
2
Background, Definitions, Concepts
The committee’s examination of breast cancer and the environment required considerations at the intersection of diverse fields, including the biology and epidemiology of breast cancer, the identification of carcinogens and cancer-promoting agents, exposure assessment, toxicity and carcinogenicity testing, and the design and interpretation of research studies. This chapter provides some brief, fundamental background on these topics as a basis for the discussions in subsequent chapters.
AN INTRODUCTION TO BREAST CANCER
The breast begins forming during the prenatal period and undergoes substantial changes during adolescence and adulthood. Breast cancer arises when abnormal cellular growth occurs in certain structures and types of cells within the breast.
Although breast cancer is often spoken of as if it were a single disease, evolving techniques of analysis of the molecular characteristics of tumors are pointing to a variety of types of potentially differing origins. Gaining a better understanding of the nature of the heterogeneity of breast cancer will be critical in helping researchers improve the design and interpretation of studies of possible risk factors, and it may influence approaches to prevention.
Described here are the basics of the anatomy of the breast and breast development, types of breast cancer, and levels and trends in the incidence of the disease, focusing primarily on experience in the United States. The mechanisms that appear to result in female breast cancers and the pathways
Approximately 1 percent of breast cancer cases occur in men, and less than 1 percent of men’s cancer diagnoses are for breast cancer (ACS, 2011b). Because it is rare, breast cancer in men has been difficult to study. Based on what is known, however, it is considered to resemble breast cancer in postmenopausal women (Korde et al., 2010).
As in women, men’s breasts respond to changes in sex hormone concentrations (both estrogens and androgens), but under normal circumstances they do not undergo the differentiation and lobular development that women’s breasts experience with puberty, pregnancy, and lactation (Johansen Taber et al., 2010). Either an excess of estrogens or deficit of androgens appears to increase risk of breast cancer in men (Korde et al., 2010). Beginning after age 20, rates rise steadily with age. Approximately 92 percent of male breast cancers are estrogen receptor positive, compared with approximately 78 percent of breast cancers in women (Anderson et al., 2010). As is the case for women, inherited mutations in BRCA1 and especially BRCA2, as well as other mutations, are associated with an increased risk of male breast cancer, but the majority of cases are not associated with a family history of the disease (Korde et al., 2010).
along which they operate are one of the main topics in Chapter 5. A brief description of breast cancer in men is provided in Box 2-1.
The Breast, Breast Development, and Breast Cancer
The development of the human female breast begins during gestation but is not complete at the time of birth. Further development and differentiation of breast tissue occurs over time and especially in response to fluctuating estrogen and other hormonal signals beginning in puberty, continuing through the reproductive years, during pregnancy and lactation, and at menopause. Monthly ovulatory cycles are accompanied by cyclical changes in the form and behavior of cells and structures in the breast, including progressive differentiation. Pregnancy and lactation trigger maximal differentiation of the breast. When pregnancy and lactation end, as well as at menopause, breast tissue regresses to a less differentiated state.
Within the breast are adipose and connective tissues that surround multiple collections of lobules in which milk is produced during lactation. Milk moves to the nipple through ductal structures. The ducts are lined by luminal epithelial cells and have an outer layer of myoepithelial cells. Popu-
lations of stem cells that can give rise to either luminal or myoepithelial cells are also found in the ductal tissue. The ducts are anchored to a basement membrane, which contributes to both the structure and the function of the ductal tissue. Connective tissue within and between the lobules, known as the stroma, further contributes to the structure of the breast and plays an important role in regulating both normal and abnormal breast cell growth and function (Arendt et al., 2010). Cell types within the stroma include (but are not limited to) fibroblasts, adipocytes, macrophages, and lymphocytes (Johnson, 2010). These cells and structures in the breast generate and respond to a diverse mix of hormones, especially estrogen, and other regulatory factors.
Certain disruptions in the complex processes that govern the structure and function of breast tissue may set the stage for breast cancer. Some carcinogenic events occur spontaneously in the course of normal biological processes and others are triggered by external factors. Although the body has efficient protective responses, such as DNA repair and immune surveillance, that can reduce the effect of such events, these protective responses are not always successful. The interval between the earliest “event” and the detection of a cancer may span several decades.
Specific mechanisms that may play a role in breast cancer are noted here but discussed further in Chapter 5. The contribution of genetic mutations to cancer is well known. They may be inherited (e.g., germline mutations in the BRCA1 or BRCA2 genes, which normally have a role in DNA repair) or develop in some cells during a person’s lifetime (somatic mutations) as a result of reactive by-products of normal biological processes, or from the effects of external exposures. Other mechanisms include epigenetic changes that can alter gene expression without changes to DNA, promotion of cell growth by estrogen and other hormones or cell-signaling proteins, and evasion of the immune system.
Types of Breast Cancer
Most commonly, breast cancers develop in the ducts, but cancers also develop in the lobules or take other forms. Several systems are used to characterize breast cancers, with the systems developed primarily to provide information on prognosis and treatment decisions. For example, breast tumors may be classified by tumor size, extent of spread beyond the tumor site (localized, regional, distant), the anatomical characteristics of the tumor cells (e.g., ductal or lobular histology), and the molecular features of the tumor cells, such as presence or absence of estrogen and progesterone receptors and human epidermal growth factor receptor 2 (HER2/neu).
The age at which a woman is diagnosed with breast cancer is associated with tumor characteristics, such as the likelihood that the breast cancer
is estrogen receptor positive or negative (ER+ or ER–). In addition, age or menopausal status also guides treatment decisions. For example, aromatase inhibitors are part of treatment for postmenopausal women who have ER+ breast cancers, but tamoxifen is used among premenopausal women. Except for reference to menopausal status, breast cancers in men are characterized in similar ways. Differences in patterns of such features as tumor histology, grade, and receptor status may distinguish between a more aggressive form of breast cancer with a generally earlier onset and a more common and less aggressive form that tends to occur at older ages (see Anderson et al., 2006b, 2007; Kravchenko et al., 2011).
Another major distinction is between invasive and noninvasive (or in situ) tumors. As the terms suggest, invasive tumors spread beyond the site at which they arise, while in situ tumors remain within the tissue where they originate, such as the epithelial cells lining the breast ducts. About 20 percent of reported tumors are noninvasive (ACS, 2011a). Ductal carcinoma in situ (DCIS) is the most common form of abnormal but noninvasive growth in the breast. Although DCIS can, in some cases, progress to an invasive cancer, the natural history of these tumors is poorly understood, and it is not yet possible to identify which ones are likely to progress (Allred, 2010). As a result, most women with in situ tumors receive treatment that is similar to the treatment for early-stage invasive tumors.
Estrogen and Progesterone Receptor Status
The molecular and genetic characteristics of breast tumors are used to guide treatment and assess prognosis. A feature for which breast tumors are now commonly evaluated is whether the cells express estrogen or progesterone receptors. Tumors that express these receptors are designated ER+ or PR+, and those that do not as ER– or PR–. In the United States, approximately 75 percent of invasive tumors for which receptor status is reported are ER+ and 65 percent are PR+ (Ries and Eisner, 2007; Kravchenko et al., 2011). ER+ and PR+ tumors have a generally better prognosis than tumors that do not express these receptors. These receptor characteristics are correlated with other tumor markers related to regulation of cell growth and proliferation and appear to reflect important differences in tumor origin (Phipps et al., 2010). Researchers are also finding that they are associated with differences in response to risk factors (e.g., Althuis et al., 2004; Yang et al., 2011).
Triple Negative Breast Cancer
Tumors lacking not only ER and PR expression but also HER2 are called triple negative breast cancers (TNBCs), and they are considered
closely related to basal-like breast cancers (Carey et al., 2006; Foulkes et al., 2010). Triple negative breast tumors are typically aggressive and are more likely to be diagnosed in women who are younger (below age 50) and are African American. These cancers in African American women tend to be more advanced and of higher grade at the time of diagnosis than tumors in other racial groups (Carey et al., 2006; Stead et al., 2009; Trivers et al., 2009). Triple negative tumors have been associated with BRCA1 and BRCA2 mutations (Armes et al., 1999; Foulkes et al., 2003; Turner et al., 2007; Atchley et al., 2008). Additionally, a large proportion of TNBCs have altered p53 levels (Carey et al., 2006; Kreike et al., 2007; Rakha et al., 2007).
Genetic Susceptibility to Breast Cancer
Genetic mutations may contribute to breast cancer by altering various critical processes such as those related to DNA repair, hormone synthesis, and metabolism of carcinogens. Two types of genetic mutations are possible. Germline mutations are genetic variants that are passed from parents to offspring and are present in all cells. Genetic changes can also occur in specific cells during a person’s lifetime; these changes, which can persist as cells divide, are called somatic mutations. They can arise by chance, as a by-product of normal processes such as cellular respiration or DNA replication, or from external exposures. Such mutations may lead to that cell becoming a cancer cell.
Inherited genetic variation is found across the population. Many of these variations, called polymorphisms, may have little or no impact on the function of a gene, but some of them are associated with increased susceptibility to disease. Common genetic variants are found in 1 percent or more of the population.
Every breast cancer contains somatic genetic changes, but only a few inherited mutations are known to convey a high risk of breast cancer in the carrier. The strongest evidence of inherited genetic susceptibility is for germline mutations in the BRCA1 and BRCA2 genes. Research suggests that a larger number of lower-risk germline variants also exist.
Hereditary Syndromes
A family history of breast cancer is an established breast cancer risk factor. This risk factor represents both inherited genetic risks as well as environmental factors that may cluster in families. Overall an inherited susceptibility to breast cancer contributes to about 10 percent of breast cancer cases, and in about 5 percent of breast cancer cases this inherited susceptibility is attributed to mutation in the BRCA1 or BRCA2 genes.
Mutations in these two genes are associated with increased susceptibility not only for breast cancer, but also for other cancers such as ovarian cancer.
BRCA1/2 mutations are high-penetrance mutations, meaning that women with these mutations have a very high lifetime risk of developing breast cancer. This risk is estimated to be at least 40 percent and possibly as high as 85 percent (Oldenburg et al., 2007). However, these mutations are rare, with substantially less than 1 percent of women in most populations carrying them (Narod and Offit, 2005). In addition to increasing the risk of breast cancer for women, they also increase risk for male breast cancer. Families in which such mutations may be present may have multiple cases of breast cancer, occurring at younger ages and in multiple generations, and a family history of ovarian cancer (Narod and Offit, 2005). Other sources of increased familial genetic risk include the Li-Fraumeni syndrome1 from germline mutations in the p53 gene (Malkin et al., 1990) and Cowden disease2 from germline mutations in the PTEN gene (Liaw et al., 1997).
Genetic testing is available to identify BRCA1 and BRCA2 mutations. Identification of a familial mutation that carries an increased risk of breast cancer allows women, and men, who carry such a mutation to seek closer monitoring of their health and to consider primary and secondary preventive measures, such as increased screening, bilateral prophylactic mastectomy and, for women, bilateral salpingo-oophorectomy (Walsh et al., 2006). Use of medications that can reduce the risk of breast cancer (i.e., tamoxifen and raloxifene) may also be appropriate for some women (USPSTF, 2002).
Breast Cancers in Women Without a Strong Family History
Most women diagnosed with breast cancer do not have a strong family history of the disease and do not carry mutations in highly penetrant cancer-susceptibility genes. They may, however, have other more common genetic variants that affect gene function and that may be responsible for a proportion of the breast cancer cases that develop. These genetic variants are called low-penetrance variants because they are associated with only a small degree of risk for breast cancer. Yet because they are common, they may contribute to the burden of disease. In addition, these variants may interact with environmental exposures such that risk is only expressed in the presence of the environment exposure (gene–environment interaction).
Two approaches have been used to identify low-penetrance genetic variants: a candidate gene approach and genome-wide association studies.
_________________
1Li-Fraumeni syndrome is characterized by a predisposition to sarcomas, lung cancer, brain cancer, leukemia, lymphoma, adrenal-cortical carcinoma, and breast cancer.
2Cowden disease is a syndrome involving mucocutaneous and gastrointestinal lesions and breast cancer.
Studies initially relied on the candidate gene approach, in which polymorphic variants of genes that plausibly influence breast cancer risk are assessed in epidemiologic studies (i.e., case–control or cohort studies) for their association with breast cancer. For example, the Breast and Prostate Cancer Cohort Consortium has conducted extensive analyses of genetic variation in large numbers of specific genes in biological pathways thought to be most relevant to breast cancer, such as the steroid hormone metabolism and insulin-like growth factor pathways (Canzian et al., 2010; Gu et al., 2010). These studies did not find an association with breast cancer risk. In general, the candidate gene approach has had limited success in consistently identifying specific variants associated with breast cancer.
Genome-wide association studies (GWAS) allow for a comprehensive and unbiased search for modest associations across the genome. The approach in these studies is to identify a relatively limited set of readily recognized single nucleotide polymorphisms (SNPs) that are highly correlated with a larger block of genetic variants and to use the limited set of “tagSNPs” in the analysis (Manolio, 2010). These studies require very large sample sizes (thousands or tens of thousands of cases and controls) because these variants tend to be associated with a small degree of risk. Because these studies make use of large numbers of statistical tests, they require extreme levels of statistical significance to identify true positive results (Hunter et al., 2008).
Results from several GWAS of breast cancer in women of European ancestry have been published (Easton et al., 2007; Hunter et al., 2007; Stacey et al., 2007; Turnbull et al., 2010), and one of women of Asian ancestry (Zheng et al., 2009). Out of the many variants studied, approximately 20 risk variants have been robustly associated with breast cancer risk, all having only modest influence on risk (relative risks in the range of 1.05–1.3 per allele). Stronger associations with common variants are unlikely to exist, but they may be possible for rarer variants (e.g., those with minor allele frequencies of <5 percent) that have not been tested with the technologies available to date. Even so, statistical modeling suggests that low-penetrance gene variants may do at least as well in predicting risk as using traditional risk factors such as age at first birth, family history of breast cancer, and history of breast biopsy(ies) (Wacholder et al., 2010). This is a rapidly evolving area of research.
BREAST CANCER INCIDENCE IN THE UNITED STATES
As noted in Chapter 1, an estimated 230,480 new cases of invasive breast cancer were diagnosed among women in the United States in 2011 and another 2,140 new cases among men (ACS, 2011a). In addition, approximately 57,650 in situ cases were diagnosed in women, of which
For data on patterns and trends in incidence and mortality for all forms of cancer in the United States, researchers generally rely on data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program. In 1973, SEER began systematic collection of data from cancer registries in sites selected to characterize the diversity of the U.S. population. The number of participating registries has increased, and as of 2005 covered approximately a quarter of the U.S. population (NCI, 2005). The SEER Program establishes standards for completeness and quality of the data provided to it, and it works with participating registries to achieve those standards. As practices change, new data elements may be collected. For breast cancer, for example, data on estrogen and progesterone receptor status of tumors were added in 1990 (Ries and Eisner, 2007). Annual reports present data and analysis on cancer incidence, mortality, survival, and trends since 1975. Datasets can also be made available to qualified researchers for independent analyses.
States also have cancer registries, but some of these registries are less than 20 years old (CDC, 2010). Through the National Program of Cancer Registries (NPCR), which was established by federal legislation in 1992 and is administered by the Centers for Disease Control and Prevention, states receive assistance to improve the quality and completeness of their cancer registries. The NPCR now produces an annual report that combines data from state registries with data from the SEER program.
about 85 percent were DCIS (ACS, 2011a). Sources of surveillance data on breast cancer are described in Box 2-2.
Age Patterns and Changes Over Time
Breast cancer can occur in women and men of any age, but it is predominantly a disease of middle and older ages. Rates of invasive cancer increase rapidly after age 35 and currently peak at approximately 432 cases per 100,000 women in the age group 75–79 years (NCI, 2011) (see Figure 2-1). Rates of in situ disease rise more slowly and increase as women reach ages at which mammographic screening becomes common. The peak rate is 99 cases per 100,000 women at ages 65–69 (NCI, 2011). Among men, cases of invasive breast cancer are found at young ages, but incidence peaks at ages 85 and older at a rate of approximately 10 cases per 100,000 men (NCI, 2011).
The incidence of breast cancer has increased since at least the mid-1970s but has dropped from its peak in 1999. Figure 2-2 shows the rates
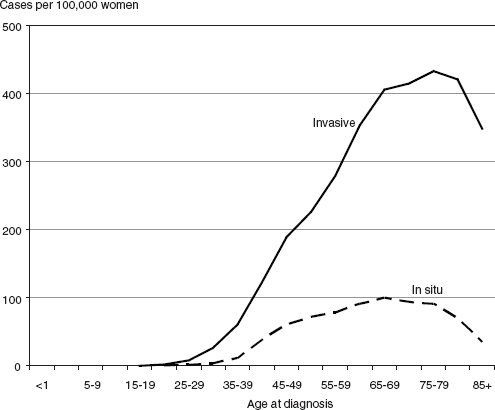
FIGURE 2-1 Age-specific incidence rates for invasive and in situ breast cancer among women in the United States, 2004–2008.
SOURCE: NCI (2011).
over time for both older (age 50 and older) and younger women (ages 20–49) and for invasive and in situ cases. Among older women, rates of invasive cancer rose during the 1980s and showed a slower increase during the 1990s. During the 1980s, use of menopausal hormone therapy had increased (Hersh et al., 2004; Glass et al., 2007). The 1980s and 1990s were also a period when use of screening mammography increased (Breen et al., 2001; Anderson et al., 2006a; Glass et al., 2007). In 1987, roughly 23 to 32 percent of women were screened, depending on their age, and by 1997, screening rates were as high as 74 percent among women ages 50–64 (Breen et al., 2001). Increased screening allowed for the earlier detection of tumors and for the detection of tumors that might never have progressed. When more tumors are detected at earlier stages, it will appear as if incidence rates are rising even if they are not, or are rising more rapidly than they actually are.
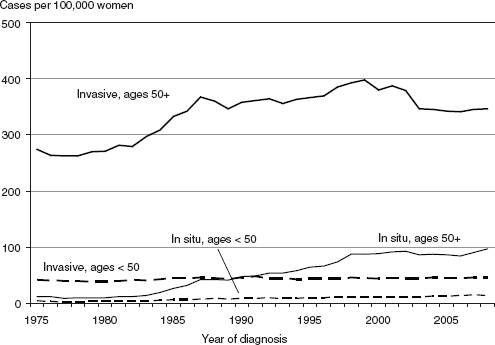
FIGURE 2-2 Age-adjusted incidence of invasive and in situ breast cancer in women, United States, 1975–2008.
SOURCE: NCI (2011).
A decline in breast cancer incidence occurred between 1999 and 2003 (Figure 2-2), principally in ER+ tumors in women ages 50–69 (Jemal et al., 2007). The decline is widely attributed to reductions in the use of hormone therapy (HT) (Clarke et al., 2006; Ravdin et al., 2007; Robbins and Clarke, 2007). In 1998, the Heart and Estrogen/Progestin Replacement Study (HERS) reported that use of combined estrogen–progestin HT failed to show an anticipated protective effect against coronary heart disease and was associated with an increase in risk for blood clots (Hulley et al., 1998). The subsequent publication of findings from the Women’s Health Initiative confirmed the lack of benefit for heart disease and also showed an increased risk for breast cancer with use of combined estrogen–progestin therapy (Writing Group for the Women’s Health Initiative Investigators, 2002). Reports from these studies were a major factor in the decline in use of HT.
As reflected in Figure 2-2, a recent analysis found that for 2003–2007 incidence rates of invasive cancer did not significantly change, although use of HT continued to decline (DeSantis et al., 2011). Use of screening mammography in 2008 remained similar to rates seen in 1997 (Breen et al., 2011). Rates of in situ cancer among older women also rose somewhat
in the 1980s and into the 1990s, but they have remained relatively stable since the late 1990s.
Although the perception is widespread that breast cancer is becoming more common among young women, the best data available indicate that invasive breast cancer incidence rates have been almost unchanged since 1975 in women ages 20–49 (Figure 2-2). What has changed is the rate of in situ breast cancer, which has been rising since the introduction of mammography screening in the 1980s (Breen et al., 2001; Kerlikowske, 2010). The perception that breast cancer is increasing in younger women may come from several factors. First, any cancer diagnosis in a young woman in her prime working and reproductive years is notable, emotionally laden, and an event that will gain attention in many settings. An analysis of vignettes about breast cancer in popular magazines found that nearly half the stories were about women who were diagnosed before age 40 (Burke et al., 2001), a group that accounts for approximately 5 percent of cases (ACS, 2011a). Second, diagnosis of cases of “carcinoma in situ,” especially DCIS, has increased, but its relation to invasive cancer can be unclear to women, at least in part because of the terminology and because of the aggressive treatment that may be recommended (De Morgan et al., 2002; Partridge et al., 2008; Liu et al., 2010). As noted, even within the research and medical communities, the natural history of DCIS is poorly understood, so the proportion of DCIS cases that would become invasive if untreated is unclear (Allred, 2010).
Race and Ethnicity
Differences can be seen in the age patterns and trends in breast cancer among the country’s racial and ethnic groups. For 2004–2008, the overall incidence of breast cancer was 136 cases per 100,000 among non-Hispanic white women, 120 per 100,000 among African American women, 94 per 100,000 among Asian and Pacific Islander women, and 78 per 100,000 among Hispanic women (who can be of any race) (NCI, 2011).3
For African American women, the lower incidence rates compared with white women are most evident at older ages (Figure 2-3). However, incidence rates are higher among African American women under age 45. At ages 30–34, for example, African American women have an incidence of breast cancer of 31.8 cases per 100,000, compared with a rate of 25.8 for
_________________
3Throughout the report, incidence rates such as these are age-adjusted using the U.S. standard population for 2000. Age adjustment applies each group’s incidence rates at specific ages to a single common population, the U.S. population for 2000 in this case. This process ensures that comparisons of rates are not affected by differences among the groups the age distribution of their populations.
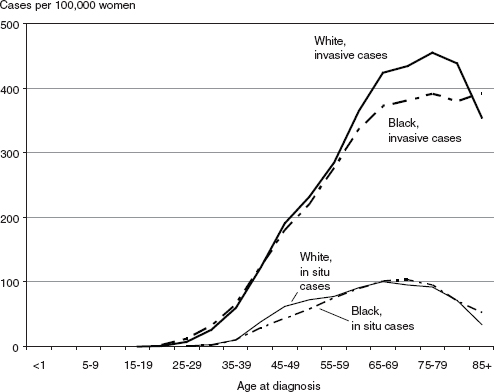
FIGURE 2-3 Age-specific incidence rates for invasive and in situ breast cancer among white and black women in the United States, 2004–2008.
SOURCE: NCI (2011).
white women in that age group (NCI, 2011). At ages 40–44 the differences are smaller; the incidence rates are 123.6 for African American women and 122.4 for white women.
Despite ongoing efforts to improve detection and treatment of breast cancer for all women, African American women continue to experience greater mortality from breast cancer compared to women from other ethnic and racial groups. Surveillance, Epidemiology, and End Results (SEER) data from the National Cancer Institute show that the 5-year survival rate for women diagnosed with breast cancer during the period 2001–2007 was 77 percent among African American women and 91 percent among white women (NCI, 2011). These differences in breast cancer survival have been attributed in part to a higher proportion of African American women being diagnosed with advanced-stage disease; only 51 percent of breast cancers among African American women are localized at diagnosis compared with 61 percent of cancers among white women (NCI, 2011). Among women diagnosed with localized cancer, the 5-year survival rate for 2001–2007 was
93 percent for African American women and 99 percent for white women (NCI, 2011), reflecting a smaller but persistent difference in outcomes. Other factors contributing to poorer survival rates for African American women may include less access to early detection and treatment services as well as differences in tumor characteristics.
Among Hispanic women, the incidence of breast cancer is consistently lower than for non-Hispanic white women or African American women, with greater differences at older ages (NCI, 2006; Hines et al., 2010; Liu et al., 2011). Data from California show that the incidence of breast cancer for the period 1988–2004 was lower among the foreign-born Hispanic women: 68.2 per 100,000 for the foreign-born, 93.8 per 100,000 for U.S.- born Hispanic women, and 125.7 per 100,000 for non-Hispanic white women (Keegan et al., 2010). Approximately 40 percent of the Hispanic population living in the United States in 2007 was born in other countries (Grieco, 2010).
Analysis of the breast cancer experience of Hispanic women is still limited and based primarily on populations in specific areas of the United States, such as California (e.g., Keegan et al., 2010; Liu et al., 2011) or the Southwest (e.g., Hines et al., 2010). Additional research will be needed to assess whether the observations in these areas are representative of the experience of Hispanic women who live in other parts of the country and whose countries of origin and history of residence in the United States may differ from those of the women in the available studies.
The incidence of breast cancer has also traditionally been lower in Asian women, compared to white and black women, as reflected in both international and U.S. surveillance data (Stanford et al., 1995; Parkin et al., 1997, 2005; Jemal et al., 2005; Joslyn et al., 2005; Miller et al., 2008). Incidence rates commonly transition to higher levels as Asian women who migrate to the United States and their descendents experience greater acculturation. This pattern of increasing incidence among immigrants is often cited as evidence for the influence of social and environmental factors in disease risk because genetic factors are unlikely to be able to account for differences from the rates in their countries of origin (Buell, 1973; Thomas and Karagas, 1987; Ziegler et al., 1993; Kolonel and Wilkens, 2006).
Evaluating breast cancer incidence in the Asian and Pacific Islander population4 is challenging because it is highly heterogeneous, with more than 60 distinct ethnicities. There is increasing evidence that the aggregate data on breast cancer incidence for these women tend to obscure large differences, including striking elevations in incidence for some subgroups (Deapen et al., 2002; Keegan et al., 2007; McCracken et al., 2007; Miller
_________________
4The Asian and Pacific Islander populations are combined as a standard reporting category for race and ethnicity for many federal data collection activities.
et al., 2008). Moreover, two studies that used different methods for assessing nativity suggest that young U.S.-born women from some Asian groups, especially women of Japanese and Filipina ancestry, are actually experiencing a higher risk for breast cancer than their white or African American contemporaries (Gomez et al., 2010; Reynolds et al., 2011).
Although Asian and Pacific Islanders, as a group, are less likely to receive an initial diagnosis of late-stage breast cancer than non-Hispanic white women (Hedeen et al., 1999; Morris and Kwong, 2004), foreign-born Asian women and some ethnic groups, including Hawaiians and South Asian Indians, are diagnosed with significantly more late-stage tumors than non-Hispanic white women (Li et al., 2003). Likewise, data from the 2001 California Health Interview Survey suggest that Asian women and Pacific Islander women have lower rates of mammography screening (67.2 percent and 63.4 percent, respectively) than non-Hispanic white women (78.1 percent) (Ponce et al., 2003a). The differences are further accentuated when disaggregated by ethnicity (53.1 percent among Korean women, 56.6 percent among Cambodian women) (Ponce et al., 2003b).
Racial and ethnic differences are also seen in terms of tumor types. The likelihood of having triple negative breast cancer, which is more difficult to treat, is significantly higher in African American women compared to women from other racial and ethnic groups (Bauer et al., 2007; Kwan et al., 2009; Stead et al., 2009). An analysis of SEER data for California found that African American women had a 1.98 percent lifetime risk of developing triple negative breast cancer, whereas Hispanic women had a 1.04 percent lifetime risk and white women had a 1.25 percent risk (Kurian et al., 2010). A high prevalence of triple negative tumors has also been reported in breast cancer cases from Nigeria and Senegal; of 507 cases, 27 percent were triple negative (Huo et al., 2009).
Reproductive Risk Factors
Several factors that are generally considered associated with increased risk for breast cancer include having a family history of the disease, particular reproductive characteristics (e.g., earlier age at menarche, later age at menopause, later age at first live birth), and certain forms of benign breast disease, as determined by breast biopsies (ACS, 2011a). Greater mammographic density, which reflects a higher proportion of connective and epithelial tissue in the breast, is a physiologic characteristic that is consistently associated with increased risk of breast cancer (Boyd et al., 2010). Studies in twins indicate that it is a heritable trait (e.g., Boyd et al., 2002; Ursin et al., 2009).
Differences in breast cancer incidence among population groups may reflect, in part, differences among them in the patterns of these types of risk
factors. For example, data from the Third National Health and Nutrition Examination Survey (NHANES III) show that the median age at menarche for non-Hispanic black girls is 12.06 years compared to 12.25 years for Mexican American girls, and 12.55 years for non-Hispanic white girls (Chumlea et al., 2003).
In a review of epidemiologic studies, Bernstein and colleagues (2003) also found differences between African American and white women in reproductive risk factor profiles. For example, African American women have a higher birth rate than white women until age 30. This is important because while there may be a short-term increase in breast cancer risk immediately following pregnancy, earlier childbearing and higher numbers of births appear to be associated with a long-term reduction in risk. Lactation has been associated with a reduced risk of developing breast cancer; it induces additional differentiation in the breast and delays the re-initiation of ovulation. Studies included in the review conducted by Bernstein et al. (2003) found that, compared to African American women, white women are about twice as likely to breastfeed, and their cumulative time spent breastfeeding is longer.
Differences in breast cancer incidence and reproductive risk factor profiles have also been reported for Hispanic and non-Hispanic white women (e.g., Hines et al., 2010). Both premenopausal and postmenopausal Hispanic women had a higher prevalence of factors that have been associated with decreased breast cancer risk, including younger age at first birth and greater parity. But they were also more likely to have a younger age at menarche and to breastfeed less, characteristics associated with greater risk.
However, some of the associations between reproductive factors and breast cancer risk may be stronger for white non-Hispanic women than for women of other races and ethnicities. Hines and colleagues (2010) found that among premenopausal Hispanic women, only late age at first birth had a statistically significant association with increased risk of breast cancer. Reproductive factors were not associated with breast cancer risk among postmenopausal Hispanic women.
The contribution of differences in patterns of reproductive factors may also be influenced by racial and ethnic differences in risk for particular subtypes of breast cancer. Some reproductive factors appear to be more closely associated with ER+/PR+ tumors (Althuis et al., 2004; Ma et al., 2006) or lobular (versus ductal) tumors (Kotsopoulos et al., 2010; Newcomb et al., 2011). The risk for ER–/PR– and triple negative breast cancers is greater for African American women than for non-Hispanic white women, and reproductive factors have a more limited influence on risk for these forms of breast cancer.
A BROAD PERSPECTIVE ON THE ENVIRONMENT
As noted in Chapter 1, the committee adopted a broad interpretation of the environment that encompasses all factors that are not directly inherited through DNA. This definition allows for the consideration of a broad range of factors that may be encountered at any time in life and in any setting: the physiologic and developmental course of an individual, diet and other ingested substances, physical activity, microbial agents, physical and chemical agents encountered at home or work, medical treatments and interventions, social factors, and cultural practices. Figure 2-4 illustrates the multiple levels of biologic and social organization through which potential environmental exposures can influence breast cancer, and Figure 2-5 illustrates one approach to integrating this socio-ecologic perspective into investigation of potential contributions to breast cancer over the life course.
Many of these environmental influences overlap. For example, the physical environment encompasses medical interventions, dietary exposures to nutrients, energy and toxicants, ionizing radiation, and chemicals from industrial and agricultural processes and from consumer products. These in turn are influenced by the social environment, because cultural and economic factors influence diet at various stages of life, reproductive choices, energy balance, adult weight gain, body fatness, voluntary and involuntary physical activity, medical care, exposure to tobacco smoke and alcohol, and
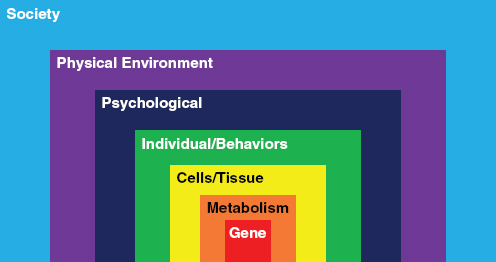
FIGURE 2-4 Multiple levels on which environmental exposures may act to influence breast cancer.
SOURCE: Personal communication, R. A. Hiatt, University of California, San Francisco, September 16, 2010.
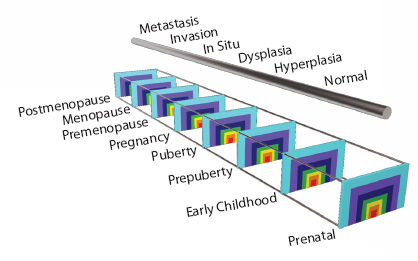
FIGURE 2-5 A schematic illustration of the potential for environmental exposures at various levels and times over the life course to influence the initiation and progression of breast cancer.
SOURCE: Personal communication, R. A. Hiatt, University of California, San Francisco, September 16, 2010.
occupational exposures, including shift work. Exposures at the tissue level are further influenced by metabolic and physiologic processes that modify the body’s internal environment.
A full appreciation of environmental influences on breast cancer calls for an analysis at multiple levels (Anderson and May, 1995), from genetic and cellular mechanisms to the influence of societal factors. Applying this perspective to research requires a transdisciplinary approach. A previous Institute of Medicine committee advanced this socio-ecologic model as a way to understand the relationship of health and disease to complex societal influences (IOM, 2000; Smedley and Syme, 2001). Social determinants then encompass various factors: social and economic conditions such as poverty; the conditions of work, and access to health care delivery; the chemical toxicants and pollutants associated with industrial development; and the positive aspects of human settlements that make active living and healthy eating possible (Hiatt and Breen, 2008). The socio-ecologic model also incorporates and augments discoveries in cancer biology and toxicology, in addition to those from the behavioral and social sciences.
Within this framework, the committee’s predominant focus was on exposure to physical and chemical toxicants, and on individual behavior related to diet and physical activity. When possible, the committee examined evidence regarding the implications of the timing of those exposures
across the life course. Although the committee recognizes that the nature of households, families, workplaces, communities, and societies in which people live play a major role in determining these exposures (Hiatt and Breen, 2008), the focus of this report was on the more proximate environmental exposures that may increase the risk of breast cancer. As understanding of the epidemiology, toxicology, and mechanisms of breast cancer continues to improve, efforts to develop effective interventions to mitigate risk may be aided by approaches that include modification of the social determinants of exposure to various risk factors.
INVESTIGATING WHETHER ENVIRONMENTAL FACTORS ARE RELATED TO BREAST CANCER
Efforts to determine whether exposure to an aspect of the environment is related to the development of breast cancer depend on many types of research, including laboratory analyses of the response of cells or tissues (in vitro testing), experimental studies of effects in laboratory animals (in vivo testing), and epidemiologic studies of human subjects. U.S. regulatory agencies, including the Environmental Protection Agency (EPA) and the Food and Drug Administration (FDA), require a variety of in vitro and animal tests for cancer and other endpoints for licensing or registering pesticides, food additives, and pharmaceuticals (NRC, 2006). In laboratory studies, exposures are determined by the researcher, but in studies of human subjects, exposure assessment becomes a crucial part of the investigation.
Reviewed briefly here are basic features of this range of studies and of exposure assessment. Chapter 4 provides discussion of the challenges in using these various research tools to study breast cancer and draw valid conclusions about environmental risk factors.
In Vitro Testing
In vitro testing makes use of artificial environments to study tissues, cells, and cellular components. In the context of breast cancer, this type of testing allows for detailed examination of behavior of specific parts of larger, more complex organisms. Increasingly, in vitro testing allows for rapid analysis of a large number of variables, such as changes in gene expression. Although in vitro testing does not capture the critical interactions of the multiple systems in an intact organism, it provides a means to explore biological processes that are otherwise difficult to isolate.
In vitro tests for genotoxicity are an integral part of screening chemicals for their potential to cause DNA damage and thereby contribute to tumor formation. Various assays are used to assess gene mutations (e.g., Ames test, mouse lymphoma TK+/– assay) and structural or numerical aberrations in
chromosomes (e.g., Chinese hamster ovary cells or mouse lymphoma TK+/– assay). Chemicals that show potential for genotoxicity are often avoided in product development programs for pesticides and pharmaceuticals.
Advances in molecular genetics, proteomics, and immunohistochemistry are fine-tuning investigations of mechanisms of action and treatment for breast cancer through studies of gene amplification, hormone receptor binding, biomolecular analysis of cells derived from tissue microdissection, and genome and transcriptional analysis (Thayer and Foster, 2007; Pasqualini, 2009). For example, such tools have led to the development of selective estrogen receptor modulators (SERMs; e.g., tamoxifen and raloxifene) and down-regulators (SERDs) that have provided both new therapeutic approaches to treating breast cancer and pharmacologic approaches to the prevention of breast cancer in some women (McDonell and Wardell, 2010). Next-generation SERMs and SERDs are now in clinical trials. Such tools will also allow a deeper understanding of the cell signaling events that are disrupted in the process of breast carcinogenesis, providing a rational basis from which to identify potential environmental influences on breast cancer risk. For example, they can aid in studying the potential role of melatonin and circadian disruption as a modulator of breast cancer risk (Blask et al., 2011). High-throughput microarray methods are used to examine various global gene expression changes related to high tumor aggressiveness, potentially leading to a new breast cancer molecular taxonomy and multigene signatures that might predict outcome and response to systemic therapies (Colombo et al., 2011).
Cell cultures from normal breast tissue and from breast tumors are being used to screen for the potential for chemicals to promote the growth of breast cancer cells or to evaluate the effectiveness of various therapeutic agents. Immortalized human breast cell lines (e.g., MCF-10F) have been established to study various aspects of tumorigenicity (e.g., Russo et al., 2002), and immortalized breast cancer cell lines (e.g., MCF-7) to study tumor progress and response to therapeutic agents (Wistuba et al., 1998; Fillmore and Kuperwasser, 2008). In vitro tests of the potential for chemicals to interact with estrogen, androgen, and thyroid hormonal systems may eventually be applied to most pesticides to generate other mechanistic information related to carcinogenicity. At present, while much has been learned about the potential for hormonal activity for some chemicals, data are limited on many others. In 2009, EPA required that about two dozen pesticides be screened for these effects (EPA, 2009).
Whole Animal (In Vivo) Studies of Carcinogenicity
Rodents have long been used to study mammary tumorigenesis. Specific rat and mouse strains have been selected for routine screening of chemi-
cals and pharmaceuticals for carcinogenic effects. This testing is generally intended to detect any indication of carcinogenicity at any site in the body; it is not designed to identify likely sites for specific human cancers, such as breast cancer. EPA’s (2005) Guidelines for Carcinogen Risk Assessment notes, however, that certain modes of action (e.g., disruption of thyroid function) will have consequences for particular tissues and that this provides a basis for anticipation of site concordance between rodents and humans in certain cases. Rodent models are also widely used by research scientists to investigate mammary carcinogenesis and the effects of timing and combinations of exposure to environmental factors. Challenges in using these models are discussed in Chapter 4.
Scope of Carcinogenicity Testing
Carcinogenicity testing in two species, typically rodents, is part of the standard battery of tests required for most pharmaceuticals, pesticides, and some food additives. Registration or licensing for marketing for products that require such approval involves establishing to the satisfaction of the appropriate government agency that the compound can be safely used under the registered use scenarios or, in the case of a pharmaceutical, that it has an adequate “risk–benefit” ratio.
Premarket testing of chemicals used in consumer products and in industry is rarely undertaken because the federal government has limited authority to require it under the Toxic Substances Control Act, which was enacted in 1976 (GAO, 2009). Only about 15 percent of the notices submitted to EPA for manufacturing or importing new industrial chemicals have any specific health or safety data (GAO, 2009). Instead, considerable reliance is placed on evaluating, qualitatively or through modeling, the similarities in structure to compounds that are carcinogenic or mutagenic (GAO, 2005; NRC, 2006). Each year, the National Toxicology Program (NTP) of the National Institute of Environmental Health Sciences conducts carcinogenicity screening for a few chemicals that would otherwise go untested. These chemicals are selected based on concern about their potential toxicity or the extent of human exposure. In 2007, the European Union began transferring responsibility for safety testing to manufacturers under the REACH program (Registration, Evaluation, Authorisation and Restriction of Chemical Substances) (European Chemicals Agency, 2007).
Carcinogenicity testing is also generally not required before new cosmetics and dietary supplements are marketed (FDA, 2005, 2009). Manufacturers are responsible for identifying ingredients and declaring that they are safe for the intended use. The FDA does have the authority to remove products from the market if they are found to be adulterated or misbranded.
NTP Carcinogenicity Study Protocols
Whole-animal studies are conducted as part of many types of academic and industry research on breast cancer and carcinogenicity. These studies can vary widely in design, depending on their purpose. For formal carcinogenicity reviews by EPA or the International Agency for Research on Cancer (IARC), the NTP study designs for whole-animal bioassays typically represent a recognized standard for carcinogenicity testing.
Under NTP (2006) protocols, carcinogenicity testing is usually based on a 2-year chronic dosing program. Testing uses three or more exposure-level groups and one unexposed control group, with separate test groups for male and female animals. Each group typically has 50 animals. The highest dose used in the assays is usually the maximally tolerated dose, with the aim of maximizing the ability to detect effects in small numbers of animals and minimizing the loss of animals from acutely toxic effects of the test substance. Dosing usually begins when the animals are 5 to 6 weeks of age. Under revised NTP (2010) study designs, rats (but not mice) may receive in utero and lactational exposure to the test substance, which will allow the testing procedures to identify adverse effects associated with exposures at the very earliest times of life.
The NTP currently uses Harlan Sprague Dawley rats, and one strain of mice, the B6C3F1 hybrid. Previously, other rat strains have been used (typically F344/N, although some chemicals were tested in Sprague Dawley and Osborne Mendel strains). Tests of similar design are required for pesticide registration (EPA, 1998) and pharmaceutical testing (FDA, 1997), although the animal strains used typically differ, and in utero testing is rarely performed (EPA, 2002).
At the end of the 2-year test period, the surviving animals are killed and necropsied. Any animals that die during the study period are also necropsied. To date, the NTP (2011) has tested more than 500 chemicals. Overall evaluation of the test results for carcinogenic hazard includes consideration of both malignant and benign tumors found anywhere in the animals.
Assessing the Process of Carcinogenesis and Susceptibility to Environmental Exposures
In addition to the use of experimental animals for standardized carcinogen bioassays, several animal models of chemically induced breast cancer have been used to evaluate (1) the cellular and molecular development and progression of breast cancer, and (2) the ability of environmental and developmental factors to modify breast carcinogenesis. The two most common models use induction of mammary tumors in rodents by the administration of N-methyl-N-nitrosourea (MNU) or 7,12-dimethylbenz[a]anthracene
(DMBA) (Russo and Russo, 1996; Thompson and Singh, 2000; Medina, 2010). In rats, these carcinogen-induced tumors arise from terminal end buds, which are similar in structure to the terminal ductal lobular unit in the human breast. Similar to human breast cancers, these chemically induced mammary carcinomas have altered expression of proteins that regulate cell growth and differentiation (e.g., HER2), and most rat mammary tumors express estrogen and progesterone receptors. For example, rat mammary tumors induced by MNU appear to be similar to low- to intermediate-grade human breast cancers that are ER+ and noninvasive (Chan et al., 2005).
Although these rodent models differ in important ways from human breast cancer (e.g., specific gene mutations, metastatic potential), they have been used extensively to explore mechanisms of mammary carcinogenesis and ways environmental factors influence that process. For example, studies have used DMBA-induction of mammary tumors in rats to demonstrate that obesity enhances tumor incidence and shortens the time to tumor development (e.g., Hakkak et al., 2005). These models make it possible to explore the impact of exposure to environmental agents at different times in life. For example, as discussed in Chapter 3, dioxins do not induce mammary tumors in rats in the 2-year chronic bioassay, but rats with prenatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) have shown altered mammary gland differentiation and an increased susceptibility to DMBA-induced mammary tumors (Jenkins et al., 2007). However, prenatal exposure of mice to TCDD delayed DMBA-induced tumor formation by 4 weeks relative to controls, and resulted in lower tumor incidence throughout the 27-week time course (Wang et al., 2011). The authors suggested that activation of the aryl hydrocarbon receptor (AhR) by TCDD slows the promotion of preneoplastic lesions to overt mammary tumors in mice. Interpreting such differences in response between rats and mice is among the challenges discussed in Chapter 4.
Another example of the use of whole animal models of carcinogen-induced mammary tumors in evaluating environmental risk factors for breast cancer was provided by La Merrill et al. (2009). Because some forms of breast cancer are associated with greater adiposity, these authors used three mouse models of breast cancer to examine the effect of prenatal TCDD exposure and high- or low-fat diet on physical characteristics associated with metabolic syndrome. The models were the DMBA mouse model and two different transgenic models of ER– breast cancer. Each model showed a different response (e.g., increase in body fat with or without changes in fasting glucose), but the TCDD exposure was associated with effects (reduced triglycerides) in only one of the models and only in the animals on the high-fat diet. The variation in response in models such as
these may help in exploring the variability in human susceptibility to factors that increase risk of breast cancer.
Epidemiologic Studies5
Case–control studies compare exposures to the factor of interest (an “exposure”) among individuals who have a disease of interest (cases) and individuals who do not have the disease (controls). The controls should come from a population that is judged comparable to the one from which the cases were identified (e.g., people with similar characteristics from the same community or the same hospital). Because of their more efficient study design, case–control studies are often done when a disease is rare or to explore a suspected association within a shorter period than a cohort approach would require. They are usually retrospective, looking back at exposure histories among the cases and controls. But assessing the timing of the exposures can be challenging. Among cases, it can difficult to be certain that the exposure preceded the disease. Studies with retrospective data collection that involves patient interviews can be subject to recall bias.6 For example, cases, who have been diagnosed with cancer and who are likely to have thought carefully about why they have it, may be more likely to recall an exposure than controls, who do not have the disease and therefore may not have thought quite as carefully about whether they may have been exposed.
Cohort studies compare the occurrence of health outcomes among groups with different levels of exposure to a factor of interest. These studies may be prospective, beginning before individuals have been diagnosed with a disease and following them for a given period of time, or retrospective, using records or interviews to collect information about past exposures and health outcomes. For example, cohorts of smokers and nonsmokers could be followed to assess the incidence of lung cancer in each group. A prospective study ensures that exposure precedes diagnosis but exposure levels are not controlled by the investigator. Collection of information on exposures that vary over time is difficult and often not carried out with sufficient detail. Cohort studies avoid the problem of recall bias, but they can be subject to other forms of bias. The time frame for prospective cohort studies may be several years or as long as decades, depending on the hypothesized nature of the relation between the exposure(s) and the disease being studied. With breast cancer, for example, the disease may become evident only many years after an exposure of interest, so cohorts must be followed long enough to
_________________
5Additional information about study design and analysis is available from sources such as Rothman (2002) and Szklo and Nieto (2004).
6Forms of bias in epidemiologic studies are discussed in Chapter 4.allow for this interval. If childhood or prenatal exposures play a role, then it could require five or more decades of follow-up. Extended follow-up of a study population can be expensive and administratively challenging. A listing of approximately 50 cohorts in the United States and other countries that have investigated breast cancer risks has been compiled by the Silent Spring Institute (2011). The listing illustrates the variation in characteristics and size of these study populations.
Controlled trials, also referred to as clinical trials, are experiments in which the investigator makes the decision as to who is assigned to receive the treatment (exposure) versus being in the comparison group. If the assignment is made at random and the sample size is adequate to ensure that confounding was minimized by the random assignment, then the result of the experiment can have a causal interpretation. For example, to determine if a medication that lowers serum cholesterol prevents heart attacks, it is possible to treat one group of individuals with a cholesterol-lowering medication and compare their cholesterol levels and incidence of heart attacks to those of a control group that did not receive the intervention. If the study is sufficiently large (in this case, takes place over a long enough time period for the number of events in the comparison group to be sufficient) and the assignment to treatment is random, then any reduction in incidence of heart attacks among the treated group, relative to the controls, can be interpreted to be a causal one. The comparison of measurements of cholesterol can also be used in drawing conclusions about the mechanism of action of the medications, although other mechanisms would also need to be taken into account. Studies that are investigating preventive care may be referred to as intervention trials. If an exposure is potentially harmful, controlled trials can examine ways to minimize or eliminate the exposure, but studies that deliberately expose participants to something expected to be harmful are not done. An optimal design of a clinical trial includes not only random assignment of study participants to the treatment or comparison group but also blinding of study participants and researchers to those assignments. Such blinding will minimize bias in the assessment of the outcomes.
Exposure Assessment
Studying the potential effects of environmental factors on risk for breast cancer requires some basis for distinguishing the women who have been exposed to the factor from those who have not. Exposure assessment is the process of establishing that an exposure has occurred and determining critical features of the exposure, including who is exposed and the magnitude, route, and timing of exposure. Errors in classifying who is more and who is less exposed (exposure misclassification) can limit the ability of a
study to determine whether the environmental factor is associated with an increase or decrease in risk for breast cancer.
The approach to exposure assessment may depend on the type of study, the nature of the environmental factor of interest, the way exposure occurs, and the tools available to measure the exposure. In clinical trials or intervention trials, the population to be exposed and the exposure are determined in advance by the researchers. Even so, study participants may deviate from their prescribed exposures. In cohort and case–control studies, exposure status can sometimes be objectively determined (e.g., by measuring weight), but it often depends on reports by study participants of past or present experience (e.g., exposure to tobacco smoke in childhood or use of specific products in the home). Researchers may also use indirect means to estimate exposures, such as residence in a particular locality or distance from a particular source of concern (e.g., an air pollution source). Exposure to some chemicals can be established with tests of biologic specimens (e.g., blood, urine), but many exposures are not detectable in this manner and collection of specimens may not be possible. Because the first steps in breast cancer may begin decades before the diagnosis, relevant exposures may occur several decades before a cancer is detected.
Historically, studies in occupational settings have been an important means for identifying chemical carcinogens. The types and amounts of chemicals used may be documented, and exposure levels may be higher than in other settings. Studies in an occupational setting may be able to draw on records of job histories, understanding of production processes and chemicals used, or data from personal or area sampling. Exposure of certain workers to some chemicals may be thousands of times greater (or more) than that experienced by the general public, while other workers with different job tasks might experience a wide range of exposures. These pronounced variations in exposure allow for firmer conclusions as to whether exposure is associated with risk of disease. When exposure levels are low, contrasts between the exposed and unexposed are smaller, and associations with differences in disease risk may be more difficult to detect. However, the relatively small number of women in industries with heavy exposures, except during World War II, has limited the opportunity to study risks for breast cancer in those settings.
A potentially hazardous environmental factor can only pose a risk when it can enter the body and interact with tissues where it can do harm. Thus, an understanding of the possible points of entry of a given substance into the body, called “routes of exposure,” is fundamental to evaluating its potential effects. These routes of exposure are inhalation, ingestion, or contact with the skin (dermal exposure). In occupational settings, inhalation is frequently the primary route of exposure, with dermal contact as a secondary route. In the general population, ingestion and dermal expo-
sure play a large role, but inhalation is highly relevant for tobacco smoke and other air pollutants. Sometimes potential routes of exposure can be overlooked. For example, when taking showers, people experience dermal exposure to chemicals in the water supply, but showers also present an opportunity to inhale (typically low levels of) any water contaminants that readily volatilize.
The potential effect of an environmental exposure is usually strongly influenced by the magnitude of that exposure—the dose. A higher dose of a hazardous exposure is generally more likely to be associated with adverse health effects than a lower dose is. Factors that influence dose include the duration and frequency of exposure and the biologic processes that govern the absorption, distribution, metabolism, excretion, and storage of a substance in the body. The results of these toxicokinetic processes differ depending on the substance introduced into the body. Some ingested chemicals, for example, are poorly absorbed and rapidly excreted, while others may be readily absorbed, transformed by metabolism into new substances, and possibly stored in body tissues such as fat. The route of exposure may influence how the body responds to a substance. Also, differences among individuals in their genetics or exposure to other risk factors can result in differing responses to equal doses of a substance.
Estimates of disease risk associated with a factor of interest—such as a personal characteristic (e.g., age), an environmental exposure (e.g., alcohol consumption or radiation exposure), or a medical treatment (e.g., a prescribed medication)—can be measured in multiple ways, including absolute risk, relative risk, hazard ratios, odds ratios, attributable risk, population attributable risk, and number needed to treat (NNT) or number needed to harm (NNH). The measure that is used depends on the study design, the available data, and in some cases the purpose for which the information is presented.7
In case–control studies, the prevalence of the factor of interest among cases and controls is compared using an odds ratio: the odds that a case is exposed compared to the odds that a control is exposed. Odds ratios of 1.0 mean that cases and controls were equally likely to have been exposed, and therefore the exposure is not associated with the disease and it is not a risk factor. An odds ratio that is statistically significantly less than 1.0 means that cases were less likely to have been exposed than controls. An odds ratio that is statistically significantly greater than 1.0 indicates that the
_________________
7Additional methodologic information is available from sources such as Rothman (2002) and Jewell (2004).
exposure is more likely to be reported among the case group than among the control group, indicating that the exposure is statistically associated with the disease, and thus is a potential risk factor for the disease.
Cohort studies typically use the measure of relative risk or the hazard ratio. Relative risk is a ratio of the absolute risk (incidence) of disease in an exposed group (or groups with different levels of exposure) to the absolute risk (incidence) of disease in an unexposed group (or some other designated comparison group). A hazard ratio incorporates information on the pace at which events (e.g., cases of breast cancer) occur over the course of a study. Clinical trials also use relative risk and hazard ratios. The relative risk is interpreted in much the same way as the odds ratio. A relative risk of 1.0 means the exposure is not associated with development of disease; a ratio that is statistically significantly less than 1.0 means that those who were exposed were less likely to develop the disease than those who were not (indicating that the exposure is protective); and a ratio that is statistically significantly greater than 1.0 means that the exposure is associated with the disease, indicating that it is potentially a risk factor for the disease.
Relative risk estimates and odds ratios represent an estimate of the strength of the association of a risk factor with breast cancer, but by themselves they do not provide insight into the underlying incidence of the disease and the absolute impact of a given factor. A relative risk of 2.0 means that a factor is associated with a doubling of the incidence of the health outcome in the exposed group compared to the unexposed. But this can mean an increase to 2 cases per 100,000 people or 200 cases per 100,000 people, depending on whether the underlying incidence is 1 case per 100,000 people or 100 cases per 100,000 people. Measures such as NNT and NNH are other ways of relating estimates of risk to absolute numbers. NNT is the number of people who would have to receive a treatment during a given time period for one person to benefit.
Other measures that are used to assess the impact of a risk factor include attributable risk (AR) and population attributable risk (PAR). The AR is defined as the percentage of cases that occur in the exposed group that are in excess of the cases in the comparison group. The PAR is a population-based measure of the percentage of excess cases associated with the exposure of interest that also takes into account the distribution of the risk factor within the population. If a risk factor is rare, it may contribute only a small proportion of a population’s disease risk, even if the incidence of the disease is much higher among those who are exposed (which would produce a high relative risk). To adequately estimate the PAR requires high-quality studies in which confounding and overlapping contributions from multiple factors are analyzed appropriately. There are numerous pitfalls in interpreting the PAR (discussed in Chapter 4) (Rockhill et al., 1998). Ideally, the PAR provides information on the percentage of disease that can
be eliminated by avoiding the exposure, but the variation in estimates of PAR underscores how difficult it is to separate the effects from multiple risk factors. Because of this problem, and because PARs for individual factors cannot simply be added together, PARs are sometimes calculated for a group of factors rather than single factors. Appendix D shows, for instance, a range of estimated PAR values (see e.g., physical activity or hormone therapy). These ranges may reflect variation in the contribution of a given factor across different populations, or variation in the degree to which the different studies adequately controlled confounding, or a combination of the two.
Overall, breast cancer becomes increasingly common as women grow older, but the patterns of the disease vary among women in different racial and ethnic groups. These differences are likely to reflect the influence of a mix of genetic and environmental factors. Although the scope of environmental influences can be understood to encompass cultural and societal factors, most of the human, animal, and mechanistic research to date has focused more narrowly on individual exposures and the related biological processes. In the following chapter, the committee examines evidence regarding a set of environmental factors that illustrate varied types of exposures that may occur and the range of evidence available to assess whether exposure is associated with increased risk of breast cancer.
ACS (American Cancer Society). 2011a. Breast cancer facts and figures 2011–2012. Atlanta, GA: ACS. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-030975.pdf (accessed October 24, 2011).
ACS. 2011b. Cancer facts and figures 2011. Atlanta, GA: ACS. http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-figures-2011 (accessed June 22, 2011).
Allred, D. C. 2010. Ductal carcinoma in situ: Terminology, classification, and natural history. J Natl Cancer Inst Monogr 2010(41):134–138.
Althuis, M. D., J. H. Fergenbaum, M. Garcia-Closas, L. A. Brinton, M. P. Madigan, and M. E. Sherman. 2004. Etiology of hormone receptor-defined breast cancer: A systematic review of the literature. Cancer Epidemiol Biomarkers Prev 13(10):1558–1568.
Anderson, L. M., and D. S. May. 1995. Has the use of cervical, breast, and colorectal cancer screening increased in the United States? Am J Public Health 85(6):840–842.
Anderson, W. F., I. Jatoi, and S. S. Devesa. 2006a. Assessing the impact of screening mammography: Breast cancer incidence and mortality rates in Connecticut (1943–2002). Breast Cancer Res Treat 99(3):333–340.
Anderson, W. F., R. M. Pfeiffer, G. M. Dores, and M. E. Sherman. 2006b. Comparison of age distribution patterns for different histopathologic types of breast carcinoma. Cancer Epidemiol Biomarkers Prev 15(10):1899–1905.
Anderson, W. F., B. E. Chen, L. A. Brinton, and S. S. Devesa. 2007. Qualitative age interactions (or effect modification) suggest different cancer pathways for early-onset and late-onset breast cancers. Cancer Causes Control 18(10):1187–1198.
Anderson, W. F., I. Jatoi, J. Tse, and P. S. Rosenberg. 2010. Male breast cancer: A population-based comparison with female breast cancer. J Clin Oncol 28(2):232–239.
Arendt, L. M., J. A. Rudnick, P. J. Keller, and C. Kuperwasser. 2010. Stroma in breast development and disease. Semin Cell Dev Biol 21(1):11–18.
Armes, J. E., L. Trute, D. White, M. C. Southey, F. Hammet, A. Tesoriero, A. M. Hutchins, G. S. Dite, et al. 1999. Distinct molecular pathogeneses of early-onset breast cancers in BRCA1 and BRCA2 mutation carriers: A population-based study. Cancer Res 59(8):2011–2017.
Atchley, D. P., C. T. Albarracin, A. Lopez, V. Valero, C. I. Amos, A. M. Gonzalez-Angulo, G. N. Hortobagyi, and B. K. Arun. 2008. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol 26(26):4282–4288.
Bauer, K. R., M. Brown, R. D. Cress, C. A. Parise, and V. Caggiano. 2007. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California Cancer Registry. Cancer 109(9):1721–1728.
Bernstein, L., C. R. Teal, S. Joslyn, and J. Wilson. 2003. Ethnicity-related variation in breast cancer risk factors. Cancer 97(1 Suppl):222–229.
Blask, D. E., S. M. Hill, R. T. Dauchy, S. Xiang, L. Yuan, T. Duplessis, L. Mao, E. Dauchy, et al. 2011. Circadian regulation of molecular, dietary, and metabolic signaling mechanisms of human breast cancer growth by the nocturnal melatonin signal and the consequences of its disruption by light at night. J Pineal Res 51(3):259–269.
Boyd, N. F., G. S. Dite, J. Stone, A. Gunasekara, D. R. English, M. R. McCredie, G. G. Giles, D. Tritchler, et al. 2002. Heritability of mammographic density, a risk factor for breast cancer. N Engl J Med 347(12):886–894.
Boyd, N. F., L. J. Martin, M. Bronskill, M. J. Yaffe, N. Duric, and S. Minkin. 2010. Breast tissue composition and susceptibility to breast cancer. J Natl Cancer Inst 102(16):1224–1237. Breen, N., D. K. Wagener, M. L. Brown, W. W. Davis, and R. Ballard-Barbash. 2001. Progress in cancer screening over a decade: Results of cancer screening from the 1987, 1992, and 1998 National Health Interview Surveys. J Natl Cancer Inst 93(22):1704–1713.
Breen, N., J. F. Gentleman, and J. S. Schiller. 2011. Update on mammography trends: Comparisons of rates in 2000, 2005, and 2008. Cancer 117(10):2209–2218.
Buell, P. 1973. Changing incidence of breast cancer in Japanese-American women. J Natl Cancer Inst 51(5):1479–1483.
Burke, W., A. H. Olsen, L. E. Pinsky, S. E. Reynolds, and N. A. Press. 2001. Misleading presentation of breast cancer in popular magazines. Eff Clin Pract 4(2):58–64.
Canzian, F., D. G. Cox, V. W. Setiawan, D. O. Stram, R. G. Ziegler, L. Dossus, L. Beckmann, H. Blanche, et al. 2010. Comprehensive analysis of common genetic variation in 61 genes related to steroid hormone and insulin-like growth factor-I metabolism and breast cancer risk in the NCI breast and prostate cancer cohort consortium. Hum Mol Genet 19(19):3873–3884.
Carey, L. A., C. M. Perou, C. A. Livasy, L. G. Dressler, D. Cowan, K. Conway, G. Karaca, M. A. Troester, et al. 2006. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295(21):2492–2502.
CDC (Centers for Disease Control and Prevention). 2010. National Program of Cancer Registries. http://www.cdc.gov/cancer/npcr/about.htm (accessed November 8, 2011).
Chan, M. M., X. Lu, F. M. Merchant, J. D. Iglehart, and P. L. Miron. 2005. Gene expression profiling of NMU-induced rat mammary tumors: Cross species comparison with human breast cancer. Carcinogenesis 26(8):1343–1353.
Chumlea, W. C., C. M. Schubert, A. F. Roche, H. E. Kulin, P. A. Lee, J. H. Himes, and S. S. Sun. 2003. Age at menarche and racial comparisons in U.S. girls. Pediatrics 111(1):110–113.
Clarke, C. A., S. L. Glaser, C. S. Uratsu, J. V. Selby, L. H. Kushi, and L. J. Herrinton. 2006. Recent declines in hormone therapy utilization and breast cancer incidence: Clinical and population-based evidence. J Clin Oncol 24(33):e49–e50.
Colombo, P. E., F. Milanezi, B. Weigelt, and J. S. Reis-Filho. 2011. Microarrays in the 2010s: The contribution of microarray-based gene expression profiling to breast cancer classification, prognostication and prediction. Breast Cancer Res 13(3):212.
De Morgan, S., S. Redman, K. J. White, B. Cakir, and J. Boyages. 2002. “Well, have I got cancer or haven’t I?” The psycho-social issues for women diagnosed with ductal carcinoma in situ. Health Expect 5(4):310–318.
Deapen, D., L. Liu, C. Perkins, L. Bernstein, and R. K. Ross. 2002. Rapidly rising breast cancer incidence rates among Asian-American women. Int J Cancer 99(5):747–750.
DeSantis, C., N. Howlader, K. A. Cronin, and A. Jemal. 2011. Breast cancer incidence rates in U.S. women are no longer declining. Cancer Epidemiol Biomarkers Prev 20(5):733–739.
Easton, D. F., K. A. Pooley, A. M. Dunning, P. D. Pharoah, D. Thompson, D. G. Ballinger, J. P. Struewing, J. Morrison, et al. 2007. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447(7148):1087–1093.
EPA (Environmental Protection Agency). 1998. Health effects test guidelines. OPPTS 870.4200: Carcinogenicity. EPA 712–C–98–211. Washington, DC: Government Printing Office. http://hero.epa.gov/index.cfm?action=search.view&reference_ID=6378 (accessed November 16, 2011).
EPA. 2002. A review of the reference dose and reference concentration processes. EPA/630/P-02/002F. Washington, DC: EPA. http://www.epa.gov/raf/publications/pdfs/rfd-final.pdf (accessed November 17, 2011).
EPA. 2005. Guidelines for carcinogen risk assessment. Washington, DC: EPA. http://www.epa.gov/osa/mmoaframework/pdfs/CANCER-GUIDELINES-FINAL-3-25-05%5B1%5D.pdf (accessed October 23, 2011).
EPA. 2009. Final list of initial pesticide active ingredients and pesticide inert ingredients to be screened under the Federal Food, Drug, and Cosmetic Act. Federal Register 74(71): 17579–17585. http://www.epa.gov/scipoly/oscpendo/pubs/final_list_frn_041509.pdf (accessed October 25, 2011).
European Chemicals Agency. 2007. REACH. http://echa.europa.eu/reach_en.asp (accessed November 8, 2011).
FDA (Food and Drug Administration). 1997. Guidance for industry: S1B testing for carcinogenicity of pharmaceuticals. Rockville, MD: FDA. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm074916.pdf (accessed November 17, 2011).
FDA. 2005. Fair Packaging and Labeling Act, Title 15—Commerce and Trade, Chapter 39—Fair Packaging and Labeling Program. http://www.fda.gov/regulatoryinformation/legislation/ucm148722.htm (accessed November 8, 2011).
FDA. 2009. Overview of dietary supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/ucm110417.htm (accessed November 8, 2011).
Fillmore, C. M., and C. Kuperwasser. 2008. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res 10(2):R25.
Foulkes, W. D., I. M. Stefansson, P. O. Chappuis, L. R. Begin, J. R. Goffin, N. Wong, M. Trudel, and L. A. Akslen. 2003. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 95(19):1482–1485.
Foulkes, W. D., I. E. Smith, and J. S. Reis-Filho. 2010. Triple-negative breast cancer. N Engl J Med 363(20):1938–1948.
GAO (Government Accountability Office). 2005. Chemical regulation: Options exist to improve EPA’s ability to assess health risks and manage its chemical review program. GAO-05-458. Washington, DC: GAO. http://www.gao.gov/new.items/d05458.pdf (accessed December 12, 2011).
GAO. 2009. Chemical regulation: Observations on improving the Toxic Substances Control Act. GAO-10-292T. Washington, DC: GAO. http://www.gao.gov/products/GAO-10-292T (accessed October 24, 2011).
Glass, A. G., J. V. Lacey, Jr., J. D. Carreon, and R. N. Hoover. 2007. Breast cancer incidence, 1980–2006: Combined roles of menopausal hormone therapy, screening mammography, and estrogen receptor status. J Natl Cancer Inst 99(15):1152–1161.
Gomez, S. L., C. A. Clarke, S. J. Shema, E. T. Chang, T. H. Keegan, and S. L. Glaser. 2010. Disparities in breast cancer survival among Asian women by ethnicity and immigrant status: A population-based study. Am J Public Health 100(5):861–869.
Grieco, E. 2010. Race and Hispanic origin of the foreign-born population in the United States: 2007. http://www.census.gov/prod/2010pubs/acs-11.pdf (accessed November 8, 2011).
Gu, F., F. R. Schumacher, F. Canzian, N. E. Allen, D. Albanes, C. D. Berg, S. I. Berndt, H. Boeing, et al. 2010. Eighteen insulin-like growth factor pathway genes, circulating levels of IGF-I and its binding protein, and risk of prostate and breast cancer. Cancer Epidemiol Biomarkers Prev 19(11):2877–2887.
Hakkak, R., A. W. Holley, S. L. Macleod, P. M. Simpson, G. J. Fuchs, C. H. Jo, T. Kieber-Emmons, and S. Korourian. 2005. Obesity promotes 7,12-dimethylbenz(a)anthracene-induced mammary tumor development in female Zucker rats. Breast Cancer Res 7(5):R627–R633.
Hedeen, A. N., E. White, and V. Taylor. 1999. Ethnicity and birthplace in relation to tumor size and stage in Asian American women with breast cancer. Am J Public Health 89(8):1248–1252.
Hersh, A. L., M. L. Stefanick, and R. S. Stafford. 2004. National use of postmenopausal hormone therapy: Annual trends and response to recent evidence. JAMA 291(1):47–53.
Hiatt, R. A., and N. Breen. 2008. The social determinants of cancer: A challenge for transdisciplinary science. Am J Prev Med 35(2 Suppl):S141–S150.
Hines, L. M., B. Risendal, M. L. Slattery, K. B. Baumgartner, A. R. Giuliano, C. Sweeney, D. E. Rollison, and T. Byers. 2010. Comparative analysis of breast cancer risk factors among Hispanic and non-Hispanic white women. Cancer 116(13):3215–3223.
Hulley, S., D. Grady, T. Bush, C. Furberg, D. Herrington, B. Riggs, and E. Vittinghoff. 1998. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA 280(7):605–613.
Hunter, D. J., P. Kraft, K. B. Jacobs, D. G. Cox, M. Yeager, S. E. Hankinson, S. Wacholder, Z. Wang, et al. 2007. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39(7):870–874. Hunter, D. J., D. Altshuler, and D. J. Rader. 2008. From Darwin’s finches to canaries in the coal mine—mining the genome for new biology. N Engl J Med 358(26):2760–2763.
Huo, D., F. Ikpatt, A. Khramtsov, J. M. Dangou, R. Nanda, J. Dignam, B. Zhang, T. Grushko, et al. 2009. Population differences in breast cancer: Survey in indigenous African women reveals overrepresentation of triple-negative breast cancer. J Clin Oncol 27(27):4515–4521.
IOM (Institute of Medicine). 2000. Promoting health: Intervention strategies from social and behavioral research. Washington, DC: National Academy Press.
Jemal, A., T. Murray, E. Ward, A. Samuels, R. C. Tiwari, A. Ghafoor, E. J. Feuer, and M. J. Thun. 2005. Cancer statistics, 2005. CA Cancer J Clin 55(1):10–30.
Jemal, A., E. Ward, and M. J. Thun. 2007. Recent trends in breast cancer incidence rates by age and tumor characteristics among U.S. women. Breast Cancer Res 9(3):R28.
Jenkins, S., C. Rowell, J. Wang, and C. A. Lamartiniere. 2007. Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod Toxicol 23(3):391–396.
Jewell, N. P. 2004. Statistics for epidemiology. Boca Raton, FL: Chapman & Hall/CRC.
Johansen Taber, K. A., L. R. Morisy, A. J. Osbahr, 3rd, and B. D. Dickinson. 2010. Male breast cancer: Risk factors, diagnosis, and management (review). Oncol Rep 24(5):1115–1120.
Johnson, M. C. 2010. Anatomy and physiology of the breast. In Management of breast diseases, edited by I. Jatoi and M. Kaufmann. Berlin, Germany: Springer-Verlag.
Joslyn, S. A., M. L. Foote, K. Nasseri, S. S. Coughlin, and H. L. Howe. 2005. Racial and ethnic disparities in breast cancer rates by age: NAACCR Breast Cancer Project. Breast Cancer Res Treat 92(2):97–105.
Keegan, T. H., S. L. Gomez, C. A. Clarke, J. K. Chan, and S. L. Glaser. 2007. Recent trends in breast cancer incidence among 6 Asian groups in the Greater Bay Area of Northern California. Int J Cancer 120(6):1324–1329.
Keegan, T. H., E. M. John, K. M. Fish, T. Alfaro-Velcamp, C. A. Clarke, and S. L. Gomez. 2010. Breast cancer incidence patterns among California Hispanic women: Differences by nativity and residence in an enclave. Cancer Epidemiol Biomarkers Prev 19(5):1208–1218.
Kerlikowske, K. 2010. Epidemiology of ductal carcinoma in situ. J Natl Cancer Inst Monogr 41:139–141.
Kolonel, L. N., and L. Wilkens. 2006. Migrant studies. In Cancer epidemiology and prevention, 3rd ed. Edited by D. Schottenfeld and J. Fraumeni. New York: Oxford University Press.
Korde, L. A., J. A. Zujewski, L. Kamin, S. Giordano, S. Domchek, W. F. Anderson, J. M. Bartlett, K. Gelmon, et al. 2010. Multidisciplinary meeting on male breast cancer: Summary and research recommendations. J Clin Oncol 28(12):2114–2122.
Kotsopoulos, J., W. Y. Chen, M. A. Gates, S. S. Tworoger, S. E. Hankinson, and B. A. Rosner. 2010. Risk factors for ductal and lobular breast cancer: Results from the Nurses’ Health Study. Breast Cancer Res 12(6):R106.
Kravchenko, J., I. Akushevich, V. L. Seewaldt, A. P. Abernethy, and H. K. Lyerly. 2011. Breast cancer as heterogeneous disease: Contributing factors and carcinogenesis mechanisms. Breast Cancer Res Treat 128(2):483–493.
Kreike, B., M. van Kouwenhove, H. Horlings, B. Weigelt, H. Peterse, H. Bartelink, and M. J. van de Vijver. 2007. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res 9(5):R65.
Kurian, A. W., K. Fish, S. J. Shema, and C. A. Clarke. 2010. Lifetime risks of specific breast cancer subtypes among women in four racial/ethnic groups. Breast Cancer Res 12(6):R99.
Kwan, M. L., L. H. Kushi, E. Weltzien, B. Maring, S. E. Kutner, R. S. Fulton, M. M. Lee, C. B. Ambrosone, et al. 2009. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res 11(3):R31.
La Merrill, M., D. S. Baston, M. S. Denison, L. S. Birnbaum, D. Pomp, and D. W. Threadgill. 2009. Mouse breast cancer model-dependent changes in metabolic syndrome-associated phenotypes caused by maternal dioxin exposure and dietary fat. Am J Physiol Endocrinol Metab 296(1):E203–E210.
Li, C. I., K. E. Malone, and J. R. Daling. 2003. Differences in breast cancer stage, treatment, and survival by race and ethnicity. Arch Intern Med 163(1):49–56.
Liaw, D., D. J. Marsh, J. Li, P. L. Dahia, S. I. Wang, Z. Zheng, S. Bose, K. M. Call, et al. 1997. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16(1):64–67.
Liu, Y., M. Perez, M. Schootman, R. L. Aft, W. E. Gillanders, M. J. Ellis, and D. B. Jeffe. 2010. A longitudinal study of factors associated with perceived risk of recurrence in women with ductal carcinoma in situ and early-stage invasive breast cancer. Breast Cancer Res Treat 124(3):835–844.
Liu, L., J. Zhang, A. H. Wu, M. C. Pike, and D. Deapen. 2011. Invasive breast cancer incidence trends by detailed race/ethnicity and age. Int J Cancer. doi: 10.1002/ijc.26004. [Epub ahead of print]
Ma, H., L. Bernstein, M. C. Pike, and G. Ursin. 2006. Reproductive factors and breast cancer risk according to joint estrogen and progesterone receptor status: A meta-analysis of epidemiological studies. Breast Cancer Res 8(4):R43.
Malkin, D., F. P. Li, L. C. Strong, J. F. Fraumeni, Jr., C. E. Nelson, D. H. Kim, J. Kassel, M. A. Gryka, et al. 1990. Germline p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250(4985):1233–1238.
Manolio, T. A. 2010. Genomewide association studies and assessment of the risk of disease. N Engl J Med 363(2):166–176.
McCracken, M., M. Olsen, M. S. Chen, Jr., A. Jemal, M. Thun, V. Cokkinides, D. Deapen, and E. Ward. 2007. Cancer incidence, mortality, and associated risk factors among Asian Americans of Chinese, Filipino, Vietnamese, Korean, and Japanese ethnicities. CA Cancer J Clin 57(4):190–205.
McDonnell, D. P., and S. E. Wardell. 2010. The molecular mechanisms underlying the pharmacological actions of ER modulators: Implications for new drug discovery in breast cancer. Curr Opin Pharmacol 10(6):620–628.
Medina, D. 2010. Of mice and women: A short history of mouse mammary cancer research with an emphasis on the paradigms inspired by the transplantation method. Cold Spring Harb Perspect Biol 2(10):a004523.
Miller, B. A., K. C. Chu, B. F. Hankey, and L. A. Ries. 2008. Cancer incidence and mortality patterns among specific Asian and Pacific Islander populations in the U.S. Cancer Causes Control 19(3):227–256.
Morris, C.R., and S. L. Kwong, eds. 2004. Breast cancer in California, 2003. Sacramento, CA: California Department of Health Services.
Narod, S. A., and K. Offit. 2005. Prevention and management of hereditary breast cancer. J Clin Oncol 23(8):1656–1663.
NCI (National Cancer Institute). 2005. SEER brochure. http://seer.cancer.gov/about/SEER_brochure.pdf (accessed November 8, 2011).
NCI. 2006. Supplemental data for the annual report to the nation (1975–2003). http://www.seer.cancer.gov/report_to_nation/1975_2003/supplemental.html (accessed November 8, 2011).
NCI. 2011. SEER cancer statistics review, 1975–2008. Edited by N. Howlader, A. M. Noone, M. Krapcho, N. Neyman, R. Aminou, W. Waldron, S. F. Altekruse, C. L. Kosary, J. Ruhl, Z. Tatalovich, H. Cho, A. Mariotto, M. P. Eisner, D. R. Lewis, H. S. Chen, E. J. Feuer, K. A. Cronin, and B. K. Edwards. Bethesda, MD: NCI. (based on November 2010 SEER data submission, posted to the SEER website, 2011). http://seer.cancer.gov/csr/1975_2008/ (accessed June 1, 2011).
Newcomb, P. A., A. Trentham-Dietz, J. M. Hampton, K. M. Egan, L. Titus-Ernstoff, S. Warren Andersen, E. R. Greenberg, and W. C. Willett. 2011. Late age at first full term birth is strongly associated with lobular breast cancer. Cancer 117(9):1946–1956.
NRC (National Research Council). 2006. Toxicity testing for assessment of environmental agents: Interim report. Washington, DC: The National Academies Press.
NTP (National Toxicology Program). 2006. Specifications for the conduct of studies to evaluate the toxic and carcinogenic potential of chemical, biological and physical agents in laboratory animals for the National Toxicology Program (NTP). http://ntp.niehs.nih.gov/?objectid=72015DAF-BDB7-CEBA-F9A7F9CAA57DD7F5 (accessed November 8, 2011).
NTP. 2010. Toxicology/carcinogenicity. http://ntp.niehs.nih.gov/?objectid=72015DAF-BDB7-CEBA-F9A7F9CAA57DD7F5 (accessed November 3, 2011).
NTP. 2011. Long-term study reports and abstracts. http://ntp.niehs.nih.gov/index.cfm?objectid=0847DDA0-F261-59BF-FAA04EB1EC032B61 (accessed November 1, 2011).
Oldenburg, R. A., H. Meijers-Heijboer, C. J. Cornelisse, and P. Devilee. 2007. Genetic susceptibility for breast cancer: How many more genes to be found? Crit Rev Oncol Hematol 63(2):125–149.
Parkin, D. M., S. L. Whelan, J. Ferlay, L. Raymond, and J. Young, eds. 1997. Cancer incidence in five continents. Vol. VII. Lyon, France: International Agency for Research on Cancer (IARC).
Parkin, D. M., F. Bray, J. Ferlay, and P. Pisani. 2005. Global cancer statistics, 2002. CA Cancer J Clin 55(2):74–108.
Partridge, A., K. Adloff, E. Blood, E. C. Dees, C. Kaelin, M. Golshan, J. Ligibel, J. S. de Moor, et al. 2008. Risk perceptions and psychosocial outcomes of women with ductal carcinoma in situ: Longitudinal results from a cohort study. J Natl Cancer Inst 100(4):243–251.
Pasqualini, J. R. 2009. Breast cancer and steroid metabolizing enzymes: The role of progestogens. Maturitas 65(Suppl 1):S17–S21.
Phipps, A. I., C. I. Li, K. Kerlikowske, W. E. Barlow, and D. S. Buist. 2010. Risk factors for ductal, lobular, and mixed ductal-lobular breast cancer in a screening population. Cancer Epidemiol Biomarkers Prev 19(6):1643–1654.
Ponce, N. A., S. H. Babey, D. A. Etzioni, B. A. Spencer, E. R. Brown, and N. Chawla. 2003a. Cancer screening in California: Findings from the 2001 Health Interview Study. Los Angeles, CA: University of California at Los Angeles, Center for Health Policy Research. http://www.healthpolicy.ucla.edu/pubs/files/Cancer_Screening_Report.pdf (accessed December 12, 2011).
Ponce, N. A., M. Gatchell, and E. R. Brown. 2003b. Cancer screening rates among Asian ethnic groups. Los Angeles, CA: University of California at Los Angeles, Center for Health Policy Research. http://www.healthpolicy.ucla.edu/pubs/files/Asian_Cancer_FactSheet.pdf (accessed December 12, 2011).
Rakha, E. A., M. E. El-Sayed, A. R. Green, A. H. Lee, J. F. Robertson, and I. O. Ellis. 2007. Prognostic markers in triple-negative breast cancer. Cancer 109(1):25–32.
Ravdin, P. M., K. A. Cronin, N. Howlader, C. D. Berg, R. T. Chlebowski, E. J. Feuer, B. K. Edwards, and D. A. Berry. 2007. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 356(16):1670–1674.
Reynolds, P., S. Hurley, D. Goldberg, T. Quach, R. Rull, and J. Von Behren. 2011. An excess of breast cancer among young California-born Asian women. Ethn Dis 21(2):196–201.
Ries, L. A. G., and M. P. Eisner. 2007. Chapter 13: Cancer of the female breast. In SEER survival monograph: Cancer survival among adults: U.S. SEER Program, 1988–2001, patient and tumor characteristics. Edited by L. A. G. Ries, J. L. Young, G. E. Keel, M. P. Eisner, Y. D. Lin, and M. J. Horner. Bethesda, MD: National Cancer Institute, SEER Program.
Robbins, A. S., and C. A. Clarke. 2007. Regional changes in hormone therapy use and breast cancer incidence in California from 2001 to 2004. J Clin Oncol 25(23):3437–3439.
Rockhill, B., B. Newman, and C. Weinberg. 1998. Use and misuse of population attributable fractions. Am J Public Health 88(1):15–19.
Rothman, K. J. 2002. Epidemiology: An introduction. New York: Oxford University Press.
Russo, J., and I. H. Russo. 1996. Experimentally induced mammary tumors in rats. Breast Cancer Res Treat 39(1):7–20.
Russo, J., Q. Tahin, M. H. Lareef, Y. F. Hu, and I. H. Russo. 2002. Neoplastic transformation of human breast epithelial cells by estrogens and chemical carcinogens. Environ Mol Mutagen 39(2–3):254–263.
Silent Spring Institute. 2011. Silent Spring Institute guide to cohort studies for environmental breast cancer research. http://www.silentspring.org/pdf/our_tools/CohortStudiesTable.pdf (accessed November 8, 2011).
Smedley, B. D., and S. L. Syme. 2001. Promoting health: Intervention strategies from social and behavioral research. Am J Health Promot 15(3):149–166.
Stacey, S. N., A. Manolescu, P. Sulem, T. Rafnar, J. Gudmundsson, S. A. Gudjonsson, G. Masson, M. Jakobsdottir, et al. 2007. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39(7): 865–869.
Stanford, J. L., L. J. Herrinton, S. M. Schwartz, and N. S. Weiss. 1995. Breast cancer incidence in Asian migrants to the United States and their descendants. Epidemiology 6(2):181–183.
Stead, L. A., T. L. Lash, J. E. Sobieraj, D. D. Chi, J. L. Westrup, M. Charlot, R. A. Blanchard, J. C. Lee, et al. 2009. Triple-negative breast cancers are increased in black women regardless of age or body mass index. Breast Cancer Res 11(2):R18.
Szklo, M., and J. Nieto. 2004. Epidemiology: Beyond the basics, 2nd ed. Seattle, WA: Jones and Bartlett.
Thayer, K. A., and P. M. Foster. 2007. Workgroup report: National Toxicology Program workshop on hormonally induced reproductive tumors—relevance of rodent bioassays. Environ Health Perspect 115(9):1351–1356.
Thomas, D. B., and M. R. Karagas. 1987. Cancer in first and second generation Americans. Cancer Res 47(21):5771–5776.
Thompson, H. J., and M. Singh. 2000. Rat models of premalignant breast disease. J Mammary Gland Biol Neoplasia 5(4):409–420.
Trivers, K. F., M. J. Lund, P. L. Porter, J. M. Liff, E. W. Flagg, R. J. Coates, and J. W. Eley. 2009. The epidemiology of triple-negative breast cancer, including race. Cancer Causes Control 20(7):1071–1082.
Turnbull, C., S. Ahmed, J. Morrison, D. Pernet, A. Renwick, M. Maranian, S. Seal, M. Ghoussaini, et al. 2010. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42(6):504–507.
Turner, N. C., J. S. Reis-Filho, A. M. Russell, R. J. Springall, K. Ryder, D. Steele, K. Savage, C. E. Gillett, et al. 2007. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 26(14):2126–2132.
Ursin, G., E. O. Lillie, E. Lee, M. Cockburn, N. J. Schork, W. Cozen, Y. R. Parisky, A. S. Hamilton, et al. 2009. The relative importance of genetics and environment on mammographic density. Cancer Epidemiol Biomarkers Prev 18(1):102–112.
USPSTF (U.S. Preventive Services Task Force). 2002. Chemoprevention of breast cancer: Recommendations and rationale. http://www.uspreventiveservicestaskforce.org/uspstf/uspsbrpv.htm (accessed August 9, 2011).
Wacholder, S., P. Hartge, R. Prentice, M. Garcia-Closas, H. S. Feigelson, W. R. Diver, M. J. Thun, D. G. Cox, et al. 2010. Performance of common genetic variants in breast-cancer risk models. N Engl J Med 362(11):986–993.
Walsh, T., S. Casadei, K. H. Coats, E. Swisher, S. M. Stray, J. Higgins, K. C. Roach, J. Mandell, et al. 2006. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295(12):1379–1388.
Wang, T., H. M. Gavin, V. M. Arlt, B. P. Lawrence, S. E. Fenton, D. Medina, and B. A. Vorderstrasse. 2011. Aryl hydrocarbon receptor activation during pregnancy, and in adult nulliparous mice, delays the subsequent development of DMBA-induced mammary tumors. Int J Cancer 128(7):1509–1523.
Wistuba, I. I., C. Behrens, S. Milchgrub, S. Syed, M. Ahmadian, A. K. Virmani, V. Kurvari, T. H. Cunningham, et al. 1998. Comparison of features of human breast cancer cell lines and their corresponding tumors. Clin Cancer Res 4(12):2931–2938.
Writing Group for the Women’s Health Initiative Investigators. 2002. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288(3):321–333.
Yang, X. R., J. Chang-Claude, E. L. Goode, F. J. Couch, H. Nevanlinna, R. L. Milne, M. Gaudet, M. K. Schmidt, et al. 2011. Associations of breast cancer risk factors with tumor subtypes: A pooled analysis from the Breast Cancer Association Consortium studies. J Natl Cancer Inst 103(3):250–263.
Zheng, W., J. Long, Y. T. Gao, C. Li, Y. Zheng, Y. B. Xiang, W. Wen, S. Levy, et al. 2009. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet 41(3):324–328.
Ziegler, R. G., R. N. Hoover, M. C. Pike, A. Hildesheim, A. M. Nomura, D. W. West, A. H. Wu-Williams, L. N. Kolonel, et al. 1993. Migration patterns and breast cancer risk in Asian-American women. J Natl Cancer Inst 85(22):1819–1827.




































