furthermore, patients and payers depend on the laboratory to determine whether a test identifies patients who have a disease or are at risk for a disease.
Today’s validation standards do not apply to multianalyte tests, much less to whole genomes. With millions of variants estimated to exist within a single genome, there are more potential backgrounds than individuals who can be used to validate the variants. It is thus a major question regarding how many patients or tumor types need to be identified for each variant detected; with the cross-genome differences among these patients, validating each variant in the same way that single-gene tests are handled is not possible, Monzon said.
No sequencing technology can validate every base pair, Monzon said, especially with variants that have never before been seen. Instead, laboratories will need to assess the concordance across results. Reference genomes will help in achieving analytically valid results. Laboratories will develop confidence in platforms based on experience, which will then generate confidence in results. Madhuri Hegde from the Emory University School of Medicine added that sequencing systems will inevitably introduce artifacts, and genomic analysis will need to separate those artifacts from real variants. As a result, some sort of confirmation, such as Sanger sequencing for the individual variant in question, will be necessary in many circumstances.
Just as there is no agreed-upon method for validating all of the variations, there currently is a lack of regulatory guidance for whole-genome tests, and routine whole-genome sequencing will not be allowed to move into clinical settings without major changes in current regulations. In setting standards for whole-genome sequencing, Monzon said, it will be important to strike the proper balance between ensuring the quality of genomic testing and allowing innovation to proceed. Heidi Rehm from Partners HealthCare Center for Personalized Genetic Medicine (PCPGM) said that she expects more involvement from the U.S. Food and Drug Administration (FDA) and other regulatory bodies, which is generally a good thing, she said. The FDA has been trying to understand its role, and so far it has not been inhibitory. There are differences in the quality of testing, which the FDA should address. “I think we would benefit from some regulation,” Rehm said. Monzon agreed that the FDA has been cautious about regulating such tests, partly because the vast majority of genetic tests have been developed by laboratories. Hegde said that different areas of testing will require different levels of oversight and that laboratories need to observe the guidelines and quality assurance documents that are currently in place.
The challenges stemming from the lack of guidance or regulation are also apparent in situations in which the physician needs to know whether test results direct clinical action. The laboratory has the responsibility of deciding which of its findings to report to the physician, but the meaning of most of what it finds is unknown. Monzon pointed out that this is especially true in cancer testing where there is an accumulation of mutations. There is no single repository for laboratory directors to find up-todate standard-of-care guidelines. Current CLIA regulations specify that the responsibility for clinical validation rests with the medical director of the laboratory. “This is a heavy burden to have,” Monzon said.
How Much Evidence Is Needed to Adopt a New Test?
As an example of many of the current regulatory and standardization issues, Monzon described a current standard-of-care test for chronic lymphocytic leukemia (CLL). Historically, fluorescent in situ hybridization (FISH) panels have been the only technique used to detect genetic aberrations in CLL that have a major impact on the behavior of the disease. These panels have many disadvantages, however. Commercial FISH panels do not detect all chromosomal deletions, cannot capture all relevant genomic lesions, and are limited in either their resolution or their breadth of coverage (Hagenkord et al., 2010). Cytogenomic arrays are beginning to gain acceptance as an alternative to the standard FISH panel for detecting these and other aberrations (Hagenkord et al., 2010). These new tests have the advantage that they can detect a condition known as genomic complexity,
which describes a condition in which there are a large number of genetic changes. However, it is not clear that this is an actionable result that should be reported. The condition has been linked to adverse outcomes, but the combined literature is limited to data from only about 160 patients. Is this enough information to start using this test in the clinic? If so, how should it be reflected in the guidelines for treatment or management of these patients? What level of evidence is needed? Does the result provide prognostic information that can be used for decision making?
To answer these and other questions, Monzon said, guidelines, regulations, and standards are needed that allow such tests to be validated. Validation is needed for technologies and platforms, not just individual tests. Furthermore, laboratories need tools to define the clinical relevance of a result and link this information to therapeutic agents.
Meeting the Demands for Actionable Results
Monzon emphasized that in the near term it will be important to accumulate data regarding which mutations drive diseases such as cancer and which mutations are passenger variants. Actionable results need to be distinguished from results that are not relevant, and this distinction may change as research progresses. Laboratory directors need to be thoroughly familiar with current research, and sophisticated laboratory information systems are essential.
Laboratories currently “do not have the resources to deal with genomic medicine,” Monzon concluded. “Access to curated information is fragmented, and there is no … sole source of information that we can go through.” Furthermore, reimbursement for these activities is virtually nonexistent today, especially when the activity involves reviewing past results in light of recent research.
The Emory Genetics Laboratory is a not-for-profit clinical testing laboratory that focuses on rare genetic disorders, Hegde said. It is a comprehensive laboratory that performs biochemical, cytogenetic, nutritional, and clinical testing. Its DNA laboratory uses a variety of technologies to conduct tests on a wide variety of genes and inherited disorders. It has been doing next-generation sequencing for more than a year and has accrued considerable experience with clinical applications. It is certified and accredited through CLIA, the College of American Pathologists (CAP), and New York State.
There is a typical life cycle for the development and use of a single-gene test, Hegde said (Figure 3-1). After a gene is reported in the literature,
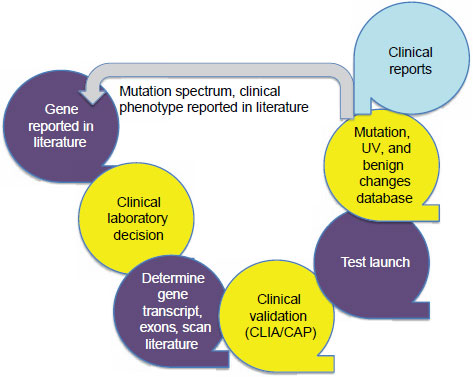
NOTE: CAP = College of American Pathologists; CLIA = Clinical Laboratory Improvement Amendments; UV = unclassified variants.
SOURCE: Hegde, IOM workshop presentation, July 19, 2011.
a clinical laboratory decides whether to investigate and test for that gene based on requests from clinicians or others. The laboratory determines the gene transcript, including such information as the number of exons contained, and the laboratory scans the literature to extract all relevant information. A clinical validation step is performed that meets CLIA and CAP standards, and then the laboratory makes the test available. Once the test is being used, the laboratory has to report its findings in a way that is understandable to a clinician. The current policy of the Emory Genetics Laboratory is to report only the data that have been requested, an approach that allows the laboratory to avoid many of the interpretation issues that Monzon described.
From the data collected from the tests it runs, the laboratory constructs databases of mutations, unclassified variants, and benign changes. These restricted-use databases can be used to determine whether a variant or group
of variants is clinically actionable. In the past not many clinical laboratories have put mutation and phenotype databases into the open literature, Hegde said, but the Emory Genetics Laboratory has produced many such reports.
Detecting Variance with Genome Test Results
The Emory Genetics Laboratory has been moving from single-gene testing to multigene panels using next-generation sequencing. The laboratory is now testing for three disorders: X-linked intellectual disability (XLID), congenital disorders of glycosylation (CDG), and congenital muscular dystrophy (CMD). The XLID panel examines 92 genes that segregate with the disease, including both genes that are syndromic and those that are non-syndromic (Nelson and Gibbs, 2009). The CDG panel tests for a group of metabolic disorders caused by a deficiency in some or all parts of the glycosylation pathways that results in an abnormal glycosylation of oligosaccharides. Just as there are many different organelles that could be affected, there is a wide range of phenotypic symptoms for this disease, ranging from death in infancy to mild involvement in adults (Jaeken, 2011). The CMD panel surveys a collection of gene mutations that result in muscle disorders that are defined by a combination of early-onset hypotonia and weakness, contractures, variable progression, normal or elevated serum creatine kinase (CK), and myopathic changes on an electromyogram and that are usually associated with a dystrophic muscle biopsy. The gene mutations considered on the CMD panel result in muscle weakness soon after birth (Peat et al., 2008).
Because of limitations in sequencing technology, some of the laboratory’s tests detect more gene variants than others. The CMD panel detects an average of 39 variants in just 13 genes, for example, while the XLID panel detects an average of 31 variants in 92 genes.
Sequencing detects many genetic changes that cannot be interpreted, Hegde noted. In response, the laboratory uses its database of known variants as an initial screening tool for determining which changes may be real and which may be artifacts. It can then continuously re-classify variants and re-contact physicians to tell them if a variant that had been detected previously but that had been considered non-actionable at the time has been re-classified. In order to help determine which panel of genes to test, the laboratory has developed a tool to allow the physician to link phenotypic symptoms with genes that are known to be related to those symptoms.
Reimbursement and Collaboration Challenges
The experience with next-generation sequencing at the Emory Genetics Laboratory has identified a number of challenges with important impli-
cations for whole-exome or whole-genome sequencing, Hegde said. The amount of reimbursement available compared with the amount of time a comprehensive analysis requires is a concern. “The reality of the situation is that the cost of the tests is not the cost of the reagents. There are lab directors. There are technicians. There are a lot of other factors that need to be accounted for when you are costing out a test.”
Whole-exome and whole-genome analysis will require a team composed of clinical and laboratory geneticists, genetic counselors, and other health care specialists, Hegde said. It will be important to get as much data as possible into databases, including the clinical presentation, in order to aid in the interpretation of the data. Futhermore, curating the variant databases will be very important in making the transition from sequencing gene panels to sequencing the whole genome.
MEANINGFUL USE OF TEST RESULTS
For genetic test results to be used in clinical settings, Rehm said, the data need to be put into useful and meaningful formats for physicians. In particular, she identified four issues for consideration: (1) structuring genomic data; (2) providing accurate and readily accessible data interpretation resources; (3) supporting the generation of high-quality, clinically relevant reports; and (4) creating systems to support re-analysis and independent interpretations of genomic data.
Structured Genomic Data
The standards group at the Laboratory for Molecular Medicine within PCPGM has been working to define a system for reporting genetic variation and test content in well-structured ways. The system includes such information as allele state, nucleotide changes, amino acid changes, and clinical classification. At Partners HealthCare this information goes into an electronic medical record (EMR) and into a research repository as structured data.
Structured data make it possible to use clinical support tools that can leverage genetic data even as algorithms and use cases change over time. Whole-genome sequence records can be accessed in EMRs as clinical symptoms arise or as adverse-event warnings are received, and proactive alerts can be generated as new clinically actionable knowledge is learned. It is important, Rehm said, that regulatory partners, including the FDA and Centers for Medicare and Medicaid Services, be engaged in order for laboratory systems and health care systems to be sure of the validation of this information.
Data Interpretation Resources
A basic challenge in using genomic data, Rehm said, is that the terminology is not standardized. Genetic variants can be referred to as mutations, polymorphisms, or SNPs (single nucleotide polymorphisms); their effects might be called pathogenic, deleterious, or disease-associated; and harmful effects can be possible, probable, or likely.
Another challenge is that most variants are rare, with the association linked in only a small population. For example, of the more than 1,400 mutations in the hypertrophic cardiomyopathy (HCM) database, twothirds have been seen in only one family. In addition to the challenges presented by the rarity of variants, other challenges arise from the fact that new clinically significant mutations continue to be identified at a substantial rate. For example, even though more than 150,000 patients already have been tested for BRCA1 (breast cancer 1, early onset) variants, tests continue to detect 10 to 20 new missense variants each week. Thus, once standards for reporting and clinical action have been created, they will need to be re-evaluated and updated continually.
As our understanding of associations is refined, it will become possible to use new models and algorithms to evaluate actions based not only on genetic variants but also on lifestyle, environmental influences, and other factors. This will require extensive information technology support to search databases for variants, and a centralized and standardized open-access variant database with standardized nomenclature and careful curation, Rehm said. Steps in that direction have already been taken by the MutaDATABASE Project and the ClinVar Project, both of which are attempting to create a single place for genetic variations to be housed and curated in standardized ways. ClinVar, for example, is seeking to build rich datasets that are updated on an ongoing basis. Another model is the work done by the International Standards for Cytogenomic Arrays consortium, which is seeking to create a single place in which data generated by clinical laboratories can be deposited. “We are going to have to continue to learn from the datasets even though we are at a limited level of understanding today,” Rehm said.
Rehm noted that many current databases contain much incorrect data. Discussions among the groups mentioned above have centered on developing quality thresholds that set standards for data, but even with thresholds there will be questions about whether data are correct or not.
Most clinical laboratories have been willing to submit their data to public databases, which Rehm said she found surprising. Their willingness demonstrates, she said, “that we all recognize, as we move to wholegenome sequencing, that none of us can interpret the entire genome in an effective way, and we will all need to rely upon everybody’s data.”
High-Quality Clinically Relevant Reports
Clinically relevant reporting of genetic data must do four things, Rehm said. It must:
• accurately interpret the impact of each variant on a gene or protein;
• accurately interpret a set of variants relevant to a single phenotype;
• accurately relay the relevance of the identified variants in the patient’s presentation; and
• determine how to apply the genetic information to the care of the patient (and the patient’s family members).
Interpreting the impact of a variant or set of variants is clearly the role of the laboratory, Rehm said. But as the complexity of an interpretation increases, this role becomes more diffuse. For example, secondary variants can modify the effects of a primary variant, and some laboratories do not analyze these data fully. Furthermore, laboratories often do not get clinical data, so putting variants into the context of a patient’s presentation can be difficult.
As interpretation becomes more complex, few laboratories will have the infrastructure needed to provide interpretations of results, especially in smaller laboratories. Rehm predicted that some entities will be good at analysis while others will be good at interpretation and that questions about interpretation will go to those with the greatest expertise. “There needs to be better recognition that not the same entity has to do both,” she said. Regardless of how the specialization of laboratories happens, interpretation will have to be a team effort because most physicians are not currently capable of determining how to apply genetic information to care, Rehm said. Hegde observed that actions need to be determined through a collaborative effort. Clinicians, patients and their families, and laboratories all need to work with each other to make optimal decisions. Furthermore, responses may be tiered, with primary care physicians dealing with certain things and other professionals being called in to deal with other findings. Hegde added that the role of genetic counselors as intermediaries between the laboratory and the clinician is very important.
PCPGM has been planning a genetic consulting service that would enlist cardiologists with genetics expertise who could combine information about a patient’s phenotype and family history with the results from the genetic report in order to generate patient and family care packages. The group has also been developing software to help laboratories generate customized reports that would allow cardiologists to integrate genomic data into patient care.
Re-analysis and Independent Interpretations
Guidelines issued by the American College of Medical Genetics is 2007 state that “the testing laboratory … should make an effort to contact physicians of previously tested patients in the event that new information changes the initial clinical interpretation of their sequence variant” (Richards et al., 2008). However, Rehm said, no effective method of updating information currently exists. For example, over the past 5 years about 300 HCM variants have been moved from one to another of the five risk categories (benign, likely benign, unknown significance, likely pathogenic, and pathogenic), and more than half of those moves represented significant changes.
PCPGM has developed an extension of its laboratory software called GeneInsight Interface that reports new results in a structured form to clinicians. When a variant is changed in the mutation database, patient reports are automatically updated. Clinicians see that a previously reported category is crossed out and a new category has been inserted. The clinician can click on the variant to read the evidence for why the classification of the variant was changed. The clinician also receives an e-mail alert that links to any patients who have the variant for which the new information is relevant. PCPGM, which has a grant to evaluate the system, has found that, for the most part, physicians have found it extremely useful.
In order to address other problems with re-contacting health care providers and patients, the health care system needs methods that will ease the laboratory’s efforts, Rehm said. She suggested developing a system that allows patients to access their genetic data when appropriate and makes patients responsible for updating their current health care providers so that updated information can be delivered to medical professionals when appropriate. This is necessary since the laboratory often does not have reliable methods for determining who is currently caring for a patient or how to reach a patient or physician.
The GeneInsight Interface can also be used to direct researchers to patients who have certain genetic variants so that they can be notified of relevant clinical trials. PCPGM is also considering ways to allow multiple knowledge sources to feed information into genetic databases. “The bigger the network, the more information can be generated and shared across it,” Rehm concluded.
In a 2008 interview, Francis Collins, who is now the National Institutes of Health (NIH) director, said, “We desperately need, in this country, a large-scale, prospective, population-based cohort study. And we need to enroll at a minimum half a million people. We would need to have their
environmental exposures carefully monitored and recorded, their DNA information recorded, their electronic medical records included, and have them consented for all sort of other follow-ups” (Collins, 2008).
The Coriell Institute for Medical Research, a large biorepository located in New Jersey, is conducting, in partnership with several medical centers, a smaller version of such a study through its Coriell Personalized Medicine Collaborative (CPMC). The goals of this collaborative are to study the use of genome-informed medicine in a real-world clinical setting to determine the best mechanisms for providing information to providers and patients, as well as to find correlations in observational data. “There are not enough resources out there to do randomized clinical trials on every bit of genome information or other quantitative information that is emerging that may be clinically relevant,” said Michael Christman of the Coriell Institute. A large observational database will be essential.
More than 5,000 people are currently enrolled in the collaborative study. In order to enter the study, participants are required to fill out extensive medical and family history and lifestyle questionnaires, which are used to report quantitative risks based on environmental or family history information. Extensive genomic analysis—although not yet whole-genome sequencing—is conducted on DNA extracted from each participant’s saliva. Only results that are deemed clinically relevant by an expert panel are reported to the patient and, depending on the arm of the study in which the patient is enrolled, also to his or her physician. Much of that information is pharmacogenomic, which is currently the most clinically actionable information and applies to the largest number of people.
Participants and physicians can view the information through a secure Web portal. When only participants get the information, they are encouraged to present it to their physicians. The collaborative study offers genetic counseling and access to pharmacists at no cost since it is a research study. The Web portal allows for continual follow-up with participants on such issues as the actions they take, their perceptions of risks and benefits, and their participation in ancillary studies. The collaborative is fully compliant with the Health Insurance Portability and Accountability Act. But Christman also said that we should not claim to provide complete security for genetic data because “we have to assume that [the data are] going to get out there. If Google can get hacked, then anyone can get hacked.” People worried about their privacy when Internet shopping was first getting under way, but the system used for such shopping generally works well, even though some people abuse it, and some people go to jail as a result. “With genetics, it would be largely the same way,” Christman concluded.
The collaborative study currently has three arms. The largest arm is a community study of otherwise healthy individuals. Participants have to be at least 18 years old and be able to use the Internet. The other two
arms focus on heart disease and cancer and use EMRs and other clinical information. For example, in a partnership with The Ohio State University the collaborative is enrolling 2,000 people with congestive heart failure or hypertension.
The Informed Cohort Oversight Board
One innovation with relevance to whole-genome sequencing was the development of the Informed Cohort Oversight Board (ICOB) (Kohane et al., 2007). CPMC uses the ICOB concept to determine which genomic data should be considered clinically relevant and thus reportable to participants. The ICOB is composed of geneticists, statisticians, ethicists, and a community member. Researchers at Coriell curate the scientific and medical literature and present evidence to the board to determine which genetic information is actionable. The board meets twice a year and approves or disapproves proposals in much the same way as a study section for NIH would.
Consent is obtained from each participant in the study cohort. New results are reported to all participants, and each participant then decides whether or not to view information about his or her genome. The information is presented as a relative risk, taking into account family history and environmental factors. “This is a dynamic group,” said Christman. “They know that they will find out new information, but they don’t know exactly what.”
Potentially actionable conditions that are currently approved to be reported by the CPMC study include several drug metabolism variants and the following complex diseases:
• Age-related macular degeneration
• Bladder cancer
• Breast cancer
• Chronic obstructive pulmonary disease
• Colon cancer
• Coronary artery disease
• Diabetes, types 1 and 2
• Hemochromatosis
• Inflammatory bowel disease
• Lupus
• Melanoma
• Obesity
• Prostate cancer
• Rheumatoid arthritis
• Testicular cancer
At this time, Christman acknowledged, groups of variants explain very little of the risk of complex diseases, but, he said, “with the advent of wholegenome sequencing and the appreciation of rare variants, we are going to be there sooner rather than later, and we have to figure out the system.”
A different ICOB advises on testing for pharmacogenomic variants. The project has developed a strength-of-evidence code for these variants that gives greater importance to variants that will affect clinical decisions. The strongest evidence code is reserved for situations in which the reference drug has shown actual clinical outcomes in a randomized clinical trial.
Study Parameters
In order to put together a representative sample, the project is trying to match the demographics of the Delaware Valley, which is about 15 percent African American and 15 percent Hispanic. Follow-up surveys conducted 3 months and 12 months after a result is reported to participants ask them what actions they have taken. For example, the collaborative study keeps track of which medical tests the participants or their physicians choose to do. It has found that the number of tests or procedures taken correlate strongly with age and with the completeness of family history information. The participants’ actions are not significantly related to the number of genetic risk variants that they have for that disease. This is good news, Christman said. “People in the study tend to understand that these are risk factors and not determinative.”
Another investigation is looking at what participants in the study perceive as the risks and benefits of participation (Gollust et al., 2011). One alarming result is that 13 percent of participants think that they will be able to change their risk with gene therapy if testing is done. “This shows the level of misunderstanding that can be out there,” Christman said. Furthermore, about one-third of participants are worried that they will find out something that they do not want to know.
The study of patients with congestive heart failure or hypertension will track physicians’ actions in addition to the patients’ actions when they receive genetic information. In this arm of the study patients have been randomized for either receiving genetic counseling or not receiving genetic counseling, although everyone in the latter group has access to counseling if requested. Anyone who wants to withdraw from the study can do so at any time and have his or her sample destroyed as well, which some have chosen to do.
Many ethical, legal, and social issues remain to be resolved, Christman said, including various genetics-related privacy issues, the anxiety associated with genetic prognoses, education of various stakeholder groups, and the fact that payers are likely to drive clinical application. But work is ongoing in each of these areas.














