
Omics-Based Clinical Discovery: Science, Technology, and Applications
Since the process of mapping and sequencing the human genome began, new technologies have made it possible to obtain a huge number of molecular measurements within a tissue or cell. These technologies can be applied to a biological system of interest to obtain a snapshot of the underlying biology at a resolution that has never before been possible. Broadly speaking, the scientific fields associated with measuring such biological molecules in a high-throughput way are called “omics.”
Many areas of research can be classified as omics. Examples include proteomics, transcriptomics, genomics, metabolomics, lipidomics, and epigenomics, which correspond to global analyses of proteins, RNA, genes, metabolites, lipids, and methylated DNA or modified histone proteins in chromosomes, respectively. There are many motivations for conducting omics research. One common reason is to obtain a comprehensive understanding of the biological system under study. For instance, one might perform a proteomics study on normal human kidney tissues to better understand protein activity, functional pathways, and protein interactions in the kidney. Another common goal of omics studies is to associate the omics-based molecular measurements with a clinical outcome of interest, such as prostate cancer survival time, risk of breast cancer recurrence, or response to therapy. The rationale is that by taking advantage of omics-based measurements, there is the potential to develop a more accurate predictive or prognostic model of a particular condition or disease—namely, an omics-based test (see definition in the Introduction)—that is more accurate than can be obtained using standard clinical approaches.
This report focuses on the the stages of omics-based test development
that should occur prior to use to direct treatment choice in a clinical trial. In this chapter, the discovery phase (see Figures 2-1 and S-1) of the recommended omics-based test development process is discussed, beginning with examples of specific types of omics studies and the technologies involved, followed by the statistical, computational, and bioinformatics challenges that arise in the analysis of omics data. Some of these challenges are unique to omics data, whereas others relate to fundamental principles of good scientific research. The chapter begins with an overview of the types of omics data and a discussion of emerging directions for omics research as they relate to the discovery and future development of omics-based tests for clinical use.
Examples of the types of omics data that can be used to develop an omics-based test are discussed below. This list is by no means meant to be comprehensive, and indeed a comprehensive list would be impossible because new omics technologies are rapidly developing.
Genomics
The genome is the complete sequence of DNA in a cell or organism. This genetic material may be found in the cell nucleus or in other organelles, such as mitochondria. With the exception of mutations and chromosomal rearrangements, the genome of an organism remains essentially constant over time. Complete or partial DNA sequence can be assayed using various experimental platforms, including single nucleotide polymorphism (SNP) chips and DNA sequencing technology. SNP chips are arrays of thousands of oligonucleotide probes that hybridize (or bind) to specific DNA sequences in which nucleotide variants are known to occur. Only known sequence variants can be assayed using SNP chips, and in practice only common variants are assayed in this way. Genomic analysis also can detect insertions and deletions and copy number variation, referring to loss of or amplification of the expected two copies of each gene (one from the mother and one from the father at each gene locus). Personal genome sequencing is a more recent and powerful technology, which allows for direct and complete sequencing of genomes and transcriptomes (see below). DNA also can be modified by methylation of cytosines (see Epigenomics, below). There is also an emerging interest in using genomics technologies to study the impact of an individual’s microbiome (the aggregate of microorganisms that reside within the human body) in health and disease (Honda and Littman, 2011; Kinros et al., 2011; Tilg and Kaser, 2011).
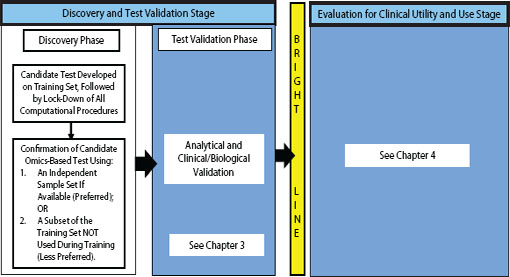
FIGURE 2-1 Omics-based test development process, highlighting the discovery phase. In the discovery phase, a candidate test is developed, precisely defined, and confirmed. The computational procedures developed in this phase should be fully specified and locked down through all subsequent development steps. Ideally, confirmation should take place on an independent sample set. Under exceptional circumstances it may be necessary to move into the test validation phase without first confirming the candidate test on an independent sample set if using an independent test set in the discovery phase is not possible, but this increases the risk of test failure in the validation phase. Statistics and bioinformatics validation occurs throughout the discovery and test validation stage as well as the stage for evaluation of clinical utility and use.
Transcriptomics
The transcriptome is the complete set of RNA transcripts from DNA in a cell or tissue. The transcriptome includes ribosomal RNA (rRNA), messenger RNA (mRNA), transfer RNA (tRNA), micro RNA (miRNA), and other non-coding RNA (ncRNA). In humans, only 1.5 to 2 percent of the genome is represented in the transcriptome as protein-coding genes. The two dominant classes of measurement technologies for the transcriptome are microarrays and RNA sequencing (RNAseq). Microarrays are based on oligonucleotide probes that hybridize to specific RNA transcripts. RNAseq is a much more recent approach, which allows for direct sequencing of RNAs without the need for probes. Oncotype DX, MammaPrint, Tissue of Origin, AlloMap, CorusCAD, and the Duke case studies described in Appendix A and B all involve transcriptomics-based tests.
Proteomics
The proteome is the complete set of proteins expressed by a cell, tissue, or organism. The proteome is inherently quite complex because proteins can undergo posttranslational modifications (glycosylation, phosphorylation, acetylation, ubiquitylation, and many other modifications to the amino acids comprising proteins), have different spatial configurations and intracellular localizations, and interact with other proteins as well as other molecules. This complexity can lead to challenges in proteomics-based test development. The proteome can be assayed using mass spectrometry and protein microarrays (reviewed in Ahrens et al., 2010; Wolf-Yadlin et al., 2009). Unlike RNA transcripts, proteins do not have obvious complementary binding partners, so the identification and characterization of capture agents is critical to the success of protein arrays. The Ova1 and OvaCheck tests discussed in Appendix A are proteomics-based tests.
Epigenomics
The epigenome consists of reversible chemical modifications to the DNA, or to the histones that bind DNA, and produce changes in the expression of genes without altering their base sequence. Epigenomic modifications can occur in a tissue-specific manner, in response to environmental factors, or in the development of disease states, and can persist across generations. The epigenome can vary substantially among different cell types within the same organism. Biochemically, epigenetic changes that are measured at high-throughput belong to two categories: methylation of DNA cytosine residues (at CpG) and multiple kinds of modifications of specific histone proteins in the chromosomes (histone marks). RNA editing
is another mechanism for epigenetic changes in gene expression, measured primarily by transcriptomic methods (Maas, 2010).
Metabolomics
The metabolome is the complete set of small molecule metabolites found within a biological sample (including metabolic intermediates in carbohydrate, lipid, amino acid, nucleic acid, and other biochemical pathways, along with hormones and other signaling molecules, as well as exogenous substances such as drugs and their metabolites). The metabolome is dynamic and can vary within a single organism and among organisms of the same species because of many factors such as changes in diet, stress, physical activity, pharmacological effects, and disease. The components of the metabolome can be measured with mass spectrometry (reviewed in Weckwerth, 2003) as well as by nuclear magnetic resonance spectroscopy (Zhang et al., 2011). This method also can be used to study the lipidome (reviewed in Seppanen-Laakso and Oresic, 2009), which is the complete set of lipids in a biological sample.
EMERGING OMICS TECHNOLOGIES AND DATA ANALYSIS TECHNIQUES
Many emerging omics technologies are likely to influence the development of omics-based tests in the future, as both the types and numbers of molecular measurements continue to increase. Furthermore, advancing bio-informatics and computational approaches are enabling improved analyses of omics data, such as greater integration of different data types. Given the rapid pace of development in these fields, it is not possible to list all relevant emerging technologies or data analytic techniques. A few illustrative developments are briefly discussed.
Advances in RNA sequencing technology are making possible a higher resolution view of the transcriptome. These new approaches could facilitate the development of more novel molecular diagnostics. In the future it may be possible to develop omics-based tests on the basis of small non-coding RNAs, RNA editing events, or alternative splice variants that were not measured using previous hybridization-based technologies such as microarrays. For example, analysis of miRNA (derived from RNA sequencing) shows great promise for clinical diagnostics (Moussay et al., 2011; Sugatani et al., 2011; Tan et al., 2011; Yu et al., 2008).
Similarly, DNA sequencing is making it possible to identify rare or previously unmeasured mutations that may have important clinical implications. Next-generation sequencing technologies hold tremendous promise for not only identification of complete DNA and RNA sequences, but also
high-throughput identification of epigenetic and posttranscriptional modifications to DNA or RNA, respectively. For instance, new sequencing technologies can monitor a wide variety of epigenetic changes at the genomic scale, in addition to sequencing information.
However, it is important to note that because next-generation RNA and DNA sequencing produces even more measurements per sample than do traditional approaches, these new technologies add to the challenge of extremely high data dimensionality and the risks of overfitting computational models to the available data (see the section on Computational Model Development and Cross-Validation for a discussion of overfitting). Large meta-analyses of sequencing datasets collected at multiple sites may prove useful for overcoming these risks and aid in developing clinically useful omics-based tests.
The field of proteomics has benefited from a number of recent advances. One example is the development of selected reaction monitoring (SRM) proteomics based on automated techniques (Picotti et al., 2010). During the past 2 years, multiple peptides distinctive for proteins from each of the 20,300 human protein-coding genes have been synthesized and their mass spectra determined. The resulting SRMAtlas is publicly available for the entire scientific community to use in choosing targets and purchasing peptides for quantitative analyses (Omenn et al., 2011). In addition, data from untargeted “shotgun” mass spectrometry-based proteomics have been collected and uniformly analyzed to generate peptide atlases for plasma, liver, and other organs and biofluids (Farrah et al., 2011).
Meanwhile, antibody-based protein identification and tissue expression studies have progressed considerably (Ayoglu et al., 2011; Fagerberg et al., 2011); the Human Protein Atlas has antibody findings for more than 12,000 of the 20,300 gene-coded proteins. The Protein Atlas is a useful resource for planning experiments and will be enhanced by linkage with mass spectrometry findings through the emerging Human Proteome Project (Legrain et al., 2011).
Recently developed protein capture-agent aptamer chips also can be used to make quantitative measurements of approximately 1,000 proteins from the blood or other sources (Gold et al., 2010). For example, Ostroff et al. (2010) recently reported generation of a 12-protein panel from analysis of 1,100 plasma proteins that was shown to have promising clinical test characteristics for diagnosis of non-small cell lung cancers.
A major bottleneck in the successful deployment of large-scale pro-teomic approaches is the lack of high-affinity capture agents with high sensitivity and specificity for particular proteins (including variants due to posttranslational modifications, alternative splicing, and single-nucleotide polymorphisms or gene fusions). This challenge is exacerbated in highly complex mixtures such as blood, where the concentrations of different proteins
vary by more than 10 orders of magnitude. One technology that holds great promise in this regard is “click chemistry” (Service, 2008), which uses a highly specific chemical linkage (generally formed through the Huisgen reaction) to “click” together low-affinity capture agents to create a single capture agent with much higher affinity. It also is feasible to combine computational algorithms for modeling protein structures and conformation to infer functional differences among alternative splice isoforms of proteins, including those involved in key cancer pathways (Menon et al., 2011).
Improving technologies for measurements of small molecules (Drexler et al., 2011) also is enabling the use of metabolomics for the development of candidate omics-based tests with potential clinical utility (Lewis and Gerszten, 2010). Promising early examples include a metabolomic analysis that identified a role for sarcosine, an N-methyl derivative of the amino acid glycine, in prostate cancer progression and metastasis (Sreekumar et al., 2009), metabolomic characterization of ovarian epithelial carcinomas (Ben Sellem et al., 2011), and an integrated metabolomic and proteomic approach to diagnosis, prediction, and therapy selection for heart failure (Arrell et al., 2011). Included within metabolomics is the emerging ability to more fully measure the lipids in a sample, a rich source of additional potential biomarkers (Masoodi et al., 2010). As with other omics data types, a lengthy, complex development path is necessary to establish a clinically relevant omics-based test from reports identifying metabolite concentration differences associated with a phenotype of interest (Koulman et al., 2009).
New technologies are emerging that will make it possible to obtain omics measurements (such as transcriptomics, proteomics) on single cells (Tang et al., 2011; Teague et al., 2010). Such detailed molecular measurements provide deep insight into the underlying biology of tissues, and potentially form a powerful basis for omics-based test development. However, as the resolution of these measurements increases, so too does the variability in the measurements due to the heterogeneity of cell states (Ma et al., 2011). Thus, while emerging omics technologies hold great potential for the development of omics-based tests, they also may exacerbate dangers of overfitting the computational model to the datasets.
Recent interest has focused on measuring multiple omics data types on a single set of samples, in order to integrate different types of molecular measurements into an omics-based test. Such multidimensional datasets have the potential to provide deep insight into biological mechanisms and networks, allowing for the development of more powerful clinical diagnostics. An encouraging example of simultaneous measurement of multiple types of omics data is the DNA-encoded antibody libraries approach (Bailey et al., 2007), which can measure DNA, RNA, and protein from the same sample. Another example is the analysis of histone modifications to identify
potential epigenetic biomarkers for prostate cancer prognosis (Bianco-Miotto et al., 2010).
Approaches that integrate multiple omics data types within the same clinical test are expected to grow in importance as the number of simultaneous measurements that can be made continues to increase. While it is relatively straightforward to increase the number of genomic and transcriptomic measurements (because DNA and RNA have complementary binding partners), increasing the number of protein measurements is more challenging because of the need for high-affinity capture agents, as discussed previously in this section.
Systems approaches that integrate multiple data types in functionally based models can be advantageous for the development of omics-based tests. For instance, the analysis of omics measurements in the context of bio molecular networks or pathways can help to reduce the number of variables in the data by constraining the possible relationships between variables, ultimately leading to more robust and clinically useful molecular tests. General approaches for using prior biological knowledge to enhance signal in omics data include removing measurements that are believed to be noise or for which there is no support in the published biological literature (filtering), using pathway databases or other sources to guide model construction, and aggregating individual measurements, often across data types, to integrate multiple sources of evidence to support conclusions (Ideker et al., 2011). For example, in a study of prion-mediated neurodegeneration, data from five mouse strains and three prion strains were used to identify the transcripts, pathways, and networks that were commonly perturbed across all genetic backgrounds (Hwang et al., 2009; Omenn, 2009).
Datasets from genome-wide association studies, in which a set of cases and controls are sampled from a large population and genotyped and each mutation identified is evaluated for association with the phenotype of interest, also can be analyzed within the context of biological pathways in order to increase identification of disease-related mutations (Segre et al., 2010). The incorporation of evolutionarily conserved gene sets can lead to the identification of often unexpected factors in disease (McGary et al., 2010). Large-scale mechanistic network models (for example, for metabolic, regulatory, or signaling networks) may be used to identify biomarkers grounded in disease mechanisms (Folger et al., 2011; Frezza et al., 2011; Gottlieb et al., 2011; Lewis et al., 2010; Shlomi et al., 2011). Genomics, transcriptomics, proteomics, and metabolomics data can be combined with structural protein analysis in order to predict drug targets or even drug off-target effects (Chang et al., 2010). While computational models of bio-molecular networks for eventual clinical use are still in their infancy, their
potential for providing stronger mechanistic underpinnings to omics-based test development is encouraging.
During the past 10 years, much of the effort to identify genes linked to disease and other conditions of biological interest has focused on genome-wide association studies. However, more recent work has successfully identified disease-causal genes using whole genome or exome sequencing (Ng et al., 2010; Roach et al., 2010). Such studies may prove to be very beneficial for the development of omics-based tests, and indeed such strategies are being used clinically today for the identification of the causal gene mutation resulting in unidentified and uncommon inherited disease states.
STATISTICS AND BIOINFORMATICS DEVELOPMENT OF OMICS-BASED TESTS
In recent years, a large number of papers have reported new omics-based discoveries and the development of new candidate omics-based tests: that is, computational procedures applied to omics-based measurements to produce a clinically actionable result. However, few of these candidate omics-based tests have progressed to clinical use (Ransohoff, 2008, 2009). Some of this discrepancy may be due to the inevitable time lapse of moving from initial identification of a candidate omics-based test to a precisely defined and validated test that can be used clinically.
However, more important are the many significant challenges in the formulation of appropriate research questions and in research design and conduct that confront the successful discovery of candidate omics-based tests, including the complexity of the data and the need for rigorous analyses, and the frequent lack of a plausible biological mechanism underpinning many of these discoveries. These challenges need to be addressed in order to realize the full clinical potential of omics research, taking into account issues specific to the field as well as broader principles of good scientific research.
Two primary scientific causes for failure of a candidate omics-based test to progress to clinical use are:
- A candidate omics-based test may not be adequately designed for answering a specific, well-defined, and relevant clinical question. This crucial point is addressed in Chapters 3 and 4.
- Omics-based discovery studies may not be conducted with adequate statistical or bioinformatics rigor, making it unlikely or even impossible that the candidate omics-based test will prove to be clinically valid or useful. This critical problem is addressed in the remainder of this chapter.
Figure 2-1 highlights the discovery and confirmation of a candidate omics-based test, the first component of the committee’s recommended test development and evaluation process. When candidate omics-based tests from the discovery phase are intended for further clinical development, several criteria should be satisfied and fully disclosed (for example, through publication or patent application) to enable independent verification of the findings (Recommendation 1), as discussed below. For the purpose of this discussion, the committee assumed that a clearly defined and clinically relevant scientific or clinical question or questions have been identified, and that an omics dataset from analyses of a set of patient samples, along with an associated clinical outcome for each patient, is available.
For example, an investigator may ask whether gene expression measurements could be used to predict recurrence in node-negative breast cancer samples in a way that is substantially more accurate than standard clinical prognostic factors, such as tumor size and grade. The investigator might have data consisting of gene expression measurements for breast cancer tissue samples obtained from patients with node-negative breast cancer, along with disease-free survival time for each patient following surgery. The goal would be to develop a defined assay method for data generation and a fully specified computational procedure1 that can be used to reliably predict, on the basis of gene expression measurements on a new patient sample, whether a patient’s cancer will recur.
Before embarking on omics-based discovery, it is worth considering whether or not the test that will eventually be developed has a reasonable chance of demonstrating clinical validity and utility. For example, the sensitivity and specificity needed, particularly in light of the prevalence of the condition in the population to be tested, should be considered (see also Appendix A, page 209, for a discussion of sensitivity and specificity needs for an ovarian cancer screening test).
Several steps need to be followed to achieve this goal: (1) data quality control; (2) computational model development and cross-validation; (3) confirmation of the computational model on an independent dataset; and (4) release of data, code, and the fully specified computational procedures to the scientific community. Each of these is discussed below.
1 All component steps of the computational procedure—namely, all data processing steps, normalization techniques, weights, parameters, and other aspects of the model, as well as the mathematical formula or formulas used to convert the data into a prediction of the phenotype of interest—are completely formulated in writing.
Step 1: Data Quality Control
As in most areas of science, data quality control is a crucial first step. Because omics datasets are typically composed of many thousands, if not millions, of measurements, data quality control is often performed computationally. For instance, an investigator might remove genes expressed across conditions near or below background levels on a microarray. The reproducibility of the measurements from run to run (the technical variance) also can be assessed. Furthermore, it may be useful to closely examine aspects of experimental design, including sample run date and other possible confounding factors such as the source of the tissue analyzed (including normal control tissue) and potential heterogeneities within the tissues, to determine if these have had an effect on the data. This is particularly important because factors such as run date or machine operator can often have a much larger effect on omics measurements than the factors of biological interest (Leek et al., 2010), such as time to disease recurrence or cancer subtype.
It is essential that such quality assessment evaluations of the data be done in a blinded fashion, without knowledge of the clinical status or treatment outcomes of the patients whose specimens were tested.
Step 2: Computational Model Development and Cross-Validation
Once investigators have determined in Step 1 that the data are of adequate quality, a candidate omics-based test associated with a phenotype of interest, such as a biologic subgroup, preclinical responsiveness to a novel therapy, or a clinical outcome, can be developed on the basis of the omics measurements. An almost unlimited number of statistical tools can be used to perform this task; therefore, they are not enumerated here. However, some key characteristics and challenges are shared by nearly all of these methods and are discussed below.
In general, omics datasets consist of thousands to millions of molecular measurements. Typically, investigators first perform feature selection, which entails selecting a subset of the measurements that appear to be associated with the characteristic or outcome or that is thought to be biologically relevant based on prior knowledge. Using just this subset of measurements, a fully defined computational model can be developed to predict the clinical outcome on the basis of the omics measurements. This reduction of required measurements can be beneficial for avoiding the later possibility that an omics-based test involving a huge number of measurements is not clinically viable for financial or technical reasons. Note that if cross-validation will be performed in order to select tuning parameters or evaluate the computational model performance, then feature selection
must be carefully performed as part of the cross-validation process, as will be explained below.
Because omics datasets typically are composed of an extremely large number of molecular measurements, and because the sample size, in general, is quite limited relative to the number of molecular measurements, overfitting 2 the data is a major concern. In fact, in the absence of adherence to proper statistical procedures, it is likely that the data will be overfit. That is, given a typical omics dataset and an associated clinical outcome, it is nearly always possible to develop a computational model that fits the data perfectly, even in the absence of any true association between the omics measurements and the clinical outcome. However, such a model will be ineffective because it will perform very poorly on an independent test data-set; a computational model’s ultimate utility is measured by its performance on future patients rather than its performance on patients comprising the original dataset used to develop the computational model.
While it is always important to increase the “power” of the statistical analysis by using the largest sample size possible, overfitting will always still be a concern in any analysis of omics data because of the vast number of feature measurements. It is unrealistic that investigators could ever have such a large sample size that overfitting would not be a concern.
The best way to avoid developing a computational model that overfits the data is to develop the model using a training set/test set approach, and to not use models with large numbers of parameters that are not justified by the sample sizes. When a limited availability of appropriate samples makes this approach infeasible, developers sometimes opt to use a less stringent process called cross-validation. First the training set/test set approach is described, followed by the cross-validation approach.
Ideally, an investigator will develop a computational model using two distinct datasets, referred to as a training set and a test set, each composed of independent samples that have been collected and processed by different investigators at different institutions. Because any computational model contains a number of possible tuning parameters, and there are multiple ways to normalize the data, each choice of tuning parameter, normalization technique, and so forth can be considered a separate computational model.
First, investigators fit each computational model under consideration on the training samples. Note that if feature selection is performed before
2 Overfitting occurs when the model-fitting process unintentionally exploits characteristics of the data that are due to noise, experimental artifacts, or other chance effects that are not shared between datasets, rather than to the underlying biology that is shared between datasets. Overfitting leads to a statistical or computational model that exhibits very good performance on the particular dataset on which it is fit, but poor performance on other datasets. Although not unique to omics research, the chance of overfitting increases when the model has a large number of measurements relative to the number of samples.
or as part of the model-fitting process, then those features must be selected on the basis of the training samples only. To get a fair estimate of the error, it is important that no information about the test set is used to fit the model on the training set. Once the model is trained, investigators then evaluate its performance on the test sample set. Investigators should be aware that if multiple models were considered, then the best test performance observed may be an overestimate of the performance (or correspondingly an underestimate of the error rate) that will be observed on future samples. The tuning parameters and normalization techniques corresponding to the model that performed best on the test samples will be used one final time to build the model on the training and test set together—mirroring precisely what was done on the training set—because the final development of the candidate omics-based test should use all available data. The error estimate for the final model learned from all the data is what was established in the test set performance (and not the apparent accuracy of this final model, which would be overly optimistic).
At this point, the fully specified computational procedures are locked down, and the investigator proceeds into Step 3, in which the chosen model is evaluated on an independent dataset. In other words, the precisely defined series of computational steps performed in processing the raw data, as well as the mathematical formula or formulas used to convert the data into a prediction of the phenotype of interest, are recorded and no longer changed (see also a more detailed discussion of computational procedure lock-down on page 56).
In many studies, however, only a limited number of samples are available, and so developing separate training and test sets is not feasible. An alternative to having designated training and test sets is “cross-validation,” a statistical method for preliminary confirmation of a model’s performance using a single dataset, by dividing the data into multiple segments, and iteratively fitting the model to all but one segment and then evaluating its performance on the remaining segment. (Cross-validation should not be confused with analytical and clinical/biological validation, as described in Chapter 3.) If performed properly, cross-validation can be expected to mitigate overfitting, but it does not necessarily eliminate it.
In greater detail, cross-validation involves splitting the samples that constitute a single dataset into K sample sets. Then, K-1 of these sample sets are used to fit a number of computational models (with different tuning parameter values and normalization techniques) and the models are evaluated on the remaining, held-out sample set. This process is repeated until each of the K sample sets has been used to evaluate the models. Then, the investigators select a single computational model to fit on the entire set of samples, using the tuning parameter values and normalization technique
that performed best, overall, on the held-out sample sets. This computational model is then locked down, and the investigator proceeds to Step 3.
Cross-validation provides a measure of the extent to which a given computational model can be expected to perform well on future observations, as opposed to having overfit the data used to train the model. Hence, a computational model that performs well in cross-validation is more likely to perform well on future patient samples than one that does not. In fact, a computational model that performs poorly in cross-validation has little chance of leading to a successful omics-based test.
Though cross-validation is a simple approach, if not performed carefully, problems can arise. For example, in some published studies, the subset of omics measurements that has the highest association with the clinical outcome of interest is identified on the basis of all of the samples in the dataset. Then cross-validation is performed using that subset of measurements. Unfortunately, the resulting cross-validation error rates grossly underestimate the true error rates because the clinical outcome of interest on the held-out samples in each cross-validation fold has already been looked at to identify the subset of omics measurements. In other words, the held-out samples are not truly “held out.” (For instance, this was the case with the cross- validation performed in the development of the MammaPrint test, which was described by Simon et al., [2003] as improperly performed “partial cross-validation,” see Appendix A.)
This can lead to substantial overfitting of the data and produces omics-based tests that appear to perform well in cross-validation, but are unlikely to perform well on future patient samples (Simon et al., 2003). To perform cross-validation correctly, all aspects of model-fitting, including feature selection and data processing, must be performed using only the K-1 sample sets used to train the computational model in each iteration of cross-validation.
Though both the training set/test set approach and the cross-validation approach provide error rates that estimate the accuracy of the computational model on independent test samples, these error rates can be highly optimistic. Cross-validation error rates tend to be overly optimistic because by randomly splitting the data it is guaranteed that the training and test sets within each cross-validation fold are drawn from the same population distribution. This means that a whole host of relevant sources of variance that affect clinical performance are ignored. If the training set/test set approach is used, then the resulting error rate might be overly optimistic because there can be similarities between the way that the training and test set samples were processed—for instance, in terms of the experimental protocol used—that might not be shared by future patient samples. Thus, neither cross-validation nor the training set/test set approach can be used in place of confirmation on an independent dataset, to be discussed in Step 3.
Occasionally, an algorithm used to develop a computational model may contain some stochastic elements, such as k-means clustering. If this is the case, random seeds should be stored, and robustness of the results from multiple runs should be reported in the publications describing the discovery phase results. Before proceeding to Step 3, the computational model needs to be fully locked down so there is no longer any element of randomness—that is, a single random seed must be selected. The failure to select a random seed and lock down the model was a significant error in the development process for tests developed at Duke University (McShane, 2010).3 An investigator should be able to fully specify and publish the computational procedures4 developed in Step 2 that will be further investigated in Step 3. In addition, investigators should define expectations for successful confirmation of a computational model before proceeding to Step 3.
Under certain circumstances, a computational model might not have shown adequate training set/test set performance or cross-validation performance. In this case, further improvement or refinement of the computational model may be necessary. After refinement, the modified computational model must once again be evaluated using the training set/ test set approach or cross-validation.
The fact that cross-validation or the training set/test set process was performed repeatedly must be reported in detail in any publications describing the candidate omics-based test, as repeated refinements of the model can contribute to overfitting and can increase the probability that the model will show a deterioration in performance when applied to independent samples in Step 3. The process of refinement/improvement should not extend into Step 3; that is, before proceeding into Step 3, the computational model must be fully defined and locked down.
Step 3: Confirmation on an Independent Dataset
As mentioned in the previous step, an error estimation approach such as cross-validation should be applied while developing a candidate omics-based test in order to avoid overfitting of the data. However, such an approach will generally yield an underestimate of the error rate that will result from applying the computational model to future patient samples. This is because when cross-validation is applied, the samples used to develop the model and the samples used to assess its performance typically share many characteristics in common, such as the patient population from which the samples are obtained or the lab in which the samples were processed.
3 McShane, L. M. 2010. Notes from June 29 meeting with Duke.
4 In this report, the computational model is referred to as fully specified computational procedures after the candidate test is locked down in Step 2. That is, the samples typically correspond to a relatively homogeneous set of patients, collected at one or a few institutions, which were run on the same machine at approximately the same point in time.
This does not correspond to the intended use case of a candidate omics-based test, which will typically involve a much more heterogeneous patient population with samples run in different laboratories at different times. This is particularly important because the variability in omics data due to differences in time, laboratories, technicians, and patient populations often exceeds the variability that is due to differences that are of scientific interest, such as those that are associated with clinical outcome (Leek et al., 2010).
Furthermore, cross-validation estimates can suffer from bias due to high variance and the fact that multiple tuning parameters and models are typically considered and only the best is selected. Therefore, cross-validation error rates provide insufficient evidence of a candidate test’s performance. To avoid the wasted time, energy, cost, and resources associated with taking a test that has little chance of success into the later phases of the development process, it is important to confirm the computational model on an independent test set in the discovery phase.
Candidate omics-based tests should be confirmed using an independent set of samples not used in the generation of the computational model and, when feasible, blinded to any outcome or phenotypic data until after the computational procedures have been locked down, and the candidate omics-based test has been applied to the samples (Recommendation 1a). It is important that no samples used to develop the computational model in Step 2, and indeed no samples that were accessed or examined by investigators before the computational model was locked down at the end of Step 2, be included in this independent dataset or in the evaluation of clinical utility (see Chapter 4). In the cases that the committee reviewed, two tests used overlapping training and confirmation datasets at some point in their development processes: MammaPrint and AlloMap (discussed in Chapter 6 and Appendix A).
The independent specimen and clinical dataset must be relevant to the intended use of the candidate omics-based test. Specifically, patients with the same type of disease, the same stage and same clinical setting for which the candidate test is intended to be used in the future must be used for the independent confirmation of the candidate omics-based test. Ideally, the specimens for independent confirmation will have been collected at a different point in time, at different institutions, from a different patient population, with samples processed in a different laboratory to demonstrate that the test has broad applicability and is not overfit to any particular situation.
If the independent set of specimens for confirmation are collected in the same laboratory, run on the same machine, processed by the same technician,
etc., then peculiarities of that data, machine, lab, etc. will be shared between the samples used to develop the computational model and the samples used to evaluate the model. As a result, even if the computational model performs well on that independent set of samples, it might not perform well on samples from patients at a different hospital, processed by a different technician in a different lab, etc. The OvaCheck case study (further discussed in Chapter 6 and Appendix A) is illustrative of the importance of independent datasets for confirmation. Most of the tissue specimens used to test the computational model were obtained from the same institution that provided the specimens used to train the computational model (Baggerly et al., 2004: Petricoin et al., 2002).
In some cases it will not be possible to obtain independent sets of specimens and associated clinical data with all of these characteristics; however, it is important to keep in mind that the quality of evidence provided by good model performance on an independent specimen set depends critically on the characteristics of that set. Hence, it is important that full descriptions of the independent specimen set are reported along with results of the computational model’s performance in the discovery phase. Below, two “levels of evidence” are presented for assessing omics-based computational model performance on an independent specimen set.
Lower Level of Evidence: Independent sets of specimens and clinical data collected at a single institution using carefully controlled protocols, with samples from the same patient population.
Under these circumstances, good performance of the locked-down computational procedure indicates that it works in the particular setting that was studied, with the protocols and the patient profile at that institution, etc. However, this candidate omics-based test might not work well with a slightly different patient population or with samples processed in a different laboratory or using a slightly different protocol.
Higher Level of Evidence: Independent sets of specimens and clinical data collected at multiple institutions.
Success in this setting strongly suggests that the omics measurements and locked-down computational procedure will work well on future patients. It provides evidence that the test is robust to the kinds of things that might change between locations: namely, aspects of the biology of the populations who tend to go to a particular hospital, sample collection and handling, and measurement techniques, etc. This is important because such differences can have large effects on the omics measurements obtained, often larger than the differences associated with the phenotypes of interest.
Once Step 3 has been initiated, further refinements to the computational model are strongly discouraged because they can lead to overfitting and consequently a very high risk that the model will not perform well in subsequent steps of the development process (unless investigators plan to get yet another independent dataset to redo Step 3). In the event that further model refinement occurs once Step 3 has been initiated, this must be clearly documented in describing the computational model development as well as its performance in Step 3.
Step 4: Release of Data, Code, and the Fully Specified Computational Procedures to the Scientific Community
Once an omics-based measurement method and the locked-down computational procedures have been shown to perform well on an independent dataset (Step 3), the candidate omics-based test is ready to proceed to the test validation phase in which analytical and clinical/biological validity are assessed, as described in detail in Chapter 3. At this time, data and metadata used for development of the candidate omics-based test should be made available in an independently managed database (e.g., the database of Genotypes and Phenotypes [dbGaP]) in standard format (Recommendation 1b). dbGaP is designed to archive and distribute data from genome-wide association studies to examine genetic associations with phenotype and disease characteristics (Mailman et al., 2007; NLM, 2006).
In addition, computer code and fully specified computational procedures used for development of the candidate omics-based test should be made sustainably available (Recommendation 1c). For publicly funded research this means that computer code and fully specified computational procedures should be made publicly available either at the time of publication or at the end of funding. For commercially developed tests, code and fully specified computational procedures will be submitted to the Food and Drug Administration (FDA) for review if the developers are seeking FDA approval or clearance. In the case of a laboratory-developed test (LDT, see Chapter 3), publication is essentially required because laboratories need to justify the claims for the test. As indicated in chapter 5, the committee also recommends that journals require authors to make data, metadata, pre-specified analysis plans, computer code, and fully specified computational procedures publicly available (Recommendation 7a[ii]). (See also the discussion on data availability and transparency in Chapter 5.)
Ideally, the computer code that is released will encompass all of the steps of computational analysis, including all data preprocessing steps, that have been described in this chapter. All aspects of the analysis need to be transparently reported. If some aspect of the code or data cannot be made available, then the reason for this omission should be documented.
Release of data, code, and the fully specified computational procedures is important for independent verification of results. This recommendation reinforces the call for transparency in reporting made in several National Research Council reports (NRC, 2003, 2005, 2006). Others have also recently called for wider access to the full data, protocols, and computer codes for published omics studies (Ince et al., 2012; Ioannidis and Khoury, 2011; Morin et al., 2012), despite the known challenges to broad disclosure (Box 2-1). In the omics setting, release of this information is particularly crucial because:
- The complex nature of the data and analyses can make it difficult to replicate results, but rigorous verification of results by the scientific community is necessary to ensure that candidate omics-based tests are scientifically and statistically valid; and
- If data are made available, then subsequent investigators can continue to conduct additional analyses to obtain future scientific insights.
Listed below are characteristics of candidate omics-based test concepts that pose risks for development of a spurious test. Some of these issues have already been mentioned. Others are not specific to omics-based test development, but are nonetheless extremely important for the development of such tests.
- High dimensionality of the data: Omics datasets are typically characterized by measurements (e.g., genes or gene products such as RNA or proteins) that are many orders of magnitude greater than the number of samples for which clinical data are available. This can lead to overfitting of the data unless proper measures, such as cross-validation combined with confirmation on an independent dataset, are performed. The initial MammaPrint studies were criticized for improper cross-validation and mixing training samples with the independent dataset used for confirmation (discussed in Appendix A).
- Biological plausibility: While lack of a known plausible biological mechanism should not, in itself, prevent an investigator from moving forward with a candidate omics-based test, such tests are more likely to endure further investigation if there is a plausible biological mechanism behind them. For instance, an effect modifier for breast cancer recurrence that is based on a set of genes known to be involved in breast cancer is more likely to hold up in further study than an effect modifier based on some set of genes not already known to be implicated in the disease. Better results can often be obtained by beginning the omics-based test development process using a subset of the omics measurements for which a plausible biological mechanism is available. For instance, there was a plausible biological mechanism behind the HER2 tests and Oncotype DX to motivate their initial clinical trials, but less so for the Duke, MammaPrint, and Ova1 tests (discussed in Appendix A and B). Bioinformatics methods to link transcript or protein expression changes to relevant signaling pathways or biological networks need to be deployed appropriately.
Considerations in Data and Information Sharing
Intellectual Property Protections
Test developers might be reluctant to share their data, code, and computational procedures out of concern that others may be able to capitalize on their research. Intellectual property protections provide one mechanism for test developers to protect their proprietary information. However, intellectual property protections have serious limitations, and test developers may be cautious about relying on these laws as the principal basis for their competitive advantage. For example, clinical trial results, customer service, price, and FDA approval are also elements of competitive advantage.
Copyright is a type of intellectual property that protects original works of authorship, including literary, dramatic, musical, and artistic works.a A 1991 U.S. Supreme Court case held that a compilation of factual information can only be copyrighted if it involves originality and creativity in its selection and arrangement, and not in the compiled facts as such.b In most cases subsequent to that decision in the United States, the copyright in collections of data has not offered much protection from re-use by not-for-profit users, such as academics and researchers, and even by commercial competitors. However, database compilers and rights holders have used other types of legal and technical protection, such as restrictive contracts coupled with digital rights management techniques, as well as various forms of online business practices (NRC, 1999).
A patent is a type of intellectual property right that gives the owner of the patent “the right to exclude others from making, using, offering for sale, or selling the invention throughout the United States or importing the invention into the United States”c in exchange for disclosing the invention. The inventor must disclose enough information to enable “a person of ordinary skill in the art”d to achieve the same result or make the same device when following the steps provided in the patent application; however, determining the level of detail needed to meet that goal is subjective. A patent can be obtained for inventions that are novel, useful, and nonobvious to fellow scientists in the same field.e Various aspects of an omics-based test could potentially be patentable, including the assay used to make molecular measurements, and the code and computational procedures used to analyze the results.
However, there are limitations to the protections offered by patents, and court decisions can change the landscape of patent protections. For example, a recent federal circuit court case regarding gene patents may increase the uncertainties that developers have about the value of patents for gene-based tests. The American Civil Liberties Union (ACLU) sued Myriad Genetics, the exclusive holder of licenses for the BRCA genes, challenging the practice of patenting genes.f The court held that it is permissible to patent isolated DNA molecules. It applied the “machine or transformation” testg to assess the patentability of the methods of the BRCA tests, which states that a method is patentable if: “(1) it is tied to a particular machine or apparatus, or (2) it transforms a particular article into a different state or thing.” The court held that the method of comparing an individual’s BRCA sequence to a reference sequence in order to identify genetic mutations
were not patentable because “they claim only an abstract mental process.” However, Myriad’s method for screening potential cancer therapeutics via changes in cell growth rates was patentable because it required the transformative step of manipulating the cells and their growth medium. The European Patent Office reached a similar decision, upholding Myriad’s patent for the BRCA test in a limited form (Siva, 2009). Applying the holding of these cases to omics-based tests, it is unclear whether the algorithms and methods for analyzing molecular measurements in omics-based tests are patentable. A U.S. court would likely apply the machine-or-transformation test to the exact methods and computational procedures used in each test and make a decision on a case-by-case basis. However, as a 2006 NRC report noted, there has been an overall trend in the courts toward an expansive interpretation of patentable subject matter (NRC, 2006).
There are several additional limitations to relying on patent protections to guard proprietary interests in omics-based tests. The patent holder must enforce his/her exclusive right to use and market the invention. For example in the Myriad case, Myriad sent a series of “cease and desist” letters to laboratories engaged in the commercial testing of the BRCA genes without a license. Myriad ultimately had to litigate the validity of its patent to maintain the exclusive right to conduct these tests. It also is unclear how extensively others must change or modify an omics-based test before they create a new patentable invention that they can market. In addition, the Supreme Court is likely to weigh in on the patentability of biological tests in the near futureh ,i and the U.S. Patent and Trademark Office is preparing a report on the patentability of genetic diagnostic testing, as required by the American Invests Act;j both actions have the potential to change this area of law.
Infrastructure Requirements
The infrastructure requirements for databases that store omics data are significant and costly. The databases must allow researchers to share DNA, amino acid sequences, and protein structure data in a manageable and searchable format; these datasets can be enormous (Quackenbush, 2009). Data curation is also necessary to ensure the quality of the information stored, but approaches to curation vary considerably. The databases also need to have sufficient security protections to guard the privacy of the information stored. Despite these obstacles, however, a number of genomic and proteomic databases exist, such as the database of Genotypes and Phenotypes (dbGaP) (NCBI, 2012), the European Molecular Biology Laboratory European Bioinformatics Institute (EBI, 2012), the National Library of Medicine’s Gene Expression Omnibus (NLM, 2012), Compendia Biosci-ence (2012), UCSC Gene Browser (UCSC, 2012), and ProteomeXchange (2012).
Data repositories have developed varying policies and procedures to protect the privacy and security of the information. For example, dbGaP is a public repository for individual-level phenotype, exposure, genotype, and sequence-based data and the associations between them. In order to protect research participant privacy, all studies in dbGaP have two levels of access: open and controlled. The open-access data, which can be browsed online or downloaded from dbGaP without prior permission or authorization, generally includes all the study documents, such as the protocol and questionnaires, as well as summary data for
each measured phenotype variable and for genotype results. Preauthorization is required to gain access to the phenotype and genotype results for each individual, and this individual-level data is coded to protect the identity of study participants (Mailman et al., 2007; NLM, 2006).
Privacy of Health Information
The laws protecting the privacy of individuals’ health information are a potential obstacle to making omics data sustainably available to other investigators. Much of the data in omics research is from human subjects and potentially could be linked to a specific individual, especially in the case of genetic data. In addition, most omics data used in the development of a clinical test need to be connected to individuals’ clinical data to be useful in that development process.
The Health Insurance Portability and Accountability Act Privacy Rule protects the privacy of personally identifiable health information (called “protected health information [PHI]”) created or received by health care professionals, health plans, or health care clearinghouses (“covered entities”). In general, the rule requires test developers to get authorization from research subjects in order to use and disclose their PHI in health research.k The rule does not require researchers to get authorization to use and disclose PHI that has been de-identified (as defined in the regulation). Until recently, there was considerable confusion about whether the Privacy Rule protected genetic information (IOM, 2009). However, the Genetic Information Nondiscrimination Act directed the U.S. Secretary of Health and Human Services to modify the Privacy Rule to explicitly recognize genetic information as PHI.l
The Common Rule provides human subjects protections in omics research that is federally funded. It protects the safety, autonomy, privacy, and fair treatment of patient-participants in federally funded research conducted on humans, and the cultural groups from which they are recruited. The Common Rule requires researchers to get informed consent from a person to use his/her private identifiable
information in research. Research that involves “anonymized data” (that is, information that is recorded in such a manner that subjects cannot be identified) is exempt from this requirement. However, an advanced notice of proposed rule-making includes the proposal to revise this aspect of the Common Rule to match the Privacy Rule’s more rigorous de-identification standards.m If this change becomes codified in the regulations, researchers may be required in many circumstances to obtain authorization and informed consent prior to sharing their research data, in order to comply with these laws, particularly as DNA sequence-based data can now be considered identifiable.
a The Copyright Act of 1976, 17 U.S.C. §§ 101-810 (2008).
b Feist Publications v. Rural Telephone Service Co., 499 U.S. 360 (1991).
c Patent Act, 35 U.S.C. § 154 (2008).
d Id. at § 103(a).
e Id. at §§ 101-103.
f The Association for Molecular Pathology, et al. v. United States Patent and Trademark Office, et al., 653 F.3d 1329 (Fed. Cir. 2011).
g Bilski vs. Kappos, 130 U.S. 3218 (2010).
h Mayo Collaborative Services v. Prometheus Laboratories, Inc., 628 F.3d 1347, (Fed. Cir. 2010), cert. granted, (U.S. Dec. 7, 2011) (No. 10-1150).
i The Association for Molecular Pathology, et al. v. Myriad Genetics, Inc. et al., petition for cert. filed (December 7, 2011).
j Leahy-Smith America Invents Act, Public Law No. 112-29 § 27(2011).
k The Secretary of Health and Human Services issued a notice of proposed rulemaking that includes potential modifications to the HIPAA Privacy Rule’s authorization requirements in response to the statutory amendments under the Health Information Technology for Economic and Clinical Health Act (the “HITECH Act”). See, Modifications to the HIPAA Privacy, Security, and Enforcement Rules Under the Health Information Technology for Economic and Clinical Health Act, 75 Fed. Reg. 40,868 (July 14, 2010).
l Genetic Information Nondiscrimination Act, Public Law No. 110-233 (2008).
m Human Subjects Research Protections: Enhancing Protections for Research Subjects and Reducing Burden, Delay, and Ambiguity for Investigators, 76 Fed. Reg. 44,512 (July 26, 2011).
- Data variability unrelated to clinical outcome of interest: Often, a computational model developed on one dataset (Step 2) performs poorly on another independent dataset (Step 3). This can occur for a number of reasons, such as variability in patient population, sample preparation, time of sample collection, operator variability, etc. Hence, evidence of a computational model’s performance based only on the dataset used to train the model, even if cross-validation is properly performed, provides little evidence of the model’s suitability for future samples. A relevant example here is the OvaCheck case study, discussed in Appendix A, in which signals obtained on one dataset did not hold up when the analysis was applied to other independent sample sets (Baggerly et al., 2004).
- Need for multiple datasets: For the reasons just described, computational models that are fit on multiple datasets in Step 2 will tend to perform better later. In other words, investigators are urged to develop a computational model on omics datasets derived from specimens and associated clinical outcomes collected at multiple laboratories at multiple institutions, rather than fitting a model on just a single dataset. For instance, the 21-Gene Recurrence Score (Oncotype DX) case study (Appendix A) was developed using multiple independent datasets (Paik et al., 2004). In that case, data were analyzed by the same investigators, but different datasets were derived from different clinical trials at multiple institutions.
- Study design and batch effects: As in all areas of biomedical research, good study design is crucial. If the dataset used in Step 2 to develop the computational model resulted from poor experimental design (e.g., if the samples from patients whose cancers recurred were processed at a different time or by a different technician or in a different laboratory) then batch effects (Leek et al., 2010) can occur. This will lead to spurious signal, potentially resulting in a computational model that performs extremely well on the data on which it was developed (Step 2), but that will perform poorly on future patient samples (Step 3). A relevant example is the OvaCheck case study, discussed in Appendix A, in which peaks in the noise regions of the proteomic spectra could distinguish samples from controls and cancer, indicating batch effects (Baggerly et al., 2004).
- Computational procedure lock-down: It is crucial that at the end of Step 2, the fully specified computational procedures be locked down before progressing into confirmation on an independent test set in Step 3. For instance, simply reporting the set of genes included in the computational model underlying a transcriptomics-based test is insufficient, because this does not constitute a fully specified computational procedure. In the original Oncotype DX study, the researchers locked down the computational model after Step 2 and reported the fully specified computational procedures in the paper (Paik et al., 2004). In the Corus CAD case study, lock-down and the fully specified computational procedures were reported in the clinical validation paper (Rosenberg et al., 2010). The fully specified computational procedures for the AlloMap test were reported in Deng et al. (2006). In contrast, in the Duke studies, the genes used in the development of the computational model were reported, but the fully specified computational procedures were not; furthermore, it is likely that the computational procedures were not ever fully locked down before proceeding into Step 3 or further stages of omics-based test development, including clinical trials (see Appendix B for details).
- Role of biostatistics and bioinformatics experts: In a relatively new and evolving field such as omics, it is not possible to predict all the possible pitfalls that investigators may face in the discovery phase. The involvement of properly trained biostatistical or bioinformatics collaborators who are fully integrated in all aspects of the discovery and evaluation process can serve as an additional safeguard. The type of biostatistical expertise that is required may vary depending on the stage or phase of test development. For example, experts in developing computational models for omics-based tests may not have sufficient expertise in clinical trial design, and vice versa. This is relevant to the Duke case study (as discussed in Appendix B), in which there was a lack of continuity in bio statistics personnel and numerous errors were identified in the statistical methodology and analyses.
COMPLETION OF THE DISCOVERY PHASE OF OMICS-BASED TEST DEVELOPMENT
A candidate omics-based test should be defined precisely, including the molecular measurements, the computational procedures, and the intended clinical use of the test, in anticipation of the test validation phase (Recommendation 1d). There are enormous opportunities in the rapidly improving suite of omics technologies to identify measurements with potential clinical utility. However, there are significant challenges in moving from the initial identification of potentially relevant differences in omics measurements to validated and robust clinical tests. Among these challenges are risks of overfitting the data in the development of the computational model and the enormous heterogeneity among different studies of ostensibly the same disease states (for both technical and biological reasons). Going forward, transparency in the reporting of all aspects of the development of an omics-based test, including the measurements made, preprocessing techniques used, and the fully specified computational procedure, is critical. The release of sufficient metadata with publication is also key to the identification of candidate omics-based tests that work across multiple sites, which is necessary to generate increasingly robust omics-based tests to enhance patient care.
In the next phase of test development (analytical and clinical/biological validation, described in Chapter 3), the methods used to obtain the omics measurements from patient samples may be changed in order to establish a clinically feasible, inexpensive, and robust assay for implementation in clinical practice. However, the fully specified computational procedures defined in the discovery stage must remain locked down and unchanged
in all subsequent test development steps. At the end of the validation phase in Chapter 3, the complete test method, including the methods for obtaining the omics measurements as well as the fully specified computational procedures, must be locked down before crossing the bright line to evaluate the test for clinical utility and use.
This chapter has outlined best practices for the discovery phase for omics-based test development. Because omics-based tests rely on interpretation of high-dimensional datasets, it is important to guard against over-fitting the data throughout the test development process. Overfitting due to lack of proper statistical methods can lead to a model that fits the training samples well, even though the model might perform poorly on independent samples not used in test development. The steps delineated in this chapter aim to prevent an overfit model from progressing to subsequent stages of test development. Cross-validation or a training set/test set approach can help reduce the risk of overfitting, but confirmation of all fully specified computational procedures and candidate omics-based tests on a blinded independent sample set is the “gold standard” for assessing the validity of any test. The importance of independent confirmation is also emphasized in the committee’s recommendations for funders (see Chapter 5), which urge funders to support this type of work. In addition, complex analyses of these large datasets highlight the need for availability of the data and code used for the discovery phase of omics-based test development, to enable independent verification of the findings. The result of the discovery process is a candidate omics-based test with locked-down computational procedures that is then moved into the test validation phase to assess analytical and clinical/biological validation, as described in Chapter 3.
RECOMMENDATION 1: Discovery Phase
When candidate omics-based tests from the discovery phase are intended for further clinical development, the following criteria should be satisfied and fully disclosed (for example, through publication or patent application) to enable independent verification of the findings:
a. Candidate omics-based tests should be confirmed using an independent set of samples, not used in the generation of the computational model and, when feasible, blinded to any outcome or other phenotypic data until after the computational procedures have been locked down and the candidate omics-based test has been applied to the samples;
b. Data and metadata used for development of the candidate omicsbased test should be made available in an independently managed database (such as dbGaP) in standard format;
c. Computer code and fully specified computational procedures used for development of the candidate omics-based test should be made sustainably available; and
d. A candidate omics-based test should be defined precisely, includ ing the molecular measurements, the computational procedures, and the intended clinical use of the test, in anticipation of the test validation phase.
Ahrens, C. H., E. Brunner, E. Qeli, K. Basler, and R. Aebersold. 2010. Generating and navigating proteome maps using mass spectrometry. Nature Reviews Molecular Cell Biology 11(11):789-801.
Arrell, D. K., J. Zlatkovic Lindor, S. Yamada, and A. Terzic. 2011. K(ATP) channel-dependent metaboproteome decoded: Systems approaches to heart failure prediction, diagnosis, and therapy. Cardiovascular Research 90(2):258-266.
Ayoglu, B., A. Haggmark, M. Neiman, U. Igel, M. Uhlen, J. M. Schwenk, and P. Nilsson. 2011. Systematic antibody and antigen-based proteomic profiling with microarrays. Expert Review of Molecular Diagnostics 11(2):219-234.
Baggerly, K. A., J. S. Morris, and K. R. Coombes. 2004. Reproducibility of SELDI-TOF protein patterns in serum: Comparing datasets from different experiments. Bioinformatics 20(5):777-785.
Bailey, R. C., G. A. Kwong, C. G. Radu, O. N. Witte, and J. R. Heath. 2007. DNA-encoded antibody libraries: A unified platform for multiplexed cell sorting and detection of genes and proteins. Journal of the American Chemical Society 129(7):1959-1967.
Ben Sellem, D., K. Elbayed, A. Neuville, F. M. Moussallieh, G. Lang-Averous, M. Piotto, J. P. Bellocq, and I. J. Namer. 2011. Metabolomic characterization of ovarian epithelial carcinomas by HRMAS-NMR spectroscopy. Journal of Oncology 2011:174019.
Bianco-Miotto, T., K. Chiam, G. Buchanan, S. Jindal, T. K. Day, M. Thomas, M. A. Pickering, M. A. O’Loughlin, N. K. Ryan, W. A. Raymond, L. G. Horvath, J. G. Kench, P. D. Stricker, V. R. Marshall, R. L. Sutherland, S. M. Henshall, W. L. Gerald, H. I. Scher, G. P. Risbridger, J. A. Clements, L. M. Butler, W. D. Tilley, D. J. Horsfall, and C. Ricciardelli. 2010. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiology, Biomarkers, & Prevention 19(10):2611-2622.
Chang, R. L., L. Xie, P. E. Bourne, and B. O. Palsson. 2010. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Computational Biology 6(9):e1000938.
Compendia Bioscience, Inc. 2012. Compendia Bioscience: Cure Cancer with Genomic Data. http://www.compendiabio.com/ (accessed February 23, 2012).
Deng, M. C., H. J. Eisen, M. R. Mehra, M. Billingham, C. C. Marboe, G. Berry, J. Kobashigawa, F. L. Johnson, R. C. Starling, S. Murali, D. F. Pauly, H. Baron, J. G. Wohlgemuth, R. N. Woodward, T. M. Klingler, D. Walther, P. G. Lal, S. Rosenberg, S. Hunt, and for the CARGO Investigators. 2006. Noninvasive discrimination of rejection in cardiac allo-graft recipients using gene expression profiling. American Journal of Transplantation 6(1):150-160.
Drexler, D. M., M. D. Reily, and P. A. Shipkova. 2011. Advances in mass spectrometry applied to pharmaceutical metabolomics. Analytical and Bioanalytical Chemistry. 399(8):2645-2653.
EBI (European Bioinformatics Institute). 2012. Data Resources and Tools. http://www.ebi.ac.uk/ (accessed February 23, 2012).
Fagerberg, L., S. Stromberg, A. El-Obeid, M. Gry, K. Nilsson, M. Uhlen, F. Ponten, and A. Asplund. 2011. Large-scale protein profiling in human cell lines using antibody-based proteomics. Journal of Proteome Research 10(9):4066-4075.
Farrah, T., E. W. Deutsch, G. S. Omenn, D. S. Campbell, Z. Sun, J. A. Bletz, P. Mallick, J. E. Katz, J. Malmström, R. Ossola, J. D. Watts, B. Lin, H. Zhang, R. L. Moritz, and R. Aebersold. 2011. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Molecular and Cellular Proteomics 10(9):M110.006353.
Folger, O., L. Jerby, C. Frezza, E. Gottlieb, E. Ruppin, and T. Shlomi. 2011. Predicting selective drug targets in cancer through metabolic networks. Molecular Systems Biology 7:501-527.
Frezza, C., L. Zheng, O. Folger, K. N. Rajagopalan, E. D. Mackenzie, L. Jerby, M. Micaroni, B. Chaneton, J. Adam, A. Hedley, G. Kalna, I. P. Tomlinson, P. J. Pollard, D. G. Watson, R. J. Deberardinis, T. Shlomi, E. Ruppin, and E. Gottlieb. 2011. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 477(7363):225-228.
Gold, L., D. Ayers, J. Bertino, C. Bock, A. Bock, E. N. Brody, J. Carter, A. B. Dalby, B. E. Eaton, T. Fitzwater, D. Flather, A. Forbes, T. Foreman, C. Fowler, B. Gawande, M. Goss, M. Gunn, S. Gupta, D. Halladay, J. Heil, J. Heilig, B. Hicke, G. Husar, N. Janjic, T. Jarvis, S. Jennings, E. Katilius, T. R. Keeney, N. Kim, T. H. Koch, S. Kraemer, L. Kroiss, N. Le, D. Levine, W. Lindsey, B. Lollo, W. Mayfield, M. Mehan, R. Mehler, S. K. Nelson, M. Nelson, D. Nieuwlandt, M. Nikrad, U. Ochsner, R. M. Ostroff, M. Otis, T. Parker, S. Pietrasiewicz, D. I. Resnicow, J. Rohloff, G. Sanders, S. Sattin, D. Schneider, B. Singer, M. Stanton, A. Sterkel, A. Stewart, S. Stratford, J. D. Vaught, M. Vrkljan, J. J. Walker, M. Watrobka, S. Waugh, A. Weiss, S. K. Wilcox, A. Wolfson, S. K. Wolk, C. Zhang, and D. Zichi. 2010. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 5(12):e15004.
Gottlieb, A., G. Y. Stein, E. Ruppin, and R. Sharan. 2011. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Molecular Systems Biology 7:496.
Honda, K., and D. R. Littman. 2011. The microbiome in infectious disease and inflammation. Annual Review of Immunology. 2011 Mar 24. [Epub ahead of print].
Hwang, D., I. Y. Lee, H. Yoo, N. Gehlenborg, J. H. Cho, B. Petritis, D. Baxter, R. Pitstick, R. Young, D. Spicer, N. D. Price, J. G. Hohmann, S. J. Dearmond, G. A. Carlson, and L. E. Hood. 2009. A systems approach to prion disease. Molecular Systems Biology 5:252.
Ideker, T., J. Dutkowski, and L. Hood. 2011. Boosting signal-to-noise in complex biology: Prior knowledge is power. Cell 144(6):860-863.
Ince, D. C., L. Hatton, and J. Graham-Cumming. 2012. The case for open computer programs. Nature 482:485-488.
Ioannidis, J. P. A., and M. J. Khoury. 2011. Improving validation practices in “omics” research. Science 334(6060):1230-1232.
IOM (Institute of Medicine). 2009. Beyond the HIPAA Privacy Rule: Enhancing Privacy, Improving Health through Research. Washington, DC: The National Academies Press.
Kinros, J. M., A. W. Darzi, and J. K. Nicholson. 2011. Gut microbiome-host interactions in health and disease. Genomic Medicine 3(3):14.
Koulman, A., G. A. Lane, S. J. Harrison, and D. A. Volmer. 2009. From differentiating metabolites to biomarkers. Analytical and Bioanalytical Chemistry 394(3):663-670.
Leek, J. T., R. B. Scharpf, H. C. Bravo, D. Simcha, B. Langmead, W. E. Johnson, D. Geman, K. Baggerly, and R. A. Irizarry. 2010. Tackling the widespread and critical impact of batch effects in high-throughput data. Nature Reviews Genetics 11(10):733-739.
Legrain, P., R. Aebersold, A. Archakov, A. Bairoch, K. Bala, L. Beretta, J. Bergeron, C. H. Borchers, G. L. Corthals, C. E. Costello, E. W. Deutsch, B. Domon, W. Hancock, F. He, D. Hochstrasser, G. Marko-Varga, G. H. Salekdeh, S. Sechi, M. Snyder, S. Srivastava, M. Uhlen, C. H. Wu, T. Yamamoto, Y. K. Paik, and G. S. Omenn. 2011. The human proteome project: Current state and future direction. Molecular & Cellular Proteomics10(7):M111. 009993.
Lewis, G. D., and R. E. Gerszten. 2010. Toward metabolomic signatures of cardiovascular disease. Circulation: Cardiovascular Genetics 3(2):119-121.
Lewis, N. E., G. Schramm, A. Bordbar, J. Schellenberger, M. P. Andersen, J. K. Cheng, N. Patel, A. Yee, R. A. Lewis, R. Eils, R. Konig, and B. O. Palsson. 2010. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nature Biotechnology 28(12):1279-1285.
Ma, C., R. Fan, H. Ahmad, Q. Shi, B. Comin-Anduix, T. Chodon, R. C. Koya, C. C. Liu, G. A. Kwong, C. G. Radu, A. Ribas, and J. R. Heath. 2011. A clinical microchip for evaluation >of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nature Medicine 17(6):738-743.
Maas, S., 2010. Gene regulation through RNA editing. Discovery Medicine 10(54):379-386.
Mailman, M. D., M. Feolo, Y. Jin, M. Kimura, K. Tryka, R. Bagoutdinov, L. Hao, A. Kiang, J. Paschall, L. Phan, N. Popova, S. Pretel, L. Ziyabari, M. Lee, Y. Shao, Z. Y. Wang, K. Sirotkin, M. Ward, M. Kholodov, K. Zbicz, J. Beck, M. Kimelman, S. Shevelev, D. Preuss, E. Yaschenko, A. Graeff, J. Ostell, and S. T. Sherry. 2007. The NCBI dbGaP database of genotypes and phenotypes. Nature Genetics 39:1181-1186.
Masoodi, M., M. Eiden, A. Koulman, D. Spaner, and D. A. Volmer. 2010. Comprehensive lipidomics analysis of bioactive lipids in complex regulatory networks. Analytical Chemistry 82(19):8176-8185.
McGary, K. L., T. J. Park, J. O. Woods, H. J. Cha, J. B. Wallingford, and E. M. Marcotte. 2010. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proceedings of the National Academy of Sciences 107(14):6544-6549.
McShane, L. M. 2010. NCI Address to Institute of Medicine Committee Convened to Review Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials. Presentation at Meeting 1, Washington, DC, December 20.
Menon, R., A. Roy, S. Mukerjee, S. Belkin, Y. Zhang, and G. S. Omenn. 2011. Functional implications of structural predictions for alternative splice proteins expressed in HER2/ neu-induced breast cancers. Journal of Proteome Research. [Epub ahead of print].
Morin, A., J. Urban, P. D. Adams, I. Foster, A. Sali, D. Baker, and P. Sliz. 2012. Research priorities. Shining light into black boxes. Science 336(6078):159-160.
Moussay, E., K. Wang, J. H. Cho, K. van Moer, S. Pierson, J. Paggetti, P. V. Nazarov, V. Palissot, L. E. Hood, G. Berchem, and D. J. Galas. 2011. MicroRNA as biomarkers and regulators in B-cell chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences 108(16):6573-6578.
NCBI (National Center for Biotechnology Information). 2012. dbGaP. http://www.ncbi.nlm.nih.gov/gap (accessed February 23, 2012).
Ng, S. B., A. W. Bigham, K. J. Buckingham, M. C. Hannibal, M. J. McMillin, H. I. Gildersleeve, A. E. Beck, H. K. Tabor, G. M. Cooper, H. C. Mefford, C. Lee, E. H. Turner, J. D. Smith, M. J. Rieder, K. Yoshiura, N. Matsumoto, T. Ohta, N. Niikawa, D. A. Nickerson, M. J. Bamshad, and J. Shendure. 2010. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature Genetics 42(9):790-793.
NLM (National Library of Medicine). 2006. NIH launches dbGAP, a database of Genome Wide Association Studies. http://www.nlm.nih.gov/news/press_releases/dbgap_launchPR06.html (accessed December 12, 2006).
NLM. 2012. GEO: Gene expression omnibus. http://www.ncbi.nlm.nih.gov/geo/ (accessed February 23, 2012).
NRC (National Research Council). 1999. A Question of Balance: Private Rights and them Public Interest in Scientific and Technical Databases. Washington, DC: National Academy Press.
NRC. 2003. Sharing Publication-Related Data and Materials: Responsibilities of Authorship in the Life Sciences. Washington, DC: The National Academies Press.
NRC. 2005. Catalyzing Inquiry at the Interface of Computing and Biology. Washington, DC: The National Academies Press.
NRC. 2006. Reaping the Benefits of Genomic and Proteomic Research: Intellectual Property Rights, Innovation, and Public Health. Washington, DC: The National Academies Press.
Omenn, G. S. 2009. A landmark systems analysis of prion disease of the brain. Molecular Systems Biology 5:254.
Omenn, G. S., M. S. Baker, and R. Aebersold. 2011. Recent workshops of the HUPO Human Plasma Proteome Project (HPPP): A bridge with the HUPO CardioVascular Initiative and the emergence of SRM targeted proteomics. Proteomics 11(17):3439-3443.
Ostroff, R. M., W. L. Bigbee, W. Franklin, L. Gold, M. Mehan, Y. E. Miller, H. I. Pass, W. N. Rom, J. M. Siegfried, A. Stewart, J. J. Walker, J. L. Weissfeld, S. Williams, D. Zichi, and E. N. Brody. 2010. Unlocking biomarker discovery: Large scale application of aptamer proteomic technology for early detection of lung cancer. PLoS One 5(12):e15003.
Paik, S., S. Shak, G. Tang, C. Kim, J. Baker, M. Cronin, F. L. Baehner, M. G. Walker, D. Watson, T. Park, W. Hiller, E. R. Fisher, D. L. Wickerham, J. Bryant, and N. Wolmark. 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New England Journal of Medicine 351(27):2817-2826.
Petricoin, E. F., A. M. Ardekani, B. A. Hitt, P. J. Levine, V. A. Fusaro, S. M. Steinberg, G. B. Mills, C. Simone, D. A. Fishman, E. C. Kohn, and L. A. Liotta. 2002. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359(9306):572-577.
Picotti, P., O. Rinner, R. Stallmach, F. Dautel, T. Farrah, B. Domon, H. Wenschuh, and R. Aebersold. 2010. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nature Methods 7(1):43-46. ProteomeXchange. 2012. Mission. http://www.proteomexchange.org/ (accessed February 23, 2012).
Ransohoff, D. F. 2008. The process to discover and develop biomarkers for cancer: A work in progress. Journal of the National Cancer Institute 100(20):1419-1420.
Ransohoff, D. F. 2009. Promises and limitations of biomarkers. Recent Results in Cancer Research 181:55-59.
Roach, J. C., G. Glusman, A. F. Smit, C. D. Huff, R. Hubley, P. T. Shannon, L. Rowen, K. P. Pant, N. Goodman, M. Bamshad, J. Shendure, R. Drmanac, L. B. Jorde, L. Hood, and D. J. Galas. 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328(5978):636-639.
Rosenberg, S., M. R. Elashoff, P. Beineke, S. E. Daniels, J. A. Wingrove, W. G. Tingley, P. T. Sager, A. J. Sehnert, M. Yau, W. E. Kraus, K. Newby, R. S. Schwartz, S. Voros, S. G. Ellis, N. Tahirkhelli, R. Waksman, J. McPherson, A. Lansky, M. E. Winn, N. J. Schork, E. J. Topol, and for the PREDICT (Personalized Risk Evaluation and Diagnosis In the Coronary Tree) Investigators. 2010. Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Annals of Internal Medicine 153(7):425-434.
Segre, A. V., L. Groop, V. K. Mootha, M. J. Daly, and D. Altshuler. 2010. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 6(8). pii: e1001058.
Seppanen-Laakso, T., and M. Oresic. 2009. How to study lipidomes. Journal of Molecular Endocrinology 42(3):185-190.
Service, R. F. 2008. Chemistry. Click chemistry clicks along. Science 320(5878):868-869.
Shlomi, T., T. Benyamini, E. Gottlieb, R. Sharan, and E. Ruppin. 2011. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Computational Biology 7(3):e1002018.
Simon, R., M. D. Radmacher, K. Dobbin, and L. M. McShane. 2003. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. Journal of the National Cancer Institute 95(1):14-18.
Siva, N. 2009. Myriad wins BRCA1 row. Nature Biotechnology 27:8.
Sreekumar, A., L. M. Poisson, T. M. Rajendiran, A. P. Khan, Q. Cao, J. Yu, B. Laxman, R. Mehra, R. J. Lonigro, Y. Li, M. K. Nyati, A. Ahsan, S. Kalyana-Sundaram, B. Han, X. Cao, J. Byun, G. S. Omenn, D. Ghosh, S. Pennathur, D. C. Alexander, A. Berger, J. R. Shuster, J. T. Wei, S. Varambally, C. Beecher, and A. M. Chinnaiyan. 2009. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457(7231):910-914.
Sugatani, T., J. Vacher, and K. A. Hruska. 2011. A microRNA expression signature of osteo-clastogenesis. Blood 117(13):3648-3657.
Tan, X., W. Qin, L. Zhang, J. Hang, B. Li, C. Zhang, J. Wan, F. Zhou, K. Shao, Y. Sun, J. Wu, X. Zhang, B. Qiu, N. Li, S. Shi, X. Feng, S. Zhao, Z. Wang, X. Zhao, Z. Chen, K. Mitchelson, J. Cheng, Y. Guo, and J. He. 2011. A five-microRNA signature for squamous cell lung carcinoma (SCC) diagnosis and Hsa-miR-31 for SCC prognosis. Clinical Cancer Research 17(21):6802-6811.
Tang, F., K. Lao, and M. A. Surani. 2011. Development and applications of single-cell transcriptome analysis. Nature Methods 8(4 Suppl):S6-S11.
Teague, B., M. S. Waterman, S. Goldstein, K. Potamousis, S. Zhou, S. Reslewic, D. Sarkar, A. Valouev, C. Churas, J. M. Kidd, S. Kohn, R. Runnheim, C. Lamers, D. Forrest, M. A. Newton, E. E. Eichler, M. Kent-First, U. Surti, M. Livny, and D. C. Schwartz. 2010. High-resolution human genome structure by single-molecule analysis. Proceedings of the National Academy of Sciences 107(24):10848-10853.
Tilg, H., and A. Kaser. 2011. Gut microbiome, obesity, and metabolic dysfunction. Journal of Clinical Investigations 121(6):2126-2132.
UCSC (University of California, Santa Cruz). 2012. UCSC genome bioinformatics. http://genome.ucsc.edu/ (accessed February 23, 2012).
Weckwerth, W. 2003. Metabolomics in systems biology. Annual Review of Plant Biology 54:669-689.
Wolf-Yadlin, A., M. Sevecka, and G. MacBeath. 2009. Dissecting protein function and signaling using protein microarrays. Current Opinion in Chemical Biology 13(4):398-405.
Yu, S. L., H. Y. Chen, G. C. Chang, C. Y. Chen, H. W. Chen, S. Singh, C. L. Cheng, C. J. Yu, Y. C. Lee, H. S. Chen, T. J. Su, C. C. Chiang, H. N. Li, Q. S. Hong, H. Y. Su, C. C. Chen, W. J. Chen, C. C. Liu, W. K. Chan, W. J. Chen, K. C. Li, J. J. Chen, and P. C. Yang. 2008. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 13(1):48-57.
Zhang, G. F., S. Sadhukhan, G. P. Tochtrop, and H. Brunengraber. 2011. Metabolomics, pathway regulation, and pathway discovery. Journal of Biological Chemistry 286(27):23631-23635.
This Page is Blank
































