
“Omics” is a term encompassing multiple molecular disciplines that involve the characterization of global sets of biological molecules such as DNAs, RNAs, proteins, and metabolites. For example, genomics investigates thousands of DNA sequences, transcriptomics investigates all or many gene transcripts, proteomics investigates large numbers of proteins, and metabolomics investigates large sets of metabolites.
Omics research generates complex high-dimensional data; these data are often generated through measurement of many more variables per sample than the total number of biological samples used to generate the dataset. These data can be used to produce a computational model1 that potentially distinguishes a health-related characteristic of clinical significance and is intended for eventual analysis of individual patient specimens in a clinical setting. High-dimensional data are particularly prone to overfitting; as a result, a computational model emerging from the research and discovery phase may function well on the samples used for the discovery research, but is inaccurate on any other sample. A carefully designed and executed series of studies is necessary to develop a clinically useful omics-based test for patient management and care, with the goal of improving patient outcomes.
Several characteristics distinguish omics-based tests from other medical technologies, including a different regulatory oversight process, the difficulty in defining the biological rationale behind a test based on multiple
______________
1 The series of computational steps performed in processing the raw data, as well as the mathematical formula or formulas used to convert the data into a prediction of the phenotype of interest, all precisely defined in written form.
individual biomarkers, the complexity of data sharing with other scientists, and the high degree of hope placed in the promise of omics-enabled technologies and medical care.
Omics-based tests, and indeed all clinical laboratory tests, are subject to a different regulatory framework than drugs. Specifically, there are more pathways for regulation of in vitro diagnostic test devices—the category under which omics-based tests fall—than there are for drugs. Tests can be developed, validated, and placed into clinical use either through review by the Food and Drug Administration (FDA) or through validation and performance in a specific laboratory, also called laboratory-developed tests (LDTs). Any clinical laboratory that reports tests for clinical management of patients falls under the purview of the Clinical Laboratory Improvement Act (a CLIA-certified clinical laboratory) that provides a baseline level of oversight with respect to test development and the quality of laboratory operations. While FDA has the authority for regulatory oversight of all tests used in patient care, it has not defined a regulatory framework that includes oversight of LDTs and has only reviewed LDT tests that it has determined to be of high complexity and therefore high risk to patients. This alternate LDT pathway is not possible for drug development, and all drugs must be approved by FDA. It is precisely this LDT pathway that allows academic medical centers to move omics-based tests from discovery to clinical use without external regulatory review of the new test, and places a new and mostly unrecognized demand on academic institutions to provide proper oversight for omics-based test development, validation, and clinical implementation. While pharmaceutical and medical device companies follow well-established medical product development pathways and have many process controls in place for strong oversight of development, clinical validation, and manufacturing, academic institutions are not as accustomed to overseeing the development of medical products.
The frequent lack of a clear biological rationale further distinguishes omics-based tests from most other clinical laboratory tests based on a single analyte. The biological rationale behind a single-analyte test is frequently quite evident: The test is useful because the gene, RNA, protein, or metabolite plays an understood role in the disease pathology or other biological process under investigation. Examples of single-analyte tests include human epidermal growth factor receptor 2 (HER2) testing of breast cancers or measuring low-density lipoprotein (LDL) cholesterol level in the blood for cardiac risk assessment. In contrast, the biological rationale for the set of biomarkers in an omics-based test frequently is not well-defined scientifically. This difference puts an additional burden on the statisticians and bioinformatics experts involved in test validation to ensure that the biological data and computational model are scientifically sound. Because of the increased risk of overfitting large datasets in the development of the
computational model, the need for rigor, validation, and accountability is even higher than for other single biomarker-based tests.
The complexity of omics research also makes data provenance more challenging and makes sharing of the complex datasets and computational models difficult, which limits the ability of other scientists to replicate and verify the findings and conclusions of omics research studies. Database repositories for genomic datasets are available, but data sharing is not routine, and without access to the datasets or a precisely defined computational model, replication and verification are more difficult than for single biomarker tests. While independent confirmation studies are expensive, the need for replication is beneficial in the omics field given the data complexities that can lead to errors, from simple data management errors to incorrectly designed computational models. This level of complexity does not exist for single-biomarker test research, development, and validation.
Despite the nearly complete identification of the human genome sequence in 2001, development of omics-based products that influence or improve patient health has been slower than expected. One possible reason for this limited progress is that there has not been a widely agreed-upon process for translation of omics discoveries into clinical omics-based tests intended to improve patient outcomes and care. Many hope that the promise that omics science holds for medicine and public health will be realized. With the creation of high-throughput measurement technologies, it is now feasible to take a snapshot of a patient’s molecular profile at specific stages in the progression of disease pathology or at a given location in the body. However, the complexity of these technologies and of the resulting high-dimensional data introduces major challenges for the scientific community, as rigorous statistical, bioinformatics, laboratory, and clinical procedures are required to develop and validate these tests and evaluate their clinical usefulness.
The failure of scientific collaboration, review processes by journals, regulatory oversight, institutional systems for protection of patient- participants, and institutional systems for management of conflicts of interest in a recent case involving the premature use of gene expression-based tests in clinical trials at Duke University led the National Cancer Institute (NCI) to request establishment of this Institute of Medicine (IOM) committee. The committee’s charge was to develop recommendations to clarify and improve the pathway from discovery to first use of omics-based tests in a clinical trial, to assess the potential for new omics-based tests to benefit patients.
STUDY SCOPE
Recent events have highlighted the lack of clarity about best practices for omics-based test validations and the failure of current oversight systems.
In July 2010, NCI Director Harold Varmus received a letter from more than 30 statisticians and bioinformatics scientists expressing concerns over several gene expression–based tests already in use in clinical trials at Duke University to predict the type of chemotherapy that individual cancer patients were most likely to benefit from. As a result, an IOM committee was convened to help clarify questions about how to effectively develop omics-based tests to enable progress toward improving patient outcomes. The IOM study was focused on making recommendations useful to investigators in the broader field of omics-based test development, rather than simply examining what went wrong in the test development process at Duke University. With support from NCI, FDA, the Centers for Disease Control and Prevention, the U.S. Department of Veterans Affairs, the American Society for Clinical Pathology, and the College of American Patholo-gists, the IOM committee’s charge was to recommend sound principles for appropriate development and evaluation in translating omics-based tests from the research laboratory into clinical trials, with the ultimate goal of guiding therapeutic decisions to improve patient outcomes. The complete charge to the committee can be found in Chapter 1.
FINDINGS, CONCLUSIONS, AND RECOMMENDATIONS
The committee considered its task in the context of the scientific processes of discovery, confirmation, validation, and evaluation for clinical use of candidate omics-based tests and in relation to the many parties responsible for the discovery and development of omics-based tests. The primary investigators, who often work in interdisciplinary teams, bear the greatest responsibility and accountability for the scientific rigor of the discovery research and test development. Academic institutions, other nonprofit research organizations, and for-profit companies that support the development of omics-based tests also bear responsibility for proper oversight of the discovery, translational, and clinical research conducted and reported by their faculty or research staff seeking to generate successful omics-based tests. Although these institutions depend on the rigor and integrity with which individual investigators perform and defend their work, they also have a significant role to play in providing necessary infrastructure, supporting scientific integrity, and organizing and conducting investigations of allegations of improper or incorrect research and reporting practices.
The evaluation process recommended in this report (Figure S-1) defines the best practices for translation of an omics-based discovery into a validated omics-based test for use in a clinical trial, and focuses on the responsibilities of the investigators (Recommendations 1-3; Box S-1), with additional recommendations for other responsible parties, particularly institutions, but also funding agencies, journals, and FDA (Recommendations 4-7; Box S-2).
Throughout its recommendations, the committee emphasized the importance of transparency in reporting—making data, metadata (information about a dataset and how it was generated), prespecified analysis plans, computer code, and fully specified computational procedures for external evaluation or confirmation. This reinforces recommendations made in several National Research Council reports (NRC, 2003, 2005, 2006).
Development and Evaluation Process
The committee’s recommended development and evaluation process for omics-based tests is summarized in Figure S-1. The two major stages of test development and evaluation entail (1) discovery and test validation phases and (2) evaluation of clinical utility and use. The discovery phase includes complete definition of the computational model to be used for data analysis in a clinical test and independent confirmation of that model. At this point, the fully specified computational procedures should be locked down— recorded and no longer changed. The candidate omics-based test from the research laboratory is then transferred to a CLIA-certified clinical laboratory for development of the clinical testing methods followed by analytical validation and clinical/biological validation. The final stage is assessment of the clinical utility and use of the validated omics-based test within a clinical trial, with multiple design options depending on the intended clinical use of the test and availability of specimens from previous clinical trials. Statistics and bioinformatics validation occurs throughout both development stages. Overfitting of statistical models derived from omics data is common and many published gene expression results have been difficult to replicate.
The committee was charged with the task of recommending an evaluation process for determining when predictive tests utilizing omics-based technologies are fit for use in clinical trials, especially those in which the assay is used to determine patient management. Review of published work and several case studies of tests—including some that have been adopted into clinical use and some that did not achieve that goal—helped the committee outline and recommend a process for developing and evaluating omics-based tests. The committee’s recommendations aim to reinforce, clarify, and build on the current regulations and processes that apply to medical devices such as in vitro diagnostic multivariate index assays and LDTs. The quality and quantity of data and information available for making this determination depends in part on the test discovery process. Because omics-based tests rely on interpretation of high-dimensional datasets, methods that avoid overfitting the data in development of the computational model should be used throughout the test development process. Overfitting due to use of improper statistical methods leads to a computational model that fits the training samples well, but will perform poorly on independent samples
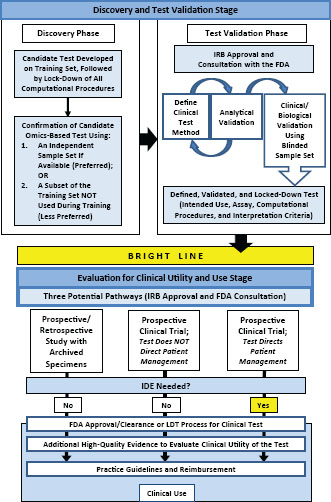
not used in the discovery phase. Confirmation of all computational models and candidate omics-based tests on an independent sample set appropriate for the intended use of the test, blinded to assay results and clinical information linked to the specimens, is the “gold standard” method to assess test validity and to avoid taking an overfit computational model into subsequent development steps. In addition, the complexities of the data management and analyses of the large omics datasets highlight the need for availability of the data and computer code used in the discovery phase of omics-based test development for external evaluation and confirmation. The result of the discovery process, outlined in Figure S-1 and Recommendation 1 (see Box S-1) and fully described in Chapter 2, is a candidate omics-based test with fully specified and locked-down computational procedures.
The candidate omics-based test is then transitioned into the test validation phase to assess analytical and clinical/biological validation. There is a wealth of existing work on best practices for the development of clinical laboratory tests, and much of this can be applied to omics-based tests. A candidate omics-based test method should be optimized for performance
FIGURE S-1 Omics-based test development process. In the first stage of omics-based test development, there are two phases: discovery and test validation. In the discovery phase, a candidate test is developed and confirmed. The fully specified computational procedures are locked down in the discovery phase and should remain unchanged in all subsequent development steps. Ideally, confirmation should take place on an independent sample set. Under exceptional circumstances it may be necessary to move into the test validation phase without first confirming the candidate test on an independent sample set if using an independent test set in the discovery phase is not possible, but this increases the risk of test failure in the validation phase. In the test validation phase, the omics-based test undergoes analytical and clinical/biological validation. The bright line signifies the point in test development where a fully defined, validated, and locked-down clinical test (analytical and clinical/biological validation) is necessary. Changes to the test after the bright line is crossed require a return to the test validation phase, approval by the Institutional Review Board, and possibly consultation with the Food and Drug Administration. In the second stage of test development, the fully defined, validated, and locked-down omics-based test undergoes evaluation for its intended clinical use. Evaluation of clinical utility and use is a process that often continues after initial adoption into clinical use. Statistics and bioinformatics validation occurs throughout the discovery and test validation stage as well as the stage of evaluation for clinical utility and use. NOTE: FDA = Food and Drug Administration, IDE = investigational device exemption, IRB = Institutional Review Board, LDT = laboratory-developed test.
Development and Evaluation Process Recommendations
Discovery and Test Validation Stage: Discovery Phase
1. When candidate omics-based tests from the discovery phase are intended for further clinical development, the following criteria should be satisfied and fully disclosed (for example, through publication or patent application) to enable independent verification of the findings:
a. Candidate omics-based tests should be confirmed using an independent set of samples, not used in the generation of the computational model and, when feasible, blinded to any outcome or other phenotypic data until after the computational procedures have been locked down and the candidate omics-based test has been applied to the samples;
b. The data and metadata used for development of the candidate omics-based test should be made available in an independently managed database (e.g., the database of Genotypes and Phenotypes [dbGaP]) in standard format;
c. The computer code and fully specified computational procedures used for the development of the candidate omics-based test should be made sustainably available;a and
d. The candidate omics-based test should be defined precisely, including the molecular measurements, the computational procedures, and the intended clinical use of the test, in anticipation of the test validation phase.
Discovery and Test Validation Stage: Test Validation Phase
2. An omics-based test consists of both the data-generating assay and the fully specified computational procedures used for analysis of the assay data. Both components of omics-based tests used to direct patient management in a clinical trial setting should be validated during the test validation phase using the following steps:
in a clinical laboratory, and then should undergo analytical validation and clinical/biological validation. The clinical test methods can be optimized based on feedback from the analytical validation performance characteristics, but must be fully defined, completely validated analytically and biologically/clinically with acceptable performance of the test, and locked down prior to assessment of the clinical utility and use of the test in a clinical trial. The optimal process for validating a candidate omics-based test is the same process used for validating any clinical test to be performed in clinical laboratories. This process is outlined in Figure S-1 and Recommendation 2 (see Box S-1) and fully described in Chapter 3.
Once the analytical and clinical/biological performance of the defined omics-based test is acceptable based on intended clinical use, the test is
a. The candidate omics-based test and its intended use should be discussed with the Food and Drug Administration (FDA) prior to the initiation of valida tion studies.
b. Test validation should be performed in a CLIA-certified clinical laboratory, beginning with a defined candidate omics-based test from the discovery phase.
c. The CLIA-certified laboratory should design, optimize, validate, and imple ment the omics-based test under current clinical laboratory standards.
d. If the omics-based test will be performed in more than one CLIA-certified laboratory for a clinical trial, analytical validation and CLIA requirements for the same omics-based test should be met by each laboratory, working with the primary laboratory.
Evaluation for Clinical Utility and Use Stage
3. For investigators conducting a clinical trial to assess the clinical utility and use of an omics-based test that has been confirmed and validated as described above (Recommendations 1-2), the committee recommends that:
a. Investigators should communicate early with FDA regarding the investigational device exemption (IDE) process and validation requirements.
b. Omics-based tests should not be changed during a clinical trial without a protocol amendment and discussion with FDA. A substantive change to the omics-based test may require that the study be restarted.
a For publicly funded research, code and fully specified computational procedures should be made publicly available either at the time of publication or at the end of funding. For commercially developed tests, this information would be submitted for FDA review if seeking approval or clearance, or would be described in a publication in the case of a laboratory-developed test.
ready for a clinical study to assess clinical utility and use. Ideally, determination of clinical utility should be derived from a prospective randomized clinical trial, but in some circumstances that may not be feasible. Figure S-1 shows several possible pathways to evaluate clinical utility and use. Depending on the choice of study design, investigators may need to obtain an investigational device exemption (IDE) from FDA. Regardless of which pathway is chosen, however, the committee strongly recommends consulting with FDA prior to initiation of a clinical trial. In the case of a trial where patient management will be influenced by the omics-based test findings, this is a legal requirement. In other cases, the committee recommends consultation with FDA because the requirement for an IDE based on the trial design is not always clear. In addition, if the test will later need
Recommendations on Appropriate Actions to Ensure Adoption and Adherence
Institutions
4.a. Institutions are responsible for establishing, supporting, and overseeing the infrastructure and research processes for omics-based test development and evaluation as well as best practices for clinical trials and observational research, including those incorporating omics technologies, and should assure that the evaluation process outlined in this report is followed for omics-based test development and evaluation at their institution.
4.b. Given the complexity of omics research and omics-based tests, the multi-disciplinary nature of omics research, and the potential for conflicts of interest in developing and evaluating tests for clinical use, institutional leaders should pay heightened attention to providing appropriate oversight and promoting a culture of scientific integrity and transparency. They should designate:
i. A specific Institutional Review Board (IRB) member(s) to be responsible for considering investigational device exemption (IDE) and investigational new drug (IND) requirements as a component of ensuring the proper conduct of clinical omics research.
ii. An institutional official who is responsible for comprehensive and timely documentation, disclosure, and management of financial and non- financial conflicts of interest, both individual and institutional.
iii. An institutional official who is responsible for establishing and managing a safe system for preventing, reporting, and adjudicating lapses in scientific integrity, to enhance patient safety.
iv. An institutional official who is responsible for establishing clear procedures for response to inquiries and/or serious criticism about the science being conducted at the institution. For example, this individual would be the responsible official for journals to contact with a serious concern about a manuscript, ensure that relevant information is provided to external scientists to help resolve issues of transparency of methods and data, and inform funders when an investigation of potential scientific misconduct is initiated.
4.c. Institutions that conduct biomedical omics research, including test development and clinical trials, should train, recognize, and support the faculty-level careers of individuals from the multiple collaborating disciplines, including biostatistics, bioinformatics, pathology, omics technologies, and clinical trial-ists, and ensure that they are:
i. Treated as equal co-investigators and co-owners of responsibility
ii. Represented on all relevant review and oversight bodies within the institutions
iii Intellectually independent, preferably reporting to an independent mentor and/or department chair as well as to the project leader
Funders
5.a. All funders of omics-based translational research should:
i. Require investigators to make all data, metadata, prespecified analysis plans, computer code, and fully specified computational procedures publicly available and readily interpretable either at the time of publication or, if not published, at the end of funding, and funders should financially support this requirement;
ii. Provide continuing support for independent repositories to guarantee ongoing access to relevant omics and clinical data;
iii. Support test validation in a CLIA-certified clinical laboratory and consider the usefulness of an independent confirmation of a candidate omics-based test prior to evaluation for clinical use;
iv. Designate an official to alert the institutional leadership when serious allegations or questions have been raised that may warrant an institutional investigation; if the funder (e.g., the National Institutes of Health) has initiated that question, then the funder and institution should communicate during the investigation; and
v. Establish lines of communication with other funders to be used when serious problems appear to involve interdependent research sponsored by another funder along the omics-based test development process.
5.b. Federal funders of omics-based translational research should have authority to exercise the option of investigating any research being conducted by a funding recipient after requesting an investigation by the institution.
FDA
6.a. In order to enable investigators and institutions to have a clear understanding of their regulatory responsibilities, the Food and Drug Administration (FDA) should develop and finalize a risk-based guidance or a regulation on:
i. Bringing omics-based tests to FDA for review; and
ii. Oversight of laboratory-developed tests (LDTs).
6.b. FDA should communicate the IDE requirements for use of omics-based tests in clinical trials to the Office of Human Research Protections (OHRP), IRBs, and other relevant institutional leadership.
Journals
7. Journal editors should:
a. Require authors who submit manuscripts describing clinical evaluations of omics-based tests to:
i. Register all clinical trials at www.clinicaltrials.gov or another trial registry acceptable to the journal.
ii. Make data, metadata, prespecified analysis plans, computer code, and fully specified computational procedures publicly available in an independently managed database (e.g., the database of Genotypes and Phenotypes [dbGAP]) in standard format.
iii. Provide the journal with the sections of the research protocol relevant to their manuscript.
iv. I dentify each author’s role in the development, conduct, analysis, writing, and editing of the manuscript. Require the lead and senior authors to attest to the integrity of the study and the coauthors to confirm shared responsibility for study integrity.
v. Use appropriate guidelines (e.g., the Consolidated Standards of Reporting Trials [CONSORT] and the REporting recommendations for tumor MARKer prognostic studies [REMARK]) and submit checklists to certify guideline use.
b. Develop mechanisms to resolve possible serious errors in published data, metadata, code, and/or computational models and establish clear procedures for management of error reports.
c. Alert the institutional leadership and all authors when a serious question of accuracy or integrity has been raised.
clearance or approval from FDA before clinical use, the study design and analysis will be subject to FDA review. A pre-IDE consultation can assist both the test developer and FDA in agreeing on the appropriate pathway for FDA review and the data necessary for FDA approval or clearance. Critical considerations for moving a fully defined omics-based test into clinical trials for assessing clinical utility and use are outlined in Figure S-1 and Recommendation 3 (see Box S-1) and fully described in Chapter 4.
Ensuring Adoption and Adherence to the Development and Evaluation Process
The committee’s recommendations to ensure adoption of appropriate research and development practices (Recommendations 4-7) are put forth in the spirit of “best practices.” There could be numerous approaches to ensuring adherence, and the optimal approach will need to be determined by each of the stakeholders involved in omics research.
Investigators and institutions that conduct omics-based research with the goal of improving patient care have responsibilities for supporting that research and test development. Both contribute to the scientific research culture in which omics-based research is conducted; investigators control the culture of individual laboratories, while institutions put policies and procedures in place that support scientific integrity and ensure sound and ethical practices for clinical research. To avoid adding new barriers to innovation in this promising field, the committee’s recommendations aim to emphasize and enhance institutional awareness of existing responsibilities to ensure the integrity of the scientific process. The committee recognized that these recommendations might increase the oversight requirements for omics research in some institutions, but agreed that these potential costs were offset by the added safeguards for the integrity of this research. If an institution does not have the infrastructure or capability to follow the recommended Test Development and Evaluation Process defined in this report, then the committee believes that institution should consider not engaging in the translation of omics-based discoveries into validated tests intended for clinical use.
As the Duke case study clearly demonstrates, existing procedures in some institutions may not adequately ensure the scientific integrity of translational omics. For example, although most institutions have clear policies and procedures for financial conflicts of interest for individuals, there is often less clarity when handling institutional conflicts, both financial and non-financial. An institution might appear so conflicted in certain situations that an outside body should be asked to take responsibility for an investigation.
The committee also addressed responsibilities of funders, FDA, and journals in ensuring rigorous development of omics-based tests (Box S-2). Funders play a leadership role in encouraging a culture of integrity and transparency in science, while they seek to accelerate progress through discovery, translation, and clinical applications. The committee highlighted the importance of funders supporting independent confirmation as well as validation in a CLIA-certified clinical laboratory of candidate omics-based tests. Funders have generally not supported such work in the past because they do not consider it to be original, innovative science. Without
this support, confirmation and validation studies will be difficult, and the field will be left with promising ideas published in journals that may be used prematurely in clinical trials. The responsibilities of journal editors with respect to the adoption and adherence to the recommended omics-based test development and evaluation process are complicated by the wide spectrum of adopted policies and resources available to individual journals, but the committee recommends several actions—all specified in Box S-2—to improve the transparency and reproducibility of published research. Finally, there are several steps that FDA should take to improve understanding of the regulatory requirements for omics-based tests, by directly communicating with investigators and academic institutions and by developing a guidance or regulation that clarifies the relevant requirements in this dynamic field.
Case Studies
The committee examined several case studies of tests whose development histories provide lessons learned and illustrate the committee’s recommendations. These include the series of gene expression–based tests used in clinical trials at Duke University; the commercial tests Oncotype Dx, MammaPrint, Ova1, AlloMap Testing, CorusCAD, and the Tissue of Origin Test; and the first OvaCheck test, which did not reach clinical use and for which errors were found in the methods used in the discovery phase. HER2 testing also is included as a case study to illustrate the challenges associated with a single-biomarker test, which could be magnified in omics-based tests. The committee was charged with presenting findings related to the gene expression–based tests used in the three Duke University clinical trials named in the statement of task. Published papers describing the development of those tests have been retracted, and, thus, it is now widely accepted that the clinical trials should not have used the omics-based tests for patient management decisions.
The events at Duke University captured the attention of biological and quantitative scientists around the world. The committee gathered information about the series of events leading to the inappropriate use of the gene expression–based tests for patient management decisions in clinical trials at Duke University. Unfortunately, multiple systems put in place by Duke University to ensure the integrity and rigor of the scientific process failed. However, Duke University is not unique. Many of these failures stemmed from problems that may exist at other institutions: unclear lines of accountability, lack of consistently strong data management, lack of confirmation of the omics discovery using an independent sample set, lack of definition or lock-down of the specific assay and computational analysis methods, lack of analytical and clinical/biological validation of the omics-based test
prior to commencing clinical trials, and individual and institutional conflicts of interest, both financial and nonfinancial. As a result, public trust in the scientific and medical systems and patient-participant safety have been put at risk.
During the 10 years since the research leading to the erroneous predictive tests was initiated, omics technologies and the regulation of omics-based tests have evolved. Institutions are better equipped now to answer investigator questions about appropriate development processes. Nonetheless, the committee identified needs for improvement. The committee believes the problems at Duke University could have been prevented had its recommendations been available and followed. Furthermore, the committee believes that scientific progress in omics-based test development will improve if these recommendations are broadly adopted because they ensure wide availability of data and computational models for the scientific community to explore, clarify the regulatory steps that must be followed along the process, and clarify responsibilities for the parties involved in this process.
The committee hopes this report will provide a guide to the entire pathway for the development of omics-based tests, from discovery to clinical trials, to assist the many parties contributing to this translational research in understanding the complete pathway and not just their focused contributions. This broader perspective may help the whole investigative team to understand the entire pathway and the pitfalls of each stage, with the hope of avoiding future problems in translating omics-based discoveries into clinical tests for the benefit of improved patient care.
The committee’s recommendations are forward-looking and directed toward the whole field of translational omics research, with the goal of improving research and accelerating progress. Envisioning the improvement of omics-based test development through the implementation of its recommendations, the committee joins patients, clinicians, and scientists in seeking revolutionary new omics-based tools for improving patient care.
REFERENCES
NRC (National Research Council). 2003. Sharing Publication-Related Data and Materials: Responsibilities of Authorship in the Life Sciences. Washington, DC: The National Academies Press.
NRC. 2005. Catalyzing Inquiry at the Interface of Computing and Biology. Washington, DC: The National Academies Press.
NRC. 2006. Reaping the Benefits of Genomic and Proteomic Research: Intellectual Property Rights, Innovation, and Public Health. Washington, DC: The National Academies Press.















