
The rise of data-intensive biology, advances in information technology, and changes in the way health care is delivered have created a compelling opportunity to improve the diagnosis and treatment of disease by developing a Knowledge Network, and associated New Taxonomy, that would integrate biological, patient, and outcomes data on a scale hitherto beyond our reach. Key enablers of this opportunity include:
- New capabilities to compile molecular data on patients on a scale that was unimaginable 20 years ago.
- Increasing success in utilizing molecular information to improve the diagnosis and treatment of disease.
- Advances in information technology, such as the advent of electronic health records, that make it possible to acquire detailed clinical information about large numbers of individual patients and to search for unexpected correlations within enormous datasets.
- A “perfect storm” among stakeholders that has increased receptivity to fundamental changes throughout the biomedical research and healthcare-delivery systems.
- Shifting public attitudes toward molecular data and privacy of healthcare information.
Scientific research, information technology, medicine, and public attitudes are all undergoing unprecedented changes. Biology has acquired the capacity to systematically compile molecular data on a scale that was unimaginable 20 years ago. Diverse technological advances make it possible to gather, integrate, analyze, and disseminate health-related biological data in ways that could greatly
advance both biomedical research and clinical care. Meanwhile, the magnitude of the challenges posed by the sheer scientific complexity of the molecular influences on health and disease are becoming apparent and suggest the need for powerful new research resources. All these changes provide an opportunity for the biomedical science and clinical communities to come together to improve both the discovery of new knowledge and health-care delivery. As discussed in this chapter, the Committee concluded that this opportunity could best be exploited through a major, long-term commitment to create an Information Commons, a Knowledge Network of Disease, and a New Taxonomy.
BIOLOGY HAS BECOME A DATA-INTENSIVE SCIENCE
Advances in DNA-sequencing technology powerfully illustrate biology’s conversion to a data-intensive science. The first papers describing practical methods of DNA sequencing were published in 1977 (Maxam and Gilbert 1977; Sanger et al. 1977). These methods required radioisotopic labeling of DNA, hand-crafting of large electrophoretic gels, and considerable expertise with biochemical and recombinant-DNA techniques. Although the impact of these early DNA-sequencing methods on biological discovery was profound, the total amount of sequence deposited in GenBank, the central depository for such data, did not pass one billion base pairs (one-third of the length of a single human genome) until 1997 (NCBI 2011a), and it only reached this landmark after a first generation of automated instruments came into widespread use (Favello et al. 1995). Since then > 300 billion base pairs (Benson et al. 2011) have been deposited, illustrating the still ongoing explosion of genomic data in the last 20 years.
The National Human Genome Research Institute estimated that the total cost of obtaining a single human-genome sequence in 2001 was $95 million (Wetterstrand 2011; see Figure 2-1). Costs subsequently dropped exponentially following a trajectory described in electronics as Moore’s Law, connoting a reduction of cost by 50 percent every two years, until the spring of 2007, at which point the estimated cost of a single human-genome sequence was still nearly $10 million. At that point, introduction of a second generation of automated DNA-sequencing instruments, based on massively parallel, miniaturized analysis, led to a collapse in costs far faster than the Moore’s Law projection. The most recent update, in January 2011, estimates the cost of a complete-genome sequence at $21,000, and the cost is still dropping rapidly, with a “$1000 genome” becoming a realistic target within a few years. (Wolinsky 2007; MITRE Corporation 2010; Mardis 2011)While whole-genome sequencing remains expensive by the standards of most clinical laboratory tests, the trend-line leaves little doubt that costs will drop into the range of many routine clinical tests within a few years. Whole-genome sequencing will soon become cheaper than many of the specific genetic tests that are widely ordered today and ultimately
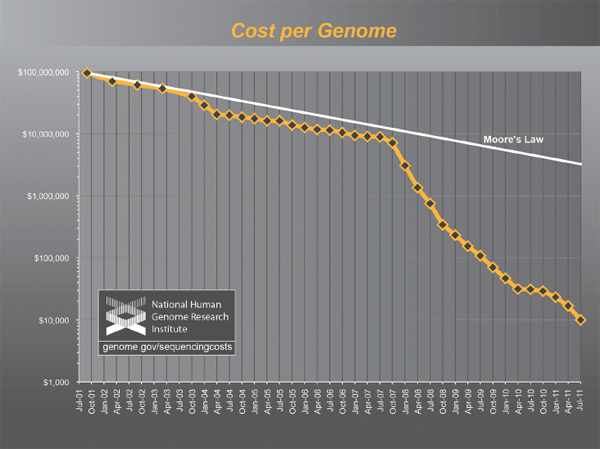
FIGURE 2-1 The plummeting cost of complete genome sequencing.
The cost of complete genome sequencing is falling faster than Moore’s Law. The cost is still dropping rapidly, with a “$1000 genome” becoming a realistic target within a few years.
SOURCE: Wetterstrand 2011.
will likely become trivial compared to the cost of routine medical care. Hence, the costs of DNA sequencing will soon cease to be a limiting factor (MITRE Corporation 2010). Instead, the clinical utility of genome sequences and public acceptance of their use will drive future developments.
Admittedly, the cost trajectory of DNA sequencing is an unusual success story, even by high-tech standards. However, it is by no means unique: parallel developments in other areas of molecular analysis, such as the analysis of large numbers of small-molecule metabolites and proteins, and the detection of single molecules, are likely to sweep away purely economic barriers to the diffusion of many data-intensive molecular methods into biomedical research and clinical medicine. These technologies will make it possible to monitor and ultimately to understand and predict the functioning of complex molecular networks in health and disease.
THE OPPORTUNITY TO INTEGRATE DATA-INTENSIVE BIOLOGY WITH MEDICINE
Human physiology is far more complex than any known machine. The molecular idiosyncrasies of each human being underlie both the exhilarating potential and daunting challenges associated with “personalized medicine”. Individual humans typically differ from each other at millions of sites in their genomes (Ng et al. 2009). More than ten thousand of these differences are known to have the potential to alter physiology, and this estimate is certain to grow as our understanding of the genome expands. All of this new genetic information could potentially improve diagnosis and treatment of diseases by taking into account individual differences among patients. We now have the technology to identify these genetic differences—and, in some instances, infer their consequences for disease risk and treatment response. Some successes along these lines have already occurred; however, the scale of these efforts is currently limited by the lack of the infrastructure that would be required to integrate molecular information with electronic medical records during the ordinary course of health care.
The human microbiome project represents an additional opportunity to inform human health care. The microorganisms that live inside and on humans are estimated to outnumber human somatic cells by a factor of ten. “If humans are thought of as a composite of microbial and human cells”, then “the human genetic landscape is an aggregate of the genes in the human genome and the microbiome, and human metabolic features” are “a blend of human and microbial traits” (Turnbaugh et al. 2007). A growing list of diseases, including obesity, inflammatory bowel disease, gastrointestinal cancers, eczema, and psoriasis, have been associated with changes in the structure or function of human microbiota. The ultimate goal of studying the human microbiome is to better understand the impact of microbial variation across individuals and populations and to use this information to target the human microbiome with antibiotics, probiotics, and prebiotics as therapies for specific disorders. While this field is in its infancy, growing knowledge of the human microbiome and its function will enable disease classification and medicine to encompass both humans and their resident microbes.
There are already compelling examples of improvements in patient care that have emerged from studies of the human genome and human microbiome:
- Some patients with high cholesterol are heterozygous for a non-functional variant of the low-density-lipoprotein-receptor gene, a genotype found in one out of every 500 individuals. Lifestyle interventions alone are ineffective in these individuals at reducing the likelihood of early-onset cardiovascular disease (Huijgen et al. 2008). Consequently, the ability to identify the patients who carry the non-functional receptor
-
makes it possible to proceed directly to the use of statin drugs at an early age, rather than first attempting to control cholesterol with diet and exercise. There is strong evidence that the early use of statin drugs in these individuals can provide a therapeutic benefit.
- In the United States it is estimated that 0.06 percent of the population carries mutations in the tumor suppressor BRCA1 and 0.4 percent of people carry mutations in BRCA2 (Malone et al. 2006). These mutations predispose to cancer, particularly breast and ovarian cancer (King et al. 2003). Women who carry these mutations can reduce their risk of death from cancer through increased cancer screening or through prophylactic surgeries to remove their breasts or ovaries (Roukos and Briasoulis 2007); until these mutations were identified it was not possible to determine who carried the mutations or to take proactive steps to manage risk.
- Lung cancer patients can now be separated by the genetic profiles of their cancers into distinct groups that benefit from different treatments (see Box 2-1).
- It is now clear that most cases of stomach ulcers, once thought to be caused by stress and other non-infectious factors, are due to colonization of the stomach lining with the Helicobacter pylori bacterium, which is very common in human populations (Atherton 2006). This finding has radically changed the treatment of this disorder. H. pylori infection also is thought to predispose to the development of stomach cancer, suggesting that treatment of this infection can both help cure gastric ulcers and also may reduce the development of cancer of the stomach. In addition, epidemiological studies and other data have raised the possibility that H. pylori infection may reduce the individual’s likelihood of developing allergic diseases or even obesity (Blaser and Falkow 2009), suggesting that the full complexity of the relationship between infection with this organism and human health and disease remains to be determined.
- In the last decade, genetic analyses have allowed a more precise diagnostic classification of type 2 diabetes. Children and young adults with mild glucose intolerance and often a strong family history of diabetes were previously categorized as having “Maturity Onset Diabetes of the Young (MODY)”. MODY is now understood to represent a series of specific genetic variants that affect pancreatic beta cell function (Fajans et al. 2001) such that the American Diabetes Association classification of type 2 diabetes has replaced the descriptive term MODY with the specific genetic defects (e.g., chromosome 7, glucokinase; chromosome 12, hepatic nuclear factor 1—alpha; etc.) A dynamic, continuously evolving Knowledge Network of Disease will be needed to accommodate future additions to this list of specific genetic predispositions to
Box 2-1
Distinguishing Types of Lung Cancer
Lung cancer is the leading cause of cancer-related death in the United States as well as worldwide, causing more than one million total deaths annually (ACS 2011). Traditionally, lung cancers have been divided into two main types based on the tumors’ histological appearance: small-cell lung cancer and non-small-cell lung cancer. Non-small-cell lung cancer is comprised of three subgroups, each of them defined by histology, including adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma.
Since 2004, knowledge of the molecular drivers of non-small-cell lung cancer has exploded (Figure 2-2). Drivers are mutations in genes that contribute to inappropriate cellular proliferation. These driver mutations are necessary for tumor formation and tumor maintenance. If the inappropriate function of the mutant protein is shut down, dramatic anti-tumor effects can ensue.
In 2004 two drugs were in development, Gefitinib and Erlotinib, which inhibited the function of certain receptor tyrosine kinases, including epidermal growth factor receptor (EGFR). These receptors were known to send signals that promote cellular proliferation and survival, and increased signaling was thought to contribute to some cancers. In early trials, the drugs were shown to produce dramatic anti-tumor effects in about 10 percent of patients with non-small-cell lung cancer (MSKCC 2005). Other patients did not appear to respond at all. However, the dramatic tumor shrinkage in some patients was enough for U.S. Food and Drug Administration approval in 2003, even though the molecular basis for the response was then unknown. Without the ability to recognize the responding patients as a biologically distinct subset, these agents were tried unsuccessfully on a broad range of lung-cancer patients, doing nothing for most patients other than increasing costs and side effects. In retrospect, some clinical trials with these agents probably failed because the actual responders represented too small a proportion of the patients in the trials (Pao and Miller 2005).
Subsequently, it was discovered that the responding patients carried mutations that activated EGFR in their cancers (Kris et al. 2003; Lynch et al. 2004; Paez et al. 2004; Pao et al. 2004). This made it possible to predict which patients would respond to the therapy and to administer the therapy only to this subset of patients. This led to the design of much more effective clinical trials as well as reduced treatment costs and increased treatment effectiveness.
Since then, many studies have further divided lung cancers into subsets that can be defined by driver mutations. Not all of these driver mutations can currently be targeted with drugs and cancer cells are quick to develop resistance to targeted drugs even when they are available. Nonetheless, this recent information makes it possible to develop new targeted therapies that can extend and improve the quality of life for cancer patients.
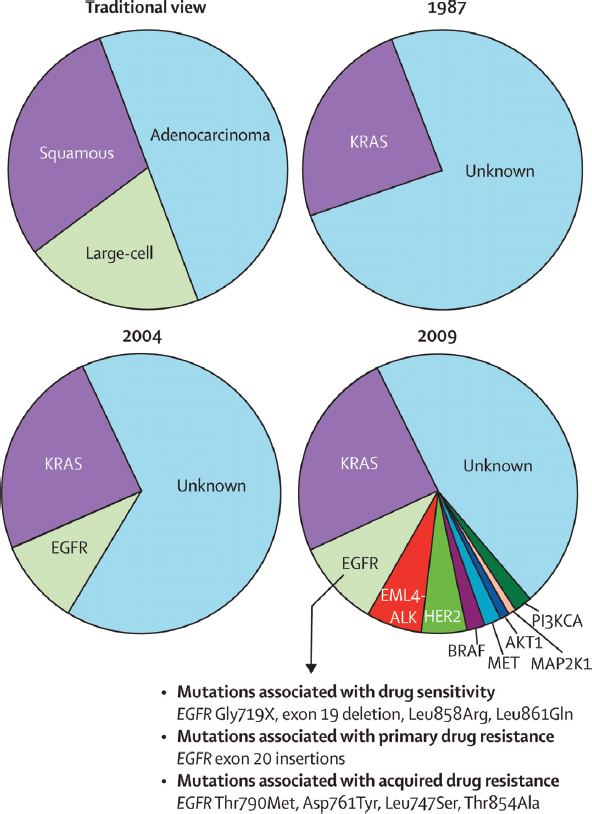
FIGURE 2-2 Knowledge of non-small-cell lung cancer has evolved substantially in recent decades.
The traditional characterization of lung cancers based on histology has been replaced over the past 20 years by classifications based on driver mutations. In 1987, this classification was rudimentary as only one driver mutation had been identified, KRAS. However, the sophistication of this system for molecular classification has improved with the advent of more genetic information and the identification of many more driver mutations. Similar approaches could improve the diagnosis, classification, and treatment of many other diseases.
SOURCE: Pao and Girard 2011.
in type 2 diabetes (with the vision of adding clarity to the diagnosis of Patient 2).
The human genome and microbiome projects are only two examples of emerging biological information that has the potential to inform health care. It is similarly likely that other molecular data (such as epigenetic or metabolomic data), information on the patient’s history of exposure to environmental agents, and psychosocial or behavioral information will all need to be incorporated into a Knowledge Network and New Taxonomy that would enhance the diagnosis and treatment of disease.
THE URGENT NEED TO BETTER UNDERSTAND PHENOTYPE-GENOTYPE CORRELATIONS
While dramatic progress in understanding the relationship between molecular features and phenotype is being made, there is an urgent need to understand these links better and to develop strategies to deal with their implications for the individual patient. BRCA1 and BRCA2 genes are a good example. The discovery of mutations in BRCA1 and BRCA2 in some people made it possible to identify individuals at increased risk of cancer, allowing them to manage their risk with increased cancer screening and prophylactic surgeries. A database cataloging BRCA1 mutations recently listed 2,136 distinct variants of the gene (NHGRI 2011). Of these, 1,167 were judged by the database’s curators as likely to be clinically significant, while most of the rest were categorized as of “unknown” clinical significance. Among the mutations that are believed to be clinically significant, some are thought to confer a higher risk of cancer than others (Gayther et al. 1995) but there remains uncertainty about the extent to which most mutations increase cancer risk (Fackenthal and Olopade 2007).
As a consequence, people with BRCA1 and BRCA2 mutations are forced to make life-altering treatment decisions with incomplete information. To what extent does their mutation increase their risks of breast and ovarian cancer and how do these risks change with age? Should they have prophylactic mastectomies or oophorectomies, and if so, when? Should they wait until after childbearing and how would that affect their risks? All of these real-life decisions carry heavy personal consequences as well as implications for health-care costs.
These treatment decisions do not need to be made based on such fragmentary information. If all patients who test positive for BRCA1 or BRCA2 mutations were tracked by their health-care providers long term, it would be possible to determine what percentage of patients with each mutation develop cancer, and which cancers. It would be possible to assess the extent to which prophylactic surgeries reduced risk. It would be possible to assess the effectiveness of increased cancer screening, the best ways to screen these patients, and the complications that arise from the inevitable false-positive results that come
from increased screening. Efforts along these lines have so far been based on modest numbers of patients or cohorts that are not fully representative of the larger population because it has not been practical to integrate genetic information, treatment decisions, and outcomes data for large numbers of unselected patients. However, recent advances in genomic and information technologies now make it possible to address systematically these issues by integrating large datasets that already exist (see Box 2-2).
BRCA1 and BRCA2 are only two of many genes in which differences between individuals have significant implications for disease risk and treatment decisions. We have approximately 20,000 genes in our genome and many of these genes may have many disease-relevant alleles, just like BRCA1. Even if only a subset of this variation has significant implications for disease risk or treatment response we have the potential to improve the detection, diagnosis, and treatment of disease dramatically by large-scale efforts to assess phenotype-genotype correlations. By integrating patient genotype with health information and outcomes data a New Taxonomy could identify many new genetic variants with significant implications for health care.
There is every reason to expect that the genetic influences on most common diseases will be complex. In each patient, variants in multiple genes will affect disease onset, progression, and response to treatment, and long-term environmental modulation of these processes will be the rule rather than the exception. While recent breakthroughs have focused on genomics as a consequence of the rapid development of technology in that area, the future may see comparable advances in our ability to understand epigenetic, environmental, microbial, and social contributions to disease risk and progression. Under these circumstances, there is an obvious need to categorize diseases with finer granularity, greater reference to the underlying biology, and in the context of a dynamic Knowledge Network that has the capacity to integrate the new information on many levels. Unraveling these diverse influences on human diseases will be a major scientific challenge of the 21st century.
DRAMATIC ADVANCES IN INFORMATION TECHNOLOGY ARE DRIVING SYSTEMIC CHANGE
The United States and other countries are currently making multibilliondollar investments to implement electronic health records (EHRs) to improve clinical care. The development of such records creates several new opportunities to integrate health-care information and biological data and to search for new links between clinical test results, patient data, and outcomes.
- The increased functionality of EHRs and the improved performance of search tools open the door to conduct large cohort studies on a wide range of diseases. Patients with characteristics of interest—for
Box 2-2
Prospective Cohort Studies—A Special Role
Much of our knowledge about “risk factors” that predispose to complex diseases comes from observational epidemiological studies, either case-control studies in which aspects of the life experience of a series of cases are compared with those of appropriate controls, or prospective cohort studies in which large numbers of people are followed over time and the life experience of those who develop a specific disease are compared with those of the much larger number who have not.
For example, much of what is known about the predictive value of biochemical factors that are measured in plasma or serum, such as the relation of cholesterol or other lipid with risk of heart attack, is derived from prospective cohort studies such as the Framingham Heart Study (FHS). Prospective studies are particularly valuable because the occurrence or treatment of disease may alter the levels of the biochemical factors so that inference based on levels measured in a series of already diagnosed cases may be biased. These biomarkers can be combined with information on lifestyle risk factors such as smoking and body mass index, and measurements that may also change after diagnosis such as blood pressure, to create a risk score such as the Framingham Risk Score, that is widely used to predict the 10-year risk of heart attack (Anderson et al. 1991). The Risk Score was based on data from slightly more than 5,500 subjects, among whom several hundred coronary heart disease (CHD) events occurred. A study of this size is adequate for relatively common disease events such as CHD, or quantitative traits such as blood pressure or bone density for which every participant has
-
example, those with rheumatoid arthritis or lacking response to antidepressants—could be selected via EHRs. Patients in these groups could then be recruited to provide samples or have their discarded clinical samples analyzed for research.
- EHRs could be used to provide additional clinical characterization or to help fill in missing details on subjects studied in a cohort or biobank. In either case, the result would be a rich clinical characterization of patients at low cost and with linkages to corresponding biological samples that can be used for molecular studies. Research questions could be addressed faster and at lower cost as compared to the current standard practice of designing large, labor-intensive prospective studies.
- EHRs permit longitudinal analyses of data from millions of people. When linked with genomic information—yielding what has been called EHR genomic research—EHRs could provide the large num-
a value, but too small to provide enough cases of less common diseases such as site-specific cancers. Larger prospective cohort studies such as the Nurses’ Health Study (Missmer et al. 2004), and the European Prospective Investigation into Cancer (EPIC) (Kaaks et al. 2005), have explored the relationship between circulating steroid hormone levels and risk of breast cancer in cohorts of tens of thousands of women, with many hundreds of cases, but the number of cases occurring in the FHS is still relatively small due to the smaller size of the cohort. For risk factors with small effects, or to study the interactions between multiple risk factors, even the largest cohort studies may have too few cases to generate statistical power, and consortia such as the NCI Breast & Prostate Cohort Consortium (Campa et al. 2005) are needed to generate the thousands of cases necessary for adequate power. For less common diseases, consortia are again needed as no single study will have enough cases.
Some countries have established very large prospective cohort studies such as the UK Biobank (about 500,000 persons) (Palmer 2007), and some have advocated for a similar study in the United States (Collins 2004). However, the cost of enrolling half a million or more persons in a such a research study in the US, tracking them over decades, and obtaining information on medical diagnoses for a research database are estimated to be several billion dollars (Willett et al. 2007), even if the many feasibility issues can be overcome.
An Information Commons and Knowledge Network with appropriate informatic and consent mechanisms could generate similar large longitudinal sample sets and data through the provision of regular medical care, rather than considering these as research studies external to the health systems.
bers of subjects and detailed information needed to resolve many of the questions that smaller cohorts cannot address.
While the use of data from EHRs faces many difficulties, none of these difficulties is insurmountable. The cost advantages and potential to advance clinical care make expanding and accelerating studies using EHRs a high priority. Several health-care systems in the United States have started accruing large EHR databases linked to clinical biosamples. Notable among the U.S. efforts are the Harvard University/Partners Healthcare i2b2 effort, the Vanderbilt BioVu effort (Roden et al. 2008), the UCSF-Kaiser collaboration (discussed in Box 2-3).
Since EHR systems cover a broad swath of human illness, they provide a unique research opportunity to explore genotype-phenotype associations across many diseases. As articulated by Jones et al. (2005) and implemented by Denny et al. (2010), all diseases can be scanned using EHRs for significant associations
with any genetic variant or set of variants. For example, a single genetic variant (e.g., a single nucleotide polymorphism or SNP) associated with diabetes in a standard genetics study can be promptly assessed for correlation with every EHR-derived phenotype such as obesity, heart disease, smoking history, and hypothyroidism. This approach has been colloquially termed PheWAS (Phenome-Wide Association Study), in contrast with the more widely developed approach
The Electronic Medical Records and Genomics (eMERGE) Network (www.gwas.org) is an NIH-funded consortium of five institutions with DNA data linked to electronic medical records. (All of the institutions agreed to contribute their genomic association results to dbGAP at the National Library of Medicine.) The goal of the consortium is to assess the utility of electronic medical records (EMRs) as resources for genomic science. The project includes an ethics component, community engagement, and the use of natural language processingto interpret EMRs. Each institution individually had proposed a genome-wide association study (GWAS) of about 3,000 subjects with a particular phenotype of interest (e.g. type 2 diabetes, cataracts, dementia, heart disease, and peripheral vascular disease) and an associated comparison group.
Several important lessons have been learned from the consortium’s experience. First, patient data, obtained during the normal course of clinical care, has proven to be a valid source for replicating genome-phenome associations that previously had been reported only in carefully qualified research cohorts. Second, although the individual institutions initially thought that they had large enough effect sizes and odds ratios to be adequately powered, in most cases, the entire network was needed to determine genome-wide association. Third, high-quality EMR-derived phenotypes require four elements: codes (including ICD codes, though codes have to be repeated multiple times to gain validity), laboratory-medicine results, medication histories, and natural language processing of physician comments. The ability to extract high-quality phenotypes from narrative text is essential along with codes, laboratory results, and medication histories to get high predictive values. Fourth, although the five electronic medical systems have widely varying structures, coding systems, user interfaces, and users, once validated at one site, the information transported across the network with almost no degradation of its specificity and precision.
Another lesson of critical importance was that the major impediments that the eMERGE Consortium has had to address are policy related, rather than technical. For instance, a particular challenge has been to achieve both meaningful data sharing and respect for patient privacy concerns, while adhering to applicable regulations and laws (Kho et al. 2011; Masys 2011; McGuire et al. 2011) (eMERGE has addressed this issue, in part, by developing a simplified Data Use Agreement—see Appendix D.)
called GWAS (Genome-Wide Association Study). Such a scan may show that the original association is either an epiphenomenon of another pathology or part of a broader pathotype (Loscalzo et al. 2007). This approach provides an opportunity to explore this broader range of pathological mechanisms across a variety of disease types, which is not possible in single phenotype studies. The power of such association studies to detect relationships between genotype and disease is limited by the granularity and precision of the current taxonomic system for disease. A knowledge-network-derived taxonomy that distinguishes diseases with different biological drivers would enhance the power of association studies to uncover new insights.
GATHERING INFORMATION FROM INFORMAL DATA SOURCES
The explosive growth of social networks, particularly in the context of healthcare issues, may also serve as a novel source of data on health and disease. Evidence is already accumulating that these alternative and “informal” sources of health-care data, including information shared by individuals from ubiquitous technologies such as smart phones and social networks, can contribute significantly to collecting disease and health data (Brownstein et al. 2008, 2009, 2010a,b).
Many data sources exist outside of traditional health-care records that could be extremely useful in biomedical research and medical practice. Informal reports from large groups of people (also known as “crowd sourcing”), when properly filtered and refined, can produce data complementary to information from traditional sources. One example is the use of information from the web to detect the spread of disease in a population. In one instance, a system called HealthMap, which crawls about 50,000 websites each hour using a fully automated process, was able to detect an unusual respiratory illness in Veracruz, Mexico, weeks before traditional public-health agencies (Brownstein et al. 2009). It also was able to track the progression and spread of H1N1 on a global scale when no particular public-health agency or health-care resource could produce that kind of a picture.
The use of mobile phones also has tremendous potential, especially with developers building apps that engage patient populations. For example, a recent app called Outbreaks Near Me allows people to use their cell phones to learn about all the disease events in their neighborhood. People also can report back to the system, putting their own health information into the system.
Many of the social networking sites built around medical conditions are patient specific and allow individuals to share unstructured information about health outcomes. Mining that information within proper ethical guidelines provides a novel opportunity to monitor health outcomes. For example, Google has mined de-identified search data to build a picture of flu trends. The advent of these inexpensive ways of collecting health information creates new oppor-
tunities to integrate information that will enhance the diagnosis and treatment of disease.
INTEGRATING CLINICAL MEDICINE AND BASIC SCIENCE
Traditionally, a physician’s office or clinic has had few direct connections with academic research laboratories. In this environment, patient-oriented research—particularly if it involved studying patients or patient-derived samples with state-of-the-art scientific techniques and experimental designs—required a major division of labor between the research and clinical settings. Typically, researchers have used informal referral networks to make contact with physicians caring for patients with diseases of special interest to the researchers. Once enrolled in a research study, the patient—or, in some cases, simply a tissue sample and a little clinical information—passed into a research setting that maintained its own infrastructure, including Institutional Review Boards (IRBs), patient coordinators, clinical evaluation centers, instrumentation, laboratory facilities, and data analysis centers. This approach often yielded descriptive and anecdotal results of uncertain relevance to larger (and more diverse) patient populations. Moreover, the patients who contributed are unlikely to remain connected to the research process or be aware of outcomes.1 This research model is ill-suited to long-term follow-up of patients since it was never designed for this purpose.
Although remarkably successful in addressing its original goals of testing clearly defined hypotheses, this traditional approach to clinical research is poorly suited to answering current questions about human health that are often more open-ended and larger in scope than those typically addressed in the past. Based on committee experience and the input from multiple stakeholders during the course of this study, including the two-day workshop, the Committee identified several reasons that current study designs are mismatched to current needs. Traditional designs:
- Require very large sample sizes—hence most studies are inevitably under-powered. As emphasized above, the number and complexity of questions inherent in genotype-phenotype correlations is virtually unbounded. Patients with particularly informative genotypes and phenotypes—often difficult or impossible to recognize in advance—will typically be rare. Identification and recruitment of such patients in sufficient numbers to acquire clinically actionable information about their
_____________
1 There are notable exceptions such as the Framingham Heart Study and Nurses’ Health Study, which were designed from the outset to follow a cohort of patients over an extended period of time. See Box 2-2: Prospective Cohort Studies—A Special Role.
diseases will be possible only if molecular and clinical information can be combined in huge patient cohorts.
- Involve high costs that are largely unnecessary because of increasing redundancy between the infrastructure present in research and clinical settings. Most of what is needed to carry out data-intensive molecular studies of huge patient populations already exists in the health-care system or, increasingly, will exist as large coordinated health-care organizations absorb increasing portions of the patient population, EHRs are more widely implemented, medical decisions are increasingly driven by molecular analyses (particularly in the realm of oncology, but increasingly in other subspecialties as well), and consent standards for treatment converge with those for both outcomes and molecular research.
- Encourage building closed rather than open research systems. Researchers who devote their careers to building the research infrastructure described above, cultivating the physician networks, and navigating the IRB process have little incentive to share patient samples and data widely. Indeed, the suite of obstacles that a young investigator must overcome to penetrate this system are a major disincentive for involvement in patient-oriented research. In addition, the many talented biomedical researchers who choose to focus their work on model organisms (such as flies, worms, and mice) have little opportunity to share insights or collaborate with clinical researchers.
- Leave most researchers and physicians living in separate, largely disconnected communities. The current biomedical training system separates researchers and physicians from the earliest stages of their education and creates silos of specialized, but limited knowledge. The insular nature of the current biomedical system does not encourage interdisciplinary collaborations and has significant negative effects on training, study design, prioritization of research efforts, and translation of new research findings.
- Are poorly suited to long-term follow-up of patients. Long-term follow-up was not required to conduct the first generation of genotype-phenotype studies. The questions under investigation were typically of the nature “Do all cystic fibrosis patients have loss-of-function mutations in the CFTR gene?” Therefore, researchers who sought to establish the causal role of genotypes in particular phenotypes only needed confidence that patients had been correctly diagnosed. However, questions such as “Do cystic fibrosis patients with particular genotypes do better over a period of decades with particular treatments?” require long-term follow-up.
- May not provide feedback on clinically relevant results for integration into a patient’s clinical care. To the extent that inherited germline
variation and/or somatic genomic patterns are predictive of prognosis or response, the feedback of results to the clinical care of the individual research subjects may or may not occur according to a complex mixture of factors including the original informed-consent documents, the logistics of re-contacting subjects, the perceived validity of the scientific results (which may change over time), the time that has elapsed between when a sample was taken and the results were generated, and whether the laboratory work was performed under protocols that permit results feedback. These limiting factors mean that most research results are not integrated into clinical care. Expert opinion on the “duty to inform” research participants of clinically relevant results vary widely. Indeed, many researchers are reluctant to contribute data to a common resource as it may expose them to questions about whether feedback to participants is necessary or desirable.
For these, and many other reasons, the project of developing an Information Commons, a Knowledge Network of disease, and a New Taxonomy requires a long-term perspective. In a sense, this challenge has parallels with the building of Europe’s great cathedrals–studies started by one generation will be completed by another, and plans will change over time as new techniques are developed and knowledge evolves. As costs in the health-care system are increasingly dominated by the health problems of a long-lived, aging population, one can imagine that studies that last 5, 10, or even 50 years can answer many of the key questions on which clinicians will look to researchers for guidance. Many patients are already put on powerful drugs in their 40s, 50s, and 60s that they will take for the rest of their lives. The very success of some cancer treatments is shifting attention from short-term survival to the long-term sequelae of treatment. For all these reasons, the era during which a genetic researcher simply needed a blood sample and a reliable diagnosis is passing.
Outcomes research is also creating new opportunities for a close integration of medicine and data-intensive biology. Cost constraints on health-care services—as well as an increasing appreciation of how often conventional medical wisdom is wrong—has led to a growing outcomes-research enterprise that barely existed a few decades ago. The requirements of outcomes researchers for access to uniform medical records of large patient populations are remarkably similar to those of molecularly oriented researchers.
MULTIPLE STAKEHOLDERS ARE READY FOR CHANGE
The tremendous recent progress in genetics, molecular biology, and information technology has been projected to lead to novel therapeutics and improved health-care outcomes with reduced overall health-care costs. However, there is little evidence that these benefits are accruing in mainstream medicine
(OECD 2011). Instead, health-care costs have steadily increased, and these increased costs have not necessarily translated into significantly better clinical outcomes (OECD 2011). This situation has created a “perfect storm” for a wide variety of stakeholders, including health-care providers, payers, regulators, patients, and drug developers. The economics of the present situation are not sustainable and demand change.
Clinical and basic researchers have learned that for their collective efforts to provide affordable improvements in health care, increased collaboration and coordination are required. Public–private collaborations are needed to combine longitudinal health outcomes data with new advances in technology and basic research. Such initiatives are essential to gain and apply the specific biological knowledge required to develop new approaches to treat and prevent disease. A dynamically evolving Knowledge Network of Disease would provide a framework in which a closer, more effective, relationship between clinical and basic researchers could thrive.
Nowhere is the need for change more evident and urgent than in the pharmaceutical and biotechnology industries. Despite a massive increase in the amount of genomic and molecular information available over the past decade, the number of effective new therapies developed each year has remained stable, while the cost of developing each successful therapy has increased dramatically (Munos 2009). While the new molecular technologies have identified a large number of novel drug targets, an inadequate biological understanding of these targets has resulted in an ever-increasing failure rate of expensive clinical trials (Arrowsmith 2011a,b). The present situation in drug development is not sustainable. The pharmaceutical and biotechnology industries are now leading proponents for developing public–private collaborations and consortia in which longitudinal clinical outcomes data can be combined with new molecular technology to develop the deep biological understanding needed to re-define disease based on biological mechanisms. Given the time scale on which private entities must seek return on investment, there is an increased willingness to regard much of this information as precompetitive. Hence, the information itself, and the costs of acquiring it, must be widely shared.
A major beneficiary of the proposed Knowledge Network of Disease and New Taxonomy would be what has been termed “precision medicine.” In precision medicine, the ultimate end point is the selection of a subset of patients, with a common biological basis of disease, who are most likely to benefit from a drug or other treatment, such as a particular surgical procedure. Today, researchers look for relatively small differences between treated and untreated patients in trials that involve unselected patients, with little insight into the biological heterogeneity among the patients or their diseases. This approach requires a much larger number of patients, more time, and greater costs to assess the effectiveness of new therapies than would more targeted study designs. By using a precision-medicine approach to focus on those patients early in the drug-development
Box 2-4
Precision Medicine for Drug Development
A successful example of the precision medicine approach to drug development involves the drug Crizotinib, an inhibitor of the MET and ALK kinases, which began clinical development in a broad population of patients with lung cancer (Kwak et al. 2010). During the early stages of the initial Crizotinib clinical trial conducted by pharmaceutical industry scientists, an independent group of academic scientists published their discovery that a particular chromosomal translocation involving the gene encoding ALK drives tumor growth in a subset of non-small cell lung cancer patients (Soda et al. 2007). Access to this knowledge allowed the pharmaceutical industry scientists to modify their clinical trial to look specifically at a cohort of patients with this translocation, and the results were dramatic. For those patients who had the translocation, the median disease-free survival with Crizotinib was a year, compared to just a few months with the standard of care. Thus, even in a trial that involved only a small number of patients that were compared to historical controls, it was obvious that the drug was active. In contrast, in an unselected patient population, most patients did not benefit from this drug and it was unclear whether the drug had any activity.
(Crizotinib is expected to receive regulatory approval for treatment of ALK translocation-positive lung cancer within the next year.)
process who are most likely to be helped, fewer side effects and reduced costs are likely to ensue. In such studies, compliance will likely be better, treatment duration longer, and therapeutic benefits more obvious than is the case with traditional designs. Greater therapeutic differences could also result in more efficient regulatory approval, and faster adoption by physicians and payers.
As illustrated in Box 2-4, data sharing is essential to the development of precision medicine. Data sharing needs to occur across companies and across academic institutions to ensure that everyone benefits from fundamental biological knowledge. Institutions need to be convinced that they gain from openness. Broad engagement of a vast array of public and private stakeholders, including university scientists, regulators, health-care providers, payers, government, and perhaps most importantly the public at large, will be required to support and sustain the changes required for development of innovative new therapies that improve health outcomes based on the proposed Knowledge Network of Disease and associated New Taxonomy.
PUBLIC ATTITUDES TOWARD INFORMATION AND PRIVACY ARE IN FLUX
Genetic privacy was a central preoccupation during the early years of genomics, which led to implementation of stringent regulatory procedures to limit the use of genetic data in patient-oriented research (Andrews and Jaeger 1991). Many privacy-related issues—ranging from insurance coverage to employment discrimination, social stigmatization, and the simple desire to be left alone—are by no means resolved, although passage of the Genetic Information Nondiscrimination Act (GINA) alleviates many concerns about such discrimination (Hudson et al. 2008). During the ensuing years, the diffusion of the internet into every corner of our lives is driving massive changes in public attitudes toward privacy. Research studies of public attitudes reveal deep ambivalence about informational privacy. In the particular arena of genetic information and health records, members of focus groups typically grasp the broad social benefits of sharing data. A consistent theme is that people who contribute their own information to public databases want to be asked for permission, to have a clear explanation of how the data will be used, and to be treated as true partners in the research process (Damschroder et al. 2007; Trinidad et al. 2010; Haga and O’Daniel 2011). Although privacy concerns remain, there is little evidence that the public has the extreme sensitivity toward genetic data that many researchers anticipated 25 years ago.
The powerful forces affecting basic biological research, information technology, clinical medicine, and public attitudes toward the privacy of health records and personal genetic information create an unprecedented opportunity to change how biomedical research is conducted and to improve health outcomes. The development of the proposed Knowledge Network of Disease and its associated New Taxonomy could take advantage of these forces to inspire revolutionary change. This Committee regards commitment to the development of these resources as a powerful unifying idea that could harness—and, to an appropriate degree, redirect—the creative energies of the key constituencies to achieve the full potential of biology to improve health outcomes.




















