
Important Points Highlighted by the Individual Speakers
• Regulation of genomic diagnostic tests can be a critical factor in the extent of use of those tests and in the competitiveness of companies.
• Standards, quality control, regulatory guidelines, and technology assessments all can facilitate the movement of a test from bench to bedside.
• Any coverage and reimbursement reform should recognize the value of advanced medical diagnostic tests, their impact on health care, and the resources needed to develop and validate them.
• Establishing the value of a test requires that its use be compared to traditional practices.
Four speakers at the workshop addressed the development of genomic diagnostic tests from the perspective of test developers. All pointed to the need to develop better evidence regarding the value of a test, which in turn can affect coverage and reimbursement. They also called for clear regulatory standards to guide test development.
REGULATORY CLARITY IN A COMPETITIVE MARKETPLACE
Quest Diagnostics is one of the world’s leading providers of diagnostic testing, information, and services. The company serves half of U.S. physicians and receives samples from half of U.S. hospitals every day. In addition to providing clinical services using both laboratory developed and FDA approved tests, Quest develops LDTs under CLIA requirements and commercializes in vitro diagnostic kits under FDA oversight. Quest also provides genetic counseling services for physicians, works closely with major pharmaceutical companies to facilitate the introduction of new therapeutics and companion diagnostics, provides electronic health records for health plans and patients, and even has a smart phone app that allows patients to receive their own test results. “We deliver high-impact, high-value, low-cost information to the health care system,” said Nicholas Conti of Quest.
Conti and his colleagues are responsible for evaluating new technologies that will become clinical diagnostic tests to be offered by Quest. They are interested in any field of medicine and in all disease states, since “genomic-based testing impacts all of them.”
Innovation and Competitiveness
Reducing costs and delivering services based on evidence of value are vital to health care in the 21st century, said Conti. Tremendous innovations are occurring not only in the technologies used to discover new biomarkers, but also in their application to clinical practice. In an increasingly global marketplace, delays or friction points involving regulation or reimbursement can compromise a company’s competitive standing and ability to create sustained high-paying jobs.
Improving Rather Than Adding Regulation
Overwrought regulation or limits to physician discretion could stifle innovation and the practice of medicine. Increased regulatory oversight and the application of evidence-based protocols are worthwhile goals, said Conti, but the issues are complex and long-standing. The current system by which LDTs are regulated by CMS under CLIA has yielded many diagnostic innovations that have radically improved the practice of medicine. HIV testing, genetic tests for mutations causing hypertrophic cardiomyopathy, and screening tests for neurological conditions, for example, are all based on LDTs.
The practice of medicine would be dramatically changed if all tests were required to obtain FDA approval, according to Conti. For certain medical specialties, the generation and scope of evidence vary greatly, par-
ticularly where the adoption cycle is quick and the peer-reviewed literature is critical. The testing pipeline in these disciplines would dry up, he said, and many LDTs that are already incorporated into practice guidelines would disappear. For example, with infectious diseases, mutations occur at a rapid pace, which infectious disease specialists use to track and treat disease. In such cases, if companies were required to go through an FDA submission process, by the time approval is gained, the product would no longer be useful.
The incentives for diagnostic companies to develop some of these tests as FDA-approved kits are also lacking, with companies citing too small a market and prohibitive study costs for many of the tests. If a good study for a low-volume test can demonstrate concordance with the published literature, developing that test and bringing it to market is not as costly an endeavor, said Conti. There is a need to balance increased regulation with its affects on medical practice and innovation. “That is really where the value of CLIA comes in.”
The important question to ask is not whether there should be more regulation, but rather is it better regulation? Can duplicative regulatory efforts be eliminated? Can the existing system be improved rather than constructing a new system? “That’s the discussion we need to have,” said Conti.
Protecting Physician Discretion
Limiting the incentive to develop or access innovative tests could hamper advances in patient management, according to Conti. The medical system in the United States is grounded on the concept that physicians are the arbiters of medical care. In the interests of their patients, they will exercise their medical discretion in ordering, interpreting, and delivering diagnostics, therapies, and other forms of care. Reference laboratories such as Quest provide physicians with access to tests for which published research indicates that the test can improve the care of patients. Potential interference with this discretion “should be considered with the greatest of caution.”
Solutions to the Problems
Conti offered several solutions to the problems currently facing the development of genomic diagnostics. First, genomic test development needs clear regulatory certainty. Federal regulation should not be duplicative, as would be the case if FDA had to clear LDTs. Instead, legislation should build on what works by modernizing CLIA. Agency decision making should be transparent, with rulemaking by notice and comment rather than through
guidances. People who are qualified in both the public and private sectors should come together to discuss problems and develop solutions.
Finally, stakeholders should be patient and allow some of the initiatives currently under way to progress. Conti cited recent work by the American Medical Association to develop CPT codes for multivariate tests (AMA, 2011), CMS’s modification to reimbursement procedures for CPT code stacking (CMS, 2012), and the development of the National Institutes of Health (NIH) Genetic Test Registry as examples (NIH, 2010).
IMPROVING THE EFFICIENCY OF TEST DEVELOPMENT
MammaPrint is a multigene index assay that uses the gene activity of a tumor sample to identify the risk of recurrence for an individual breast cancer patient, said Laura van ‘t Veer of the University of California, San Francisco. She drew upon her experience in developing the MammaPrint test to discuss increasing the efficiency of bringing a test from research discovery to clinical use.
The movement of a genomic test from bench to bedside has two important end points, van ‘t Veer said. One is the use of the test in clinical trials and the second is the commercialization of the test. The path from discovery to clinical trials involves discovery, confirmation of research, independent validation, quality assurance, regulatory oversight, and the initiation of a trial with a clinical trial group. To commercialize a test, additional steps are needed which include technology assessment, the development of guideline recommendations, a determination of cost-effectiveness, and agreement by the health care system to reimburse the use of the test.
Standardization of validation protocols will facilitate the efficient development of genomic-based tests, stated van ‘t Veer. She and her colleagues early on began working with clinical trial groups to conduct independent validation using external audits and a predefined statistical protocol in the development of MammaPrint (Buyse et al., 2006). The purpose of the initial validation was to demonstrate the robustness of the risk assessment and to establish a background for the subsequent clinical trial. Many of the data used in the independent validation were separately evaluated by experts in informatics, clinical data, pathology, and statistics. Particularly important are predefined acceptance criteria, which show that a test is being validated and reviewed by external parties. “A lot of the literature that is currently around on validation of genomic tests doesn’t include all the independent steps, and independent review, as we’ve learned over the years, is very crucial.”
A second opportunity for improved efficiency lies in quality control. Quality assessment for clinical trials involves a number of technical features, including precision, reproducibility, repeatability, accuracy, sensitiv
ity, and robustness. Software validation is also critical, van ‘t Veer observed, before a test is used with patients.
Oversight of Genomic Tests
In order to ensure good quality control and regulatory oversight, genomic tests used in clinical trials should be required to obtain an investigational device exemption (IDE) from FDA, said van ‘t Veer, and in the case of companion diagnostics this should be included as part of the Investigational New Drug (IND) application. Institutional review boards (IRBs) also need guidance on how to review genomic tests. “There are large differences between IRBs of different institutions in how they review these tests. Some metrics—what they should look at—should be established.” In cases of local hospital trials, where broader oversight is not available, entities should be established to review genomic tests for clinical trials. Meanwhile, FDA oversight of in vitro diagnostic tests “is working,” according to van ‘t Veer. Postmarket surveillance and medical device reporting create much more standardized reporting around the use of these tests, which is important for patients.
Educational Needs for Decision Making
There is a great need for education about the clinical use and clinical impact of genomic tests. Many require the use of new technology by patients and physicians, making their assessment essential to facilitating proper use and understanding. Even for tests that are currently available, use is not 100 percent. Provision of more information over the Internet or through some other means could help move the field forward, van ‘t Veer said. For reimbursement agencies, cost-effectiveness studies and technology assessments which review logistical processes in hospitals should be included as part of their procedures for gaining information of clinical utility. van ‘t Veer also stressed that guidelines committees and regulatory bodies need to come together and harmonize their definitions of clinical utility, which currently vary from one group to another and between situations.
OVERCOMING OBSTACLES TO TEST DEVELOPMENT
Diagnostic expenditures account for only about 2 percent of health care costs in the United States, but are used to direct 70 percent of clinical decision making (West, 2011), said Russell Enns of Cepheid while speaking on behalf of Advamed Diagnostics. Molecular diagnostics in particular has been the fastest-growing sector of the diagnostics industry and it continues to grow at a faster pace than other areas of traditional laboratory medicine.
Enns discussed the three biggest obstacles that need to be overcome in the development and implementation of these tests.
Establishing Safety and Effectiveness
Novel technologies present great challenges for FDA’s premarket review paradigm, said Enns. What is needed is a modernized, risk-based regulatory approach for all diagnostics that would support public health, encourage innovation, improve the transparency of the FDA decision process, and focus review resources on the products with the highest or most unknown risk.
Enns called particular attention to class II devices, where it has become more and more difficult to grandfather a device to pre-1976 standards. However, Enns has cleared 34 class II 510(k) molecular diagnostic devices through FDA. “My takeaway is that the FDA system does work,” he said. “It has worked for me not only with infectious disease diagnostics, but also for cancer and genetic tests. I would say that it’s been just as successful using the system in breast cancer and bladder cancer.” However, the system needs refinements if test developers are to remain strong in the United States and continue to provide high-paying jobs for researchers, drug developers, and state-of-the-art manufacturing personnel.
In the European Union, the IVD Medical Devices Directive allows most products to be introduced into the market through the self-declaration of a compliance process with standards issued by the International Organization for Standardization. “This system has been in place for about 10 years and has served Europe well,” said Enns. “Perhaps Congress can take a closer look at the EU system to better assess global competition while maintaining product safety and effectiveness standards.” However, Enns cautioned that drug development standards should not be overlaid onto diagnostics products.
Establishing the Value of Diagnostic Tests
Establishing the medical necessity or the value added by a diagnostic test would help overcome what Enns termed “the largest obstacle to [the] successful introduction of new molecular diagnostic tests and platforms”— reimbursement coverage. Payment reform is needed to recognize the value of advanced medical diagnostic tests, their impact on treatment and management decisions for patients, and the resources needed to develop and validate tests. The current reimbursement rates for diagnostic tests are based on an outdated, flawed fee schedule that has not even kept pace with inflation, according to Enns. Inadequate payment affects innovation, as well as patient access to new tests. “It’s much simpler to make major medical
discoveries and advances in medical diagnostics than it is to obtain reasonable and timely reimbursement in coverage decisions.” A legislative solution to address payment reform may be needed to ensure the development of “reliable and transparent procedures open to public review and debate by all stakeholders,” said Enns.
Establishing Performance Standards
Reliable and accurate performance standards and practice guidelines for new genomic tests need to be established, stressed Enns. Patients deserve standardized, consistent test results regardless of where or when tests are performed. “Patients need to be able to go to any cancer institute in this country and [have] a blood sample drawn for an accurate and reliable test.” Many organizations develop performance standards for such attributes as analytical sensitivity and specificity, interference, precision, reproducibility, and clinical sensitivity and specificity. Enns described in particular the Clinical and Laboratory Standards Institute, where he has volunteered for more than 25 years. Over the past two decades, the institute has developed 16 different molecular standards for laboratory medicine, many of which have been officially recognized by FDA as performance standards.
Enns concluded by saying that the United States can keep doing what it has been doing or it can keep up with changes in laboratory medicine by modulating regulatory requirements and professional practice standards. “Since the United States now represents less than 5 percent of the world’s population, we can stick our proverbial heads in the sand and watch the world pass us by. [But] there’s no need for that to happen. . . . Let’s stay competitive, let’s continue to solve disease problems, and let’s further improve the quality of life and the length of productive lives by working together on solutions to these obstacles.”
Cancer kills more than a half-million people in the United States each year (Siegel et al., 2012), yet it is often still treated with products of limited clinical utility and a one-size-fits-all approach, said Steven Shak of Genomic Health. As a result, “we punish the many to benefit the few.” For example, the classic B-20 study of the National Surgical Adjuvant Breast and Bowel Project, which looked at chemotherapy plus tamoxifen versus tamoxifen alone in the treatment of patients with auxiliary lymph node–negative, estrogen receptor–positive breast cancer, showed that only 4 out of every 100 women benefit from chemotherapy (Figure 3-1) (Fisher et al., 1997).
At the end of the 20th century, a new generation of technologies was developed which was used to sequence the human genome. These tech-
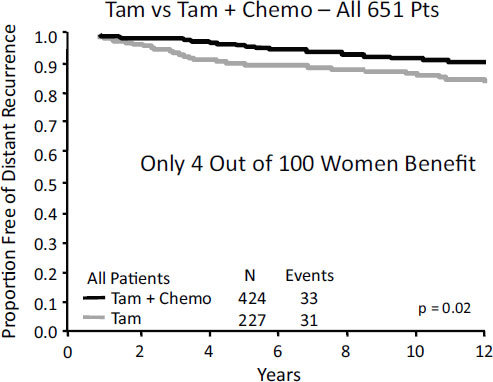
NOTE: Tam, tamoxifen; Chemo, chemotherapy.
SOURCE: As adapted by Shak from Fisher et al., 1997.
niques have revealed the underlying complex biological systems involved in cancer and suggested many new drug candidates. However, efforts to codevelop drugs and diagnostics have not been very productive, Shak observed, with just a few exceptions. The directed use of trastuzumab for metastatic breast cancer patients that test positive for HER2 (Slamon et al., 2001) has saved tens of thousands of lives, but other tests linked to biologic therapies have not emerged from clinical evaluation, said Shak.
Oncotype DX
Genomic Health initiated an effort in 2000 to develop and commercialize molecular diagnostic tests that would empower cancer patients and their physicians to be able to select the right treatment based on the underlying biology and on reliable evidence of clinical utility. “That was a tall goal,” said Shak, but the company’s Oncotype DX breast cancer assay, which was
developed as an LDT under the provisions of CLIA, has now been clinically validated in 13 studies involving more than 4,000 patients and it has been used in more than a quarter million patients since becoming available in 2004. The test is reimbursed in the United States by Medicare and all the major payers, is provided for patients in more than 65 countries, and has been incorporated in published treatment guidelines.
Shak described several key principles on which success in the biomarker field is based. First, a test needs to deliver what patients, physicians, regulators, and payers need and this has to be considered at the very beginning prior to development. Most important, it should be “fit to purpose,” with evidence relevant to that specific purpose. It should give consistent results across multiple, well-designed studies. And the test must be shown to have value beyond traditional measures to all stakeholders. To do that, the test needs to be compared head-to-head with what has been used traditionally, so that comparative effectiveness is built into the strategy from the beginning. “The short version of this is that you need to bring the rigor of drug development to the development of diagnostics, but also fit for purpose,” said Shak.
Shak laid out a roadmap to establish clinical utility that consists of the following steps:
• Definition of purpose
• Technical feasibility
• Development studies
• Analytical methods finalization
• Analytical methods finalization and validation
• Clinical validation studies, including comparative effectiveness
• Treatment decision studies
• Health economic analysis
Shak pointed out that diagnostics can have a major impact on treatment decisions. Seven studies of the Oncotype DX recurrence score in 912 patients showed that treatment decisions changed 30 percent of the time compared with what would have been done without the recurrence score. This is one way of addressing the question asked by Hayes: Is a test being used appropriately?
The second principle Shak listed is that technical innovation needs to be brought to standardized implementation. This requires that all assay methods and procedures be defined prior to clinical validation studies in such areas as specimen eligibility, reagent qualification, instrument validation, controls and calibrators, and linearity, precision, and reproducibility.
CLIA is built on regulations and principles of laboratory medicine “that have been in existence now for decades and really work,” said Shak.
He cited the CLIA-certified reference laboratory process accredited by the College of American Pathologists (CAP) for the 21-gene recurrence score, which uses more than 150 standard operating procedures, 94 forms, and an information technology system that looks at every reagent and ensures that the appropriate quality control is used. “An inspector can come into the Genomic Health [laboratory]—and we’ve had 10 inspections—and say, ‘On March 7 we want you to look at the tenth test that you did that day and then pull out the quality control metrics for every reagent that was used in that particular case.’ We monitor that and can do that.”
The third and final principle Shak listed is that the development of diagnostic tests requires collaborations, clinical research funding, and the skills, processes, resources, and incentives to do it right. “Sometimes the hardest thing isn’t the technology; it’s people.”
One particular obstacle Shak mentioned is the potential need to address all of the payers individually, of which there may be 100 or more in the United States and abroad. Innovative systems need to be developed by payers to gauge the value of the diagnostic in their own system. He described a method implemented by Clalit, which is the largest payer in Israel, to document the value of Oncotype Dx use. Clalit created a simple form that physicians filled out in order to get access to the test, in which they indicated what they would have done without it. “Since they’re the payer, they could collect what was done. They could rapidly document for themselves the impact of the test in their clinical practice.” Another innovative system implemented by CareFirst and Highmark was to pilot a program which provided greater reimbursement at a higher rate for the appropriate use of Oncotype Dx. “We need to be innovative in the way that we think about capturing data in clinical practice … and providing incentives around it,” said Shak.
He also listed obstacles and potential solutions to the development of diagnostic tests, some of which were also described by other speakers:
• There is a knowledge and experience gap among those working in and assessing this new field which needs to be closed through continued education.
• Current incentives have the effect of encouraging individual rather than team science. Leadership, teamwork, and collaboration needs to be incentivized and rewarded.
• Reimbursement is uncertain, which points to the continued need to move to pricing based on value.
• Regulation is uncertain, which points to the need for regulations that are fit to purpose and suitable for continued rapid introductions of new and improved tests.
Shak concluded by observing that his company has been working in recent years on next-generation sequencing technologies that have made it possible to investigate not just candidate genes but the entire transcriptome. “I never would have dreamed in my lifetime that it’s now possible to see … over 50 million reads from a single sample. How do we analyze that data? What are the bioinformatics? How do we apply and harness this technology? The principles that we’re talking about here are what is needed to make it possible to actually bring these advances in a responsible way to patients.”












