
This appendix reviews the case studies that the committee examined, including six commercially available omics-based tests, an early single-marker test, and an omics-based test that did not advance to clinical use. These case studies appear in the following order:
- Human epidermal growth factor receptor 2 (HER2)
- Oncotype DX
- MammaPrint
- Tissue of Origin
- OVA1
- OvaCheck
- AlloMap
- Corus CAD
The case study focusing on several omics-based tests developed by a Duke University laboratory to predict sensitivity to chemotherapeutic agents appears in Appendix B.
HER2
HER2 is one of the earliest biomarker tests for guiding therapeutic decisions, and is widely used in clinical practice. The development of HER2 as an effect modifier biomarker has transformed breast cancer treatment by identifying the 20-30 percent of patients with overexpression of the HER2 oncogene who are likely to benefit from therapy targeting HER2 (De et
al., 2010; Phillips et al., 2009). At least seven tests to detect HER2 gene amplification and protein overexpression have Food and Drug Administration (FDA) approval for use as effect modifier markers for tumor response to trastuzumab (reviewed by Allison, 2010, and Shah and Chen, 2010). In addition, some companies have received FDA clearance for imaging analysis tools accompanying FDA-approved HER2 tests (FDA, 2011a). However, despite more than 20 years of research and development, difficulties remain in defining optimal implementation of this single-marker test (De et al., 2010), illustrating some of the profound challenges confronting developers of multianalyte, omics-based tests.
These difficulties include the number of modalities for evaluating HER2 (IHC [immunohistochemistry], FISH [fluorescent in situ hybridization], and others), the subjectivity of test results, lab-to-lab variability (central or reference laboratory versus smaller laboratories), laboratory errors leading to false positives and false negatives, differences in cut-off recommendations, and some uncertainty regarding clinical benefit of trastuzumab for patients with borderline HER2-positive results. Accurate selection of patients for therapy targeting HER2, or conversely, identification of those patients who are not likely to benefit from HER2-targeted therapy, depends on reliable HER2 testing and appropriate cut-off criteria (Kroese et al., 2007).
HER2 Testing in Clinical Practice
In 2007, a panel established by the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) recommended HER2 status determination for all invasive breast cancers (Wolff et al., 2007a,b) and clarified some of the technical limitations of both IHC and FISH (Schmitt, 2009). In 2002, substantial discordance was reported for both IHC and FISH results performed in community laboratories versus a central reference laboratory in the course of two clinical trials (Paik et al., 2002; Roche et al., 2002). In response, the ASCO/CAP panel issued recommendations for the HER2 testing process (e.g., ways to reduce lab-based errors) and interpretation (Wolff et al., 2007a,b). These guidelines alleviated some lab effects within a single HER2 testing modality, though interlab reproducibility continues to be an area of substantial concern for HER2 testing.
The choice of HER2 testing modality is also debated (Sauter et al., 2009; Schmitt, 2009). Historically, IHC has been the primary method for HER2 testing, and FISH has been used to confirm these findings, when IHC testing is equivocal. However, some assert that FISH should be the primary HER2 testing platform (Sauter et al., 2009), while others have advised that IHC alone should never be relied on for selecting anti-HER2 treatment (De et al., 2010). The ASCO/CAP recommendations did not recommend one
method for HER2 testing over another, and the National Cancer Institute (NCI) website currently states that “limitations in assay precision make it inadvisable to rely on a single method to rule out potential Herceptin benefit” (NCI, 2011). New methodologies are also in development for HER2 testing, including quantitative real-time reverse-transcriptase poly-merase chain reaction (qRT-PCR)-based detection of HER2 gene overexpression, which has been presented as the most quantitative platform to date (Baehner et al., 2010).
Differences in cut-off recommendations and equivocal HER2 test results also present challenges. For example, a tumor in which 10 percent of tumor cells show +3 IHC immunoreactivity, and another in which 99 percent of tumor cells display intermediate +2 immunoreactivity might both respond to the same treatment (De et al., 2010). Recently published highly exploratory studies from two of the largest randomized trials of the anti-HER2 therapy trastuzumab have suggested that patients who have some HER2 expression (but below the established cut-off points and not amplified in a FISH test) might benefit from adjuvant trastuzumab (Paik et al., 2008; Perez et al., 2010). A prospective randomized trial (National Surgical Adjuvant Breast and Bowel Project [NSABP] B-47) aims to address this question by randomizing patients with HER2 IHC scores of 1+ or 2+ (but not amplified according to FISH) to chemotherapy plus or minus trastuzumab.
False-positive and false-negative results remain a significant concern for HER2 testing as well. False negatives result in a potentially life-saving anti-HER2 therapy being withheld from a patient. False positives result in treatment with anti-HER2 therapies in the adjuvant or neoadjuvant setting, despite a small chance of benefiting from such treatment. This is a concern given trastuzumab’s association with cardiotoxicity, as well as the expense of treatment ($800-$1,000/week for 26-52 weeks) (Sauter et al., 2009).
Case Highlights
The development of HER2 testing and HER2 targeted therapy represents a significant advance in the treatment of breast cancer and the field of molecularly targeted medicine, but the challenges in implementing HER2 testing in practice have been substantial. Different testing modalities, subjectivity of test results, lab-to-lab variability, false-positive and false-negative results, differences in cut-off recommendations, and some uncertainty regarding clinical benefit of trastuzumab for patients with borderline HER2-positive results make it difficult to determine how best to conduct HER2 testing. There is not yet complete consensus on the standardization of HER2 testing, and as new testing methodologies emerge, new questions about HER2 testing will arise. The challenges involved in developing a single-analyte test such as HER2 are informative as the community is moving
toward the development of multianalyte, omics-based tests in which these challenges may be magnified.
ONCOTYPE DX
Oncotype DX (Genomic Health Inc.) is a multigene expression test developed to predict the risk of recurrence for node-negative, estrogen-receptor-positive breast cancer. Oncotype DX estimates the likelihood of distant recurrence at 10 years, and classifies individuals at low (scores less than 18), intermediate (18-30) and high (31-100) risk of breast cancer recurrence, assuming the use of adjuvant endocrine therapy, such as tamoxifen and/or an aromatase inhibitor, without chemotherapy. Developed as a laboratory-developed test (LDT), the test has not been submitted to FDA for clearance or approval; however, Genomic Health indicated that the company benefited from prior interaction with FDA and the extensive background material FDA provides on its website about assay validation.1 Two ongoing prospective studies (the TAILORx and RxPONDER trials, see section below on Clinical Utility) direct patient management on the basis of Oncotype DX Recurrence Score. For both trials, information required for approval of investigational use of Oncotype DX in the trial was submitted as part of an investigational new drug application to FDA.2
Developers of Oncotype DX sought to identify a subgroup of patients who were at such a low risk of recurrence that even if chemotherapy is active, the risks of chemotherapy would outweigh the benefit. Large randomized trials had previously demonstrated the benefit of adding chemotherapy to tamoxifen therapy for patients with estrogen-receptor-positive tumors (Berry et al., 2005; EBCTCG, 2005; Fisher et al., 1989, 1997, 2004). Adjuvant chemotherapy studies have generally demonstrated that the relative risk reduction from chemotherapy is constant across risk groups, but studies by the developers of Oncotype DX in patients with node-negative and node-positive estrogen-receptor-positive early breast cancer randomized to chemotherapy suggested that the relative risk reduction of chemotherapy in women with low Recurrence Scores was lower (Albain et al., 2010; Paik et al., 2006). This suggested that the absolute benefit of chemotherapy is lowest for those with the smallest risk of recurrence, and many women treated with tamoxifen alone are likely to remain free of distant recurrence with minimal, if any, benefit from the addition of chemotherapy. In this regard, Oncotype DX is used as a prognostic factor.
______________
1 Personal communication, Steven Shak, Genomic Health, December 13, 2011.
2 Personal communication, Lisa McShane, National Cancer Institute, February 9, 2012.
Discovery Phase
The discovery of Oncotype DX is described by Paik et al. (2004) and the Oncotype DX website (Genomic Health, 2011c). Investigators optimized the methods using a high-throughput, RT-PCR assay for quantifying RNA expression in formalin-fixed, paraffin-embedded (FFPE) tissue (Cronin et al., 2004). Two hundred and fifty candidate genes were selected for assay development based on microarray expression data and information from genomic databases, published literature, and experiments in molecular and cell biology. The relationship between gene expression and recurrence was analyzed in archival tissue from 447 breast cancer patients in three separate clinical studies (see Table A-1). Investigators generated a 21-gene panel (16 cancer-related genes and 5 reference genes) and computational model for determining the Recurrence Score. Five steps were used to develop the final gene list and Recurrence Score computational model. First, univarible analysis of each gene was performed separately for the three studies. Second, 16 cancer-related genes were selected based on their performance in predicting recurrence across all three studies. Third, based on coexpression by cluster and principal component analysis, 13 of the 16 genes were put into four gene groups (proliferation, estrogen receptor, HER2, and invasion). Fourth, martingale residual analysis was used to identify linear or non-linear functions for each of the gene groups. Fifth, regression analysis performed on each of the three studies was used to select the coefficients for each of the four gene groups and the remaining three individual genes.3 Additional analyses indicated that inclusion of additional cancer-related genes beyond 16 did not increase the robustness of prediction across the three datasets and that inclusion of fewer than 16 reduced the robustness (Paik et al., 2004). Many, but not all, of the 16 cancer-related genes in Oncotype DX were previously well established in the cancer literature for their association with prognosis (Kim and Paik, 2010).
Test Validation Phase
More than 150 standard operating procedures (SOPs) were developed for the 5-step 21-Gene Recurrence Score, including SOPs for equipment, histopathology, information technology, pre- and postanalytical methods, production and quality control, and quality assurance (Shak, 2011).
______________
3 Personal communication, Steven Shak, Genomic Health, December 13, 2011.
TABLE A-1 Archival Tissue Used in the Development of Oncotype DX Computational Model and Gene List
| Study | Paik et al., 2003 | Cobleigh et al., 2005 | Esteban et al., 2003 |
| Tissue source | Tamoxifen arm of NSABP B-20 | Rush Presbyterian-St. Luke’s Hospital | Providence St. Joseph’s Hospital |
| Sample # | 233 | 78 | 136 |
| Lymph-node status | Negative | > 10 positive nodes | Positive or negative |
| Estrogen-receptor status | Positive | Positive and negative | Positive and negative |
| Treatment | Tamoxifen (100%) | Tamoxifen (54%) Chemotherapy (80%) | Tamoxifen (41%) Chemotherapy (39%) |
| NOTE: NSABP = National Surgical Adjuvant Breast and Bowel Project. SOURCE: Shak (2011). |
|||
Analytical Validation
The analytical validity of Oncotype DX was assessed in the Agency for Healthcare Research and Quality (AHRQ) report, Impact of Gene Expression Profiling Tests on Breast Cancer Outcomes (AHRQ, 2008). The report noted there is evidence about Oncotype DX’s assay performance and laboratory characteristics as well as some limited information on its reproducibility. Cronin et al. (2007) found that Oncotype DX met acceptable operational performance ranges with minimal assay imprecision due to instrument, operator, reagent, and day-to-day baseline variation. Investigators also conducted technical feasibility studies during assay development, including analysis of preanalytical factors such as variability in preparation, tumor block age, and dissection (Shak, 2011). Paik and colleagues (2004) measured and reported the reproducibility within and between blocks in the clinical validation study.
Statistical and Bioinformatics Validation
The computational procedures used to determine the Recurrence Score are published, and there is public information that provides an overview of how the computational model was generated (see Discovery Phase) (Paik et al., 2004). The supplementary materials of Paik et al. (2004) note that the investigators weighted the NSABP B-20 results most heavily in selecting the final gene list and developing the computational model because investigators planned to clinically validate the test in similar archival tissue from NSABP B-14 patients. However, more detailed information on model
development and the RT-PCR and clinical data used in the development of 21-Gene RS are not publicly available.4
The test was locked down prior to clinical validation. The investigators reported that “the prospectively defined assay methods and endpoints were finalized in a protocol signed on August 27, 2003, and RT-PCR data were transferred to the NSABP for analysis on September 29, 2003” (Paik et al., 2004, p. 2820). Genomic Health was blinded to the clinical outcome data until the RT-PCR data were locked and transferred to NSABP.5
Clinical/Biological Validation
Archival tissue from breast cancer patients in three studies was used to clinically validate the prognostic value of Oncotype DX (Table A-2). Paik et al. (2004) found that the Recurrence Score quantified the likelihood of distant recurrence in tamoxifen-treated patients with lymph-node-negative, estrogen-receptor-positive breast cancer. Investigators prospectively defined the endpoints for validation and prespecified the cut-off values for low, intermediate, and high risk of recurrence. They had a large number of patient samples on which to clinically validate the prognostic value of the Recurrence Score, and did not use samples from the discovery phase in the validation studies. Although this study was not a true prospective clinical validation, many assert the prospective–retrospective study design has evidentiary value close to a prospective study (AHRQ, 2008; Harris et al., 2007; Simon et al., 2009).
The second study, Habel et al. (2006), assessed the prognostic value of the Recurrence Score using archival tissue from patients treated within the Northern California Kaiser Permanente health plan. Investigators found that the Recurrence Score was associated with the risk of breast cancer death among patients with estrogen-receptor-positive breast cancer who were treated with tamoxifen or were not treated with systemic adjuvant therapy.
The third study, Esteva et al. (2005), used a smaller number of archival tissue samples and did not find an association between the Recurrence Score and risk of distant recurrence. Investigators hypothesized that this result could be due to potential selection bias or confounding factors. However, investigators did find a high degree of concordance between RT-PCR and immunohistochemical assays for estrogen receptor, progesterone receptor, and HER2.
Chemotherapy benefit In an exploratory analysis designed to assess the test’s ability to predict the benefit of chemotherapy treatment, investigators
______________
4 Personal communication, Steven Shak, Genomic Health, December 13, 2011.
5 Personal communication, Steven Shak, Genomic Health, December 13, 2011.
TABLE A-2 Clinical/Biological Validation Studies for Oncotype DX
|
|
|||
| Study | Paik et al. (2004) | Habel et al. (2006) | Esteva et al. (2005) |
|
|
|||
| Tissue source | NSABP B-14 | Kaiser Permanente | MD Anderson Cancer Center |
| Study question | Does 21-Gene RS correlate with likelihood of distant recurrence? | Does 21-Gene RS predict risk of breast cancer-specific mortality in women treated and not treated with tamoxifen? | Does 21-Gene RS predict risk of recurrence in women not treated with systemic therapy? |
| Study design | Prospective–retrospective | Retrospective; matched case control | Retrospective with case inclusion criteria |
| Cases = patients who died from breast cancer Controls = breast cancer patients individually matched to cases alive at the date of death of their matched case |
|||
| Patient characteristics | Lymph-node-negative; ER+ | Lymph-node-negative; ER +/– | Lymph-node-negative; ER +/– |
| Treatment | Tamoxifen; no chemotherapy | +/–Tamoxifen; no chemotherapy | No systemic therapy |
| Sample # | 668 | 220 cases, 570 controls | 149 |
| Blinding | Yes | Yes | Yes |
| Independence | Different specimens than used in discovery | Different specimens than used in discovery | Different specimens than used in discovery |
| NSABP control of clinical outcome data | Kaiser Permanente control of clinical outcome data | MD Anderson control of clinical outcome data | |
| Study | Paik et al. (2004) | Habel et al. (2006) | Esteva et al. (2005) |
| Results | Rate of recurrence significantly lower (p < 0.001) with low-risk RSs compared to high-risk RSs; RS provided significant predictive power independent of age and tumor size; RS was predictive of overall survival | RS associated with risk of breast cancer death in ER+, tamoxifen-treated and tamoxifen-untreated patients (p = 0.003 and p = 0.03, respectively | No association between RS and distant recurrence-free survival in ER+/– patients with no adjuvant systemic therapy |
NOTE: ER = estrogen receptor, NSABP = National Surgical Adjuvant Breast and Bowel Project, RS = Recurrence Score.
used archival tissue from the NSABP B-20 study, in which patients were randomized to tamoxifen or tamoxifen plus chemotherapy. There were two chemotherapy arms: the cyclophosphamide, methotrexate, and fluorouracil (CMF) arm and the methotrexate and fluorouracil (MF) arm (Paik et al., 2006). In this study, there appeared to be a relative treatment modifier effect independent of the prognostic role of Oncotype DX (p = 0.038 for interaction between Recurrence Score and chemotherapy treatment). Prognosis was more favorable in patients with a low Recurrence Score who received only tamoxifen, and chemotherapy did not appear to be active in this subgroup. In contrast, patients with a high risk of recurrence based on their Recurrence Score had a worse prognosis if treated with tamoxifen only, and achieved a large benefit from chemotherapy. Tissue from the tamoxifen plus chemotherapy arms were not previously used in the development of Oncotype DX, but tissue from the tamoxifen-only arm had been previously used in the test discovery phase. As noted in Chapter 2, the use of discovery phase tissue samples to assess test performance is not ideal because it can lead to overfitting. The TAILORx trial (see below) will provide higher quality evidence to assess the benefit from chemotherapy treatment in a subset of patients with Recurrence Scores of 11-25 because it will prospectively evaluate the impact of Oncotype DX on treatment outcome within a large, randomized clinical trial population and will not use tissue samples from the discovery phase.
Lymph-node-positive patients Tumor samples from lymph node-positive patients were used to help develop Oncotype DX (Cobleigh et al., 2005),
and a recent prospective–retrospective analysis of a large trial found that the Recurrence Score is prognostic for tamoxifen-treated patients with positive nodes and, as expected, their prognosis was worse than for patients with negative lymph nodes (Albain et al., 2010). The analysis also evaluated the effect modifier role of Oncotype DX and indicated that node-positive patients with low Recurrence Scores did not benefit from chemotherapy treatment, but node-positive patients with high Recurrence Scores had an improvement in disease-free survival when treated with chemotherapy. The relative effects of chemotherapy rose with increasing Recurrence Scores. The RxPONDER trial (see below) will provide more information on the clinical utility of Oncotype DX in lymph-node-positive patients.
Clinical Utility
TAILORx Trial
NCI initiated the Trial Assigning IndividuaLized Options for Treatment (TAILORx) in 2006 (now fully accrued) to assess the performance of Oncotype DX in a large, prospective, randomized clinical trial. The primary objective of TAILORx is to assess the effect of chemotherapy, in addition to hormonal therapy, in women with Recurrence Scores between 11 and 25.6 The benefit of chemotherapy for women in this mid-range risk group is currently unclear. The study involves more than 10,000 women recently diagnosed with estrogen-receptor-positive and/or progesterone-receptor-positive, HER2-negative breast cancer without lymph node involvement at 900 sites in the United States, Canada, and several other countries outside of North America. Women with a low Recurrence Score received hormonal therapy alone while women with a high Recurrence Score received hormonal therapy and chemotherapy. Women with Recurrence Scores in the mid-range risk group were randomized to receive either chemotherapy plus hormonal therapy or hormonal therapy alone. Women will be studied for 10 years, with additional follow-up 20 years after initial therapy (NCI, 2006, 2010a,b).
______________
6 While the Oncotype validation studies prespecified cut-off values of 0-18, 18-30, and 31 and above for low, intermediate, and high risk of recurrence, the TAILORx investigators defined a mid-range risk of recurrence as scores of 11-25 to roughly correlate with a 10 to 20 percent risk of distant recurrence at 10 years. In TAILORx, patients classified at mid-range risk will be randomized to receive either hormonal therapy or hormonal therapy and chemotherapy, while patients at very low risk (below 11) will be assigned to hormonal therapy only and those at high risk (above 25) will receive hormonal therapy and chemotherapy.
RxPONDER Trial
Oncotype DX is also being evaluated in lymph node-positive patients in a prospective trial, RxPONDER (Rx for POsitive NoDe, Endocrine Responsive breast cancer), which will recruit 4,000 patients with Recurrence Scores of 25 or less who have estrogen-receptor-positive tumors and 1-3 positive lymph nodes. Patients will be randomly assigned to treatment with chemotherapy plus hormonal therapy or hormonal therapy alone. The trial seeks to determine whether these women may safely forego chemotherapy treatment and whether there is an optimal Recurrence Score cut-point for recommending chemotherapy or not (SWOG, 2011).
Clinical Use
As an LDT, Oncotype DX is performed in Genomic Health’s clinical laboratory that is certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). Oncotype DX has been incorporated into guidelines from ASCO and the National Comprehensive Cancer Network (NCCN). The ASCO 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer stated that “Oncotype DX may be used to identify patients who are predicted to obtain the most therapeutic benefit from adjuvant tamoxifen and may not require adjuvant chemotherapy. In addition, patients with high recurrence scores … appear to achieve relatively more benefit from adjuvant chemotherapy (specifically [C]MF) than from tamoxifen” (Harris et al., 2007, p. 5299). The guidelines specify that there are insufficient data to suggest whether these conclusions can be generalized to other hormonal therapies (e.g., aromatase inhibitors) or other chemotherapy regimens. However, a recent study using specimens from the Arimidex, Tamoxifen, Alone or in Combination (ATAC) trial found that the Recurrence Score was an independent predictor of distant recurrence in women with node-negative and node-positive, hormone-receptor-positive patients treated with anastrozole (Arimidex), an aromatase inhibitor (Dowsett et al., 2010). NCCN guidelines note that Oncotype DX is an option when evaluating certain patients with breast cancer, and assert that “the Recurrence Score should be used for decision-making only in the context of other elements of risk stratification for an individual patient” (NCCN, 2011a, p. 85).
More than 7,500 physicians have ordered the Oncotype DX test for more than 175,000 patients (Genomic Health, 2011a), with 55,000 Oncotype DX tests ordered in 2011.7 Oncotype DX is covered by almost all private insurers and is a covered benefit for Medicare beneficiaries and some Medicaid beneficiaries
______________
7 Personal communication, Steven Shak, Genomic Health, December 13, 2011.
(Genomic Health, 2011b). The Blue Cross Blue Shield Technology Evaluation Center (TEC) found that the use of Oncotype DX meets the TEC criteria (BCBS, 2008). The current list price for the assay is $4,175.8
A meta-analysis of 912 patients found that physicians using Oncotype DX in clinical practice altered their treatment decisions in more than one third of patients, leading to a 28 percent reduction in the use of chemotherapy (Hornberger and Chien, 2010).
Case Highlights
According to a review by AHRQ, Oncotype DX is one of the more well-established omics-based breast cancer tests due to its validation pathway (AHRQ, 2008). In a presentation to the committee, Steven Shak, chief medical officer of Genomic Health, stated that the company had a clearly articulated development plan that involved a multistep, multistudy approach.
The NSABP B-14 clinical validation was a large, blinded, prospective– retrospective study that provided evidence for the test’s ability to discriminate among low, intermediate, and high risk of distant recurrence among a well-defined patient cohort with node-negative, estrogen-receptor-positive cancer who were treated with tamoxifen, but not with chemotherapy. Although this was not a true prospective clinical validation study, many assert that this study design has an evidentiary value close to a prospective study (AHRQ, 2008; Harris et al., 2007; Simon et al., 2009). The Kaiser clinical validation study demonstrated that the Recurrence Score was associated with risk of breast cancer death among a population-based sample. Ongoing clinical trials will further inform clinical use of Oncotype DX, including the benefit of chemotherapy among women with intermediate Recurrence Scores and women with 1-3 positive lymph nodes.
Oncotype DX was developed as an LDT without FDA review. The computational model for Oncotype DX was published, but several aspects of test development are not specified in detail in the published literature, including how the 250-gene list was selected during test discovery and how the archival tissue from three clinical studies was used in test training and development of the computational model. Gene expression and clinical data from the discovery phase are not publicly available.
MAMMAPRINT
MammaPrint is a prognostic test designed to predict the risk of recurrence of distant breast cancer following surgery for patients with both
______________
8 Personal communication, Steven Shak, Genomic Health, December 13, 2011.
estrogen-receptor-positive and estrogen-receptor-negative tumors. MammaPrint uses a 70-gene RNA expression signature to classify individuals as having either high or low risk of recurrence. MammaPrint was developed by investigators at the Netherlands Cancer Institute, who founded a spin-off company, Agendia, to develop the commercial test.
Agendia first met with FDA in 2005, when a pre-IDE was submitted. In 2006, FDA approved Agendia’s IDE. The IDE clarified the process and requirements for the de novo 510(k) and provided useful information to the company. Agendia subsequently submitted a draft 510(k) premarket notification in June 2006 and a full de novo 510(k) submission in September 2006.9 In 2007, MammaPrint became the first FDA-cleared molecular test profiling genetic activity (FDA, 2007b).
Discovery Phase
The 70-gene signature was developed using archival samples of primary invasive breast tissue from 78 breast cancer patients (34 patients developed distant metastases within 5 years, 44 patients were disease-free after 5 years) (van ‘t Veer et al., 2002). All patients were lymph-node-negative, under age 55, and had tumors of less than 5 centimeters. Only 6 percent of patients received adjuvant systemic therapy.
RNA was isolated from snap-frozen tissue. Each RNA sample underwent 2 hybridizations on microarrays with 25,000-gene sequences. An intensity ratio was calculated using a reference RNA pool containing equal amounts of RNA from each tissue sample.
Investigators used an unsupervised hierarchical clustering algorithm that identified approximately 5,000 genes with expression significantly increased or decreased relative to random chance in more than 3 tumors out of the 78.
Using supervised classification, investigators found that 231 genes were significantly associated with disease outcome. Subsets of five genes were sequentially added to evaluate their power in correct classification using the leave-one-out method for cross-validation.10 The optimal gene expression signature was composed of 70 genes and correctly predicted whether the patient was still recurrence free or not at 5 years for 65 of 78 patients (83 percent). To reduce the number of false negatives (patients who actually had recurred but who were identified by the classifier as having a good prognosis), investigators set the threshold so that no more than 10 percent
______________
9 Personal communication, Laura van ‘t Veer, Agendia, November 28, 2011.
10 Leave-one-out cross-validation is a statistical method used to assess the generalizability of an analysis to an independent dataset. One observation is removed from the dataset to use as the test set, and the remaining observations are used as the training set. This process is repeated for all observations, and the results of all iterations are averaged.
of patients with a poor prognosis were misclassified. This optimized sensitivity (as opposed to optimized specificity) threshold resulted in 15 mis-classifications (81 percent correct) (van ‘t Veer et al., 2002).
Test Validation Phase
Analytical Validation
The test platform was converted into a new microarray, MammaPrint, containing the 70 genes identified in the discovery phase (Glas et al., 2006). Investigators reanalyzed RNA from 162 patient samples from the discovery and clinical validation (described in the next section) of the 70-gene signature to confirm that the results from the commercial microarray were consistent with the results generated on the discovery phase microarray. Investigators reported that the original analyses and reanalysis on the commercial MammaPrint microarray showed high correlation of prognosis prediction (p ![]() 0.0001), with seven discordant cases.
0.0001), with seven discordant cases.
High intralaboratory and interlaboratory reproducibility was reported for MammaPrint in three different laboratories when RNA from four different patient samples was assessed (Ach et al., 2007). A report from AHRQ, Impact of Gene Expression Profiling Tests on Breast Cancer Outcomes, noted that evidence to support analytic validity was obtained from a limited number of patients and a moderate number of replication experiments, and the impact of RNA labeling variation on risk classification was not thoroughly investigated (AHRQ, 2008).
FDA accepted a modification to Agendia’s 510(k) premarket notification, in which the specimen type was switched from fresh frozen tissue to fresh tissue stored in an RNA preservative solution (FDA, 2007a). Investigators showed that shipment in the RNA preservative did not affect the MammaPrint test results, with no statistically significant difference in MammaPrint risk group assignment or index between fresh frozen and preserved tissue (FDA, 2007a).
Statistical and Bioinformatics Validation
Discovery microarray data and clinical information are available and reported in van ‘t Veer et al. (2002), which was uncommon at the time of its publication.11 Although the method used to derive the MammaPrint
______________
11 Microarray data were first hosted at http://www.rii.com/publications/default.htm, as mentioned in the van ‘t Veer et al. (2002) paper and are now available through the Netherlands Cancer Institute website (http://bioinformatics.nki.nl/data.php) and the Stanford open access micro array site. Personal communication, Laura van ‘t Veer, Agendia, November 1, 2011.
computational model is described in the supplementary materials, the final computational procedures are not reported, and some of the details needed for independent replication are unclear from the supplementary materials, including the methodology for gene selection and details about the statistical analysis.
Several statistical problems in the early development studies were reported in the published literature (Ransohoff, 2003; Simon et al., 2003). There was overlap of 61 discovery samples used in the second clinical validation study, which was acknowledged by the authors in the van de Vijver paper (van de Vijver et al., 2002). Model performance was assessed using the same data that were used in development of the 70-gene signature, and Simon et al. (2003) asserted that an incomplete cross-validation process was performed in van ‘t Veer et al. (2002) because the method did not include reselection of the differentially expressed genes. According to Simon and colleagues, this resulted in a biased underestimate of the error rate that, when combined with the small number of samples used in discovery and validation, likely led to overfitting and overstatement of the accuracy of the 70-gene signature (Simon et al., 2003). A subsequent study was performed in acknowledgment of some of these shortcomings (Buyse et al., 2006).
Microarray data and clinical information from the third clinical validation study are publicly available (Buyse et al., 2006).12 A statistician independently applied the MammaPrint computational model to reproduce the MammaPrint risk classification, and independent statisticians assessed the concordance (100 percent) between risk classification produced by Agendia and the external statistician. Independent auditors visited clinical centers to carry out source data verification; statistical analyses were conducted by the International Drug Development Institute; and clinical, pathologic, and microarray data were centralized at the TRANSBIG Secretariat.
The MammaPrint test was locked down twice. The 70-gene signature research assay was locked down after its development described in van ‘t Veer et al. (2002). The commercial microarray developed by Agendia was also locked down (Glas et al., 2006).13
Clinical/Biological Validation
Table A-3 lists the clinical validation studies that have been performed to assess MammaPrint. The first validation, published as part of the discovery phase paper, assessed 19 patient samples. The authors reported that the
______________
12 Accession number E-TABM-77 (European Bioinformatics Institute ArrayExpress database).
13 Personal communication, Laura van ‘t Veer, University of California, San Francisco, November 28, 2011.
TABLE A-3 Clinical/Biological Validation Studies for MammaPrint
|
|
|||
| Study | van’t Veer et al. (2002) | van de Vijver et al. (2002) | Buyse et al. (2006) |
|
|
|||
| Tissue source | Netherlands Cancer Institute | Netherlands Cancer Institute | 5 European centers |
| Test platform | Experimental microarray |
Experimental microarray |
MammaPrint (Glas et al.,2006) |
| Study question(s) |
Does 70-gene expression signature show comparable performance to development tissues? | Does 70-gene signature confirm results from previous validation study in lymph- node-negative patients? What is the performance of signature in lymph-node-positive patients? | Does MammaPrint have prognostic value in a group of independent patients, beyond clinical risk classifications? |
| Study design | Retrospective | Retrospective | Retrospective |
| Patient characteristics |
Under 55 years old Lymph-node- negative Tumor size |
Under 53 years old Lymph-node- negative and lymph-node-positive Tumor size |
Under 61 years old Lymph-node-negative Tumor size |
| Treatment | No adjuvant systemic therapy | 10 lymph-node-negative patients and 120 lymph- node-positive patients received adjuvant chemotherapy (n = 90), hormonal therapy (n = 20), or both (n = 20) | No adjuvant systemic therapy |
| Sample # | 19 | 295 | 307 |
|
|
|||
| Study | van’t Veer et al. (2002) | van de Vijver et al. (2002) | Buyse et al. (2006) |
|
|
|||
| Blinding | Not stated | Rosetta Inpharmatics carried out microarray analysis; all raw data were available to all investigators | Data centralized at the TRANSBIG Secretariat; 100% concordance between risk classification by Agendia and Swiss Institute of Bioinformatics; statistical analysis carried out by IDDI |
| Independence | Different specimens than used in discovery Not conducted by a separate group |
Included 61 samples from discovery phase Not conducted by a separate group |
Different specimens than used in discovery Involvement of the Swiss Institute of Bioinformatics, TRANSBIG Secretariat, IDDI, and independent auditors |
| Results | Disease outcome was predicted by gene signature in 17 of 19 (Fisher’s exact test for association, p = 0.0018) | Estimated HR for distant metastases in poor vs. good signature groups: 5.1 (95% CI 2.9-9.0; p Multivariate model Poor prognosis signature of 70-gene test the strongest predictor of the likelihood of distant metastases (HR = 4.6; 95% CI 2.3-9.2), with only tumor size and lack of adjuvant chemotherapy remaining in the model. |
Distant metastasis: Unadjusted HR = 2.32 (95% CI 1.35-4.00) Adjusted HR ranged from 2.13-2.15 after adjustment for various estimates of clinical risk Overall survival: Unadjusted HR = 2.79 (95% CI 1.60-4.87) Adjusted HR ranged from 2.63-2.89 after adjustment for various estimates of clinical risk |
|
|
|||
| NOTE: CI = confidence interval, HR = hazard ratio, IDDI = International Drug Development Institute, TRANSBIG = a consortium of the Breast International Group (BIG). | |||
70-gene signature predicted disease outcome better than traditional clinical prognostic factors (van ‘t Veer et al., 2002).
A second study evaluated the 70-gene signature in 295 patients, which, as noted previously, included 61 samples that were used in the discovery phase of test development (representing 78 percent of the tumor samples in the development of the signature) (van de Vijver et al., 2002). Investigators asserted that leaving out these patient samples would have resulted in selection bias because the signature was developed using a disproportionately large number of patients in whom distant metastases developed within 5 years (van de Vijver et al., 2002). However, critics do not consider this a true validation because of the overlap between training and validation datasets (Kim and Paik, 2010; Ransohoff, 2003, 2004; Simon et al., 2003).
A third validation study (Buyse et al., 2006) was performed using archived samples not collected as part of a prospective clinical trial protocol from 307 patients in 5 European centers. The authors concluded that MammaPrint outperformed traditional prognostic factors in predicting distant metastases, overall survival, and disease-free survival. However, they also noted that the hazard ratios (HRs)14 reported in the earlier study (van de Vijver et al., 2002) were much higher than those reported in Buyse et al. (2006) (see Table A-3), echoing earlier concerns “that the inclusion in the [van de Vijver et al. 2002 study] of patients whose data were used in the development of the 70-gene signature may have inflated the discriminatory power of the signature in that study, even though analytic measures had been taken to limit this effect” (Buyse et al., 2006, p. 1190). The authors suggested that the longer period of follow up in the third study may have also contributed to this difference in hazard ratios.
Expanding the eligibility age for MammaPrint Investigators assessed MammaPrint performance in a retrospective study of 131 patients with node-negative breast cancer who were older than 55 years and not treated with adjuvant therapy (FDA, 2009). The clinical sensitivity and specificity of the test in older women were comparable to previous data submitted in support of using the test in younger women. FDA expanded the intended use of the test to include breast cancer patients of all ages (FDA, 2009). In an analysis of a predominantly postmenopausal cohort of 100 women, MammaPrint correctly identified 100 percent of women at low risk for distant metastases at 5 years (Wittner et al., 2008). However, the positive predictive value of MammaPrint (women who develop distant metastases
______________
14 A hazard ratio is an expression of the risk of an event in one arm of a study as compared to the risk of the event happening in the other arm over time. This differs from the relative risk ratio, which is a proportion of the number of events that occur in one arm of the study as compared to the other arm.
who are classified as having a poor prognosis) was lower than previously observed (12 percent versus 52 percent) which, according to the investigators, was unexpected because older women are generally thought to have a lower risk of recurrence.
Lymph-node-positive patients Mook et al. (2008) used a retrospective analysis with 241 patients to assess whether MammaPrint could identify patients with one to three positive lymph nodes who have a good prognosis. Investigators found that MammaPrint was significantly better than traditional prognostic factors in predicting breast cancer-specific survival with a multivariate HR = 7.17 (95% CI 1.81-28.43; p = 0.005).
Chemotherapy benefit MammaPrint was assessed for its value in predicting chemotherapy benefit in a retrospective analysis evaluating 541 patient samples from a pool of 1,637 patients (Knauer et al., 2010).15 Investigators concluded that patients with poor prognosis, defined by MammaPrint, derive a significant benefit from the addition of chemotherapy. The MINDACT trial (see below) will provide higher quality evidence to assess the benefit of chemotherapy because data will be collected prospectively within a large, randomized clinical trial population.
Clinical Utility
The European Organisation for Research and Treatment of Cancer, with partial support from Agendia, is conducting a large multicenter, prospective randomized trial to compare MammaPrint with common clinico pathological criteria (using Adjuvant! Online) in selecting patients for adjuvant chemotherapy. The trial, “Microarray In Node negative and 1-3 positive lymph node Disease may Avoid ChemoTherapy” (MINDACT), will randomly assign chemotherapy to women who have discordant prognosis (either good prognosis with MammaPrint and high risk of recurrence based on clinicopathologic factors or poor prognosis with MammaPrint and low risk of recurrence based on clinicopathologic factors). All other women will be assigned to either (1) chemotherapy if they are at high risk for recurrence based on both their MammaPrint signature and clinicopathologic characteristics or (2) no chemotherapy if they are classified at low risk based on both. The trial has fully accrued more than 6,600 women who will be followed to assess the primary outcomes of distant metastasis-free
______________
15 The pooled sample included patients from the original discovery phase study (van ‘t Veer et al., 2002), clinical/biological validation studies (Buyse et al., 2006; van de Vijver et al., 2002; van ‘t Veer et al., 2002), Mook et al. (2008), and several community-based registries (Knauer et al., 2010).
TABLE A-4 FDA 510(k) Clearances for MammaPrint
|
|
|||
| Decision Date | 510(k) Number | Summary | |
|
|
|||
| 02/06/2007 | K062694 | Original clearance for MammaPrint test | |
| • | Single laboratory | ||
| • | Fresh frozen tissue | ||
| • | Patients less than 61 years old | ||
| • | Includes laboratory procedures, software, and computational procedures | ||
| Modifications | |||
| 06/22/2007 | K070675 | • | Changed specimen type to fresh tissue stored in specific RNA preservative |
| • | Change in software version | ||
| 07/21/2008 | K080252 | • | Addition of a second scanner |
| • | Replacement of low-density microarray with high-density microarray | ||
| 12/11/2009 | K081092 | • | Modified intended use by adding 5-year prognostic information for breast cancer patients 61 years and older |
| 01/28/2011 | K101454 | • | Added 2 scanners, 2 bioanalyzers, and a new laboratory for MammaPrint testing |
|
|
|||
SOURCE: FDA, 2011a.
survival and disease-free survival (Clinicaltrials.gov, 2011b). First results are expected in 2015.
Clinical Use
The MammaPrint test is conducted in Agendia’s two CLIA-certified laboratories. Since its 2007 FDA clearance, MammaPrint has undergone several modifications to the test over time, which have been documented by FDA (Table A-4) (Agendia, 2009). In the United States, MammaPrint is available for patients of all ages with invasive Stage I or II breast cancer, who are lymph-node-negative, have tumors of less than 5 cm, and are either estrogen-receptor-positive or estrogen-receptor-negative. The intended use statement accompanying the FDA clearance specifies that “[t]he MammaPrint® result is indicated for use by physicians as a prognostic marker only, along with other clinicopathological factors” (FDA, 2011b, p.1). In addition, the special conditions for use statement notes that “MammaPrint® is not intended for diagnosis, or to predict or detect response to therapy, or to help select the optimal therapy for patients” (FDA, 2011b, p. 1). According to Agendia, insurance coverage for MammaPrint is available from the Centers for Medicare & Medicaid Services, private health insurers, and
third-party payers. More than 14,000 MammaPrint test results have been reported (Agendia, 2011b).
Guidelines from ASCO on the use of tumor markers specify that the clinical utility and appropriate application of MammaPrint is under investigation, and further state that: “MammaPrint profiling does appear to identify groups of patients with very good or very poor prognosis. However, due to the nature of the study design, it is difficult to tell if these data pertain to an inherently favorable outcome in untreated patients, to patients whose prognosis is favorable because of the therapy, or to those with poor outcomes in the absence of treatment or despite treatment” (Harris et al., 2007, p. 5301). NCCN is awaiting the results from the MINDACT trial before determining its recommendations for use (NCCN, 2011a). The Blue Cross Blue Shield Medical Advisory Panel determined that MammaPrint did not meet the TEC criteria (BCBS, 2008). However, the 2009 update of the St. Gallen International Expert Consensus stated that “the Panel agreed that validated multigene tests, if readily available, could assist in deciding whether to add chemotherapy in cases where its use was uncertain after consideration of conventional markers” (Goldhirsch et al., 2009, p. 1324). According to Agendia, MammaPrint has been included in the 2008 update to guidelines for the Dutch Institute for Healthcare Improvement (Agendia, 2011a).
Case Highlights
Initial statistical approaches in MammaPrint test validation, including overlap between discovery and validation datasets and assessment of model performance using an incomplete cross-validation procedure that led to overfitting, were criticized in the literature (Ransohoff, 2003, 2004; Simon et al., 2003). A subsequent study (Buyse et al., 2006) provided a clinical validation with patients who had not participated in the discovery and validation studies, and confirmed MammaPrint as a prognostic test. However, the hazard ratios between the poor and good prognosis groups reported in Buyse et al. (2006) were lower than those reported in van de Vijver et al. (2002).
Discovery microarray and clinical data are available for MammaPrint, but the fully specified computational procedures are not published, and several details needed for independent replication are unclear from the supplemental materials. MammaPrint received FDA clearance, and more than 14,000 MammaPrint results have been reported, but some technology assessment groups have asserted that more information on MammaPrint is needed to determine how it should be used in clinical practice (AHRQ, 2008; BCBS, 2008; Harris et al., 2007; NCCN, 2011a). It is hoped that the prospective validation trial, MINDACT, will provide this information,
especially in determining if MammaPrint can accurately predict chemotherapy benefit.
TISSUE OF ORIGIN
The Tissue of Origin test (Pathwork® Diagnostics) is a gene expression-based test designed to identify the primary tissue of origin for tumors that are difficult to classify, including metastatic, poorly differentiated, and undifferentiated tumors. Only about 20-25 percent of patients with tumors of unknown origin receive a primary tumor diagnosis, despite extensive clinical and pathological assessments and advanced imaging (Hillen, 2000; Pavlidis and Merrouche, 2006; Pavlidis et al., 2003). This is problematic because identifying the tissue of origin can have important ramifications for treatment decisions. The Tissue of Origin test aims to assist in the classification of such tumors by comparing the similarity of gene expression in a tumor with unknown origin to a panel of gene expression data from 15 common tumors16 (Dumur et al., 2008).
FDA has cleared two versions of the Tissue of Origin test—the test for frozen tissue in 2008 and the test for FFPE tissue in 2010. Pathwork Diagnostics consulted with FDA on several occasions, including a pre-IDE meeting at which FDA determined that an IDE was not required because the proposed study design involved an analysis of archived samples.17
Discovery Phase
Published information on gene discovery and computational model development for the Tissue of Origin test is limited. The Tissue of Origin test for frozen and FFPE specimens include the same 15 tumor types, but they use different computational procedures and processing methods (Pillai et al., 2011). The test uses two computational procedures, one for standardization and one for classification. The standardization computational procedures were developed from analysis of more than 5,000 tissue samples. The classification computational procedures for FFPE specimens were developed from more than 2,000 frozen and 100 FFPE tissue specimens. Training specimens were assigned a tissue of origin diagnosis according to standard clinical and pathological practices, and the test set consisted of FFPE specimens only. Investigators state that “[m]achine learning techniques guided
______________
16 The tumor types include bladder, breast, colorectal, gastric, hepatocellular, kidney, mela noma, non-small cell lung, non-Hodgkin’s lymphoma, ovarian, pancreatic, prostate, sarcoma, testicular germ cell, and thyroid. Pathwork Diagnostics is also in the process of developing gene expression panels for endometrial and head and neck cancers.
17 Personal communication, Ed Stevens, Pathwork Diagnostics, October 18, 2011.
selection of the 2,000-gene profile and the optimal model needed to classify the tumor” for FFPE tissue specimens (Pillai et al., 2011). In comparison, the optimal procedures for frozen tissue specimens included 1,550 genes in the computational model (FDA, 2008b). The Tissue of Origin test produces a set of 15 similarity scores that describe the probability that the gene expression of a tumor of unknown origin is comparable to gene expression of the 15 tumor types included in the test.
Test Validation Phase
Analytical Validation
Information on analytical validity is available for both versions of the Tissue of Origin test, for frozen tissue specimens (Dumur et al., 2008; FDA, 2008b), and FFPE specimens (FDA, 2010; Pillai et al., 2011). The standardization computational procedures used in both versions of the Tissue of Origin test correct for variations in RNA quality, sample storage and preparation, operators, and microarray procedures (Pillai et al., 2011).
Dumur et al. (2008) evaluated the same 60 frozen tissue specimens of poorly differentiated and undifferentiated tumors from 15 tumor types with established origin in 4 different laboratories. Blinded microarray data were sent to Pathwork for generation of Tissue of Origin scores, which were then sent to pathologists blinded to the original tissue type diagnosis from the surgical reports for their use in generating an interpretation of tissue of origin based on predetermined cut-offs for the test. Investigators found that Tissue of Origin results were highly reproducible across all four laboratories (Dumur et al., 2008). One potential limitation of this analysis was that 57 of the 60 tissue specimens were obtained from the same biospecimen bank, and therefore it is possible that only some aspects of preanalytical variability were considered. The 2008 510(k) decision summary reports additional information on analytical performance for the Tissue of Origin test for frozen samples (FDA, 2008b).
The 2010 510(k) decision summary reports analytical performance information for the FFPE version of the Tissue of Origin Test, including assay precision and reproducibility, quality controls, detection limits, analytical specificity, and assay cut-off values (FDA, 2010). Pillai et al. (2011) conducted a multisite reproducibility study that showed 89.3 percent concordance among three laboratories.
Statistical and Bioinformatics Validation
Information about the methods used to develop the computational model has not been made available in the peer-reviewed literature, and the
proprietary computational procedures are not available.18 Data used in computational model development include publicly available information (GEO accession number 2109), commercial data sources, and private correspondence (Pillai et al., 2011).
The assay was locked down prior to the validation and reproducibility studies, and this information was documented in the design control procedures submitted to FDA (Monzon et al., 2009; Pillai et al., 2011).19
Clinical/Biological Validation
Table A-5 lists the clinical validation studies for the Tissue of Origin test. The clinical validation for the Tissue of Origin test for frozen samples was a blinded, multicenter study that found overall sensitivity of 87.8 percent and specificity of 99.4 percent (Monzon et al., 2009). The site of origin (called the reference diagnosis) was known for all tissue specimens, but a limitation noted by the authors was the “inability to independently verify the reference diagnosis used to assess the accuracy of the test” (Monzon et al., 2009) because the reference diagnoses originated from the surgical pathology report that accompanied the banked specimen. Investigators also considered the possibility that an unknown primary tumor might originate from a tissue site that was not covered by the panel.
The second clinical validation, for the FFPE version of the Tissue of Origin test, was a blinded study that used only specimens that were not used in discovery. Pillai et al. (2011) analyzed 462 specimens and found that overall agreement of the test result to reference diagnosis was 88.5 percent.
The Tissue of Origin test was also assessed in several small studies. In a study of 15 fresh frozen metastatic brain cancer specimens, a correct diagnosis was given in 12 out of 13 viable specimens (92.3 percent) (Wu et al., 2010). In another study of 21 fresh frozen tumor samples, Monzon et al. (2010) used the Tissue of Origin test to classify specimens from patients with a carcinoma of unknown primary (CUP). Investigators determined that the test yielded a clear identification of the primary site for 76 percent of specimens, but noted that the major limitation of the study was that CUP specimens, by definition, lack a reference diagnosis. Grenert et al. (2011) found that 35 of 37 viable FFPE specimens (95 percent) from an academic pathology department agreed with the reference diagnosis. The Tissue of Origin test was also assessed using FFPE cell blocks of cytologic body fluid specimens; investigators found that 16 of 17 viable samples, or 94.1 percent, were in agreement with reference diagnosis (Stancel et al., 2011). Dumur and colleagues (2011) assessed the Tissue of Origin test
______________
18 Personal communication, Ed Stevens, Pathwork Diagnostics, October 18, 2011.
19 Personal communication, Ed Stevens, Pathwork Diagnostics, October 18, 2011.
TABLE A-5 Clinical/Biological Validation Studies for the Tissue of Origin Test
|
|
||
| Study | Monzon et al., 2009 | Pillai et al., 2011 |
|
|
||
| Tissue source(s) | 2 academic, 3 commercial biospecimen banks; electronic microarray files for 271 tumors obtained from the International Genomics Consortium | 7 tissue banks |
| Specimen preparation | Frozen | Formalin-fixed paraffin-embedded (FFPE) tissue |
| Study purpose | Determine the performance characteristics of the frozen Tissue of Origin test in specimens representative of those likely to be classified as uncertain primary cancers | Determine the performance characteristics of the FFPE Tissue of Origin test in specimens representative of those likely to be classified as uncertain primary cancers, and to assess interlaboratory reproducibility |
| Study design | Retrospective | Retrospective |
| Tumor characteristics | No fewer than 25 specimens for each tumor type | 25-57 specimens for each tumor type |
| Approximately half of specimens were metastatic | ||
| Sample # | 547 | 462 |
| Blinding | Yes | Yes |
| Independence | Different specimens than used in discovery | Different specimens than used in discovery |
| Not conducted by a separate group | Not conducted by a separate group | |
| Results | Overall sensitivity (positive percentage agreement with reference diagnosis) = 87.8% (95% CI 84.7-90.4%) | Overall sensitivity (positive percentage agreement with reference diagnosis) = 88.5% (95% CI 85.3-91.3%) |
| Overall specificity (negative percentage agreement with reference diagnosis) = 99.4% (95% CI 98.3-99.9%) | Negative percentage agreement with reference diagnosis = 99.1% (no CI reported) | |
| Performance on metastatic tumors was slightly lower than on poorly differentiated and undifferentiated tumors | ||
|
|
||
on 43 poorly differentiated and undifferentiated frozen tumor specimens, including 6 tumor samples that are not represented in the Tissue of Origin panel of 15 tumors (off-panel) and 7 CUP specimens. Investigators found 97 percent agreement between the Tissue of Origin test result and diagnosis, but noted that for CUP and off-panel specimens, the tissue type and cell type may be confounded by the Tissue of Origin test.
Clinical Use
Pathwork transitioned the Tissue of Origin Test for FFPE tissue, originally offered through its CLIA-certified laboratory as an LDT, to an FDA-cleared in vitro diagnostic (IVD) (Pathwork Diagnostics, 2010). Currently, all testing is performed in the Pathwork Diagnostics laboratory, but the company plans to make an IVD kit available for pathologists to run in their own clinical laboratories (Pathwork Diagnostics, 2011b).
The intended use statement in the FDA clearance decision summary specifies that the Tissue of Origin test “measure[s] the degree of similarity between the RNA expression patterns in a patient’s … tumor and the RNA expression patterns in a database of fifteen tumor types (poorly differentiated, undifferentiated and metastatic cases) that were diagnosed according to the current clinical and pathological practice. This test should be evaluated by a qualified physician in the context of the patient’s clinical history and other diagnostic test results” (FDA, 2010, p. 1). The limitation statement in the FDA decision summary notes that Tissue of Origin is not intended to:
- Establish origin for tumors that cannot be diagnosed according to current practices;
- Subclassify or modify classification of tumors that can be diagnosed by current practice;
- Predict disease course, survival, or treatment efficacy, or distinguish between primary and metastatic tumors; and
- Distinguish tumors that are not in the test’s database.
As of 2011, the Tissue of Origin test had not been incorporated into ASCO or NCCN guidelines; NCCN deemed the test an exciting new area of molecular profiling, but concluded that further clinical trials were still necessary before incorporation into its guidelines (Mulcahy, 2010).
In a preliminary analysis of 59 patients, Hornberger et al. (2011) reported that oncologists changed treatment plans in 53 percent of patients with difficult to diagnose tumors after using the Tissue of Origin test. Another analysis of 284 consecutive cases found the Tissue of Origin test suggested a change in diagnosis in 81 percent (95% CI 76-85%) of cases
and confirmed a suspected primary tumor site for 15 percent of cases (95% CI 12-20%) (Laouri et al., 2011). Studies have not yet evaluated whether clinical outcomes improve following an altered course of patient management based on the Tissue of Origin test.
In 2011, the company announced that the Tissue of Origin test would be covered by Medicare and that it is working to secure additional insurance coverage for the test (Pathwork Diagnostics, 2011a). As a private company, Pathwork Diagnostics does not release information on the number of tests ordered per year, but a company spokesperson stated that the company has processed results for thousands of Tissue of Origin tests in 2011.20
Case Highlights
There is limited publicly available information on Tissue of Origin test discovery, and the computational procedures are proprietary. Pathwork initially offered its Tissue of Origin test for FFPE as an LDT through its CLIA-certified laboratory, but in 2010 received FDA clearance for this version of the test. Clinical validation studies blinded Pathwork to reference diagnoses, specified that the test was locked down, and involved tissue specimens from a number of biospecimen banks. The effect of the Tissue of Origin test on treatment decisions is under evaluation. Information on how use of the test affects patient outcomes has not been published.
OVA1
OVA1® (Vermillion, Inc.) is a test that measures five proteins in serum (CA-125-II, beta-2-microglobulin, transferrin, apolipoprotein A1, and transthyretin) to generate a score reflecting the likelihood of ovarian malignancy in patients with an adnexal mass for whom surgery is planned. The test is intended to help physicians determine which patients are more likely to have cancer and thus should be referred to a gynecologic oncologist for surgery.
OVA1 was FDA cleared in September 2009. Investigators met with FDA several times: before starting clinical trials, during clinical trials, and during the submission process.21
Initially, investigators intended to develop a screening test for ovarian cancer, but abandoned this goal because it would have required large studies, exceeding time and budgetary constraints, and necessitated a level of clinical specificity that would have been difficult to achieve (Fung, 2010). Indeed, AHRQ’s evidence report on ovarian cancer detection noted that
______________
20 Personal communication, Ed Stevens, Pathwork Diagnostics, October 18, 2011.
21 Personal communication, Scott Henderson, Vermillion, Inc., October 26, 2011.
model simulations suggest that frequent screening for ovarian cancer, even with a highly specific test, would result in a very low positive predictive value because the test would identify a large number of false positives for ovarian cancer (AHRQ, 2006). Investigators then sought to develop a diagnostic test to aid patient management decisions for women in whom an ovarian mass had already been identified.
Discovery Phase
An early study in the discovery process for OVA1 aimed to identify serum biomarkers for the detection of early-stage ovarian cancer (Zhang et al., 2004). Proteomic profiles were generated by mass spectrometry from 645 archived serum samples from healthy women and patients with ovarian cancer. The investigators identified three proteins (apolipoprotein A, trans-thyretin, and inter-α-trypsin inhibitor heavy chain 4) that, combined with CA-125, appeared to provide a modest improvement over CA-125 alone in identifying women with ovarian cancer. In subsequent studies, investigators identified seven candidate proteomic markers of ovarian cancer in addition to CA-125: the three from the initial study plus four more, including beta-2-microglobulin and transferrin (Fung, 2010; Zhang and Chan, 2010). According to Vermillion, this biomarker panel was further refined in a series of studies encompassing more than 2,000 subjects, but detailed information describing the discovery and development process that led to the panel of 5 biomarkers comprising the OVA1 computational model have not been made available in the peer-reviewed literature.22
Test Validation Phase
Analytical Validation
The investigators determined that reproducibility on the mass spectrometry platform was not adequate for routine clinical use (Fung, 2010), so the test platform was changed to immunoassays, which were already available for several proteins in the panel. Data regarding the precision and reproducibility of the test were reported to FDA (FDA, 2011c). Variability in results was measured across runs, over time, across serum lots, on different machines, and in different labs. Stability of specimens and reagents was also assessed under various conditions, such as storage temperature.
______________
22 Personal communication, Scott Henderson, Vermillion, Inc., October 26, 2011.
______________
Statistics and Bioinformatics Validation
The FDA 510(k) clearance states that the OVA1 computational model was derived using two independent training datasets. The first consisted of 284 preoperative serum samples from women with adnexal masses obtained from the University of Kentucky (175 benign disease and 109 malignancies). The second consisted of 125 evaluable specimens from a randomly selected subset of 146 preoperative serum samples that were set aside in the clinical validation trial (89 benign disease, 36 malignancies). This information has not been published in the peer-reviewed literature, but details of the training methodology were made available to FDA. The computational procedures are proprietary.23
Clinical/Biological Validation
The clinical validation study was a prospective, double-blind study involving 27 subject enrollment sites (Miller et al., 2011; Ueland et al., 2011). Enrollment was limited to women who had a documented pelvic mass following physical and clinical examination and planned surgical intervention; 743 patients were enrolled in the validation study, and 146 patient samples were randomly selected and set aside for the training set described above. Seventy-four specimens were eliminated due to missing information or an unevaluable sample, resulting in 524 evaluable patient samples of which 516 were evaluated by physician assessment. According to the FDA clearance decision summary, the OVA1 test was informative for both premenopausal and postmenopausal patients. Use of the test in conjunction with clinical presurgical assessment increased sensitivity for malignancy from 72 percent to 92 percent (Table A-6) (FDA, 2011c).
In a comparison of physician assessment with OVA1, the test correctly identified 70 percent of malignancies missed by physician assessment among gynecologists and gynecologic oncologists, and 95 percent of malignancies missed by physician assessment among gynecologic oncologists (Ueland et al., 2011).
Clinical Use
The OVA1 test is available through Quest Diagnostics, which has exclusive rights to offer the test in the clinical reference laboratory market and is subject to CLIA certification (PR Newswire, 2009; Quest Diagnostics, 2011). As described in the FDA clearance decision summary (FDA, 2011c), the test consists of the OvaCalc Software as well as the instruments, assays,
______________
23 Personal communication, Scott Henderson, Vermillion Inc., October 26, 2011.
TABLE A-6 Performance Characteristics for OVA1 Applied to Pre- and Postmenopausal Subjects Evaluated by Non-Gynecologic Oncologist Physicians
|
|
|||
| Performance Measure | Presurgical Clinical Assessment | OVA1 Test | Dual Assessment (Clinical Assessment and OVA1 Test) |
|
|
|||
| Sensitivity | 72.2% (52/72) | 87.5% (63/72) | 91.7% (66/72) |
| Specificity | 82.7% (163/197) | 50.8% (100/197) | 41.6% (82/197) |
| Positive Predictive Value | 60.5% (52/86) | 39.4% (63/160) | 36.5% (66/181) |
| Negative Predictive Value | 89.1% (163/183) | 91.7% (100/109) | 93.2% (82/88) |
| Prevalence | 26.8% (72/269) | ||
|
|
|||
| SOURCE: FDA, 2011c. | |||
and reagents recommended by Vermillion, which are sold separately from the OvaCalc Software. For example, CA-125 levels are assessed with a Roche Elecsys 2010, while the remaining four proteins are detected by a Siemens BN II (FDA, 2011c). To determine the OVA1 score, a user manually enters the results for the five protein analytes into an Excel spreadsheet together with the headers from the OvaCalc Software to generate a numerical score from 0 to 10 from the 5 analyte results (with 10 indicating the highest probability of cancer). Dialogue with FDA prompted the use of different OVA1 score cut-offs depending on menopausal status (5.0 for premenopausal women and 4.4 for post-menopausal women). The test is intended for use only as an adjunctive test to complement, not replace, other diagnostic and clinical procedures.
The American College of Obstetricians and Gynecologists and Society of Gynecologic Oncologists (SGO) issued a committee opinion in March 2011 that OVA1 “appears to improve the predictability of ovarian cancer in women with pelvic masses” but noted that the clinical utility of this test has not been established (ACOG and SGO, 2011). As of 2011, OVA1 had not been incorporated into guidelines from ASCO. NCCN guidelines note that the “SGO and [FDA] have stated that the OVA-1 test should not be used as a screening tool to detect ovarian cancer. The OVA-1 screening test uses 5 markers … to assess who should undergo surgery by an experienced gynecologic oncologist and who can have surgery in the community. Based on data documenting an increased survival, the NCCN panel recommends that all patients should undergo surgery by an experienced gynecologic oncologist” (NCCN, 2011b).
According to Vermillion, the company is working to secure coverage and reimbursement for OVA1 (Vermillion, 2011). Currently, the test is
covered under Medicare, 22 Blue Cross Blue Shield plans, and a number of other private U.S. health plans.
Case Highlights
Few details of the discovery and development of the OVA1 test, including information on the computational model, are available in the peer reviewed literature. The clinical/biological validation study is published (Miller et al., 2011; Ueland et al., 2011), and OVA1 is FDA-cleared. Input from FDA prompted the use of two different OVA1 cut-off values based on menopausal status.
OVACHECK
Ovarian cancer is the leading cause of gynecologic cancer deaths in the United States, and its high mortality rate is largely due to failure to detect the cancer at an early stage (ACS, 2011). In the late 1990s, Drs. Emanuel Petricoin and Lance Liotta, investigators from FDA and NCI, collaborated with researchers at the bioinformatics company Correlogic to develop a proteomics-based approach for ovarian cancer screening using serum samples. Investigators used data from mass spectrometry analysis of serum proteins to develop a computational model for identifying spectral patterns that could discriminate between healthy patients and those with ovarian cancer, and published their findings in the Lancet (Petricoin et al., 2002). Based on this proof of concept, Correlogic began developing a test for clinical use called OvaCheck, using a different form of mass spectrometry.24
Both the Lancet publication and the development of the first OvaCheck test garnered public attention as well as early controversy (H. Con. Res. 385, 107th Cong., 2nd sess., 2002; Check, 2004; Pollack, 2004; Wagner, 2004). The two laboratories licensed by Correlogic to perform the OvaCheck test, Quest Diagnostics and LabCorp, had planned to begin marketing the test in 2004 (Pollack, 2004). Before a validation study for OvaCheck had been published and prior to commercial availability, marketing materials for the test were distributed at an SGO conference (Wagner, 2004). Around the same time, FDA sent Correlogic a letter specifying that the agency “has determined that the OvaCheck test is subject to FDA regulation under the
______________
24 See http://www.correlogic.com/about/history.php. After the collaboration between the FDA/NCI program and Correlogic was terminated, the company continued its efforts to develop a blood test to detect ovarian cancer, with a later focus on immunoassays (Amonkar et al. 2009; www.correlogic.com/research-areas/ovarian-cancer.php). Correlogic filed for bankruptcy in 2010 and was acquired by Vermillion, Inc (the maker of OVA1) in 2011; that company has no intentions of commercializing an OvaCheck test (Bonislawski, 2011).
device provisions of the Federal Food, Drug, and Cosmetic Act.”25 In Correlogic’s reply to FDA, the company said that it “respectfully do[es] not agree … with FDA’s position that the software used to provide the OvaCheck testing service is a medical device … subject to FDA premarket review” (Correlogic, 2004). As a result of the FDA action, the unvalidated Ova-Check test was not made available to the public. In addition, independent analyses of the Lancet article by Petricoin et al. (2002), made possible by the willingness of the investigators to make data publicly available, uncovered problems with the data and methods (Baggerly et al., 2004a, 2005; Sorace and Zhan, 2003; reviewed by Diamandis, 2004).
Discovery and Validation Process
In 2002, Petricoin, Liotta, and investigators at Correlogic published their findings in the Lancet (Petricoin et al., 2002). The investigators analyzed serum samples from 100 women with ovarian cancer and 116 women who were healthy or had nonmalignant disorders. The cases and most of the controls were obtained as frozen samples from the National Ovarian Cancer Early Detection Program (NOCEDP); 17 additional controls were obtained at the Simone Protective Cancer Institute (SPCI) (Petricoin et al., 2002).
Samples were analyzed by surface-enhanced laser desorption/ ionization time-of-flight (SELDI-TOF) mass spectrometry using a hydro-phobic interaction protein chip. SELDI-TOF mass spectrometry involves spotting a biological sample on a chip surface coated with specific chemicals that bind to a subset of the proteins. Proteins that remain bound after washing are then ionized by a laser and time of flight is measured, allowing investigators to determine mass to charge (m/z) ratios and corresponding intensities. Spectra generated by SELDI-TOF were analyzed by combining “genetic algorithms” (a set of features “survives” if it can discriminate affected cases from controls in a training set, and feature sets that cannot survive this test are discarded) with “cluster analysis” (cases and controls are used to form clusters so that an unknown sample can be classified by its similarity to a cluster set) (Petricoin et al., 2002). The spectra from 50 cancer and 50 control samples were used to train the computational model, and the remaining samples were used as a test set for the resulting computational model. None of the samples used in training was used to test the model. The proprietary computational models were not described in the publication and the final form (i.e., the equations) of the computational model developed on the training data was not provided. In addition, little information was provided about data pre-processing,
______________
25 FDA 2004. Letter Re: OvaCheck to Peter J. Levine from Steven I. Gutman, July 12, 2004.
and it is unclear whether the model was locked down in the development process.
After the Lancet paper was published, two additional datasets were made publicly available by NCI and FDA through the Clinical Proteomics Program databank. The original dataset described in the initial paper (Petricoin et al., 2002) was derived with a Ciphergen H4 protein chip array, and was baseline corrected to allow for the removal of background “noise” or unnecessary peaks by running a blank set of samples that was subtracted from the data. The second set used the same samples as in dataset 1 and was also baseline corrected but was run on the Ciphergen WCX2 protein chip array. The third dataset contained new samples—91 controls and 162 with cancer—that were prepared robotically, rather than by hand, and was not baseline corrected. The dataset was derived using the Ciphergen WCX2 protein chip array, as in dataset 2 (Baggerly et al., 2004a).
Independent investigators began looking into mass spectrometry profiling of serum samples for ovarian cancer because of its clinical importance, interest of the scientific community, and potential progression into clinical use (Baggerly et al., 2004a,b, 2005a,b; Sorace and Zhan, 2003).26,27 Using the three publicly available datasets, statisticians Keith Baggerly, Jeff Morris, and Kevin Coombes concluded that there were numerous problems with the statistical and experimental methods, including inadvertent changes in protocol mid-experiment, and that the clustering approach outlined by Petricoin and colleagues (2002) would not work (Baggerly et al., 2004a). For example, the m/z values reported by Petricoin et al. (2002) suggested that no external calibration was applied (Baggerly et al., 2004a). Even when calibration methods are used, results may vary from lab to lab if differing calibrants are employed, rendering an assay that is inconsistent across settings (Baggerly et al., 2004a). Thus, lack of calibration would be a serious flaw.
The investigators also concluded that the published proteomic patterns were attributable to batch effects, or “artifacts of sample processing, not to the underlying biology of cancer” (Baggerly et al., 2004a) because the analysis identified structure in the noise regions of the spectra that could distinguish controls and cancer (Baggerly et al., 2004a). Another independent analysis by external investigators using routine statistical methods also identified peaks in the noise spectra that were able to classify samples as cancer or controls, indicative of a significant non-biological source of bias in the data (Sorace and Zhan, 2003). These artifacts in the spectra may have stemmed from differences in the way that samples from patients with
______________
26 Personal communication, Keith Baggerly, December 8, 2011.
27Petricoin and Liotta, along with some of their coauthors, have disputed the criticisms of their work on ovarian cancer (Liotta et al., 2004, 2005; Petricoin et al., 2004).
cancer and normal controls were prepared and processed, or from other differences in experimental protocol (Leek et al., 2010). Ideally, an entire validation set would have been obtained independently at a separate institution (Diamandis, 2004).
Case Highlights
The OvaCheck case study provides lessons about the dangers of batch effects and the potential sources of bias that may result from improper experimental design. Variability in specimen quality or handling can impact the ability to train and test a computational model; when specimens are “inherently different or are handled in a way that systematically introduces a signal into the data for one of the compared groups,” bias can ensue (Ransohoff, 2005). Randomizing samples, properly calibrating instruments, and revalidating results after every shift in protocol can help to ensure that the data are unbiased and that the resulting computational model will generalize to independent datasets (Baggerly et al., 2004a). Any protocol changes that occur during the experiment should be carefully documented to address potential sources of bias. These include a shift between chip types and routine maintenance of instruments mid-experiment that require recalibration of formulas, etc. In this case, the training and test sets suffered from batch effects that caused the noise region of the spectra to differ between the cases and controls. If the test samples had been obtained independently, preferably drawn from another institution at a different point in time, and had been prepared and run through the mass spectrometer separately, then the risk of such batch effects would have been greatly reduced. The study by Sorace and Zhan (2003) also demonstrated that use of a simpler statistical analysis may have illuminated deficiencies in experimental design at an early stage of research.
This case study demonstrates the benefits of making data publicly available to allow for independent assessment of the data and computational model. It also underscores the importance of consulting with FDA as recommended by the committee, because FDA action prevented the clinical implementation of the unvalidated OvaCheck test.
Problems in the development of ovarian proteomic-based tests are not unique to OvaCheck. In 2008, Yale University investigators (Visintin et al., 2008) reported the results for a six-biomarker combination (including CA-125) for ovarian cancer detection. The test, OvaSure, became available on the market in June 2008 as an LDT offered by LabCorp (Pollack, 2008a). In August, FDA sent a notice to the company that the agency believed OvaSure had not undergone adequate clinical validation (FDA, 2008c), and then sent the company a warning letter (FDA, 2008d) that
specified that the test required FDA oversight. LabCorp stopped sale of the OvaSure test, but disagreed with FDA’s position (Pollack, 2008b).
ALLOMAP
AlloMap® Molecular Expression Testing (XDx Expression Diagnostics) was developed to aid identification of heart transplant recipients who have a low risk of moderate or severe acute cellular rejection28 (ACR) at the time of testing. AlloMap is a blood test that measures RNA expression of 11 genes to obtain a single score on a scale of 0 to 40, with a lower score reflecting lower probability of ACR at the time of testing. Prior to AlloMap, the standard of care was a more invasive method for monitoring heart transplant patients for ACR. Endomyocardial biopsy (EMB) is an invasive procedure that can cause rare, but potentially serious, complications (Baraldi-Junkins et al., 1993) and is also subject to inter-observer variability in histologic evaluation (Marboe et al., 2005; Nielsen et al., 1993).
XDx met with FDA for a pre-IDE meeting to discuss the Cardiac Allograft Rejection Gene Expression Observational (CARGO) study, AlloMap development, and analytical and clinical validation procedures. At this time, it was determined that an IDE was not needed because the AlloMap test would not be directing patient management decisions in the CARGO study.29 FDA informed XDx that AlloMap would be classified as a Class II device using the de novo 510(k) process.
Discovery Phase
The data used to develop the gene expression profile for AlloMap were generated from patients enrolled in the CARGO study (Deng et al., 2006). After heart transplantation, all patients were followed prospectively with EMB at each subsequent clinical visit using standard techniques and grading by local pathologists according to International Society for Heart and Lung Transplantation (ISHLT) guidelines (Deng et al., 2006). At the same time, blood was drawn to isolate RNA from peripheral blood mononuclear cells. A subset of biopsies were graded by three independent pathologists blinded to the clinical information before selecting samples for test discovery, computational model development, and test validation.
A custom microarray representing 7,370 genes was used for the discovery phase of test development. Statistical analyses were used to select 97
______________
28 Acute cellular rejection (ACR) occurs when a transplanted organ is rejected by the immune system of the organ recipient. In the AlloMap case study, ACR refers to a transplanted heart being rejected by the immune system of the transplant recipient.
29 Personal communication, Mitch Nelles, XDx, October 12, 2011.
candidate genes from the microarray expression data. A literature review identified an additional 155 genes based on molecular pathways implicated in transplant rejection. The expression levels of the 252 candidate genes were further assessed by qRT-PCR to identify 68 genes whose expression correlated with moderate or severe rejection (as determined by EMB). Six genes were eliminated due to variation in expression based on blood sample processing time. From these remaining 62 genes, statistical modeling of gene expression correlations yielded a 20-gene classifier (11 informative genes, 9 control/normalization genes). The 11 informative genes, 6 derived from the literature and 5 from the microarray analysis, were used to calculate the AlloMap test score. Known functions of these genes include roles in hematopoiesis, platelet activation, T lymphocyte activation and migration, and response to steroids.
Test Validation Phase
Analytical Validation
Analytical validation for AlloMap was documented in the 510(k) decision summary (FDA, 2008a). XDx reported the results for the following variabilities: run-to-run, operator-to-operator (interoperator), within operator (intraoperator), lot-to-lot, plate-to-plate within a lot, and section-to- section within a plate. The range of RNA purity that is acceptable for testing was determined and reported. XDx also reported that test performance was not compromised by presence of the following in blood samples: immunosuppressants, cytomegalovirus, heparin, hemoglobin, acetylsalicylic acid, acetaminophen, triglyceride, bilirubin, and genomic DNA.
Statistics and Bioinformatics Validation
Discovery-phase microarray data are available in a publicly accessible database (GEO, accession number GSE2445). Raw data from qRT-PCR training were provided to FDA as part of premarket notification processes but were not reported in Deng et al. (2006).
All samples for training and validation originated from the CARGO study. Samples used in the primary clinical validation study did not overlap with samples used in the discovery phase. The secondary clinical validation reused all 63 patient samples from the primary validation as well as some samples that had been used in the discovery phase (Figure A-1). Deng et al. (2006) noted that the secondary validation “may provide improved power but may be biased to the extent that a longitudinal set of samples from
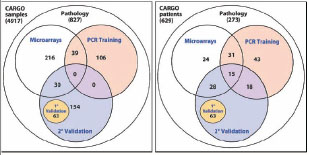
FIGURE A-1 Venn diagrams illustrating overlap in patient blood samples used for AlloMap development. A total of 4,917 samples were drawn from 629 patients: 827 biopsy samples were analyzed by centralized pathology, 285 samples from 98 patients were analyzed by microarray and the data were used in the discovery phase, 145 samples from 107 patients were analyzed for PCR training, 63 samples from 63 patients were analyzed in the primary clinical validation (1° Validation), 63 patients from the primary clinical validation plus 61 patients analyzed in the microarray and PCR training steps of the discovery phase were analyzed in a secondary clinical validation (2° Validation).
NOTE: CARGO = Cardiac Allograft Rejection Gene Expression Observational, PCR = polymerase chain reaction.
SOURCE: Deng et al., 2006.
an individual patient are not completely independent with respect to gene expression.”
Details of the computational model development were provided in the Deng et al. (2006) supplemental material and provided to FDA as part of the 510(k) submission. The test was locked down after discovery, prior to final validation.30
Clinical/Biological Validation
AlloMap was evaluated in three clinical validation studies and a prevalent population study (Table A-7) (Deng et al., 2006; FDA, 2008a). The histological characteristics of cardiac transplant rejection based on an expert panel reading of the individual EMB were used as the clinical endpoint. In the primary clinical validation study, samples from 63 patients were blinded
______________
30 Personal communication, Mitch Nelles, XDx, October 12, 2011.
TABLE A-7 Clinical/Biological Validation Studies for AlloMap
|
|
||||
| Study | CARGO Primary Clinical Validation Study (Deng et al., 2006) | CARGO Secondary Clinical Validation Study (Deng et al., 2006) | CARGO Prevalent Population Study (Deng et al., 2006) | CARGO Dataset Used for FDA Clearance (k073482) |
|
|
||||
| Tissue source | CARGO trial | CARGO trial | CARGO trial | CARGO trial |
| Test platform | qRT-PCR and EMB | qRT-PCR and EMB | qRT-PCR and EMB | qRT-PCR and EMB |
| Study question | Does AlloMap distinguish rejection from no rejection? | Does AlloMap distinguish rejection from no rejection? | Does AlloMap distinguish rejection from no rejection in samples representing expected clinical population? | Does AlloMap distinguish rejection from no rejection in samples representing the expected clinical population? |
| Study design | Prospective Not marker directed | Prospective Not marker directed | Prospective Not marker directed | Prospective Not marker directed |
| Patient characteristics | Time posttransplant not stated | Time posttransplant not stated | ≥1 year posttransplant | > 55 days posttransplant, > 30 days post rejection treatment |
| Sample number | 63 samples; 63 patients | 184 samples; 124 patients | 281 samples; 166 patients | 300 samples; 154 patients |
| Blinding | Yes | Yes | Yes | Yes |
| Independence | Different specimens than used in discovery | All specimens used in primary clinical validation plus some specimens used in discovery | Different specimens than used in discovery | Different specimens than used in discovery |
| Development involved XDx and transplant cardiologists directing CARGOa | Development involved XDx and transplant cardiologists directing CARGO | Development involved XDx and transplant cardiologists directing CARGO | Development involved XDx and transplant cardiologists directing CARGO | |
|
|
||||
| Study | CARGO Primary Clinical Validation Study (Deng et al., 2006) | CARGO Secondary Clinical Validation Study (Deng et al., 2006) | CARGO Prevalent Population Study (Deng et al., 2006) | CARGO Dataset Used for FDA Clearance (k073482) |
|
|
||||
| Results | At threshold score of 20, test correctly classified 84% (95% CI 66-94%) of patients with rejection and 38% (95% CI 22-56%) of patients with no rejection (p = 0.0018) | At threshold score of 20, test correctly classified 76% (95% CI 63-85%) of patients with rejection and 41% (95% CI 32-50%) of patients with no rejection (p = 0.0001) | At threshold score of 30, PPV for rejection was 6.8%, NPV was 99.6% (Deng et al., 2006 | AUC of 0.67 (95% CI 0.56- 0.78). At threshold scoreb of 34 at 2-6 months post- transplant PPV for rejection = 5.0%, NPV = 98.2%. At threshold score of 34 at > 6 months posttransplant PPV for rejection = 4.1%, NPV = 98.9% |
|
|
||||
| a Personal communication, Mitch Nelles, XDx, December 9, 2011. b Starling et al., 2006. NOTE: AUC = area under the receiver operating curve, CARGO = Cardiac Allograft Rejection Gene Expression Observational, CI = confidence interval, EMB = endomyocardial biopsy, NPV = negative predictive value, PPV = positive predictive value, qRT-PCR = quantitative reverse-transcriptase polymerase chain reaction. |
||||
and prospectively evaluated. Samples from these patients had not previously been introduced into any phase of test development. In this primary clinical validation, AlloMap distinguished between patients with moderate or severe rejection and those with no rejection (p = 0.0018). With the pro-spectively defined threshold score of 20 or greater as indicative of rejection, the investigators reported that the test correctly classified 84 percent (95% CI 66-94%) of patients with rejection and 38 percent (95% CI 22-56%) of patients with no rejection.
The secondary clinical validation study was performed to confirm the results in a larger sample set of CARGO patients. This clinical validation study evaluated 184 samples from 124 patients, which included the 63 samples used in the primary clinical validation study as well as samples that were used in the discovery phase of development (see Figure A-1). In this secondary clinical validation, AlloMap correctly classified 76 percent (95%
CI 63-85%) of patients with rejection and 41 percent (95% CI 32-50%) of patients with no rejection (p = 0.0001).
In the prevalent population study, investigators evaluated AlloMap’s performance among patients likely to be seen in clinical practice. This analysis included 281 CARGO samples from 166 patients who were at least a year posttransplant. None of these samples had been used in AlloMap test discovery or training. At a threshold score of 30, which was selected to maximize the negative predictive value (NPV), the positive predictive value (PPV) for rejection was 6.8 percent, the NPV was 99.6 percent, and 68 percent of tests were below this value (Deng et al., 2006).
For FDA clearance (FDA, 2008a), an analysis of the prevalent population study was evaluated. The analysis included 300 CARGO samples from 154 patients who were at least 55 days posttransplant and more than 30 days beyond a rejection episode. None of these samples had been used in the discovery or training phases of AlloMap test development. The area under the receiver operating curve (AUC) developed from the full dataset was 0.67 (95% CI 0.56-0.78)
Clinical Utility
Monitoring Risk of Future ACR
In a prospective evaluation of 104 CARGO patients who were at least 30 days past a heart transplantation and whose blood samples had not been used to develop the AlloMap computational model, results suggested that the gene expression score used to determine rejection at the time of testing may determine the likelihood of ACR in the subsequent 12 weeks (Mehra et al., 2007, 2008). The score (mean ± standard deviation) for patients with rejection within the following 12 weeks was 27.4 ± 6.3 (n = 39) and for patients with no rejection in the following 12 weeks was 23.9 ± 7.1 (p = 0.01). During this time, no samples from patients experiencing rejection within the following 12 weeks had scores of less than 20 (Mehra et al., 2007).
International Population
The CARGO II trial was designed to evaluate the correlation between AlloMap and presence or absence of ACR as determined by EMB in a mostly European cohort of heart transplant patients. The study was completed in February 2009, but the results were not yet available in 2011 (ClinicalTrials.gov, 2011a). Of the 17 transplant centers participating in the study, 4 were in North America and 13 were in Europe.
Noninferiority of AlloMap to EMB for Clinical Management of Heart Transplant Patients
The IMAGE (Invasive Monitoring Attenuation through Gene Expression) trial demonstrated that AlloMap was non-inferior to EMB for monitoring posttransplant patients for ACR and reduced the number of biopsies that needed to be performed on heart transplant patients (Pham et al., 2010). Patients were randomly assigned to monitoring for rejection by either AlloMap testing or EMB. The two groups were compared with respect to a composite primary outcome of rejection with hemodynamic compromise, graft dysfunction due to other causes, death, or retransplantation. The 2-year cumulative rates of the primary outcome were 14.5 percent for patients monitored with AlloMap and 15.3 percent for patients monitored with EMB (p = 0.86). The rates of death from any cause were 6.3 percent and 5.5 percent, respectively (p = 0.82). The frequency of biopsy per patient year of follow-up was 0.5 and 3.0, respectively (p ![]() 0.001).
0.001).
Clinical Use
The FDA 510(k) decision summary (2008a) states that the intended clinical use of AlloMap testing is “to aid in the identification of heart transplant recipients with stable allograft function who have a low probability of moderate/severe acute cellular rejection (ACR) at the time of testing in conjunction with standard clinical assessment.” AlloMap testing is performed at the XDx CLIA-certified laboratory. Since AlloMap was initially marketed in 2005, more than 32,000 commercial tests have been performed in U.S. heart transplant patients. In 2010, 7,147 tests were performed.31
ISHLT guidelines recommend that AlloMap can be used in low-risk patients who are 6 months to 5 years post heart transplantation for ruling out presence of ACR (ISHLT, 2010). The California Technology Assessment Forum determined that AlloMap “meets Technology Assessment Criteri[a] 1 through 5 for safety, effectiveness and improvement in health outcomes when used to manage heart transplant patients at least one year post- transplant” (CTAF, 2010, p. 15). However, the Blue Cross Blue Shield Association Technology Evaluation Center decided in September 2011 that AlloMap did not meet its utility criteria as a method to monitor cardiac allograft rejection because the clinical validation studies were small and the cut points defining a positive test had not been independently validated (BCBSA, in press).
Currently, those who provide coverage and/or regular payment for the AlloMap test include Medicare, MediCal, New York Medicaid, United
______________
31 Personal communication, Mitch Nelles, XDx, October 12, 2011.
Healthcare, Anthem Wellpoint, Aetna, Kaiser, and several other private insurers.32
Case Highlights
In the CARGO trial, there was no overlap between patient samples used for discovery and those used for the primary clinical validation study, but there was overlap between patient samples used for discovery and those used in a secondary clinical validation study. XDx indicated that overlap could not be avoided due to the difficulty in obtaining the requisite number of blood samples from heart transplant recipients associated with a consensus biopsy reading of ACR.33
XDx met with FDA early in the process to determine the appropriate pathway for developing AlloMap, and the test was cleared in 2008. The test was locked down prior to final validation. Some additional performance characterization has been conducted with the goal of making better use of the output from the test, but no changes have been made to the computational procedures that generate the AlloMap scores.34
CORUS CAD
Corus® CAD (CardioDx, Inc.) was developed as a less invasive method than angiography to identify obstructive coronary artery disease (CAD).35 Corus CAD is a blood test that measures the expression level of 23 genes to get a score on a scale of 1 to 40. The score is used by primary care clinicians and cardiologists for determining whether a non-diabetic patient’s symptoms of cardiovascular disease are due to CAD.
CardioDx met with FDA for a pre-IDE meeting prior to derivation of the final computational model for Corus CAD,36 but this test has not been submitted to FDA for clearance or approval. CardioDx sought the LDT pathway to market and noted that at that time, “FDA had draft guidance for IVDMIAs and did not require 510(k) clearance or PMA.”37
______________
32 Personal communication, Mitch Nelles, XDx, December 9, 2011.
33 Personal communication, Mitch Nelles, XDx, October 21, 2011.
34 Personal communication, Mitch Nelles, XDx, October 12, 2011.
35 CAD is the damage to the heart caused by atherosclerotic constriction of arteries supplying blood to the heart.
36 Personal communication, Steve Rosenberg, Cardio Dx, October 21, 2011.
37 Personal communication, Steve Rosenberg, Cardio Dx, October 21, 2011.
Discovery Phase
Initial proof-of-concept work in two retrospective cohorts undergoing coronary angiography demonstrated a set of genes that differentiated between patients with obstructive CAD and those without (Wingrove et al., 2008). The discovery process to develop Corus CAD entailed two microarray gene expression analyses (Elashoff et al., 2011; Rosenberg et al., 2010). Gene expression was measured in whole-blood cells, which were collected from patients prior to coronary angiography. The first microarray gene expression analysis was a retrospective study of samples from the repository of the Duke University CATHGEN registry,38 which contained blood samples from patients with and without diabetes (Elashoff et al., 2011). Microarray analysis of 195 patient samples suggested 2,438 CAD-associated genes. Eighty-eight genes that had the greatest statistical significance and biological relevance were selected for confirmation by RT-PCR in these same 195 samples. Diabetes was the clinical factor that had the most significant effect on gene expression (p = 0.0006). Analysis in non-diabetic and diabetic subsets (n = 124 and 71, respectively) showed expression of 42 and 12 significant CAD genes, respectively (p ![]() 0.05), with no intersection (Elashoff et al., 2011). Therefore, the authors limited further work to patients without diabetes. A second microarray analysis to further define genes that could be a hallmark of CAD, and all subsequent work to develop Corus CAD, was performed in the prospective clinical trial PREDICT (Personalized Risk Evaluation and Diagnosis in the Coronary Tree). The development of the computational model entailed a three-step approach. Patients from 39 U.S. medical centers were assigned to a group based on date of study enrollment. Blood samples from 198 PREDICT patients were used for the second microarray gene expression analysis. The analysis suggested 5,935 CAD-associated genes. There were 655 genes that overlapped with the CATHGEN microarray results.
0.05), with no intersection (Elashoff et al., 2011). Therefore, the authors limited further work to patients without diabetes. A second microarray analysis to further define genes that could be a hallmark of CAD, and all subsequent work to develop Corus CAD, was performed in the prospective clinical trial PREDICT (Personalized Risk Evaluation and Diagnosis in the Coronary Tree). The development of the computational model entailed a three-step approach. Patients from 39 U.S. medical centers were assigned to a group based on date of study enrollment. Blood samples from 198 PREDICT patients were used for the second microarray gene expression analysis. The analysis suggested 5,935 CAD-associated genes. There were 655 genes that overlapped with the CATHGEN microarray results.
A total of 113 genes was selected based on biological relevance, statistical significance, and CAD-associated gene expression measured with RT-PCR in 640 PREDICT patient samples. Gene expression correlation clustering and cell-type analyses of these genes were used to determine the final 23 genes in the computational model (20 CAD-related genes and 3 normalization genes). These genes have functions in neutrophil activation and apoptosis, natural killer cell activation, innate immunity, cell necrosis, and adaptive immune response (Rosenberg et al., 2010).
______________
38 See http://cathgen.duhs.duke.edu/modules/cath_about/index.php?id=1 (accessed January 18, 2012).
Test Validation Phase
Analytical Validation
Detailed information regarding analytical validation for Corus CAD is not publicly available. The following preanalytical clinical and demographic variables were measured in patients used in the microarray studies: sex, age, race, body mass index, current smoker, systolic blood pressure, diastolic blood pressure, hypertension, dyslipidemia, neutrophil count, and lymphocyte count (Elashoff et al., 2011).
Statistics and Bioinformatics Validation
Discovery-phase microarray data are available in a publicly accessible database (GEO, accession number GSE20686). PCR data used in the development and validation sets are not publicly available, although Cardio Dx has indicated they would be available upon request by qualified investigators.39 Analysis of the RT-PCR results from the validation study was performed at Scripps Translational Science Institute.40
Blood samples from 640 patients were used to develop the computational model (Elashoff et al., 2011); blood samples from another 526 patients were used for validation. There was no overlap in patient samples used in each phase of the study.
Details regarding development of the computational model were published (Rosenberg et al., 2010). The model was locked down prior to the start of the validation study. CardioDx funded the study and was involved in the design and conduct of the study.
Clinical/Biological Validation
Rosenberg et al. (2010) reported statistical analyses only for the validation group. The investigators predefined the primary endpoint as the receiver-operating characteristic (ROC) curve area for prediction of disease status by the test score. In a set of 526 PREDICT patients not used for gene discovery or computational model development, the AUC for ROC was 0.70 ± 0.02 (p ![]() 0.001). At a threshold score of 14.75, which corresponded to a 20 percent likelihood of obstructive CAD, the sensitivity and specificity were 85 percent and 43 percent, respectively. This yielded an NPV of 83 percent and a PPV of 46 percent, with 33 percent of patient scores below this threshold (Rosenberg et al., 2010).
0.001). At a threshold score of 14.75, which corresponded to a 20 percent likelihood of obstructive CAD, the sensitivity and specificity were 85 percent and 43 percent, respectively. This yielded an NPV of 83 percent and a PPV of 46 percent, with 33 percent of patient scores below this threshold (Rosenberg et al., 2010).
______________
39 Personal communication, Steve Rosenberg, Cardio DX, October 21, 2011; December 12, 2011.
40 Personal communication, Steve Rosenberg, Cardio DX, October 21, 2011.
Clinical Utility
The COMPASS (Coronary Obstruction Detection by Molecular Personalized Gene Expression) trial prospectively evaluated use of Corus CAD in patients referred for myocardial perfusion imaging (MPI) due to suspected CAD; 431 patients were analyzed. In the primary analysis of 63 cases, Corus CAD AUC was 0.79 (p ![]() 0.001), while MPI AUC was 0.59 (p
0.001), while MPI AUC was 0.59 (p ![]() 0.001). At a threshold score of 15, Corus CAD sensitivity and NPV were 89 percent and 96 percent, respectively; MPI sensitivity and NPV were 27 percent and 88 percent, respectively. In the secondary case analysis, Corus CAD AUC was also higher than MPI (0.77 vs. 0.64, p
0.001). At a threshold score of 15, Corus CAD sensitivity and NPV were 89 percent and 96 percent, respectively; MPI sensitivity and NPV were 27 percent and 88 percent, respectively. In the secondary case analysis, Corus CAD AUC was also higher than MPI (0.77 vs. 0.64, p ![]() 0.01) (Thomas et al., 2011). The estimated study completion date was March 2012 (ClinicalTrials.gov, 2011a).
0.01) (Thomas et al., 2011). The estimated study completion date was March 2012 (ClinicalTrials.gov, 2011a).
Clinical Use
As an LDT, Corus CAD is performed in the company’s CLIA-certified laboratory. Approximately 13,000 tests were ordered between October 2010 and September 2011.41
Corus CAD is a very newly developed test, which may account for why it has not been incorporated into any guidelines. Multiple insurance plans currently pay for the test on a patient-by-patient basis.42 As noted on the CardioDx website, “CardioDx is actively pursuing third-party payer reimbursement for Corus CAD.”
Case Highlights
This study highlights the importance of publishing detailed information regarding derivation of the computational model used for the diagnostic test. The work by Rosenberg and colleagues has been described as having an “elegant design” and being “at the vanguard of clinical genetics in cardiovascular care,” but “the report offers too little information about the derivation of the algorithm for readers to determine whether the screening tool provides internally valid results” (Arnett, 2010, p. 473). Six months after the publication of the clinical work by Rosenberg et al. (2010), Elashoff et al. (2011) published more detailed information regarding the derivation of the computational model.
______________
41 Personal communication, Steve Rosenberg, Cardio DX, October 24, 2011.
42 Personal communication, Steve Rosenberg, Cardio DX, December 12, 2011.
REFERENCES
Ach, R. A., A. Floore, B. Curry, V. Lazar, A. M. Glas, R. Pover, A. Tsalenko, H. Ripoche, F. Cardoso, M. S. d’Assignies, L. Bruhn, and L. J. van ‘t Veer. 2007. Robust interlaboratory reproducibility of a gene expression signature measurement consistent with the needs of a new generation of diagnostic tools. BMC Genomics 8(148):10.1186/1471-2164-8-148.
ACOG and SGO (American College of Obstetricians and Gynecologists and Society of Gynecologic Oncologists). 2011. Committee opinion no. 477: The role of the obstetrician-gynecologist in the early detection of epithelial ovarian cancer. Obstetrics and Gynecology 117(3):742-746.
ACS (American Cancer Society). 2011. What Are the Key Statistics about Ovarian Cancer? http://www.cancer.org/Cancer/OvarianCancer/DetailedGuide/ovarian-cancer-key-statistics. (accessed September 8, 2011).
Agendia. 2009. FDA Broadens Clearance for Agendia’s MammaPrint. http://www.agendia.com/pages/press_release/70.php?aid=90 (accessed March 16, 2011).
Agendia. 2011a. International Recognition for Pioneering Work in Translation Research and Personalized Medicine for Breast Cancer. http://www.agendia.com/pages/awards_and_recognition/97.php (accessed September 21, 2011).
Agendia. 2011b. MammaPrint Has Extensive International Clinical Validation. http://www.agendia.com/pages/validation/32.php (accessed March 27, 2011).
AHRQ (Agency for Healthcare Research and Quality). 2006. Genomic Tests for Ovarian Cancer Detection and Management. Rockville, MD: AHRQ.
AHRQ. 2008. Impact of Gene Expression Profiling Tests on Breast Cancer Outcomes. Rockville, MD: AHRQ.
Albain, K. S., W. E. Barlow, S. Shak, G. N. Hortobagyi, R. B. Livingston, I.-T. Yeh, P. Ravdin, R. Bugarini, F. L. Baehner, N. E. Davidson, G. W. Sledge, E. P. Winer, C. Hudis, J. N. Ingle, E. A. Perez, K. I. Pritchard, L. Sheperd, J. R. Gralow, C. Yoshizawa, D. C. Allred, C. K. Osborne, and D. F. Hayes. 2010. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: A retrospective analysis of a randomized trial. Lancet Oncology 11(1):55-65.
Allison, M. 2010. The HER2 testing conundrum. Nature Biotechnology 28(2):117-119.
Amonkar, S. D., G. P. Bertenshaw, T. H. Chen, K. J. Bergstrom, J. Zhao, P. Seshaiah, P. Yip, and B. C. Mansfield. 2009. Development and preliminary evaluation of a multivariate index assay for ovarian cancer. PLoS One 4(2):e4599.
Arnett, D. K. 2010. Gene expression algorithm for prevalent coronary artery disease: A first step in a long journey. Annals of Internal Medicine 153(7):473-474.
Baehner, F. L., N. Achacoso, T. Maddala, S. Shak, C. P. Quesenberry, Jr., L. C. Goldstein, A. M. Gown, and L. A. Habel. 2010. Human epidermal growth factor receptor 2 assessment in a case-control study: Comparison of fluorescence in situ hybridization and quantitative reverse transcription polymerase chain reaction performed by central laboratories. Journal of Clinical Oncology 28(28):4300-4306.
Baggerly, K. A., J. S. Morris, and K. R. Coombes. 2004a. Reproducibility of SELDI-TOF protein patterns in serum: Comparing datasets from different experiments. Bioinformatics 20(5):777-785.
Baggerly, K. A., S. R. Edmonson, J. S. Morris, and K. R. Coombes. 2004b. High-resolution serum proteomic patterns for ovarian cancer detection. Endocrine-Related Cancer. 11(4):583-584.
Baggerly, K. A., K. R. Coombes, and J. S. Morris. 2005a. Bias, randomization, and ovarian proteomic data: A reply to “producers and consumers.” Cancer Informatics 1(1):9-14.
Baggerly, K. A., J. S. Morris, S. R. Edmonson, and K. R. Coombes. 2005b. Signal in noise: Evaluating reported reproducibility of serum proteomic tests for ovarian cancer. Journal of the National Cancer Institute 97(4):307-309.
Baraldi-Junkins, C., H. R. Levin, E. K. Kasper, B. K. Rayburn, A. Herskowitz, and K. L. Baughman. 1993. Complications of endomyocardial biopsy in heart transplant patients. Journal of Heart and Lung Transplantation 12(1 Pt 1):63-67.
BCBS (Blue Cross and Blue Shield Association). 2008. Gene expression profiling of breast cancer to select women for adjuvant chemotherapy. Technology Evaluation Center 22(13):1-51.
BCBS. In Press.
Gene expression profiling as a noninvasive method to monitor for cardiac allograft rejection. Technology Evaluation Center.
Berry, D. A., C. Cirrincione, I. C. Henderson, M. L. Citron, D. R. Budman, L. J. Goldstein, S. Martino, E. A. Perez, H. B. Muss, L. Norton, C. Hudis, and E. P. Winer. 2005. Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. Journal of the American Medical Association 295(14):1658-1667.
Bonislawski, A. 2011. Vermillion Buys Correlogic’s Assets for $435K; Correlogic Settles with LabCorp, Quest. http://www.genomeweb.com/proteomics/vermillion-buys-correlogics-assets-435k-correlogic-settles-labcorp-quest.
Buyse, M., S. Loi, L. J. van ‘t Veer, G. Viale, M. Delorenzi, A. M. Glas, M. S. d’Assignies, J. Bergh, R. Lidereau, P. Ellis, A. Harris, J. Bogaerts, P. Therasse, A. Floore, M. Amakrane, F. Piette, E. T. Rutgers, C. Sortiriou, F. Cardoso, and M. J. Piccart. 2006. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. Journal of the National Cancer Institute 98(17):1183-1192.
Check, E. 2004. Proteomics and cancer: Running before we can walk? Nature 429(6991): 496-497.
ClinicalTrials.gov. 2011a. Cardiac Allograft Rejection Gene Expression Observational (CARGO) II Study (CARGO II). http://www.clinicaltrials.gov/ct2/show/NCT00761787?term=CARGO&rank=1 (accessed November 15, 2011).
Clinicaltrials.gov. 2011b. Genetic Testing or Clinical Assessment in Determining the Need for Chemotherapy in Women with Breast Cancer That Involves No More Than 3 Lymph Nodes. http://clinicaltrials.gov/ct2/show/NCT00433589?term=mindact&rank=1 (accessed March 27, 2011).
Cobleigh, M. A., B. Tabesh, P. Bitterman, J. Baker, M. Conin, M. L. Liu, R. Borchik, J. M. Mosquera, M. G. Walker, and S. Shak. 2005. Tumor gene expression and prognosis in breast cancer patients with 10 or more positive lymph nodes. Clinical Cancer Research 11(24 Pt 1):8623-8631.
Correlogic. 2004. Re: Correlogic Systems Inc. Reference Laboratory—OvaCheck Testing Service. http://www.correlogic.com/pdfs/July14SteveGutmanLetter.pdf (accessed December 2, 2011).
Cronin, M., M. Pho, D. Dutta, J. C. Stephans, S. Shak, M. C. Kiefer, J. M. Esteban, and J. Baker. 2004. Measurement of gene expression in archival paraffin-embedded tissues: Development and performance of a 92-gene reverse transcriptase-polymerase chain reaction assay. American Journal of Pathology 164(1):35-42.
Cronin, M., C. Sangli, M.-L. Liu, M. Pho, D. Dutta, A. Nguyen, J. Jeong, J. Wu, K. C. Langone, and D. Watson. 2007. Analytical validation of the Oncotype DX genomic diagnostic test for recurrence prognosis and therapeutic response prediction in node-negative, estrogen receptor-positive breast cancer. Clinical Chemistry 53(6):1084-1091.
CTAF (California Technology Assessment Forum). 2010. Gene Expression Profiling for the Diagnosis of Heart Transplant Rejection. http://ctaf.org/content/assessment/detail/1208 (accessed January23, 2012).
De, P., B. R. Smith, and B. Leyland-Jones. 2010. Human epidermal growth factor receptor 2 testing: Where are we? Journal of Clinical Oncology 28(28):4289-4292.
Deng, M. C., H. J. Eisen, M. R. Mehra, M. Billingham, C. C. Marboe, G. Berry, J. Kobashigawa, F. L. Johnson, R. C. Starling, S. Murali, D. F. Pauly, H. Baron, J. G. Wohlgemuth, R. N. Woodward, T. M. Klingler, D. Walther, P. G. Lal, S. Rosenberg, and S. Hunt. 2006. Noninvasive discrimination of rejection in cardiac allograft recipients using gene expression profiling. American Journal of Transplantation 6(1):150-160.
Diamandis, E. 2004. Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: Opportunities and potential limitations. Molecular and Cellular Proteomics 3(4):367-378.
Dowsett, M., J. Cuzick, C. Wale, J. Forbes, E. A. Mallon, J. Salter, E. Quinn, A. Dunbier, M. Baum, A. Buzdar, A. Howell, R. Bugarini, F. L. Baehner, and S. Shak. 2010. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: A TransATAC study. Journal of Clinical Oncology 28(11):1829-1834.
Dumur, C. I., M. Lyons-Weiler, C. Sciulli, C. T. Garrett, I. Schrijver, T. K. Holley, J. Rodriguez-Paris, J. R. Pollack, J. L. Zehnder, M. Price, J. M. Hagenkord, C. T. Rigl, L. J. Buturovic, G. G. Anderson, and F. A. Monzon. 2008. Interlaboratory performance of a microarray-based gene expression test to determine tissue of origin in poorly differentiated and undifferentiated cancers. Journal of Molecular Diagnostics 10(1):67-77.
Dumur, C. I., C. E. Fuller, T. L. Blevins, J. C. Schaum, D. S. Wilkinson, C. T. Garrett, C. N. Powers. 2011. Clinical verification of the performance of the Pathwork Tissue of Origin test. American Journal of Clinical Pathology 136(6):924-933.
EBCTCG (Early Breast Cancer Trialists Collaborative Group). 2005. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 365(9472):1687-16717.
Elashoff, M. R., J. A. Wingrove, P. Beineke, S. E. Daniels, W. G. Tingley, S. Rosenberg, S. Voros, W. E. Kraus, G. S. Ginsburg, R. S. Schwartz, S. G. Ellis, N. Tahirkheli, R. Waksman, J. McPherson, A. J. Lansky, and E. J. Topol. 2011. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Medical Genomics 4(1):26.
Esteban, J., J. Baker, M. Cronin, M. L. Liu, M. G. Llamas, M. G. Walker, R. Mena, and S. Shak. 2003. Tumor gene expression and prognosis in breast cancer: Multi-gene RT-PCR assay of paraffin-embedded tissue. Proceedings of the American Society of Clinical Oncology 22:Abstract 3416.
Esteva, F. J., A. A. Sahin, M. Cristofanilli, K. Coombes, S.-J. Lee, J. Baker, M. Cronin, M. Walker, D. Watson, S. Shak, and G. N. Hortobagyi. 2005. Prognostic role of a multigene reverse transcriptase-PCR assay in patients with node-negative breast cancer not receiving adjuvant systemic therapy. Clinical Cancer Research 11(9):3315-3319.
FDA (Food and Drug Administration). 2007a. 510(k) Substantial Equivalence Determination Decision Summary (k070675). http://www.accessdata.fda.gov/cdrh_docs/reviews/K070675.pdf (accessed September 20, 2011).
FDA. 2007b. FDA Clears Breast Cancer Specific Molecular Prognostic Test. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108836.htm (accessed March 14, 2011).
FDA. 2008a. 510(k) Substantial Equivalence Determination Decision Summary Assay and Instrument Combination Template (k073482). http://www.accessdata.fda.gov/cdrh_docs/reviews/K073482.pdf (accessed November 23, 2011).
FDA. 2008b. 510(k) Substantial Equivalence Determination Decision Summary (K080896). http://www.accessdata.fda.gov/cdrh_docs/reviews/K080896.pdf (accessed November 15, 2011).
FDA. 2008c. OvaSure Manufacturer Letter. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/IVDRegulatoryAssistance/ucm125130.htm (accessed November 23, 2011).
FDA. 2008d. Laboratory Corporation of America 29-Sep-08. http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2008/ucm1048114.htm (accessed November 23, 2011).
FDA. 2009. 510(k) Substantial Equivalence Determination Decision Summary (K081092). http://www.accessdata.fda.gov/cdrh_docs/reviews/K081092.pdf (accessed September 20, 2011).
FDA. 2010. 510(k) Substantial Equivalence Determination Decision Summary (K092967). http://www.accessdata.fda.gov/cdrh_docs/reviews/K092967.pdf (accessed November 16, 2011).
FDA. 2011a. 510(k) Premarket Notification CDRH SuperSearch. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm (accessed October 18, 2011).
FDA. 2011b. 510(k) Substantial Equivalence Determination Decision Summary (K101454). http://www.accessdata.fda.gov/cdrh_docs/reviews/K101454.pdf (accessed September 19, 2011).
FDA. 2011c. Substantial Equivalence Determination Decision Summary (K081754). http://www.accessdata.fda.gov/cdrh_docs/reviews/K081754.pdf (accessed October 11, 2011).
Fisher, B., J. Costantino, C. Redmond, R. Poisson, D. Bowman, J. Couture, N. V. Dimitrov, N. Wolmark, D. L. Wickerham, and E. R. Fisher. 1989. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. New England Journal of Medicine 23(320):479-484.
Fisher, B., J. Dignam, N. Wolmark, A. DeCillis, B. Emir, D. L. Wickerham, J. Bryant, N. V. Dimitrov, N. Abramson, J. N. Atkins, H. Shibata, L. Deschenes, and R. G. Margolese. 1997. Tamoxifen and chemotherapy for lymph node-negative, estrogen receptor-positive breast cancer. Journal of the National Cancer Institute 89(22):1673-1682.
Fisher, B., J. Jeong, J. Bryant, S. Anderson, J. Dignam, E. R. Fisher, and N. Wolmark. 2004. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: Long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 364(9437):858-868.
Fung, E. T. 2010. A recipe for proteomics diagnostic test development: The OVA1 test, from biomarker discovery to FDA clearance. Clinical Chemistry 56(2):327-329.
Genomic Health. 2011a. Oncotype DX Breast Cancer Assay. http://www.oncotypedx.com/en-US/Breast/HealthcareProfessional/Overview.aspx (accessed January 20, 2011).
Genomic Health. 2011b. Oncotype DX Breast Cancer Assay: Insurance Information. http://www.oncotypedx.com/en-US/Breast/HealthcareProfessional/InsuranceInformation.aspx (accessed January 31, 2011).
Genomic Health. 2011c. The Development and Clinical Validation of Oncotype DX. http://www.oncotypedx.com/en-US/Breast/HealthcareProfessional/Overview.aspx (accessed July 21, 2011).
Glas, A. M., A. Floore, L. Delahaye, A. T. Wittereveen, R. C. F. Pover, N. Bakx, J. S. T. Lahti-Domenici, T. J. Bruinsma, M. O. Warmoes, R. Bernards, L. F. A. Wessels, and L. J. van ‘t Veer. 2006. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics 7(278).
Goldhirsch, A., J. N. Ingle, R. D. Gelber, A. S. Coates, B. Thurlimann, and H.-J. Senn. 2009. Thresholds for therapies: Highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Annals of Oncology 20(8):1319-1329.
Grenert, J. P., A. Smith, W. Ruan, R. Pillai, and A. H. Wu. 2011. Gene expression profiling from formalin-fixed, paraffin-embedded tissue for tumor diagnosis. Clinica Chimica Acta 412(15-16):1462-1464.
H. Con. Res. 385, 107th Cong., 2nd sess. (July 22, 2002). Expressing the sense of the Congress that the Secretary of Health and Human Services should conduct or support research on certain tests to screen for ovarian cancer, and Federal health care programs and group and individual health plans should cover the tests if demonstrated to be effective, and for other purposes.
Habel, L. A., S. Shak, M. Jacobs, A. Capra, C. Alexander, M. Pho, J. Baker, M. Walker, D. Watson, J. Hackett, N. T. Blick, D. Greenberg, L. Fehrenbacher, B. Langholz, and C. P. Quesenberry. 2006. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Research8(3):R25.
Harris, L., H. Fritsche, R. Mennel, L. Norton, P. Ravdin, S. E. Taube, M. R. Somerfield, D. F. Hayes, and R. C. Bast, Jr. 2007. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. Journal of Clinical Oncology 25(33):5287-5312.
Hillen, H. F. 2000. Unknown primary tumours. Postgraduate Medical Journal 76(901):690-693.
Hornberger, J., and R. Chien. 2010. P2-09-06: Meta-Analysis of the Decision Impact of the 21-Gene Breast Cancer Recurrence Score in Clinical Practice. Poster presented at the 33rd Annual San Antonio Breast Cancer Symposium, San Antonio, Texas, December 8-12.
Hornberger, J. C., M. Amin, G. R. Varadhachary, W. D. Henner, and J. S. Nystrom. 2011. Effect of a gene expression-based tissue of origin test’s impact on patient management for difficult-to-diagnose primary cancers. Journal of Clinical Oncology 29(Suppl 4; abstr 459).
ISHLT (International Society of Heart and Lung Transplantation). 2010. The International Society of Heart and Lung Transplantation Guidelines for the Care of Heart Transplant Recipients. Task Force 2: Immunosuppression and Rejection. http://www.ishlt.org/ContentDocuments/ISHLT_GL_TaskForce2_110810.pdf (accessed January 23, 2012).
Kim, C., and S. Paik. 2010. Gene-expression-based prognostic assays for breast cancer. Nature Reviews 7(6):340-347.
Knauer, M., S. Mook, E. J. Rutgers, R. A. Bender, M. Hauptmann, M. J. van de Vijver, R. H. T. Koornstra, J. Bueno-de-Mesquita, S. C. Linn, and L. J. van ‘t Veer. 2010. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Research and Treament 120(3):655-661.
Kroese, M., R. L. Zimmern, and S. E. Pinder. 2007. HER2 status in breast cancer—an example of pharmacogenetic testing. Journal of the Royal Society of Medicine 100(7):326-329.
Laouri, M., M. Halks-Miller, W. D. Henner, and S. Nystrom. 2011. Potential clinical utility of gene expression profiling in identifying tumors of uncertain origin. Personalized Medicine 8(6):615-622.
Leek, J. T., R. B. Scharpf, H. C. Bravo, D. Simcha, B. Langmead, W. E. Johnson, D. Geman, K. Baggerly, and R. A. Irizarry. 2010. Tackling the widespread and critical impact of batch effects in high-throughput data. Nature Reviews Genetics 11(10):733-739.
Liotta L. A., E. F. Petricoin, III, T. D. Veenstra, and T. P. Conrads. 2004. High-resolution serum proteomic patterns for ovarian cancer detection. Endocrine-Related Cancer 11(4): 585-587.
Liotta, L. A., M. Lowenthal, A. Mehta, T. P. Conrads, T. D. Veenstra, D. A. Fishman, and E. F. Petricoin, III. 2005. Importance of communication between producers and consumers of publicly available experimental data. Journal of the National Cancer Institute 97(4):310-314.
Marboe, C. C., M. Billingham, H. Eisen, M. C. Deng, H. Baron, M. Mehra, S. Hunt, J. Wohlgemuth, J. Prentice, and G. Berry. 2005. Nodular endocardial infiltrates (quality lesions) cause significant variability in diagnosis of ISHLT grade 2 and 3A rejection in cardiac allograft recipients. Journal of Heart and Lung Transplant 24(7 Suppl.): S219-226.
Mehra, M., J. Kobashigawa, M. Deng, K. Fang, T. Klingler, P. Lal, S. Rosenberg, P. Uber, R. Starling, and S. Murali. 2007. Transcriptional signals of T-cell and corticosteroid-sensitive genes are associated with future acute cellular rejection in cardiac allografts. Journal of Heart and Lung Transplantation 26(12):1255-1263.
Mehra, M. R., J. A. Kobashigawa, M. C. Deng, K. C. Fang, T. M. Klingler, P. G. Lal, S. Rosenberg, P. A. Uber, R. C. Starling, S. Murali, D. F. Pauly, R. Dedrick, M. G. Walker, A. Zeevi, and H. J. Eisen. 2008. Clinical implications and longitudinal alteration of peripheral blood transcriptional signals indicative of future cardiac allograft rejection. Journal of Heart and Lung Transplantation 27(3):297-301.
Miller, R. W., A. Smith, C. P. DeSimone, L. Seamon, S. Goodrich, I. Podzielinski, L. Sokoll, J. R. van Nagell, Jr., Z. Zhang, and F. R. Ueland. 2011. Performance of the American College of Obstetricians and Gynecologists’ ovarian tumor referral guidelines with a multivariate index assay. Obstetrics & Gynecology 117(6):1298-1306.
Monzon, F. A., M. Lyons-Weiler, L. J. Buturovic, C. T. Rigl, W. D. Henner, C. Sciulli, C. I. Dumur, F. Medeiros, and G. G. Anderson. 2009. Multicenter validation of a 1,550-gene expression profile for identification of tumor tissue of origin. Journal of Clinical Oncology 27(15):2503-2508.
Monzon, F. A., F. Medeiros, M. Lyons-Weiler, and W. D. Henner. 2010. Identification of tissue of origin in carcinoma of unknown primary with a microarray-based gene expression test. Diagnostic Pathology 5(3):10.1186/1746-1596-5-3.
Mook, S., M. K. Schmidt, G. Viale, G. Pruneri, I. Eekhout, A. Floore, A. M. Glas, J. Bogaerts, F. Cardoso, M. J. Piccart-Gebhart, E. T. Rutgers, and L. J. van ‘t Veer. 2008. The 70-gene prognosis-signature predicts disease outcome in breast cancer patients with 1-3 positive lymph nodes in an independent validation study. Breast Cancer Research and Treament 116(2):295-302.
Mulcahy, N. 2010. NCCN Guideline on Occult Cancer Show Immunohistochemistry Is “Rapidly Changing.” http://www.medscape.com/viewarticle/718870 (accessed September 26, 2011).
NCCN (National Comprehensive Cancer Network). 2011a. NCCN Guidelines Version 2.2011 Breast Cancer. http://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed August 30, 2011).
NCCN. 2011b. NCCN Guidelines Version 2.2012 Ovarian Cancer. http://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed December 17, 2011).
NCI (National Cancer Institute). 2006. Personalized Treatment Trial for Breast Cancer Launched. http://www.cancer.gov/newscenter/pressreleases/TAILORxRelease (accessed January 27, 2011).
NCI. 2010a. Phase III Randomized Study of Adjuvant Combination Chemotherapy and Hormonal Therapy Versus Adjuvant Hormonal Therapy Alone in Women with Previously Resected Axillary Node-Negative Breast Cancer with Various Levels of Recurrence (TAILORx Trial). http://www.cancer.gov/clinicaltrials/ECOG-PACCT-1 (accessed January 27, 2011).
NCI. 2010b. TAILORx: Testing Personalized Treatment for Breast Cancer. http://www.cancer.gov/clinicaltrials/noteworthy-trials/tailorx (accessed January 27, 2011).
NCI. 2011. FDA Approval for Trastuzumab. http://www.cancer.gov/cancertopics/druginfo/fda-trastuzumab (accessed September 26, 2011).
Nielsen, H., F. B. Sorensen, B. Nielsen, J. P. Bagger, P. Thayssen, and U. Baandrup. 1993. Reproducibility of the acute rejection diagnosis in human cardiac allografts. The Stanford Classification and the International Grading System. Journal of Heart and Lung Transplantation 12(2):239-243.
Paik, S., J. Bryant, E. Tan-Chiu, E. Romond, W. Hiller, K. Park, A. Brown, G. Yothers, S. Anderson, R. Smith, D. L. Wickerham, and N. Wolmark. 2002. Real-world performance of HER2 testing—National Surgical Adjuvant Breast and Bowel Project experience. Journal of the National Cancer Institute 94(11):852-854.
Paik, S., S. Shak, G. Tang, C. Kim, J. Baker, M. Cronin, R. Baehner, M. Walker, D. W atson, and T. Park. 2003. Multi-Gene RT-PCR Assay for Predicting Recurrence in Node Negative Breast Vancer Patients—NSABP Studies B-20 and B-14: Abstract #16. Paper presented at San Antonio Breast Cancer Symposium, San Antonio, TX.
Paik, S., S. Shak, G. Tang, C. Kim, J. Baker, M. Cronin, F. L. Baehner, M. G. Walker, D. Watson, T. Park, W. Hiller, E. R. Fisher, D. L. Wickerham, J. Bryant, and N. Wolmark. 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New England Journal of Medicine 351(27):2817-2826.
Paik, S., G. Tang, S. Shak, C. Kim, J. Baker, W. Kim, M. Cronin, F. L. Baehner, D. Watson, J. Bryant, J. Costantino, C. E. Geyer, Jr., D. L. Wickerham, and N. Wolmark. 2006. Gene expression and benefit of chemotherapy in node-negative, estrogen receptor-positive breast cancer. Journal of Clinical Oncology 24(23):3726-3734.
Paik, S., C. Kim, and N. Wolmark. 2008. HER2 status and benefit from adjuvant trastuzumab in breast cancer. New England Journal of Medicine 358(13):1409-1411.
Pathwork Diagnostics. 2010. Pathwork Tissue of Origin Test for FFPE Cleared by U.S. Food and Drug Administration. http://www.pathworkdx.com/News/M129_FDA_Clearance_Final.pdf (accessed November 17, 2011).
Pathwork Diagnostics. 2011a. Pathwork Reimbursement Assistance Program (RAP). http://www.pathworkdx.com/patient_information/reimbursement1/ (accessed November 22, 2011).
Pathwork Diagnostics. 2011b. The Pathwork Tissue of Origin Test. http://www.pathworkdx.com/TissueOfOriginTest/IVDKit/ (accessed November 16, 2011).
Pavlidis, N., and Y. Merrouche. 2006. The importance of identifying CUP subsets. In Carcinoma of an Unknown Primary Site, edited by K. Fizazi. New York: Taylor & Francis Group.
Pavlidis, N., E. Briasoulis, J. Hainsworth, and F. A. Greco. 2003. Diagnostic and therapeutic management of cancer of unknown primary. European Journal of Cancer 39(14):1990-2005.
Perez, E. A., M. M. Reinholz, D. W. Hillman, K. S. Tenner, M. J. Schroeder, N. E. Davidson, S. Martino, G. W. Sledge, L. N. Harris, J. R. Gralow, A. C. Dueck, R. P. Ketterling, J. N. Ingle, W. L. Lingle, P. A. Kaufman, D. W. Visscher, and R. B. Jenkins. 2010. HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. Journal of Clinical Oncology 28(28):4307-4315.
Petricoin, E. F., A. M. Ardekani, B. A. Hitt, P. J. Levine, V. A. Fusaro, S. M. Steinberg, G. B. Mills, C. Simone, D. A. Fishman, E. C. Kohn, and L. A. Liotta. 2002. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359(9306): 572-577.
Petricoin, E. F., III, D. A. Fishman, T. P. Conrads, T. D. Veenstra, and L. A. Liotta. 2004. Proteomic pattern diagnostics: Producers and consumers in the era of correlative science. Comment on Sorace and Zhan. BMC Bioinformatics (http://www.biomedcentral.com/1471-2105/4/24/comments).
Pham, M. X., J. J. Teuteberg, A. G. Kfoury, R. C. Starling, M. C. Deng, T. P. Cappola, A. Kao, A. S. Anderson, W. G. Cotts, G. A. Ewald, D. A. Baran, R. C. Bogaev, B. Elashoff, H. Baron, J. Yee, and H. A. Valantine. 2010. Gene-expression profiling for rejection surveillance after cardiac transplantation. New England Journal of Medicine 362(20):1890-1900.
Phillips, K. A., D. A. Marshall, J. S. Haas, E. B. Elkin, S. Y. Liang, M. J. Hassett, I. Ferrusi, J. E. Brock, and S. L. Van Bebber. 2009. Clinical practice patterns and cost effectiveness of human epidermal growth receptor 2 testing strategies in breast cancer patients. Cancer 115(22):5166-5174.
Pillai, R., R. Deeter, C. T. Rigl, J. S. Nystrom, M. H. Miller, L. Buturovic, and W. D. Henner. 2011. Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens. Journal of Molecular Diagnostics 13(1):48-56.
Pollack, A. 2004. New cancer test stirs hope and concern. New York Times. http://www.nytimes.com/2004/02/03/science/new-cancer-test-stirs-hope-and-concern.html?src=pm (accessed November 23, 2011).
Pollack, A. 2008a. Cancer test for women raises hope, and concern. New York Times. http://www.nytimes.com/2008/08/26/health/26ovar.html?pagewanted=all (accessed November 23, 2011).
Pollack, A. 2008b. Sales of test for ovarian cancer halted. New York Times. http://www.nytimes.com/2008/10/25/business/25cancer.html (accessed November 23, 2011).
PR Newswire. 2009. U.S. Food and Drug Administration clears Vermillion’s OVA1(TM) test to determine likelihood of ovarian cancer in women with pelvic mass. http://www.prnewswire.com/news-releases/us-food-and-drug-administration-clears-vermillions-ova1tm-test-to-determine-likelihood-of-ovarian-cancer-in-women-with-pelvic-mass-62150212.html (accessed December 17, 2011).
Quest Diagnostics. 2011. Licenses and Accreditation. http://www.questdiagnostics.com/brand/company/b_comp_licenses.html (accessed November 21, 2011).
Ransohoff, D. F. 2003. Gene-expression signatures in breast cancer. New England Journal of Medicine 348(17):1716.
Ransohoff, D. F. 2004. Rules of evidence for cancer molecular-marker discovery and validation. Nature Reviews Cancer 4(4):309-314.
Ransohoff, D. F. 2005. Lessons from controversy: Ovarian cancer screening and serum proteomics. Journal of the National Cancer Institute 97(4):315-319.
Roche, P. C., V. J. Suman, R. B. Jenkins, N. E. Davidson, S. Martino, P. A. Kaufman, F. K. Addo, B. Murphy, J. N. Ingle, and E. A. Perez. 2002. Concordance between local and central laboratory HER2 testing in the breast intergroup trial N9831. Journal of the National Cancer Institute 94(11):855-857.
Rosenberg, S., M. R. Elashoff, P. Beineke, S. E. Daniels, J. A. Wingrove, W. G. Tingley, P. T. Sager, A. J. Sehnert, M. Yau, W. E. Kraus, L. K. Newby, R. S. Schwartz, S. Voros, S. G. Ellis, N. Tahirkheli, R. Waksman, J. McPherson, A. Lansky, M. E. Winn, N. J. Schork, and E. J. Topol. 2010. Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Annals of Internal Medicine 153(7):425-434.
Sauter, G., J. Lee, J. M. Bartlett, D. J. Slamon, and M. F. Press. 2009. Guidelines for human epidermal growth factor receptor 2 testing: Biologic and methodologic considerations. Journal of Clinical Oncology 27(8):1323-1333.
Schmitt, F. 2009. HER2+ breast cancer: How to evaluate? Advanced Therapeutics 26(Suppl 1):S1-S8.
Shah, S., and B. Chen. 2010. Testing for HER2 in breast cancer: A continuing evolution. Pathology Research International 2011:903202.
Shak, S. 2011. Case Study: Oncotype DX Breast Cancer Assay. Presentation at Meeting 2 of the Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials, Washington, DC, March 30.
Simon, R., M. D. Radmacher, K. Dobbin, and L. M. McShane. 2003. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. Journal of the National Cancer Institute 95(1):14-18.
Simon, R. M., S. Paik, and D. F. Hayes. 2009. Use of archived specimens in evaluation of prognostic and predictive biomarkers. Journal of the National Cancer Institute 101(21):1446-1452.
Sorace, J. M., and M. Zhan. 2003. A data review and re-assessment of ovarian cancer serum proteomic profiling. BMC Bioinformatics 4(24):10.1186/1471-2105-4-24.
Stancel, G. A., D. Coffey, K. Alvarez, M. Halks-Miller, A. Lal, D. Mody, T. Koen, T. Fairley, and F. A. Monzon. 2011. Identification of tissue of origin in body fluid specimens using a gene expression microarray assay. Cancer Cytopathology doi: 10.1002/cncy.20167.
Starling, R. C., M. Pham, H. Valantine, L. Miller, H. Eisen, E. R. Rodriguez, D. O. Taylor, M. H. Yamani, J. Kobashigawa, K. McCurry, C. Marboe, M. R. Mehra, A. Zuckerman, M. C. Deng, and Working Group on Molecular Testing in Cardiac Transplantation. 2006. Molecular testing in the management of cardiac transplant recipients: Initial clinical experience. Journal of Heart and Lung Transplantation 25(12):1389-1395.
SWOG (Southwest Oncology Group). 2011. Spotlight: RxPONDER Trial Will Evaluate Whether Gene Expression Test Can Drive Chemotherapy Choice. http://swog.org/visitors/newsletters/2011/04/index.asp?a=spotlight (accessed May 5, 2011).
Thomas, G. S., S. Voros, J. A. McPherson, A. J. Lansky, F. L. Weiland, S. C. Cheng, S. A. Bloom, H. Salha, M. R. Elashoff, B. O. Brown, H. D. Lieu, A. Johnson, S. E. Daniels, and S. Rosenberg. 2011. The Compass trial (NCT01117506): A prospective multi-center, double-blind study assessing a whole blood gene expression test for the detection of obstructive coronary artery disease In symptomatic patients referred for myocardial perfusion imaging. Abstract presented at American Heart Association Meeting, November 15, 2011.
Ueland, F. R., C. P. Desimone, L. G. Seamon, R. A. Miller, S. Goodrich, L. Podzielinski, L. Sokoll, A. Smith, J. R. van Nagell, and Z. Zhang. 2011. Effectiveness of a multivariate index assay in the preoperative assessment of ovarian tumors. Obstetrics and Gynecology 117(6):1289-1297.
van de Vijver, M. J., Y. D. He, L. J. van ‘t Veer, H. Dai, A. A. M. Hart, D. W. Voskuil, G. J. Schreiber, J. L. Peterse, C. Roberts, M. J. Marton, M. Parrish, D. Atsma, A. Witteven, A. M. Glas, L. Delahaye, T. van der Velde, H. Bartelink, S. Rodenhuis, E. T. Rutgers, S. F. Friend, and R. Bernards. 2002. A gene-expression signature as a predictor of survival in breast cancer. New England Journal of Medicine 347(25):1999-2009.
Van ‘t Veer, L. J., H. Dai, M. J. van de Vijver, Y. D. He, A. A. M. Hart, M. Mao, H. L. Peterse, K. van der Kooy, M. J. Marton, A. T. Wittereveen, G. J. Schreiber, R. M. Kerkoven, C. Roberts, P. S. Linsley, R. Bernards, and S. F. Friend. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(31):530-536.
Vermillion. 2011. Payor Information. http://ova-1.com/resources/payor-information (accessed October 11, 2011).
Visintin, I., Z. Feng, G. Longton, D. C. Ward, A. B. Alvero, Y. Lai, J. Tenthorey, A. Leiser, R. Flores-Saaib, H. Yu, M. Azori, T. Rutherford, P. E. Schwartz, and G. Mor. 2008. Diagnostic markers for early detection of ovarian cancer. Clinical Cancer Research 14(4):1065-1072.
Wagner, L. 2004. A test before its time? FDA stalls distribution process of proteomic test. Journal of the National Cancer Institute 96(7):500-501.
Wingrove, J. A., S. E. Daniels, A. J. Sehnert, W. Tingley, M. R. Elashoff, S. Rosenberg, L. Buellesfeld, E. Grube, L. K. Newby, G. S. Ginsburg, and W. E. Kraus. 2008. Correlation of peripheral-blood gene expression with the extent of coronary artery stenosis. Circulation Cardiovascular Genetics 1:31-38.
Wittner, B. S., D. C. Sgroi, P. D. Ryan, T. J. Bruinsma, A. M. Glas, A. Male, S. Dahiya, K. Habin, R. Bernards, D. A. Haber, L. J. van ‘t Veer, and S. Ramaswamy. 2008. Analysis of the MammaPrint breast cancer assay in a predominantly postmenopausal cohort. Clinical Cancer Research 14(10):2988-2993.
Wolff, A. C., M. E. Hammond, J. N. Schwartz, K. L. Hagerty, D. C. Allred, R. J. Cote, M. Dowsett, P. L. Fitzgibbons, W. M. Hanna, A. Langer, L. M. McShane, S. Paik, M. D. Pegram, E. A. Perez, M. F. Press, A. Rhodes, C. Sturgeon, S. E. Taube, R. Tubbs, G. H. Vance, M. van de Vijver, T. M. Wheeler, and D. F. Hayes. 2007a. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Journal of Clinical Oncology 25(1):118-145.
Wolff, A. C., M. E. Hammond, J. N. Schwartz, K. L. Hagerty, D. C. Allred, R. J. Cote, M. Dowsett, P. L. Fitzgibbons, W. M. Hanna, A. Langer, L. M. McShane, S. Paik, M. D. Pegram, E. A. Perez, M. F. Press, A. Rhodes, C. Sturgeon, S. E. Taube, R. Tubbs, G. H. Vance, M. van de Vijver, T. M. Wheeler, and D. F. Hayes. 2007b. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Archives of Pathology and Laboratory Medicine 131(1):18-43.
Wu, A. H., J. C. Drees, H. Wang, S. R. VandenBerg, A. Lal, W. D. Henner, and R. Pillai. 2010. Gene expression profiles help identify the tissue of origin for metastatic brain cancers. Diagnostic Pathology 5(26):10.1186/1746-1596-5-26.
Zhang, Z., and D. W. Chan. 2010. The road from discovery to clinical diagnostics: Lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiology, Biomarkers, and Prevention 19(12):2995-2999.
Zhang, Z., R. C. Bast Jr., Y. Yu, J. Li, L. J. Sokoll, A. J. Rai, J. M. Rosenzweig, B. Cameron, Y. Y. Wang, X. Y. Meng, A. Berchuck, C. Van Haaften-Day, N. F. Hacker, H. W. de Bruijn, A. G. van der Zee, I. J. Jacobs, E. T. Fung, and D. W. Chan. 2004. Three bio-markers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Research 64(16):5882-5890.
























































