
4
Chloroform1
Acute Exposure Guideline Levels
PREFACE
Under the authority of the Federal Advisory Committee Act (FACA) P.L. 92-463 of 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous Substances (NAC/AEGL Committee) has been established to identify, review, and interpret relevant toxicologic and other scientific data and develop AEGLs for high-priority, acutely toxic chemicals.
AEGLs represent threshold exposure limits for the general public and are applicable to emergency exposure periods ranging from 10 minute (min) to 8 hour (h). Three levels—AEGL-1, AEGL-2, and AEGL-3—are developed for each of five exposure periods (10 and 30 min and 1, 4, and 8 h) and are distinguished by varying degrees of severity of toxic effects. The three AEGLs are defined as follows:
AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general population, including susceptible individuals, could experience notable discomfort, irritation, or certain asymptomatic, nonsensory
![]()
1 This document was prepared by the AEGL Development Team composed of Robert Young (Oak Ridge National Laboratory), Gary Diamond (Syracuse Research Corporation), Chemical Manager Steven Barbee (National Advisory Committee [NAC] on Acute Exposure Guideline Levels for Hazardous Substances), and Ernest V. Falke (U.S. Environmental Protection Agency). The NAC reviewed and revised the document and AEGLs as deemed necessary. Both the document and the AEGL values were then reviewed by the National Research Council (NRC) Committee on Acute Exposure Guideline Levels. The NRC committee has concluded that the AEGLs developed in this document are scientifically valid conclusions based on the data reviewed by the NRC and are consistent with the NRC guidelines reports (NRC 1993, 2001).
effects. However, the effects are not disabling and are transient and reversible upon cessation of exposure.
AEGL-2 is the airborne concentration (expressed as ppm or mg/m3) of a substance above which it is predicted that the general population, including susceptible individuals, could experience irreversible or other serious, long-lasting adverse health effects or an impaired ability to escape.
AEGL-3 is the airborne concentration (expressed as ppm or mg/m3) of a substance above which it is predicted that the general population, including susceptible individuals, could experience life-threatening health effects or death.
Airborne concentrations below the AEGL-1 represent exposure concentrations that could produce mild and progressively increasing but transient and nondisabling odor, taste, and sensory irritation or certain asymptomatic, nonsensory effects. With increasing airborne concentrations above each AEGL, there is a progressive increase in the likelihood of occurrence and the severity of effects described for each corresponding AEGL. Although the AEGL values represent threshold concentrations for the general public, including susceptible subpopulations, such as infants, children, the elderly, persons with asthma, and those with other illnesses, it is recognized that individuals, subject to idiosyncratic responses, could experience the effects described at concentrations below the corresponding AEGL.
SUMMARY
Chloroform is a volatile liquid with a pleasant, nonirritating odor. The chemical is miscible with organic solvents but is only slightly soluble in water. Chloroform is produced and imported in large quantities for use in chemical syntheses, as a solvent, and in the manufacture of some plastics. It was used in the past as an anesthetic and in pharmaceutical preparations, but such uses are no longer allowed in the United States.
Human data on acute exposure to chloroform are from older studies that tested various exposure regimens (680-7,200 ppm for 3-30 min); effects included detection of a strong odor, headaches, dizziness, and vertigo. Published reports of surgical patients anesthetized with chloroform lack precise exposure details, but suggest that exposure to high concentrations (generally greater than 13,000 ppm) might produce cardiac arrhythmias and transient hepatic and renal toxicity. Quantitative data on human fatalities after acute inhalation exposure to chloroform were not available.
Only a few animal studies on the lethality from acute exposure to chloroform were available. Quantitative data include a 4-h LC50 (lethal concentration, 50% lethality) of 9,780 ppm in rats and a 7-h LC50 of 5,687 ppm in mice. Other data indicate notable lethality after exposures ranging from 5 min at “saturated” concentration (approximately 25,000 ppm) to 12 h at 726 ppm. Nonlethal effects of chloroform in laboratory animals include biochemical (elevated serumenzyme
activity) and histopathologic indices of hepatic toxicity. Data on the reproductive and developmental toxicity of chloroform in animals are equivocal. One study reported evidence of fetotoxicity in rats after gestational exposure to chloroform at 30 ppm, but another study found no evidence of such toxicity with gestational exposures at 2,232 ppm.
There are no inhalation exposure studies demonstrating carcinogenic responses to chloroform, but oral exposure has been shown to cause tumors in rats (kidney tumors in male) and mice (hepatocarcinomas in male and female mice). Data on the mechanism of toxicity and tumorigenicity of chloroform suggest that the tumorigenic response occurs at concentrations that cause cell death and proliferative cellular regeneration. Thus, a linear low-dose extrapolation for cancer risk might not be appropriate. For this reason and because the inhalation slope factor for chloroform is based on oral-exposure studies, the AEGL values for chloroform are based on noncarcinogenic effects.
Metabolism and disposition studies have affirmed that metabolism of chloroform to phosgene is mediated by the enzyme cytochrome P-450 IIE1, and that phosgene along with the depletion of glutathione and the formation of trichlorocarbon-radical intermediates are responsible for the toxicity of chloroform. Data from several studies indicate that the metabolism and, therefore, the rate of production of reactive metabolites are greater in rodents than in humans.
AEGL-1 values for chloroform were not recommended. Attempts to identify a critical effect consistent with the AEGL-1 definition were considered tenuous and uncertain. Exposures of humans to chloroform at concentrations approaching those inducing narcosis or possibly causing hepatic and renal effects (AEGL-2 effects) are not accompanied by overt signs or symptoms. Furthermore, chloroform is not irritating and its odor is not unpleasant.
AEGL-2 values for chloroform were based on embryotoxicity and fetotoxicity observed in rats when dams were exposed to chloroform at 100 ppm for 7 h/day on gestation days 6-15 (Schwetz et al. 1974). An assumption was made that the effects could be caused by a single 7-h exposure. Because data on the metabolism and kinetics of chloroform indicate that rodents are more sensitive than humans to the toxic effects of chloroform, an uncertainty factor for interspecies differences was not applied. An intraspecies uncertainty factor of 3 was applied to account for variability in metabolism and disposition among individuals and to protect more susceptible individuals (e.g., individual exposed to other inducers of P-450 monooxygenase, such as alcohol). Additional reduction of the AEGL-2 values was not warranted because the critical effect and the assumption of a single-exposure scenario provided a conservative point of departure. The concentration-time relationship for many irritant and systemically acting vapors and gases may be described by the equation Cn × t = k, where the exponent n ranges from 0.8 to 3.5 (ten Berge et al. 1986). In the absence of data with which to empirically derive a chloroform-specific scaling exponent (n), temporal scaling was performed using default values of n = 3 when extrapolating to shorterexposure durations or n = 1 when extrapolating to longer-exposure durations.
AEGL-3 values for chloroform were based on a mouse 560-min LCt50 of 4,500 ppm. Because no data were available for estimating a lethality threshold, the LC50 was reduced by a factor of 3 to 1,500 ppm, a concentration unlikely to cause lethality based on comparisons with other human and animal data. An uncertainty factor of 3 to protect sensitive individuals was applied. As with the AEGL-2 derivations, an intraspecies uncertainty factor of 3 was selected because it is unlikely that induction of metabolism would increase toxic effects by an order of magnitude. Rodents appear to metabolize chloroform at a greater rate than humans, resulting in the production of reactive, toxic intermediates at a greater rate. Results of physiologically-based pharmacokinetic (PBPK) model studies have shown that rodents, especially mice, are considerably more susceptible to the lethal effects of chloroform than humans. Therefore, the AEGL-3 values were increased 3-fold by a weight-of-evidence adjustment factor of 1/3. This adjustment is further justified by data on the use of chloroform as a surgical anesthesia, which show that cumulative exposures to chloroform at concentrations >675,000 ppm/min or at 22,500 ppm for up to 120 min resulted in surgical anesthesia and cardiac irregularities but not death. Time scaling was performed using n = 3 to extrapolate from the 560-min duration (the point of departure) to the shorter AEGL-time periods. To minimize uncertainties with extrapolating a 560-min exposure to a 10-min exposure, the 30-min AEGL-3 value of 4,000 ppm was adopted for the 10-min AEGL value.
Carcinogenic potential after a single, acute exposure to chloroform was assessed, and AEGLs values calculated. AEGL-2 values based on noncancer toxicity were slightly greater than those based on cancer risks. However, the carcinogenic response to chloroform appears to be a function of necrosis and subsequent regenerative cellular proliferation, which are not relevant to a single acute exposure.
TABLE 4-1 Summary of AEGL Values for Chloroform
| Classification | 10 min | 30 min | 1 h | 4 h | 8 h | End Point (Reference) |
| AEGL-1 (nondisabling) | NRa | NRa | NRa | NRa | NRa | |
| AEGL-2 (disabling) | 120 ppm (580 mg/m3) | 80 ppm (390 mg/m3) | 64 ppm (312 mg/m3) | 40 ppm (195 mg/m3) | 29 ppm (141 mg/m3) | Embryotoxicity and fetotoxicity in rats exposed for 7 h/day on gestation days 6-15 (Schwetz et al. 1974); single exposure assumed. |
| AEGL-3 (lethal) | 4,000 ppm (19,000 mg/m3) | 4,000 ppm (19,000 mg/m3) | 3,200 ppm (16,000 mg/m3) | 2,000 ppm (9,700 mg/m3) | 1,600 ppm (7,800 mg/m3) | Estimated lethality threshold for mice; 3-fold reduction of 560-min LC50 of 4,500 ppm to 1,500 ppm (Gehring 1968). |
a Not recommended; data were insufficient to develop AEGL-1 values and AEGL-1 effects unlikely to occur in the absence of notable toxicity.
1. INTRODUCTION
Chloroform is a volatile liquid with a pleasant, nonirritating odor. The chemical is miscible with organic solvents but is only slightly soluble in water. Chloroform is produced and imported in large quantities (93-350 million pounds/year) and used in chemical syntheses, for refrigeration, as a solvent, and in the manufacture of polytetrafluoroethylene plastics (DeShon 1978; Li et al. 1993). It was used in the past as an anesthetic and in pharmaceutical preparations, but such uses are no longer allowed in the United States. Chloroform is also a byproduct of wood-pulp chlorination for production of paper products. Chemical and physical data on chloroform are presented in Table 4-2.
AIHA (1989) reported an odor threshold for chloroform of 192 ppm based on the geometric mean of acceptable values (133-276 ppm). An odor detection concentration of 6.1 ppm was reported by EPA (1992).
2. HUMAN TOXICITY DATA
2.1. Acute Lethality
Quantitative data on acute inhalation exposures to chloroform resulting in death were not available.
TABLE 4-2 Chemical and Physical Data for Chloroform
| Parameter | Value | Reference | ||
| Synonyms | Trichloromethane; methenyl chloride; methyl trichloride | DeShon 1978 | ||
| CAS registry no. | 67-66-3 | Budavari et al. 1996 | ||
| Chemical formula | CHCl3 | Budavari et al. 1996 | ||
| Molecular weight | 119.39 | Budavari et al. 1996 | ||
| Physical state | Liquid | Budavari et al. 1996 | ||
| Vapor pressure | 159.6 mm Hg at 20°C | DeShon 1978 | ||
| Density | 1.484 at 20°C | Budavari et al. 1996 | ||
| Boiling/melting point | 61-62°C/-63.5°C | Budavari et al. 1996 | ||
| Solubility | 1 mL/200 mL water at 20°C | Budavari et al. 1996 | ||
| Conversion factors in air | 1 ppm = 4.88 mg/m3 1 mg/m3 = 0.21 ppm |
NIOSH 2011 | ||
2.2. Nonlethal Toxicity
Several reports are available to qualitatively characterize the human health effects from acute inhalation exposure to chloroform. Hutchens and Küng (1985) reported nausea, appetite loss, transitory jaundice, cardiac arrhythmias, arterial hypotension, mild intravascular hemolysis, and unconsciousness in an individual after intentional, nonsuicidal inhalation of chloroform.
Lehmann and Hasegawa (1910) conducted controlled exposure studies on human subjects. The results of this study showed that a 3-min exposure to chloroform at 920 ppm induced vertigo and dizziness and a 30-min exposure at 680 ppm produced a moderately strong odor. A 30-min exposure at 1,400 ppm produced lightheadedness, giddiness, lassitude, and headache; at 3,000 ppm, gagging and pounding of the heart occurred. Chloroform at 4,300-5,100 ppm for 20 min or at 7,200 ppm for 15 min produced light intoxication and dizziness. These data appeared to be from only three subjects, and the methods of exposure and measurements were unavailable. The signs and symptoms of exposure described in this report appear to be consistent with early stages of narcosis.
Lehmann and Flury (1943) reported that chloroform at 389 ppm for 30 min was tolerated in humans without complaint, but that a concentration of 1,030 ppm caused dizziness, intracranial pressure, and nausea within 7 min and headache that persisted for several hours.
Whitaker and Jones (1965) analyzed the clinical effects of chloroform anesthesia in 1,502 surgery patients. Although the duration of anesthesia varied from <30 min to over 2 h, chloroform concentration never exceeded 2.25% (22,500 ppm). For most of the cases (1,164), anesthesia was for less than 30 min. Clinical observations included tachypnea, bradycardia, cardiac arrhythmias, hypotension, one case of transient jaundice, and one death (this case was complicated by renal insufficiency and could not necessarily be attributed to chloroform). The duration required to attain anesthesia was not specified, but it probably occurred within a few minutes. These observations demonstrate that a short exposure to chloroform at 22,500 ppm will induce a surgical plane of anesthesia concurrent with various physiologic responses.
The clinical effects associated with chloroform-induced anesthesia were also studied by Smith et al. (1973). However, the use of these data for AEGL development is compromised by confounders, including the use of premedication with diazepam and pentobarbital or with hydroxyzine and pentobarbital. The concentration of chloroform inspired appeared to vary between 0.85% (8,500 ppm) and 1.3% (13,000 ppm), and the average duration of anesthesia was 112.0 ± 60.38 min among the 58 surgical patients. Forty-six percent of the patients receiving chloroform experienced nausea and vomiting. Clinical assessment of liver function and toxicity indicated transient alterations. One ventricular tachycardia occurred that necessitated pharmacologic correction. Data from a single patient indicated that chloroform at 8,500 ppm would induce anesthesia.
McDonald and Vire (1992) examined the possible health hazards associated with chloroform use in endodontic procedures. Two industrial hygiene monitors were used to sample the air in the treatment operatory and additional sampling devices were attached to the dentist and the dental assistant. The concentrations of chloroform in the operatory area samples were <0.57 ppm for a 5.5-h period, and concentrations in individual air samples were <0.88 ppm over a 150-min period. Health screening tests of the dentist and assistant revealed no signs of liver, kidney, or lung damage 5 h postexposure or 1 year after the study.
Although specific data were not presented, Snyder and Andrews (1996) reported that humans might tolerate chloroform at up to 400 ppm for 30 min without complaint, but might experience dizziness and gastrointestinal upset at 1,000 ppm for 7 min and narcosis at 14,000 ppm (no duration specified).
2.2.1. Epidemiologic Studies
Several epidemiologic studies on occupational exposure to chloroform have been conducted. These studies involve worker populations exposed to chloroform for periods of time in excess of what would be considered acute exposure, and are not directly applicable to developing AEGL values. They do, however, provide some insight regarding the relationship between the AEGL values and health effects that might be associated with long-term exposures.
Challen et al. (1958) evaluated workers in a pharmaceutical manufacturing process that involved exposure to chloroform vapor. The exposure groups were described as eight “long-service operators” (3-to 10-year exposures) exposed at 77-237 ppm; nine “short-service operators” (10-to 24-month exposures), who were replacements for the long-service operators and were exposed at 23-71 ppm; and five controls, who were not exposed to processes involving chloroform. All of the workers were women whose ages ranged from 34 to 60 years. Some long-service operators had been observed staggering about the work area. All long-service workers experienced alimentary effects (e.g., nausea, flatulence, thirst), increased micturition and urinary discomfort, and behavioral effects (e.g., depression, irritability, poor concentration ability, motor deficiencies) during employment. All experienced nausea and stomach upset on smelling chloroform after leaving their employment. Two of nine short-service operators reported no effects from chloroform exposure, five reported dryness of the mouth and throat while at work, two had similar experiences as the long-service operators, and several reported lassitude.
Bomski et al. (1967) studied workers in a Polish pharmaceutical factory and gave special emphasis to chloroform-induced susceptibility to viral infection. Chloroform exposures were 2-205 ppm, but the frequency of sampling was not specified. The incidence of viral hepatitis was greater in chloroform-exposed workers than in nonexposed subjects, so the authors postulated that chloroforminduced hepatic damage might have predisposed the workers to the viral infection.
Increased incidences of spleen and liver enlargement were also found in the chloroform-exposed workers.
Li et al. (1993) conducted surveys of chloroform-producing facilities in Shanghai, China. Most of the workers exposed to chloroform were involved in the production of perspex (polymethylmethacrylate) and chemical synthesis. In the three facilities sampled (where no effective preventive equipment or measures were in place), chloroform concentrations were 4.27-147.91 mg/m3 (0.88-31.06 ppm), with a geometric mean of 21.38 mg/m3 (4.49 ppm) for 119 samples. Chloroform concentrations were <20 mg/m3 (4.20 ppm) in 45.5% of the samples. Exposure groups were classified as Exposure I (13.49 mg/m3 [2.83 ppm]; 1-15 years of exposure) and Exposure 2 (29.51 mg/m3 [6.20 ppm]; 1-15 years of exposure). The exposure groups and control group (no obvious chloroform or other hazardous exposures) included males and females as well as smokers and nonsmokers; all groups had an average age of approximately 36 years. The investigators concluded that long-term exposure to chloroform at 29.51 mg/m3 (6.20 ppm) resulted in functional liver damage, as determined by changes in various serum enzymes (alanine aminotransferase [ALT], gamma-glutamyl transferase, and adenosine deaminase), prealbumin, serum transferrin, and blood urea nitrogen.
2.3. Reproductive and Developmental Toxicity
Wennborg et al. (2000) studied a cohort of Swedish women who had worked in laboratory or nonlaboratory jobs for 1 year or more in 1990-1994. The investigators obtained data from questionnaires sent to 763 women (66 were excluded for various reasons) that assessed reproductive history, health status, time-to-pregnancy, personal habits, specific work, and exposure to various agents and specific times at which those exposures occurred. The data were compared with respective birth information from the Swedish Medical Register. Parameters examined included spontaneous abortion, birth weight, preterm delivery, small-for-gestation age, large-for-gestation age, and congenital deformities. A number of confounding variables were considered (e.g., high blood pressure, smoking, gynecologic and chronic disease, sexually transmitted infectious diseases, father’s work and potential exposures during time of conception, previous abortions). Information about consumption of alcohol, tea, and coffee and stress levels was not included. The analysis included 869 pregnancies but did not involve specific-exposure concentrations, and did not account for exposures to other chemicals. There was no association between laboratory work and spontaneous abortions. A weak association between women who had worked with chloroform before conceiving and spontaneous abortions was found, but there was no significant association between chloroform exposure and small-forgestational age or body weight.
2.4. Genotoxicity
No studies were found on the genotoxicity of chloroform in humans.
2.5. Carcinogenicity
Although epidemiology studies have been conducted to assess the carcinogenic potential of chloroform in drinking water, no studies are available on the carcinogenic potential of chloroform in humans following inhalation exposure. In 1987, EPA (2012) developed an inhalation slope factor of 6.1 × 10-3 per mg/kg/day based on an increased incidence of renal tumors in male rats after long-term exposure to chloroform in drinking water (Jorgenson et al. 1985). Route-to-route extrapolation was required for calculating the slope factor because inhalation data were not available.
2.6. Summary
Quantitative data on human lethality after acute exposure to chloroform are unavailable. Although they lack quantitative data and often pertain to oral exposures, clinical reports affirm the hepatotoxicity and renal toxicity of chloroform, as well as its neurologic effects. The available data on nonlethal responses indicate that acute inhalation of chloroform might result in narcosis and might be preceded by signs and symptoms characteristic of early stages of anesthesia. Early reports in which the effects of chloroform inhalation were observed in human subjects have uncertainties related to the concentration measurements but do provide information on the human experience that does not appear to be inconsistent with other data. A summary of data relevant to acute, nonlethal exposure of humans to chloroform is presented in Table 4-3.
3. ANIMAL TOXICITY DATA
3.1. Lethal Toxicity
3.1.1. Rats
Results of preliminary range-finding experiments for a large number of chemicals were reported by Smyth et al. (1962). Chloroform vapor (concentration not specified but presumably a saturated concentration of approximately 25,000 ppm) killed all 6 of the albino rats (strain not specified) exposed for 5 min. A 4-h exposure at 8,000 ppm (nominal concentration; no analytical determination) killed 5 of 6 albino rats.
TABLE 4-3 Nonlethal Effects of Chloroform in Humans after Acute Inhalation Exposure
| Number of Subjects | Concentration, ppm | Duration, min | Effect | Reference |
| 3 | 920 | 3 | Vertigo | Lehmann and Hasegawa 1910 |
| 3 | 680 | 30 | Strong odor | Lehmann and Hasegawa 1910 |
| 3 | 1,400 | 30 | Light headedness, lassitude, headache | Lehmann and Hasegawa 1910 |
| 3 | 3,000 | 30 | Pounding heart, gagging | Lehmann and Hasegawa 1910 |
| NA | 4,300-5,100 | 20 | Intoxication, dizziness | Lehmann and Hasegawa 1910 |
| NA | 7,200 | 15 | Intoxication, dizziness | Lehmann and Hasegawa 1910 |
| NA | 389 | 30 | No complaints | Lehmann and Flury 1943 |
| NA | 1,030 | 7 | Dizziness, intracranial pressure, nausea, persistent headache | Lehmann and Flury 1943 |
| 1,502 | 22,500 | <30 - >120 (most <30) | Surgical anesthesia, cardiac irregularities | Whitaker and Jones 1965 |
| 58 | 8,500-13,000 | 113 (mean duration) | Surgical anesthesia | Smith etal. 1973 |
| 2 | <0.5 | 330 | No effectsa | McDonald and Vire 1992 |
| 2 | <0.88 | 150 | No effectsa | McDonald and Vire 1992 |
a Health screening conducted at 5 h postexposure and at one year after exposure. Abbreviations: NA, not available
The results of an inhalation study in rats were briefly described in report to E. I. du Pont de Nemours and Co. (Haskell Laboratory 1964). The study, designed to assess the toxicity of Freon TC® and Freon-113®, also included experiments with chloroform (a component of Freon TC®). Mortality in rats (sex and strain not specified) exposed to chloroform at concentrations of 5,000, 3,700, or 3,000 ppm for 4 h was 3/4, 3/4, and 0/4, respectively. Deaths occurred 2-3 days after exposure; the four rats in the 3,000-ppm group were killed 14-days postexposure. No information was provided about the methods for measuring chloroform concentrations (atmosphere produced by heating chloroform and injection into the chamber via a nebulizer); only nominal exposure concentrations were reported. No histopathology data were provided on the chloroform-treated rats.
In experiments of the effect of chloroform on barbiturate metabolism and narcosis, Puri et al. (1971) exposed male Sprague-Dawley rats at 726 ppm for up to 48 h (continuous exposure). One group of rats was exposed to chloroform alone. On the basis data presented in graphs, continuous 12-h exposure to chloroform resulted in at least 10 deaths. It is unclear if any deaths occurred before 12 h.
Lundberg et al. (1986) reported a 4-h LC50 of 47,702 mg/m3 (9,780 ppm) for female Sprague-Dawley rats. Groups of 10 rats were exposed to a series of chloroform concentrations (specific-exposure concentrations for the series were not provided but were reported as being equivalent to 1/2, 1/4, 1/8, 1/16, or 1/32 of the LC50 or the saturation concentration). Mortality was determined 24 h after exposure. The exposure concentrations were measured by infrared detection in a suitably designed apparatus.
3.1.2. Mice
The results of studies with mice exposed to chloroform were reported by Fühner (1923). Groups of mice (sex and strain not reported; 30 mice total) were exposed to chloroform at 12-38 mg/L (2,458-7,782 ppm). Each mouse was exposed in a 10-L bottle in which chloroform was vaporized to achieve the desired concentration. Concentrations were not determined analytically. Five mice exposed at 2,458-5,120 ppm exhibited reflex loss after 48-215 min, but no deaths occurred. Exposure at 4,710-5,529 ppm resulted in reflex loss after 30-90 min; 12 of 18 animals recovered and 6 died. Deaths occurred within 71-175 min of exposure. Six of seven mice exposed to chloroform at 6,758-7,782 ppm exhibited reflex loss after 13-46 min and one mouse died after a 35-min exposure (reflex loss occurred at 8 min). The absence of validated exposure concentrations limits the quantitative validity of these data. Four additional mice were exposed at 5,585 ppm for 120 or 135 min. For the three mice exposed for 120 min, death occurred 105, 130, and 140 min after the start of exposure, and the one mouse exposed for 135 min died 95 min after exposure. Under the conditions of these experiments, the findings suggest that exposure concentrations in the vicinity of 4,710 ppm might represent a lethal threshold for mice after 1-2 h of exposure.
A 7-h LC50 of 5,687 ppm for mice was reported by von Oettingen et al. (1949). These experiments used 20 adult white mice (strain and sex not specified) exposed to chloroform in a bell jar. Chloroform concentrations were calculated on the basis of the amount of test material volatilized over time and the volume of air passed through the chamber. The concentrations were also determined by chemical analysis. The graphic presentation of the experimental results indicated an LC30 of 5,529 ppm and an LC90 of 6,963 ppm. At the concentrations tested (4,915-7,372 ppm), the mice exhibited progressive central nervous system depression followed by rapidly occurring narcosis. Deaths started occurring after 3-5 h.
In a study by Deringer et al. (1953), the nephrotoxic and lethal effects of inhaled chloroform were examined using male and female C3H mice. Groups of 2-month old mice (six of each sex) were exposed to chloroform at concentrations of 3.38-5.4 mg/L (693-1,106 ppm) for 1, 2, or 3 h. Groups of 8-month old mice (six of each sex) were also exposed similarly. Twenty-two male and 20 female mice served as untreated controls. Mice were observed daily for deaths or morbidity, and were examined weekly for tumors or other abnormal conditions. Necropsies were performed on all moribund or dead mice and any female mice with mammary tumors. Regardless of the exposure duration or concentration, all of the male mice (except one) exposed to chloroform exhibited evidence of kidney damage. Within 11 days after exposure, 15 of 18 8-month old males and 7 of 18 2-month old males died. The remainder of the 8-month old males survived 5-7 months, and the remainder of the 2-month old male mice survived 14-18 months. Generally, the deaths occurred earlier in mice exposed for 2-3 h than in those exposed for only 1 h; specific data, however, were not provided. Histologic findings in mice that died included necrosis and calcification of the proximal and distal convoluted tubules of the kidney. Necrosis appeared to be more severe with earlier deaths. Additionally, hepatic necrosis was also observed in mice exposed at 942-1,106 ppm that died within 6 days. For male mice surviving longer and in all female mice, hepatic damage was not notable. The results of this study show that a 3-h exposure of male C3H male mice to chloroform at 692 ppm or a 1-h exposure at 921 ppm resulted in severe renal damage and death.
The influence of sex-hormone status on gender-specific chloroforminduced nephrotoxicity in mice was studied by Culliford and Hewitt (1957). Although the primary objective of the study was to verify the influence of androgens on chloroform-induced nephrotoxicity, the initial results of the study provided evidence of nearly complete tubular necrosis in two strains of male mice after a 2-h inhalation exposure. Male Westminster Hospital (in-house, uniform heterozygous) mice exposed to chloroform at 3.3-7.0 mg/L (676-1,434 ppm) and male CBA mice exposed at 1.2-5 mg/L (246-1,024 ppm) all exhibited complete tubular necrosis 24 h after exposure. Female mice of these strains did not exhibit any evidence of renal damage. The study also showed that administration of estrogen to male mice abolished the susceptibility to the nephrotoxic response, and that the administration of testosterone to female mice increased susceptibility. The chloroform concentrations were calculated on the basis of the amount of chloroform added to the 6-L exposure chamber, and the assumption of complete vaporization at 80°F and uniform dispersal. No analytic measurements were made, thereby imparting some uncertainty about the chamber concentrations.
In studying the hepatotoxicity of chlorinated hydrocarbons, Gehring (1968) calculated a 4,500-ppm LCt50 for chloroform of 560 min (540-585 min, 95% confidence interval [CI]) for female Swiss-Webster mice, a 4,500-ppm ECt50 of 35 min (31.0-39.6 min, 95% CI) for narcosis, and a 4,500-ppm ECt50 of
2.3 min (1.9-2.8 min, 95% CI) for increased serum glutamic pyruvic transaminase (SGPT) activity. Groups of mice (10/group for narcosis determination and 20/group for lethality determination) were exposed to chloroform at 4,500 ppm. The control group consisted of 254 mice representing a composite group of controls for all of the chlorinated hydrocarbons tested. Chloroform concentrations were attained by metering it into a heated tube for vaporization. Actual concentrations were measured by continuous flow of the atmosphere through an infrared spectrophotometric cell. The experiment was repeated if the chloroform concentration varied by more than 7%. Mortality at 4,500-ppm ranged from approximately 5% after 400 min to 80% after 700 min. The exposure-response relationship for narcosis exhibited the same slope. These data suggest that, at a chloroform concentration of 4,500 ppm, there is approximately a 16-fold difference between the time-to-narcosis (35 min) and the time-to-death (560 min) for mice exposed under the conditions of this study. Increased SGPT was also reported but exhibited a notably different exposure-response relationship (see Section 3.2.2.).
3.1.3. Dogs
The effect of chloroform-induced anesthesia in dogs was studied by Whipple and Sperry (1909). Details of the exposure concentrations are limited to notation of the amount of chloroform (in ounces) used on each dog. Anesthesia duration varied from 1.5 to 2.5 h and chloroform amounts varied from <1 to 3 ounces. Some of the dogs died. It was not possible to ascertain a definitive doseresponse relationship from the data.
von Oettingen et al. (1949) studied the effects of chloroform (15,000 ppm nominal; 14,376 ppm determined) in 10 beagles (sex not specified) that had been surgically prepared with a tracheal cannula and carotid-and femoral-artery cannulae to which pressure transducers were attached. After recovering from the pentothal-induced surgical anesthesia (beginning of voluntary movement and “lively” reflex), the dogs were exposed continuously to the chloroform. The average survival time was 202 min with a range of 60-285 min.
3.1.4. Summary of Lethal Toxicity in Animals
The lethality of inhaled chloroform in laboratory animals is summarized in Table 4-4. With the exception of a rat 4-h LC50 value (9,780 ppm) reported by Lundberg et al. (1986) and a mouse LCt50 (4,500 ppm; 560 min) reported by Gehring (1968), the data are more qualitative in nature. Data from older studies lack details about the generation and measurement of exposure atmospheres of chloroform. The available data do not present a clear delineation of the lethality of acute inhalation exposure to chloroform.
TABLE 4-4 Lethal Toxicity of Chloroform in Laboratory Animals after Acute Inhalation Exposure
| Species | Exposure Concentration (ppm) | Exposure Duration (min) | Effect | Reference |
| Rat | 9,780 | 240 | 4-h LC50a | Lundberg et al. 1986 |
| Rat | 3,000 | 240 | 100% mortality | Haskell Laboratory 1964 |
| Rat | 3,700 | 240 | 75% mortalityb | Haskell Laboratory 1964 |
| Rat | 5,000 | 240 | 75% mortalityb | Haskell Laboratory 1964 |
| Rat | 8,000 | 240 | ≈80% mortality | Smyth etal. 1962 |
| Rat | “Saturated concentration” | 5 | 100% mortality | Smyth et al. 1962 |
| Rat | 726 | 720 | Lethality (no specifics provided) | Purietal. 1971 |
| Mouse | 5,529 | 420 | 7-h LC30 | von Oettingen et al. 1949 |
| Mouse | 5,687 | 420 | 7-h LC50 | von Oettingen et al. 1949 |
| Mouse | 6,963 | 420 | 7-h LC90 | von Oettingen et al. 1949 |
| Mouse | 4,710-5,529 | 71-175 | 66% mortality | Fuhner 1923 |
| Mouse | 6,758-7,782 | 35 | 14% mortality | Fuhner 1923 |
| Mouse | 2,458-5,120 | 48-215 | No deaths | Fuhner 1923 |
| Mouse | 5,585 | 120 | 75% mortalityc | Fuhner 1923 |
| Mouse | 4,500 | 560 min | LCt50 | Gehring 1968 |
a Mortality occurred 24-h postexposure.
b Deaths determined 2-3 days postexposure.
c Deaths occurred 105-140 min postexposure.
3.2. Nonlethal Toxicity
3.2.1. Rats
In experiments by Brown et al. (1974b), groups of 3-9 male Sprague-Dawley rats were used to assess the effect of P-450 induction by phenobarbital on chloroform-induced reductions in glutathione (GSH). Both induced and noninduced (control) rats were exposed for 2 h to chloroform at concentrations of 0.5 or 1.0% (5,000 or 10,000 ppm). Induced rats in the 5,000-and 10,000-ppm groups had a decrease in GSH of approximately 70% and 83%, respectively. Control rats exhibited no decrease in GSH concentrations, which suggests that GSH concentrations in rats are more than sufficient for scavenging reactive intermediates of chloroform metabolism at the concentrations tested.
Brondeau et al. (1983) examined the effect of chloroform on serumenzyme activities in rats. Groups of eight male Sprague-Dawley rats were exposed (whole-body) to chloroform at concentrations of 0, 137, 292, 400, 618, 942, or 1,075 ppm for 4 h. Chamber atmospheres were analyzed by gas chromatography (sample loop was compared with a known concentration standard) and
by analysis of a solid absorbent (activated charcoal or silica gel subjected to appropriate solvent extraction and gas-liquid chromatography). Exposure to the lowest concentration failed to significantly alter the activity levels of any the tested enzymes (glutamate dehydrogenase [GLDH], glutamic oxaloacetic transaminase [GOT], glutamic pyruvic transaminase [GPT], and sorbitol dehydrogenase [SDH]). Even at the highest concentration there was only a <2-to 7-fold increase in serum-enzyme activities. Statistically significant increases in GLDH and SDH were observed in rats exposed at 292 ppm. GLDH and SDH appeared to be most affected, although none of the changes in activity levels demonstrated a definitive exposure-response relationship. Although some of the increases were statistically significant (p <0.05 for GLDH; p <0.02 for SDH), the toxicologic relevance of these changes is uncertain. In a second phase of the study, rats were exposed to chloroform at 301 ppm (considered by the investigators to be a threshold for alteration in serum-enzyme activity based on the 4-h experiments) for two 6-h or four 6-h exposures. GLDH and SDH activities were somewhat greater after four 6-h exposures than after a single 4-h exposure or two 4-h exposures.
Statistically significant increases in serum SDH activity were also reported in female Sprague-Dawley rats exposed to chloroform for 4 h at concentrations as low as 153 ppm (1/64 of the LC50 for chloroform) (Lundberg et al. 1986). Although useful as biomarkers of exposure, increases in serum-enzyme activity in the absence of clinical correlates is limited use as an end point for AEGL derivation.
Ikatsu and Nakajima (1992) studied the interaction between carbon tetrachloride and chloroform in ethanol-treated rats. Controls groups of four rats were exposed only to chloroform at concentrations of 0, 50, or 100 ppm for 8 h. Concentrations in the dynamic air flow chamber were monitored every 15 to 30 min by gas chromatography. Hepatotoxicity was determined by assessing changes in serum glutamic oxaloacetic transaminase (SGOT), SGPT, liver malondialdehyde (MDA), and plasma MDA. Only marginal and statistically insignificant changes were detected for these indices in chloroform-treated rats, thereby indicating that an 8-h exposure at 50 or 100 ppm was without appreciable effect. Histopathologic examination revealed only negligible fat deposits in the liver of rats exposed at 100 ppm. These findings are consistent with those of Brondeau et al. (1983). In rats pretreated with ethanol (2 g ethanol/80 mL liquid diet fed daily for 6 weeks), only SGOT levels were increased significantly (3-fold; p <0.05) after exposure to chloroform at 50 ppm. Exposure of ethanoltreated rats to chloroform at 100 ppm chloroform, however, resulted in significant (p <0.05) increases in SGOT (7.5-fold) and SGPT (14-fold). There was no effect on liver MDA or plasma MDA. In ethanol-treated rats, ballooned hepatocytes in midzonal areas of the liver were observed only in rats exposed to chloroform at 100 ppm. The results of this study indicate that 8-h exposure of rats to chloroform at 50 or 100 ppm produce only minor effects that are more indicative of exposure rather than toxicity. Ethanol pretreatment followed by an 8-h exposure to chloroform at 100 ppm produced notable signs of toxicity.
In a study of the hepatotoxicity and renal toxicity of inhaled chloroform, male F344 rats (5 animals per group) were exposed at 1, 3, 10, 30, 100, or 300 ppm for 6 h/day for 7 days (Larson et al. 1994). Effects on nasopharyngeal tissue were also examined (Méry et al. 1994). Cage-side observations indicated no signs of toxicity during the exposure period, although there was a significant dose-dependent decrease in body weight gain at concentrations of 10 ppm and greater. Mild centrilobular vacuolation was observed only in the 300-ppm group, and histopathologic changes (hyperplasia) were found in the groups exposed at 10 ppm and greater. Two treatment-related lesions were observed in the nasal region of the chloroform-exposed rats. An increase in the size of goblet cells of the nasopharyngeal meatus was observed in rats exposed at 100 or 300 ppm. Also, region-specific changes were observed in the olfactory mucosa and bone of the ethmoid turbinates of rats exposed to chloroform at 10 ppm or greater. Although these data are not appropriate for deriving AEGL values, they may be used to evaluate the protectiveness of the AEGL values.
In studies to assess the effect of ethanol on the metabolism and toxicity of chloroform by various routes of administration, Wang et al. (1994) described nonlethal effects in male Wistar rats exposed by inhalation to chloroform alone (50, 100, or 500 ppm for 6 h). Indices of hepatotoxicity (GOT, GPT, and GSH) were evaluated in groups of five rats. Rats exposed to chloroform at 50 or 100 ppm exhibited no significant changes in any serum-enzyme activities. Both GOT and GPT were significantly (p < 0.05) increased after a 6-h exposure to chloroform at 500 ppm (about 1.6-and 1.2-fold, respectively), but were not considered indicative of severe hepatotoxicity. Ethanol pretreatment resulted in enzyme activities that were approximately 2-fold greater than from chloroform alone, and failed to alter GSH levels.
3.2.2. Mice
As described in Section 3.1.2, Fühner (1923) exposed mice to chloroform at 12-38 mg/L (2,458-7,782 ppm). In addition to lethality, nonlethal effects were observed. Mice exposed at 2,458-5,120 ppm exhibited reflex loss after 48-215 min of exposure, but no deaths occurred. Exposure at 4,710-5,529 ppm resulted in reflex loss after 30-90 min; 12 of 18 animals recovered and 6 died. Deaths occurred with 71-175 min of exposure. For mice exposed at 6,758-7,782 ppm, six mice exhibited reflex loss after 13-46 min and one mouse died after a 35-min exposure (reflex loss occurred after 8 min). The absence of validated exposure concentrations limits the quantitative validity of these data.
Kylin et al. (1963) reported on hepatotoxicity in mice after a single inhalation exposure to chloroform. A pilot study to determine the exposure duration needed to reach a maximum increase in serum ornithine carbamyl transferase (OCT) was conducted using groups of five female albino mice exposed to chloroform at 3,000 ppm for 4 h. Mice were killed after 1, 2, 4, 8, or 16 days. In the main study, groups of 10 female albino mice were exposed to chloroform at 0,
100, 200, 400, or 800 ppm for 4 h. Histopathologic examination of the liver and measurements of serum OCT were conducted 24 and 72 h after exposure and used as indices of effect. Chloroform was vaporized before injection into the constant-flow chamber, but no information was provided about whether concentrations were measured in the chamber. In the pilot study, serum OCT concentrations reach a maximum 4 days postexposure. In the main study, fatty infiltration of the liver was observed 1 day after exposure to chloroform at 100 ppm. At higher concentrations, the extent and severity of fatty degeneration increased. The authors concluded that the minimum concentration of chloroform to produce fatty infiltration of the liver in mice after a 4-h exposure was <100 ppm. Histologic changes (fatty infiltration and necrosis) also appeared to be greater after 24 h than after 72 h.
Gehring (1968), in addition to examining indices of lethality (see Section 3.1.2), determined 4,500-ppm ECt values for narcosis and for significant increases in SGPT in female Swiss-Webster mice. Groups of mice (10/group for narcosis determination and 8-10/group for SGPT determination) were exposed to chloroform at 4,500 ppm and the response rate was evaluated relative to exposure duration. The control group consisted of 254 mice representing a composite group of controls for all of the chlorinated hydrocarbons tested. SGPT increases greater than 54 Reitman-Frankel (R-F) units were considered statistically significant (control values were 24.4 ± 14.7 R-F units). Chloroform concentrations were attained by metering the chloroform into a heated tube for vaporization. Actual concentrations were measured by continuous flow of the atmosphere through an infrared spectrophotometric cell. The experiment was repeated if the chloroform concentration varied by more than 7%. The 4,500-ppm ECt50 for narcosis was 35 min (31.0-39.6 min, 95% CI), with a 10% response occurring after 15 min and 80% response occurring after 40 min. The 4,500-ppm ECt50 for a significant increase in SGPT was 13.5 min (10.1-18.1 min, 95% CI). A 20% response was observed after about 6 min, and a 90% response was observed after 20 min. The exposure-response relationship for SGPT increases was notably different than that observed for narcosis and lethality. The authors noted that elevation of SGPT activity occurred much earlier than narcosis or lethality; therefore, chloroform was induced liver damage before the onset of narcosis.
The hepatotoxicity and renal toxicity of inhaled chloroform was studied in female B6C3F1 mice (5 animals per group) exposed to chloroform at 1, 3, 10, 30, 100, or 300 ppm for 6 h/day for 7 d (Larson et al. 1994). The effects on nasopharyngeal tissue were also examined (Méry et al. 1994). Centrilobular hepatocyte necrosis and severe vacuolation in centrilobular hepatocytes were observed in mice exposed at 100 and 300 ppm. Mild to moderate vacuolar changes were observed in the 10-ppm and 30-ppm groups. Notable renal toxicity was observed only in the 300-ppm group. Histologic changes in the nasal region of female mice included increased cell proliferation at concentrations of 10 ppm and greater, and a slight indication of new bone growth in the endoturbinate of one mouse in the 300-ppm group. In a later report, however, it was noted that
the nasal lesions induced in female mice after exposure to chloroform at 10, 30, or 90 ppm for 6 h/day were transient and not sustained in mice similarly exposed for up to 13 weeks (Larson et al. 1996).
3.2.3. Dogs
Renal toxicity in dogs exposed by inhalation to chloroform was reported by Whipple and Sperry (1909). Details of the experimental protocol, including the exposure conditions, were lacking. The report provided only qualitative information regarding clinical signs (vomiting, diarrhea, and lassitude). Gross pathologic and histopathologic evidence of hepatotoxicity and renal toxicity was reported for dogs on successive days after being exposed to 1-2 ounces of chloroform over 1-2 h.
von Oettingen et al. (1949) described the effects of chloroform on various physiologic functions in dogs surgically prepared for monitoring of respiration and blood pressure (see Section 3.1.3.). Continuous exposure to chloroform at 15,000 ppm resulted in the death of all 10 dogs (6-285 min). The dogs exhibited notable cardiovascular responses (decreased arterial blood pressure), decreased respiratory rate and body temperature, and depression of voluntary and involuntary reflexes within 35 min. Although it is uncertain whether deaths would have been prevented by ending the exposure at or before 35 min, the data provide a qualitative description of the response in dog to very high concentrations of chloroform.
3.2.4. Cats
Nonlethal effects in cats exposed to chloroform were described by Lehmann and Schmidt-Kehl (1936). In this study, adult cats were exposed to chloroform at concentrations of 7,200 or 22,000 ppm. Concentrations were determined by chemical reaction (hydrolysis with alkali in alcohol). At 7,500 ppm, cats exhibited light narcosis after 78 min and deep narcosis after 93 min. Light and deep narcosis were induced after 10 min and 13 min, respectively, at 22,000 ppm. Irritation of the eyes, mouth, and nose were also found at that concentration.
3.2.5. Summary of Nonlethal Toxicity in Animals
The nonlethal toxicity of chloroform in laboratory animals (rats, mice, and cats) after acute inhalation exposure is summarized in Table 4-5. As would be expected of a hepatotoxicant, many of the nonlethal effects reported were indices of liver damage. Acute exposures (1-4 h) to chloroform at concentrations of 100-292 ppm have resulted in some degree of hepatic injury, as indicated by increased serum-enzyme activities and histopathologic examination. Without
histopathologic correlates, however, marginal increases (although statistically significant) in serum enzyme activities might not be indicative of a serious toxic response. Renal toxicity also has been demonstrated in mice at exposures that are relatively low (e.g., 246-665 ppm for 2 h or 693 ppm for 1 h) compared with those inducing narcosis (e.g., 4,500 ppm for 35 min). Data in cats are from studies that involved high, narcosis-inducing exposures.
TABLE 4-5 Nonlethal Effects of Chloroform in Laboratory Animals after Acute Inhalation Exposure
| Species | Exposure Concentration (ppm) | Exposure Duration | Effect | Reference |
| Rat | 500 | 6 h | Statistically significant increase in serum-enzyme activity | Wang et al. 1994 |
| Rat | 10 | 6 h/d for 7d | Histopathologic changes in the liver | Larson et al. 1994 |
| Rat | 50 | 8 h | No increase in liver weight | Ikatsu and Nakajimal992 |
| Rat | 100 | 8 h | Marginal, biologically insignificant increase in serum-en2yme activity | Ikatsu and Nakajima 1992 |
| Rat | 153 | 4 h | Increased serum-enzyme activity | Lundberg et al. 1986 |
| Rat | 292 | 4 h | Increased serum-enzyme activity | Brondeauetal. 1983 |
| Rat | 10,000 | 2 h | No effect on hepatic GSHa | Brown et al. 1974b |
| Mouse | 2,458-5,120 | 48 min | Reflex loss | Fiihner 1923 |
| Mouse | 100 | 4 h | Fatty infiltration of liver | Kylin et al. 1963 |
| Mouse | 693 | 1 h | Renal toxicity | Deringeretal. 1953 |
| Mouse | 246 | 2 h | Renal tubular necrosis | Culliford and Hewitt 1957 |
| Mouse | 665 | 2 h | Renal necrosis in males | Culliford and Hewitt 1957 |
| Mouse | 4,500 | 35 min | 50% narcosis (ECt50) | Gehring 1968 |
| Mouse | 4,500 | 13.5 min | 50% significantly increased SGPT(ECt50)b | Gehring 1968 |
| Cat | 7,500 | 78 min | Light narcosis | Lehmann and Schmidt-Kehl 1936 |
| Cat | 22,000 | 10 min | Narcosis; irritation of eyes, mouth, and nose | Lehmann and Schmidt-Kehl 1936 |
a Narcosis and significant reduction in glutathione was found in phenobarbital-induced rats exposed to chloroform at 5,000 ppm for 2 h.
b Approximately 2.2-fold increase relative to unexposed controls; considered by investigators to be statistically significant.
3.3. Developmental and Reproductive Toxicity
3.3.1. Rats
The embryotoxicity and fetotoxicity of chloroform in Sprague-Dawley rats was studied by Schwetz et al. (1974). Pregnant rats were exposed to chloroform at 30 ppm (22 dams), 100 ppm (23 dams), or 300 ppm (3 dams) for 7 h/day on gestation days 6-15; control rats (68) were exposed to filtered air (see Table 4-6). The exposure concentrations were subanesthetic and varied <5% from the target concentrations. Concentrations were monitored three times per day using an infrared spectrophotometer. The 300-ppm group had marked anorexia at the end of the treatment period, although a comparison with a pair-fed control group (8 dams) later showed that inanition was not a contributor to the observed embryotoxicity and fetotoxicity. Chloroform at 30 ppm induced some evidence of embryotoxicity and fetotoxicity, while the 100-and 300-ppm exposures caused significant toxicity (see Table 4-6).
The investigators concluded that chloroform produced minor effects on the embryo and fetus at 30 ppm, was highly embryotoxic and fetotoxic at 100 ppm, and was embryocidal, embryotoxic, and fetotoxic at 300 ppm. At 100 ppm, frank teratogenic effects (imperforate anus and acaudia [missing tail]) were observed in three litters. The observed effects could not be correlated with maternal toxicity or inanition.
TABLE 4-6 Embryotoxicity and Fetotoxicity of Chloroform in Rats Exposed During Gestation
| Parameter | Control | Pair-fed Control | 30ppm | 100 ppm | 300 ppm |
| % Pregnanecy (pregnant/bred) | 88 (68/77) | 100 (8/8) | 71(22/31) | 82 (23/28) | 15 (3/20)a |
| Corpora lutea/damb | 14 ± 2 | 14 ± 2 | 16 ± 3b | 14 ± 2 | 14 ± 1 |
| Live fetuses/litterb | 10 ± 4 | 10 ± 4 | 12 ± 2 | 11 ± 2 | 4 ± 7a |
| % Reabsorptions/implantations | 8 (63/769) | 7 (6/87) | 8(24/291) | 6(16/278) | 61 (20/33)a |
| Fetal body weight (g)c | 5.69 ± 0.36 | 5.19 ± 0.29a | 5.51 ± 0.20 | 5.59 ± 0.24 | 3.42 ± 0.02a |
| Fetal crown-rump length (mm)c | 43.5 ± 1.1 | 42.1 ± 1.1a | 42.5 ± 0.6a | 43.6 ± 0.7 | 36.9 ± 0.2a |
| Total gross anomaliesd | 1/68 | 0/8 | 0/22 | 3/23a | 0/3 |
| Total skeletal anomaliesd | 46/68 | 3/8 | 20/22a | 17/23 | 2/3 |
| Total soft-tissue anomaliesd | 33/68 | 3/8 | 10/22 | 15/23 | 1/3 |
a Significantly different from control; p <0.05.
b Mean ± standard deviation.
c Mean of litter means ± standard deviation.
d Litters affected/litters examined.
Source: Adapted from Schwetz et al. 1974.
Newell and Dilley (1978) conducted experiments in which Sprague-Dawley rats were exposed to chloroform at 942, 2,232, or 4,117 ppm for 1 h/day on gestation days 7-14. Controls were exposed to clean air. The number of resorptions was increased (45% relative to controls) and the average fetal body weight was decreased in the high-exposure group. No notable effects were found in the low-or mid-exposure groups. No evidence of teratogenic effects was found.
A series of experiments (two preliminary studies and one main study) to assess developmental toxicity of chloroform in Wistar rats were conducted by Baeder and Hoffman (1988). In one preliminary study, time-mated Wistar rats (4-6/group) were exposed to chloroform for 6 h/day at concentrations of 0, 10, 30, or 100 ppm on gestation days 7-11 and 14-16. At 10 ppm, two dams had no fetuses and a single implantation site. At 30 ppm, one dam had only one fetus and three empty implantation sites. No such effects were reported for the 100-ppm group. In the second preliminary experiment, Wistar rats exposed at 100 and 300 ppm (6 h/day) on gestation days 7-16 exhibited decreased feed consumption and body weight loss. Fetal weights in two litters in the 100-ppm group were slightly decreased. The 300-ppm group had three dams with normally developed fetuses, one dam with totally resorbed fetuses, and one dam had only empty implantation sites. In the main study, groups of 20-23 timemated Wistar rats were exposed to chloroform at concentrations of 0, 30, 100, or 300 ppm (7 h/day, gestation days 7-16). During exposure, chloroform-exposed rats exhibited decreased feed consumption and body-weight gain (p <0.05 for all exposure groups, except for body-weight gain for 30-ppm group exposed on gestation day 21) relative to controls. Litter data for the main study are summarized in Table 4-7. Although fetal weight was significantly decreased in the 300-ppm group and crown-rump length was significantly decreased in all chloroform-exposed groups, these effects might be a function of maternal feed consumption and body weight effects. Incidences of external and internal malformations and skeletal abnormalities were not statistically significant.
TABLE 4-7 Litter Data from Study of Wistar Rats Exposed to Chloroform During Gestation
| Parameter | Concentration(ppm) | |||
| 0 | 30 | 100 | 300 | |
| No. pregnant/no. sperm plugs | 20/20 | 20/20 | 20/21 | 20/23 |
| No. lost litters | 0 | 2 | 3 | 8 |
| No. live litters | 20 | 18 | 17 | 12 |
| Resorptions/live litter (mean) | 0.75 | 0.22 | 0.53 | 0.92 |
| Live fetuses/litter (mean) | 12.4 | 12.8 | 12.8 | 13.4 |
| Fetal weight (g)b | 3.19 ± 0.30 | 3.16 ± 0.19 | 3.13 ± 0.21 | 3.00 ± 0.19a |
| Fetal crown-rump length (cm)b | 3.52 ± 0.17 | 3.38 ± 0.12a | 3.39 ± 0.10a | 3.39 ± 0.12a |
a Significantly different from control group; p <0.5.
b Mean ± standard deviation.
Source: Baeder and Hoffman 1988.
A follow-up study was conducted by Baeder and Hoffman (1991) in which groups of 20 time-mated Wistar rats were exposed to chloroform (0, 3, 10, or 30 ppm, 7 h/day) on gestation days 7-16. Feed consumption during the first week of exposure was significantly decreased (p <0.05) and remained so for the 30-ppm group to the end of the study. Body weight of the 3-ppm group was unaffected but an exposure-dependent decrease was detected by gestation day 17. Body weights remained lower than controls on gestation day 21 for the 10-ppm and 30-ppm groups. Analysis of litter data by the investigators revealed a significant decrease in fetal weight and crown-rump length in the 30-ppm group (see Table 4-8). Significantly increased incidences of ossification variations were observed, especially for the 30-ppm group (see Table 4-9).
TABLE 4-8 Litter Data from Follow-up Study of Wistar Rats Exposed to Chloroform during Gestation
| Parameter | Concentration(ppm) | |||
| 0 | 30 | 100 | 300 | |
| Number pregnant | 20 | 20 | 20 | 20 |
| Number lost litters | 0 | 0 | 0 | 1 |
| Number live litters | 20 | 20 | 20 | 19 |
| Resorptions/live littera | 0.55 ± 0.89 | 0.40 ± 0.60 | 0.75 ± 1.02 | 0.84 ± 1.42 |
| Live fetuses/littera | 12.4 ± 2.4 | 12.4 ± 3.5 | 12.9 ± 3.0 | 12.5 ± 1.9 |
| Fetal weight (g)a | 3.4 ± 0.3 | 3.2 ± 0.3 | 3.2 ± 0.3 | 3.2 ± 0.3bb |
| Fetal crown-rump length (mm)a | 35.8 ± 2.0 | 35.5 ± 2.1 | 34.4 ± 2.6 | 34.0 ± 1.9b |
a Litter mean ± standard deviation.
b Significantly different from control group; p <0.05.
Source: Baeder and Hoffman 1991.
TABLE 4-9 Skeletal and Ossification Variations in Wistar Rats Exposed to Chloroform During Gestation
| Parameter | Concentration(ppm) | |||
| 0 | 30 | 100 | 300 | |
| Number live litters | 20 | 20 | 20 | 19 |
| Poorly ossified cranial bonesa | 42/14 | 47/17 | 48/16 | 60b /17 |
| Ossification of less than 2 caudal vertebraea | 4/3 | 14b /5 | 16b /6 | 14b /8 |
| Non- or weakly ossified sternebraea | 7/3 | 32b /13b | 35b /14b | 18b /11b |
| Wavy or thickened ribsa | 10/6 | 11/5 | 22b /10 | 15/4 |
a Number of affected fetuses/number of litters with affected fetuses.
b Significantly different from control group; p <0.05.
Source: Baeder and Hoffman 1991.
3.3.2. Mice
Murray et al. (1979) examined the developmental toxicity of chloroform in CF-1 mice after gestational exposure. Groups of 34-40 pregnant mice were exposed to chloroform at 100 ppm for 7 h/day on gestation days 6-15, 1-7, or 8-15. Controls were exposed to filtered room air. Chloroform concentrations were monitored by infrared spectrophotometry and varied by <3% from the target concentration. Maintenance of pregnancy was significantly decreased (p <0.05) in the dams exposed on gestation days 1-7 (44% vs. 74% in controls) and 6-15 (43% vs. 91% in controls), but not for those exposed on days 8-15 (decreased, but not significantly). The significant developmental toxicity findings are shown in Table 4-10. It was reported that delayed ossification of the skull bones was significantly increased in all of the chloroform-treated groups and that, with the exception of the group treated on days 6-16 of gestation, delays in the ossification of sternebrae were significantly more frequent in the treated groups compared with controls. However, these data were not presented in the report’s tables. There was also evidence of hepatotoxicity in chloroform-exposed dams as demonstrated by significantly increased absolute and relative liver weights and by elevated SGPT activity. The investigators concluded that exposure of pregnant mice to chloroform at 100 ppm (7 h/day) on gestation days 1-7 or 6-15 decreased the ability to maintain pregnancy but was not teratogenic. Exposure on gestation days 8-15 did not affect pregnancy maintenance but resulted in an increased incidence of cleft palate.
TABLE 4-10 Developmental Toxicity of Chloroform in Mice Exposed During Gestation
| Parameter | Days 1-7 | Days 6-15 | Days 8-15 | |||
| Control | 100 ppm | Control | 100 ppm | Control | 100 ppm | |
| Litters examined | 22 | 11 | 29 | 12 | 24 | 18 |
| Resorptions/littera | 2 ± 2 | 4 ± 5b | 2 ± 2 | 1 ± 1 | 2 ± 2 | 2 ± 2 |
| Fetal body weight (g)c | 1.02 ± 0.10 | 0.92 ± 0.07b | 0.99 ± 0.11 | 0.95 ± 0.13 | 1.00 ± 0.12 | 0.85 ± 0.17b |
| Fetal crown-rump length (mm)c | 24.7 ± 1.0 | 23.6 ± 1.2a | 23.7 ± 1.3 | 23.2 ± 1.1 | 24.1 ± 1.1 | 22.9 ± 2.2a |
| Cleft palate (number fetuses [no. litters]) | 3[1] | 0 | 0 | 0 | 1[1] | 10 [4]d |
a Mean ± standard deviation.
b Significantly different from control (p < 0.05).
c Mean of the litter means ± standard deviation.
d Six fetuses in one litter exhibited cleft palate.
Source: Adapted from Murray et al. 1979.
Land et al. (1981) studied the morphologic changes in spermatozoa of C57B1/C3H mice exposed to chloroform. The mice were observed 28 days after exposure to chloroform at 0.1 or 0.05 of the minimal alveolar concentration (4 h/day for 5 days). Chloroform was delivered via calibrated vaporizers and the concentration was monitored by gas chromatography. Mice were killed 28 days after the last exposure and spermatozoa (1,000/slide) were examined independently by two pathologists. On the basis of data from groups of five mice, the percentage of abnormal spermatozoa was 1.42 ± 0.08, 2.76 ± 0.31, and 3.48 ± 0.66 for the control (clean air), 0.5-and 1.0-ppm groups, respectively. Both treatment groups were significantly different (p <0.01) from the controls. Abnormalities included flattened spermatozoa, amorphous spermatozoa, and spermatozoa with abnormal hook formation.
3.4. Genotoxicity
Numerous genotoxicity assays have been performed with chloroform (ATSDR 1997). Generally, the results of these bioassays indicate chloroform to be a weak mutagen with low potential for interaction with DNA.
3.5. Carcinogenicity
Renal and hepatic tumors have been reported in rodents following chronic oral administration of chloroform (reviewed in ATSDR 1997). The results of cancer bioassays appear to be substantially influenced by the method of administration (gavage vs. drinking water) and by the vehicle (corn oil vs. water). Inhalation exposure studies of the tumorigenic potential of chloroform include a 90-day study in F344 rats by Templin et al. (1996a), a short-term exposure study by Templin et al. (1996b), and a long-term inhalation study by Yamamoto et al. (1994).
In the 90-day study by Templin et al. (1996a), male and female F344 rats were exposed to chloroform at 0, 2, 10, 20, 30, 90, or 300 ppm for 6 h/day for 7 days/week. Groups of rats (15-60/group) were subjected to different exposure protocols: male rats were exposed for 4 days or 3, 6, or 13 weeks, and female rats were exposed for 3 or 13 weeks. Exposure atmospheres were monitored by infrared gas analysis. Average analytically-determined concentrations were always within 4.5% of the target concentration. Results of the study indicate that the primary targets of toxicity are the liver, kidneys, and nasal passages. Cytolethality and regenerative cell proliferation were significant at 300 ppm. Although long-term exposure at 300 ppm would probably induce a tumorigenic response, this concentration was considered by the investigators to be highly cytotoxic (in excess of the maximum tolerated dose [MTD]) and not relevant for extrapolating to low-dose responses. Statistically significant body-weight loss was observed in male rats exposed for 4 days but kidney lesions were seen only in rats exposed at 30 (1 of 5 rats), 90 (3 of 5 rats), or 300 ppm (5 of 5 rats).
Templin et al. (1996b) conducted studies in BDF1 mice to affirm the role of cytotoxicity and regenerative cell proliferation in the tumorigenic response to chloroform. Groups of male and female mice were exposed to chloroform at 0, 0.3, 5, 30, or 90 ppm 6 h/day for 4 days. Bromodeoxyuridine (BrdU) was administered by osmotic pumps implanted 3.5 days before necropsy and served to provide a labeling index for S-phase cells. Additional groups of mice were exposed to chloroform at 30 or 90 ppm for 5 days/week for 2 weeks. Degenerative lesions and a 7-to 10-fold increase in the labeling index were observed in the kidneys of male but not female mice exposed at 30 or 90 ppm. Liver lesions and an increased hepatocyte labeling index were observed in male mice exposed at 30 and 90 ppm and in female mice exposed at 90 ppm. Lethality was 40 and 80% in the 30-and 90-ppm groups, respectively, exposed for 2 weeks; severe kidney damage was evident in the animals. These findings show that in the twoyear assays, chloroform exposures actually exceeded the MTD and were tolerated only because of the step-wise exposure protocol that allowed the animals to accommodate metabolically to the high concentrations. Templin et al. (1996b) questioned the validity of low-dose extrapolation from tumor data of this type (e.g., nongenotoxic-cytotoxic mechanism that is secondary to organ-specific toxicity).
In a preliminary report of a 2-year cancer bioassay, Yamamoto et al. (1994) observed no increase in tumor incidences in male and female F344 rats exposed to chloroform at 10, 30, or 90 ppm for 5 days/week. No further details are available on this study.
Several issues, however, are relevant to the carcinogenic potential of chloroform. These are especially relevant regarding the estimation of carcinogenic risk after a single acute exposure. As reviewed by Conolly (1995) and Golden et al. (1997), the tumorigenic dose-response of mice and rats to chloroform appears to be nonlinear and is secondary to cytotoxicity (i.e., cell necrosis and subsequent cellular regeneration) following exposures that induce frank toxicity in tissues that are tumor sites and at concentrations that often exceed the MTD. Additionally, both in vivo and in vitro genotoxicity data indicate the absence of a genotoxic mechanism for chloroform.
The significance of regenerative cell proliferation in chloroform-induced cancer was also examined by Butterworth et al. (1995) and Wolf and Butterworth (1997). An analysis of the available data indicates that chloroform acts through a nongenotoxic, cytotoxic mechanism. In rodent studies, toxicity is observed only when chloroform is metabolized to reactive metabolites at a rate sufficient to cause cytolethality. As such, a linearized extrapolation from high concentrations that produce tumors to very low concentrations is considered inappropriate. Additionally, the current inhalation cancer risk is 2.3 × 10-5 (μg/m3)-1 (EPA 2012) and is based on a tumorigenic response (hepatocellular carcinomas) in B6C3F1 mice administered chloroform by gavage (NCI 1976) and, therefore, involves the uncertainties associated with route-to-route extrapolation.
Butterworth et al. (1995) and Wolf and Butterworth (1997) compared the results of cancer risk assessments performed using the linearized multistage model for low-dose extrapolation with the results based on a threshold response (cytolethality and cellular regeneration). The resulting outcomes are remarkably different. Application of the linearized multistage model to tumor incidence data from a gavage study with mice (NCI 1976) resulted in a virtually-safe concentration (relative to a 1 × 10-6 cancer risk) of 8 × 10-6 ppm. However, a virtuallysafe concentration of 0.01 ppm is obtained when uncertainty factors are applied (three factors of 10 for interspecies differences, intraspecies variability, and use of a subchronic study) to 10 ppm, a concentration that did not produce cytolethality or cellular regeneration in inhalation studies with rodents. The investigators justify their approach by citing the apparent need for cytolethality and cellular regeneration in the tumorigenic response.
Melnick et al. (1998) provided data and alternate interpretations regarding the relevance of cytolethality and proliferative cellular regeneration to the tumorigenic response observed in rodents following oral administration of chloroform in corn oil. Following gavage dosing of female B6C3F1 mice (10/group) with chloroform (5 times/week for 3 weeks at doses of 55, 110, 238, or 477 mg/kg), biochemical indices of toxicity (ALT, SDH) and labeling index (BrdU) for S-phase hepatocytes were measured and histopathologic examination were performed to ascertain the relationship between regenerative hyperplasia and tumor induction. As expected, a dose-related response was observed for liver-tobody weight ratio, increase in ALT and SDH activity, severity and incidence of hepatocyte hydropic degeneration, and labeling index. The investigators compared the dose-response curves for tumor incidence (using data from previous cancer bioassays) and hepatocyte labeling index and reported that the processes are not causally related. In other words, an elevated labeling index resulting from cellular proliferation is not required for a tumorigenic response.
4. SPECIAL CONSIDERATIONS
4.1. Metabolism and Disposition
The metabolism of chloroform has been thoroughly studied (reviewed in ATSDR 1997). Although metabolism via cytochrome P-450 IIE1 is wellestablished, a minor anaerobic pathway also exists resulting in a dichloromethyl radical intermediate. Phosgene, formed by P-450-mediated dehydrochlorination, may react with cellular proteins or be converted to hydrochloric acid and carbon dioxide (Pohl et al. 1981). Phosgene may also react with GSH to form diglutathionyl dithiocarbonate which is then metabolized to 2-oxothiazolidine-4-carboxylic acid (Mansuy et al. 1977; Pohl et al. 1977; Branchflower et al. 1984).
Brown et al. (1974a) studied the metabolism of orally administered [14C]-chloroform (60 mg/kg) in male Sprague-Dawley rats; male CBA, CF/LP, and C57 mice; and squirrel monkeys. In all test species, 14CO2 was a major excretory
product but species-dependent variability was observed in its elimination. Eighty percent of the administered dose was excreted as14CO2 in all three strains of mice, whereas 60% and 20% was eliminated in rats and squirrel monkeys, respectively.
Fry et al. (1972) reported that 17.8-66.6% of an oral dose of radiolabeled chloroform (500 mg) was expired unchanged by eight human volunteers over an 8-h period. Maximum excretion of chloroform occurred 40 min to 2 h after administration. Carbon-dioxide excretion was measured in one male and one female volunteer. Over a 450-min period, 48% (woman) and 50% (man) of the dose was expired as carbon dioxide. The investigators also reported decreased excretion of chloroform by obese subjects and suggested that resulted from uptake of chloroform by greater amounts of adipose tissue. Peak blood concentrations ( ≈ μg/mL) occurred about 45 min after dosing. Elimination of chloroform from the blood appeared to be biphasic: an initial rapid clearance within an hour and a slower clearance over the next 6 h. As chloroform concentration in the blood increased, pulmonary excretion increased.
Corley et al. (1990) developed a PBPK model for chloroform based on a kinetic constant from in vivo studies with rats and mice, in vitro enzymatic studies with human tissue samples, and physiologically-based estimates for absorption, distribution, metabolism, and excretion processes. Macromolecular binding was considered a measure of internal dose. The model was validated by comparing predicted values with experimental data from mice, rats, and humans. Human metabolic and macromolecular-binding constants for VmaxC (15.7 mg/hr/kg) and Km (0.448 mg/L) were derived. It was also shown that metabolic activation of chloroform to reactive intermediates, such as phosgene, was greatest in mice. Metabolic activation was less in rats and lowest in humans. Therefore, it was estimated that exposure to equivalent concentrations of chloroform would result in a lower delivered dose in humans than in laboratory animals. Species variability was also reported by Brown et al. (1974a), who reported that conversion of chloroform to carbon dioxide was highest in mice (80%) and lowest in squirrel monkeys (18%). In rats and mice, [14C]-urea was detected in the urine along with two unidentified metabolites, and parent compound was found in the bile of the squirrel monkeys. In mice, radioactivity in the blood peaked 1 h after dosing and decreased gradually over the next 24 h.
The chloroform PBPK model developed by Corely et al. (1990) was used by Delic et al. (2000) to develop models for humans and rats to compare rates of metabolism in the context of assessing the validity of uncertainty factors used to determine occupational exposure limits. The study also utilized Monte Carlo analysis to determine the extent of variability within human and animal-model populations. The results demonstrated that even at the most extreme ranges within the human population, concentrations of toxic metabolites necessary to induce a toxic response would not be generated at rates comparable to that in rats. Specifically, the model showed that the mean peak rate of metabolism of inhaled chloroform (at the mouse no-observed-adverse-effect level of 10 ppm) is approximately 78-fold lower in humans and that the chloroform concentration
required to achieve a peak metabolism rate in humans would be 65-fold higher than that in mice. Monte Carlo analysis of population variability also indicated that chloroform metabolism rates between mice and humans varied by 25-to 50-fold. Overall, the work clearly demonstrated that considerably higher concentrations of chloroform are required to induce a toxic response in humans compared with mice.
Data regarding the distribution of chloroform among brain, lung, and liver tissue of humans was obtained by Gettler and Blume (1931) from suicide victims or deaths from chloroform anesthesia. The brain and lungs consistently had the highest concentrations of chloroform (60-480 mg/g in brain; 24-485 mg/g in lung), whereas liver tissue tended to have lower concentrations (24-238 mg/g). These values reflect tissue burdens after high exposures to chloroform.
The distribution of [14C]-chloroform in pregnant C57BL mice after a single 10-min inhalation exposure (approximately 16 mmoles based on specific activity) was studied by Danielsson et al. (1986). Assessments were conducted at 0.5, 4, and 24 h. At all time points, radioactivity was greatest in the lungs, liver, and kidneys. Radioactivity in the respiratory tract was associated with epithelial tissue (nasal mucosa, trachea, and bronchi). Radioactivity was also found in the fetus and placenta at all time points, peaking at 0.5 h and gradually decreasing over the 24-h time frame. In addition to total radioactivity, the investigators also determined bound radioactivity in various tissues and found that the respiratory tract and centrilobular portion of the liver contained bound radioactivity, which possibly indicates on-site production of reactive metabolites.
Wang et al. (1997) reported on the effects of ethanol pretreatment (2 g/rat/day for 3 weeks) on the metabolism and hepatotoxicity of chloroform in rats following administration of chloroform by various routes (intraperitoneal, perioral, and inhalation). Ethanol pretreatment increased cytochrome P-450 from 0.74 nmol/mg to 1.10 nmol/mg and increased the metabolism of inhaled chloroform 7-fold in rats exposed to chloroform at 500 ppm for 6 h, but did not increase the metabolism of chloroform in rats exposed at 50 ppm for 6 h. Hepatotoxicity, as determined by GPT, GOT, and GSH activity, was unaffected in the 50-ppm group and increased approximately 6-fold in the 500-ppm group.
4.2. Mechanism of Toxicity
The noncarcinogenic and carcinogenic mechanisms of chloroform have been previously reviewed (Butterworth et al. 1995; Conolly 1995; Templin et al. 1996a,b; ATSDR 1997; Golden et al. 1997; Wolf and Butterworth 1997). Chloroform toxicity may be generally categorized as effects on the central nervous system, liver, kidneys, and heart (primarily the result of myocardial sensitization to epinephrine).
The precise mechanism of chloroform on neural activity is unknown. It is generally assumed that general anesthetics act by influencing synaptic transmission (e.g., potentiating transmitter release at inhibitory synapses or inhibiting
release at excitatory synapses). These actions may be the result of interaction with protein-lipid interfaces (Kennedy and Longnecker 1996).
The underlying mechanism of chloroform’s hepatic and renal toxicity is the binding of reactive intermediates, such as phosgene (Pohl et al. 1977), to cellular macromolecules, the depletion of these macromolecules, and subsequent cell death.
Brown et al. (1974b) exposed phenobarbital-treated rats for 2 h to chloroform at 0.5% (5,000 ppm) or 1.0% (10,000 ppm) and found a 70% and 83% reduction in hepatic GSH (p <0.001), respectively. At these concentrations, however, noninduced rats exhibited no significant change in GSH activity.
The importance of GSH depletion was also demonstrated by Docks and Krishna (1976), who showed that administration of chloroform (80 mg/kg, intraperitoneal) to phenobarbital-treated rats decreased GSH and resulted in massive liver necrosis. Docks and Krishna (1976) postulated that chloroformmediated decreases in GSH was not from the trichlorocarbon radical, because depletion of GSH was greater from chloroform than by halomethanes known to be metabolized to the trichlorcarbon radical.
The mechanism of chloroform toxicity in isolated rat hepatocytes was studied by el-Shenawy and Abdel-Rahman (1993). The results support the contention of Docks and Krishna (1976) that depletion of GSH is a causative precursor for cytotoxicity. Isolated rat hepatocytes exposed to chloroform at concentrations of 1, 10, 100, or 1,000 ppm exhibited a concentration-dependent decrease in viability (p <0.05 at all concentrations). Leakage of serum aspartate aminotransferase (AST) occurred at all the concentrations, but was significant only at 1 ppm after 60 min and at 10 ppm after 30 min. Leakage of ALT was significant at 100 and 1,000 ppm after 60 min and 30 min, respectively. GSH was significantly decreased between 15-120 min after hepatocytes were incubated with chloroform at 1,000 ppm. At 100 ppm and 10 ppm, GSH depletion became significant at 30 min and 120 min, respectively.
4.3. Structure-Activity Relationships
Assessment of structure-activity relationships was not instrumental is deriving AEGL values for chloroform.
4.4. Other Relevant Information
4.4.1. Species Variability
Strain, species, and gender variability in the metabolism and toxicity of chloroform has been demonstrated. As previously noted, male mice exhibit both renal toxicity and hepatotoxicity after exposure to chloroform, whereas female mice exhibit only hepatotoxicity. This has been shown to be from hormonespecific cytochrome P-450 in the kidneys of male mice. By examining differences in the biotransformation of chloroform to phosgene, Pohl et al. (1984)
demonstrated strain and sex differences in chloroform-induced renal toxicity. The differences could be attributed to strain-and gender-dependent differences in the rate of phosgene production by microsomal and mitochondrial fractions from the kidneys. A notable difference was observed between sensitive male DBA/2J mice and less sensitive C57BL/6J mice. Male mice formed phosgene at a rate nearly an order of magnitude more rapid than female mice. Additionally, based on results of PBPK model studies using metabolism and disposition data, humans appear to be less sensitive than rodent species, and the mouse appears to be the most sensitive.
4.4.2. Concurrent Exposure Issues
Because the biotransformation of chloroform to reactive intermediates is mediated by cytochrome P-450 IIE1, exposures to chemicals that induce P-450 might increase the toxic response of chloroform. From a practical standpoint, alcohol consumption would be a special concern.
5. DATA ANALYSIS FOR AEGL-1
5.1. Summary of Human Data Relevant to AEGL-1
Human exposure data on chloroform consistent with AEGL-1 effects include studies by Lehmann and Hasegawa (1910) of human volunteers and by McDonald and Vire (1992) of dental workers. Lehmann and Hasegawa (1910) reported that exposure to chloroform at 920-1,100 ppm for 2-3 min resulted in vertigo and that concentrations as high as 1,400 ppm for 15-30 min produced lassitude, vertigo, and headache. Some individuals exposed at 620 ppm for 30 min reported only the sensation of a not unpleasant odor and no neurologic symptoms. Because vertigo could affect escape from a potentially hazardous condition, concentrations of chloroform inducing this condition are inappropriate for developing AEGL-1 values. The study by Lehmann and Hasegawa (1910) lacks details on exposure methods and validation of exposure measurements. The McDonald and Vire (1992) study involved exposure at low concentrations during endodontic procedures (<0.57 ppm for 5.5 h and <0.88 ppm for over 150 min). These exposures did not result in any signs or symptoms even after clinical screening at 4 h and 1 year after exposure. No additional human data consistent with the AEGL-1 definition were available.
5.2. Summary of Animal Data Relevant to AEGL-1
Animal data consistent with AEGL-1 effects include alterations in clinical chemistry determinations (specifically serum ALT, AST, GLDH, and SDH activity) and minor histopathologic findings in the liver and kidneys of rats and mice. Increased serum-enzyme activities were observed in rats exposed to chloroform
at 153 ppm for 4 h (Lundberg et al. 1986) or at 292 ppm (Brondeau et al. 1983). Exposure of rats to chloroform at 500 ppm for 6 h produced statistically significant increases in serum-enzyme activity (Wang et al. 1994). Rats exposed at 50 ppm for 8 h had no increase in liver weight, but rats exposed at 100 ppm had a slight increase in serum-enzyme activity (Ikatsu and Nakajima 1992). Although statistically significant increases in serum-enzyme activities were reported in several studies, they were not necessarily indicative of biologicallyrelevant hepatic damage (some of the enzyme activities were increased only 2-fold and histologic correlates were negligible) and, therefore, would not be appropriate as AEGL-1 end points.
5.3. Derivation of AEGL-1
Human data sets for determining AEGL-1 values have poorly described methodology and inadequate characterizations of exposure. Animal data consistent with AEGL-1 effects have better defined exposure data, but are limited to clinical chemistry findings that are more indicative of biologic indices of exposure than overt toxicity. Concentrations of chloroform that do not produce overt signs of toxicity in humans are neither irritating nor have an unpleasant odor. Thus, it would be difficult to identify exposures that would produce notable discomfort or mild sensory irritation without approaching concentrations that might be near a threshold for narcosis. As a result, AEGL-1 values are not recommended.
6. DATA ANALYSIS FOR AEGL-2
6.1. Summary of Human Data Relevant to AEGL-2
In an assessment of 1,502 surgical patients anesthetized with chloroform (concentrations never greater than 22,500 ppm) for <30 min to >120 min, Whitaker and Jones (1965) reported cardiac irregularities in some patients (bradycardia in 8.1%; arrhythmias in 1.3%). Protection against narcosis even in the absence of toxic effects would appear to be at least one goal of the AEGL-2 values, so these data would be an inappropriate basis for AEGL-2 values. Lehmann and Hasegawa (1910) reported “intoxication and dizziness” in human subjects exposed to chloroform at 4,300-5,100 ppm for 20 min or at 7,200 ppm for 15 min. Three volunteers reported pounding heart and experienced gagging during a 30-min exposure at 3,000 ppm, and “light-headedness” and lassitude after 30 min at 1,400 ppm. Smith et al. (1973) evaluated surgical patients anesthetized with chloroform (8,500-13,000 ppm; concentration never exceeded 2% [20,000 ppm]) for a mean duration of 112.96 min. Cardiac arrhythmias of various types were detected in 1-17 of the patients. With the exception of a slight elevation of lactate dehydrogenase, serum enzyme values (SGPT, SGOT, and alkaline phosphatase) were not altered by chloroform. Nausea and vomiting occurred in 46% of the patients.
6.2. Summary of Animal Data Relevant to AEGL-2
Several studies in rats indicate that signs of hepatotoxicity (fatty infiltration) and renal damage (tubular necrosis) might occur at cumulative exposures of 400-1,330 ppm-h that encompass exposure durations of 1-4 h and concentrations of 100-693 ppm (Deringer et al. 1953; Culliford and Hewitt 1957; Kylin et al. 1963). Exposure of pregnant rats during gestation (7 h/day on gestation days 6-15) to chloroform at 30 ppm produced minor effects in the embryo and fetus, and exposure at 100 ppm was significantly embryotoxic and fetotoxic (Schwetz et al. 1974). Newell and Dilley (1978), however, found that gestational exposure of rats to chloroform at concentrations as high as 2,232 ppm (1 h/day on gestation days 7-14) did not cause developmental effects, although exposure at 4,117 ppm increased resorptions by 45% and decreased fetal body weight.
6.3. Derivation of AEGL-2
Severe hepatic toxicity, renal toxicity, and narcosis appear to be critical effects for the development of AEGL-2 values for chloroform. Human data suggest that exposures to chloroform at 8,500 ppm will induce anesthesia. The duration of exposure required is unknown, but is assumed to be on the order of a minute. Human data reported by Lehmann and Hasegawa (1910) suggest that exposure to chloroform at 7,500 ppm for 15 min or at 4,300-5,100 ppm for 20 min approached narcosis-inducing concentrations, as determined by signs and symptoms of dizziness and “intoxication.” These data and the anesthesia data of Whitaker and Jones (1965) are, however, compromised by the uncertainties with the determination of exposure concentrations and the specific concentration-duration relationships. Alternatively, the fetotoxicity reported by Schwetz et al. (1974) in rats exposed to chloroform at 100 ppm (7 h/day) on gestation days 6-15 was considered a sensitive critical effect and was selected as the point of departure for developing AEGL-2 values. It was assumed that the reported fetotoxic effects could result from a single 7-h exposure. This assumption is not without precedent, as has been shown by analyses of developmental toxicity data for other chemicals (van Raaij et al. 2003). An intraspecies uncertainty factor of 3 was applied to account for individual variability in metabolism and disposition of chloroform. No adjustment was made for interspecies variability because metabolism and kinetics data and PBPK models (Corley et al. 1990) indicate that humans are less sensitive than laboratory species to chloroform. The attenuated uncertainty factors were justified by the sensitive end point selected for AEGL-2 development and the results of another study (Newell and Dilley 1978) that showed gestational exposure of rats to chloroform at concentrations as high as 2.232 ppm (1 h/day on gestation days 7-14) was without effect.
The concentration-time relationship for many irritant and systemicallyacting vapors and gases may be described by the equation Cn × t = k, where the
exponent n ranges from 0.8 to 3.5 (ten Berge et al. 1986). In the absence of chemical-specific data to determine an empirical value for n, default values of n = 1 for extrapolation from shorter to longer durations and n = 3 for extrapolation from longer to shorter durations were used. AEGL-2 values for chloroform are presented in Table 4-11 and Appendix A.
7. DATA ANALYSIS FOR AEGL-3
7.1. Summary of Human Data Relevant to AEGL-3
Definitive lethality data for humans are not available. Although the weight of evidence indicates that acute exposure to high concentrations of chloroform might result in narcosis and subsequent death, the precise exposure concentrations and durations for such exposures are unavailable. Human data generally suggest that concentrations greater than 10,000 ppm are required for an unspecified, although short, exposure duration for surgical anesthesia. In an analysis of surgical patients anesthetized with chloroform, Whitaker and Jones (1965) reported that a concentration of 22,500 ppm also produced evidence of potentially serious cardiovascular effects. Although these data may appear compelling for development of AEGL values, it is not possible to quantify an exposure-duration relationship. Additionally, chloroform concentrations probably varied during anesthesia. This is not unexpected; anesthesia procedures with chloroform start with very high concentrations (25,000-30,000 ppm) of very short duration (2-3 min) for the purpose of inducing unconsciousness, but then lower concentrations are used to maintain surgical anesthesia (NRC 1984; ATSDR 1997). Therefore, it is unlikely that patients were exposed at the highest concentrations for AEGLspecific durations. Arrhythmias were also reported by Smith et al. (1973) in some patients anesthetized for 113 min at concentrations (at least initially) of 8,500-13,000 ppm. The available data suggest that surgical narcosis would occur at or above 8,500 ppm after short-duration exposure. It is not feasible to extrapolate to an exposure duration that would result in death.
7.2. Summary of Animal Data Relevant to AEGL-3
Data on lethality in animals after acute inhalation exposure to chloroform include studies of rats and mice. Exposure to chloroform at 3,000-8,000 ppm for 4 h resulted in 75-100% mortality in rats (lethality determined 2-3 days postexposure) (Smyth et al. 1962; Haskell Laboratory 1964), and a 4-h LC50 of 9,780 ppm was reported by Lundberg et al. (1986). For mice, mortality was 75% at 5,585 ppm for 120-min, 66% at 4,710-5,529 ppm for durations of 71-175 min, and 14% at 6,758-7,782 ppm for 35 min (Fühner 1923). However, no deaths occurred with chloroform at 2,458-5,120 ppm for 48-215 min (Fühner 1923). If
TABLE 4-11 AEGL-2 Values for Chloroform
| 10 min | 30 min | 1h | 4h | 8h |
| 120ppm (580 mg/ m3) |
80ppm (390 mg/ m3) |
64ppm (312 mg/ m3) |
40ppm (195 mg/ m3) |
29ppm (141 mg/ m3) |
the aforementioned results are converted to consider cumulative exposures, inconsistencies in the data become apparent. For example, no deaths were observed at concentrations of 2,458-5,120 ppm for 48-215 min (a maximum of 1,100,800 ppm-min); yet 66% mortality was observed at concentrations of 4,710-5,529 ppm for durations of 71-175 min (a minimum of 334,410 ppmmin). A well-conducted study by Gehring (1968) reported a 4,500-ppm LCt50 of 560 min (540-585 min, 95% CI) for female Swiss-Webster mice.
7.3. Derivation of AEGL-3
The available data do not identify a definitive lethality threshold in humans from acute exposure to chloroform. Data regarding chloroform as an anesthetic for humans suggest that very high concentrations (greater than 8,500 ppm) are tolerated for brief durations, although quantitative concentration-time data are lacking. These limitations preclude the use of the human data in the estimation of a lethality threshold for humans.
Animal data are inconsistent regarding the lethality from acute inhalation exposure to chloroform. Data on mice are highly variable, but this species appears to be the most sensitive and is affirmed by PBPK models. Exposure to chloroform at 3,000-8,000 ppm for 4 h reportedly produced 75-100% mortality in rats (Smyth et al. 1962; Haskell Laboratory 1964). Assuming the mouse to be the most sensitive species, the 560-min LC50 of 4,500 ppm reported by Gehring (1968) appears to be a valid basis for development of the AEGL-3 values. A 3-fold reduction in this value for an estimate of the lethality threshold for mice results in a point of departure of 1,500 ppm. Consistent with the Standing Operating Procedures (SOP) for developing AEGLs (NRC 2001), an exponent of 3 was applied for time scaling (Cn × t = k) because data were insufficient for empirically deriving a value for n. Because the point of departure was based on a 560-min exposure duration, the 10-min AEGL-3 value was set equivalent to the 30-min AEGL-3 value to avoid uncertainties inherent in extrapolating from 560 min to 10 min. An uncertainty factor of 3 was applied to account for potentially sensitive individuals, such as those exposed to inducers of cytochrome P-450 IIE1 (e.g., consumers of ethanol). No interspecies uncertainty factor was applied because laboratory species metabolize chloroform more rapidly than humans and are, therefore, more susceptible to the toxic effects of the more rapidly formed toxic intermediates. PBPK models (Corley et al. 1990) support this contention. Further, human anesthesia data show that cumulative exposures considerably greater than those associated with the AEGL-3 values are not lethal. A more recent study using the PBPK model to compare the metabolism of chloroform
in mice and humans demonstrated the overwhelmingly greater sensitivity of mice (primarily from a 25-to 50-fold difference in the rate of metabolism of chloroform) and the overly protective nature of typically applied uncertainty factors. These findings and the overall weight of evidence indicating the greater sensitivity of rodents to chloroform-induced toxicity justified further adjustment of the AEGL-3 values. This adjustment, applied as a weight-of evidence factor of 1/3, effectively increases the AEGL-3 values. The resulting AEGL-3 values are shown in Table 4-12 and Appendix A.
8. SUMMARY OF PROPOSED AEGLS
8.1. AEGL Values and Toxicity End Points
The AEGL values for chloroform are presented in Table 4-13.
AEGL-1 values for chloroform were not recommended because an exposure consistent with the AEGL-1 definition could not be determined. The properties of chloroform are such that the odor is not unpleasant and it is not irritating even at concentrations approaching levels inducing narcosis.
AEGL-2 values were developed using embryotoxicity and fetotoxicity in rats as the critical effect. These were considered very sensitive end points, especially with the assumption of a single-exposure response (fetotoxic effects resulting from 7-h exposures on gestation days 6-15 were assumed possible following only one 7-h exposure).
AEGL-3 values were based on an estimate of the lethality threshold for chloroform in mice.
TABLE 4-12 AEGL-3 Values for Chloroform
| lO min | 30 min | 1 h | 4 h | 8s h |
| 4,000 ppm (19,000 mg/m3) |
4,000 ppm (19,000 mg/m3) |
3,200 ppm (16,000 mg/m3) |
2,000 ppm (9,700 mg/m3) |
1,600 ppm (7,800 mg/m3) |
TABLE 4-13 AEGL Values for Chloroform
| Classification | 10 min | 30 min | 1 h | 4 h | 8 h |
| AEGL-1 (nondisabling) | NRa | NR | NR | NR | NR |
| AEGL-2 (disabling) | 120 ppm (580 mg/m3) |
80 ppm (390 mg/m3) |
64 ppm (312 mg/m3) |
40 ppm (195 mg/m3) |
29 ppm (141 mg/m3) |
| AEGL-3 (lethal) | 4,000 ppm (19,000 mg/m3) |
4,000 ppm (19,000 mg/m3 ) |
3,200 ppm (16,000 mg/m3 ) |
2,000 ppm (9,700 mg/m3 ) |
1,600 ppm (7,800 mg/m3 ) |
aNot recommended.
AEGL values were developed using an uncertainty factor of 3 for protection of sensitive individuals. Because chloroform is metabolized to toxic intermediates (phosgene) by cytochrome P-450 IIE1, induction of this enzyme (by inducers such as ethanol) potentially increases susceptibility, although it does not appear to do so by an order of magnitude (e.g., Brown et al. [1974b] reported a 2.6-fold increase in P-450 levels after induction by phenobarbitol, a more effective P-450 inducer than ethanol). Furthermore, dose rate appears to be a relevant factor in toxicity outcomes following exposure to halogenated hydrocarbons such as chloroform, a factor that might justify the application of an intraspecies uncertainty factor of less than an order of magnitude. Because of effects on P-450 and GSH levels, single exposures result in toxic outcomes that are different from those for repeated exposures. Available data and application of pharmacokinetic modeling indicate that rodents metabolize chloroform more rapidly than humans. Therefore, the application of an interspecies uncertainty factor was minimized. Furthermore, human data indicate that cumulative exposures of >675,000 ppm-min and exposures at 22,500 ppm for up to 120 min resulted in surgical anesthesia and cardiac irregularities but not death. These data suggest that the AEGL-3 values represent a no-observed-adverse-effect level for lethality.
When compared with occupational exposure data reported by Challen et al. (1958) for pharmaceutical workers, the AEGL values appear to be sufficiently protective. Workers exposed to chloroform at 71 ppm (4 h/day for 10-24 months) experienced mild symptoms (dryness of mouth and throat) whereas workers exposed at 77-232 ppm over a period of 3-10 years exhibited notable signs of exposure (staggering). These findings are the result of repeated exposures to chloroform, and the study did not specify if any of the workers represented a sensitive population.
8.2. Comparison with Other Standards and Guidelines
Standards and guidance values for workplace and community exposures to chloroform are presented in Table 4-14. The cancer notation for some of the criteria was not considered appropriate for AEGL values.
8.3. Data Quality and Research Needs
Much of the human data on chloroform are from older studies that lacked information on the analytic techniques used to determine exposure concentrations. Human anesthesia data focus on initial concentration and duration of anesthesia, and were not sufficient for developing AEGL values.
The most obvious data deficiency regarding development of AEGL values for chloroform is the lack of data with which to determine a lethality threshold. There is also a paucity of reliable data demonstrating definitive concentrationresponse relationships. Human data are deficient in exposure-time relationships
or are unreliable and difficult to validate. The animal data are variable. Acute exposure studies providing exposure-response data for specific toxicity end points (e.g., hepatotoxicity, renal toxicity, narcosis threshold, lethality) in two or more species would be desirable.
TABLE 4-14 Extant Standards and Guidelines for Chloroform
|
|
|||||
| Exposure Duration | |||||
| Guideline | 10 min | 30 min | l h | 4 h | 8 h |
|
|
|||||
| AEGL-1 (Nondisabling) | NR | NR | NR | NR | NR |
| AEGL-2 (Disabling) | 120 ppm | 80 ppm | 64 ppm | 40 ppm | 29 ppm |
| AEGL-3 (Lethal) | 4,000 ppm | 4,000 ppm | 3,200 ppm | 2,000 ppm | 1,600 ppm |
| ERPG-1 (AIHA)a | NA | ||||
| ERPG-2 (AIHA) | 50 ppm | ||||
| ERPG-3 (AIHA) | 5,000 ppm | ||||
| EEL (NRC)b | 200 ppm (30 ppm, 24 h) | ||||
| IDLH (NIOSH)c | 500 ppm | ||||
| TLV-TWA (ACGIH)d | 10 ppm | ||||
| REL-TWA (NIOSH)e | 2 ppm (60min) | ||||
| PEL-C (OSHA)f | 50 ppm | ||||
| MAK(Germany)g | 0.5ppm | ||||
| MAC (the Netherlands)h | 5ppm (15 min) | 1 ppm | |||
|
|
|||||
aERPG (emergency response planning guideline, American Industrial Hygiene Association (AIHA 2010)
ERPG-1 is the maximum airborne concentration below which it is believed nearly all individuals could be exposed for up to 1 h without experiencing other than mild, transient adverse health effects or without perceiving a clearly defined objectionable odor.
ERPG-2 is the maximum airborne concentration below which it is believed nearly all individuals could be exposed for up to 1 h without experiencing or developing irreversible or other serious health effects or symptoms that could impair an individual’s ability to take protection action.
ERPG-3 is the maximum airborne concentration below which it is believed nearly all individuals could be exposed for up to 1 h without experiencing or developing lifethreatening health effects.
b EEL (emergency exposure guidance level, National Research Council) (NRC 1984) is a ceiling concentration that will not cause irreversible harm or prevent performance of essential tasks, such as closing a hatch or using a fire extinguisher, during a rare emergency situation usually lasting 1-24 h.
cIDLH (immediately dangerous to life or health, National Institute for Occupational Safety and Health, NIOSH 1994) represents the maximum concentration from which one could escape within 30 min without any escape-impairing symptoms, or any irreversible health effects. IDLH carries a cancer notation.
dTLV-TWA (threshold limit value - time weighted average, American Conference of Governmental Industrial Hygienists) is the time-weighted average concentration for a normal 8-h workday and a 40-h workweek, to which nearly all workers may be repeatedly exposed, day after day, without adverse effect (ACGIH 2011).
eREL-TWA (recommended exposure limit - time weighted average, National Institute for Occupational Safety and Health) is defined analogous to the TLV-TWA, with cancer notation (NIOSH 2011).
fPEL-C (permissible exposure limit-ceiling, Occupational Health and Safety Administration)is a value that must not be exceeded during any part of the workday (NIOSH 2011).
gMAK (maximale argeitsplatzkonzentration [maximum workplace concentration], German Research Association) (DFG 2005) is defined analogous to the ACGIH TLV-TWA. Cancer category 4 noted.
hMAC (maximaal aanvaaarde concentratie [maximal accepted concentration]), Dutch Expert Committee for Occupational Standards, The Netherlands(MSZW 2004) is defined analogous to the ACGIH TLV-TWA.
9. REFERENCES
ACGIH (American Conference of Government Industrial Hygienists). 2011. TLVs® and BEIs®. American Conference of Government Industrial Hygienists, Cincinnati, OH.
AIHA (American Industrial Hygiene Association). 1989. Odor Thresholds for Chemicals with Established Occupational Health Standards. Akron, OH: American Industrial Hygiene Association.
AIHA (American Industrial Hygiene Association). 2010. Emergency Response Planning Guidelines: Chloroform. Fairfax, VA: AIHA Press.
ATSDR (Agency for Toxic Substances and Disease Registry) 1997. Toxicological Profile for Chloroform. Update. U.S. Department Health and Human Services, Agency for Toxic Substances and Disease Registry, Atlanta, GA.
Baeder, C., and T. Hoffman. 1988. Initial Submission: Inhalation Embryotoxicity Study of Chloroform in Wistar Rats (Final Report) with Attachment and Cover Letter Dated 2/21/92. Pharma Research Toxicology and Pathology, Hoechst Aktiengesellschaft, Frankfurt. Submitted to U.S. Environmental Protection Agency by Occidental Chemical Corporation. EPA Document No. 88-920001208.
Baeder, C., and T. Hoffman. 1991. Initial Submission. Chloroform: Supplementary Inhalation Embryotoxicity Study in Wistar Rats (Final Report) with Attachment and Cover Letter Dated 12/24/91. Hoechst Aktiengesellschaft, Frankfurt. EPA Document No. 88-92000566.
Bomski, H., A. Sobolewska, and A. Strakowski. 1967. Toxic damage of the liver by chloroform in chemical industry workers [in German]. Int. Arch. Arbeitsmed. 24(2):127-134.
Branchflower, R.V., D.S. Nunn, R.J. Highet, J.H. Smith, J.B. Hook, and L.R. Pohl. 1984. Nephrotoxicity of chloroform: Metabolism to phosgene by the mouse kidney.Toxicol. Appl. Pharmacol. 72(1):159-168.
Brondeau, M.T., P. Bonnet, J.P. Guenier, and J. De Ceaurriz. 1983. Short-term inhalation test for evaluating industrial hepatotoxicants in rats. Toxicol. Lett. 19(1-2):139-146.
Brown, D.M., P.F. Langley, D. Smith, and D.C. Taylor. 1974a. Metabolism of chloroform-I. The metabolism of [14C]-chloroform by different species. Xenobiotica 4(3):151-163.
Brown, B.B., I.G. Sipes, and A.M. Sagalyn. 1974b. Mechanisms of acute hepatic toxicity: Chloroform, halothane, and glutathione. Anesthesiology 41(6):554-561.
Budavari, S., M.J. O'Neil, A. Smith, P.E. Heckelman, and J.F. Kinneary, eds. 1996. Chloroform. P. 360 in The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 12th Ed. Whitehouse, NJ: Merck.
Butterworth, B.E., M.V. Templin, S.J. Borghoff, R.B. Conolly, G.L. Kedderis, and D.C.Wolf. 1995. The role of regenerative cell proliferation in chloroform-induced cancer. Toxicol. Lett. 82/83:23-26.
Challen, P.J., J. Bedford, and D.E. Hickish. 1958. Chronic chloroform intoxication. Br. J. Ind. Med. 15(4):243-249.
Conolly, R.B. 1995. Cancer and non-cancer risk assessment: Not so different if you consider mechanisms. Toxicology 102(1-2):179-188.
Corley, R.A., A.L. Mendrala, F.A. Smith, D.A. Staats, M.L. Gargas, R.B. Conolly, M.E. Andersen, and R.H. Reitz. 1990. Development of a physiologically based pharmacokinetic model for chloroform. Toxicol. Appl. Pharmacol. 103(3):512-527.
Crump, K.S., and R.B. Howe. 1984. The multistage model with a time-dependent dose pattern: Applications to carcinogenic risk assessment. Risk Anal. 4(3):163-176.
Culliford, D., and H.B. Hewitt. 1957. The influence of sex hormone status on the susceptibility of mice to chloroform-induced necrosis of the renal tubules. J. Endocrinol. 14(4):381-393.
Danielsson, B.R., H. Ghantous, and L. Dencker. 1986. Distribution of chloroform and methyl chloroform and their metabolites in pregnant mice. Biol. Res. Pregnancy Perinatol. 7(2):77-83.
Delic, J.I., P.D. Lilly, A.J. MacDonald, and G.D. Loizou. 2000. The utility of PBPK in the safety assessment of chloroform and carbon tetrachloride. Regul. Toxicol. Pharmacol. 32(2):144-155.
Deringer, M.K., T.B. Dunn, and W.E. Heston. 1953. Results of exposure of strain C3H mice to chloroform. Proc. Soc. Exp. Biol. Med. 83(3):474-479.
DeShon, H.D. 1978. Chloroform. Pp. 693-703 in Kirk-Othmer Encyclopedia of Chemical Technology, 3rd Ed., M. Grayson, and D. Eckroth, eds. New York: Wiley.
DFG (Deutsche Forschungsgemeinschaft). 2005. List of MAK and BAT Values 2005.
Maximum Concentrations and Biological Tolerance Values at the Workplace Report No. 41. Weinheim, Federal Republic of Germany: Wiley VCH.
Docks, E., and G. Krishna. 1976. The role of glutathione in chloroform-induced hepatotoxicity. Exp. Mol. Pathol. 24(1):13-22.el-Shenawy, N., and M.S. Abdel-Rahman. 1993. The mechanism of chloroform toxicity in isolated rat hepatocytes. Toxicol. Lett. 69(1):77-85.
EPA (U. S. Environmental Protection Agency). 1992. Reference Guide to Odor Thresholds for Hazardous Air Pollutants Listed in the Clean Air Act Amendments of 1990. EPA/600/R-92/047. Office of Research and Development, U. S. Environmental Protection Agency, Washington, DC. March 1992 [online]. Available: http://www.epa.gov/ttn/atw/odorguide1992.pdf [accessed Feb. 13, 2012].
EPA (U. S. Environmental Protection Agency). 2001. Chloroform (CASRN 67-66-3): Carcinogenicity Assessment for Lifetime Exposure. Integrated Risk Information
System, U. S. Environmental Protection Agency [online]. Available: http://www.epa.gov/iris/subst/0025.htm [accessed Feb. 15, 2012].
EPA (U. S. Environmental Protection Agency). 2012. Chloroform Quickview (CARN 67-66-3). Integrated Risk Information System, U. S. Environmental Protection Agency [online]. Available: http://cfpub.epa.gov/ncea/iris/index.cfm?fuseaction=iris,showQuickView&substance_numbr=0025#carc [accessed Feb. 15, 2012].
Fry, B.J., T. Taylor, and D.E. Hathway. 1972. Pulmonary elimination of chloroform and its metabolite in man. Arch. Int. Pharmacodyn. Ther. 196(1):98-111.
Fühner, H. 1923. Relative potencies of chloroform and carbon tetrachloride [in German]. Arch. Exp. Pathol. 97:86-112.
Gehring, P.J. 1968. Hepatotoxic potency of various chlorinated hydrocarbon vapours relative to their narcotic and lethal potencies in mice. Toxicol. Appl. Pharmacol. 13(3):287-298.
Gettler, A.O., and H. Blume. 1931. Chloroform in the brain, lungs and liver: Quantitative recovery and determination. Arch. Pathol. 11:554-560.
Golden, R.J., S.E. Holm, D.E. Robinson, P.H. Julkunen, and E.A. Reese. 1997. Chloroform mode of action: Implications for cancer risk assessment. Regul. Toxicol. Pharmacol. 26(2):142-155.
Haskell Laboratory. 1964. Inhalation Toxicity Study on Freon-113, Freon TC, and Chloroform. Haskell Laboratory Report No. 135-64. EPA Document No. 86-870000965. Microfiche No. OTS0514867.
Hutchens, K.S., and M. Küng. 1985. “Experimentation” with chloroform. Am. J. Med. 78(4):715-718.
Ikatsu, H., and T. Nakajima. 1992. Hepatotoxic interaction between carbon tetrachloride and chloroform in ethanol treated rats. Arch. Toxicol. 66(8):580-586.
Jorgenson, T.A., E.F. Meierhenry, C.J. Rushbrook, R.J. Bull, and M. Robinson. 1985. Carcinogenicity of chloroform in drinking water to male Osborne-Mendel rats and female B6C3F1 mice. Fundam. Appl. Toxicol. 5(4):760-769.
Kennedy, S.K., and D.E. Longnecker. 1996. History and principles of anesthesiology. Pp. 295-306 in Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 9th Ed., J.G. Hardman, L.E. Limbird, P.B. Molinoff, R.W. Ruddon, and A.G. Gilman, eds. New York: McGraw-Hill.
Kylin, B., H. Reichard, I. Sümegi, and S. Yllner. 1963. Hepatotoxicity of inhaled trichloroethylene, tetrachloroethylene and chloroform. Single exposure. Acta Pharmacol. Toxicol. 20:16-26.
Land, P.C., E.L. Owen, and H.W. Linde. 1981. Morphologic changes in mouse spermatozoa after exposure to inhalation anesthetics during early spermatogenesis. Anesthesiology 54(1):53-56.
Larson, J.L., D.C. Wolf, K.T. Morgan, S. Méry, and B.E. Butterworth. 1994. The toxicity of 1-week exposures to inhaled chloroform in female B6C3F1 mice and male F-344 rats. Fundam. Appl. Toxicol. 22:431-446.
Larson, J.L., M.V. Templin, D.C. Wolf, K.C. Jamison, J.R. Leininger, S. Méry, K.T. Morgan, B.A. Wong, R.B. Conolly, and B.E. Butterworth. 1996. A 90-day chloroform inhalation study in female and male B6C3F1 mice: Implications for cancer risk assessment. Fundam. Appl. Toxicol. 30(1):118-137.
Lehmann, K.B., and F. Flury, eds. 1943. Chloroform (trichloromethanes). Pp. 138-145 in Toxicology and Hygiene of Industrial Solvents. Baltimore, MD: Williams and Wilkins.
Lehmann, K.B., and D. Hasegawa. 1910. Studies on the absorption of chlorinated hydrocarbons in animals and humans. Arch. Hyg. 72:327-342.
Lehmann, K.B., and L. Schmidt-Kehl. 1936. The thirteen most important chlorinated aliphatic hydrocarbons from the standpoint of industrial hygiene [in German]. Arch. Hyg. 116:131-268.
Li, L.H., X.Z. Jiang, Y.X. Liang, Z.Q. Chen, Y.F. Zhou, and Y.L. Wang. 1993. Studies on the toxicity and maximum allowable concentration of chloroform. Biomed. Environ. Sci. 6(2):179-186.
Lundberg, I., M. Ekdahl, T. Kronevi, V. Lidums, and S. Lundberg. 1986. Relative hepatotoxicity of some industrial solvents after intraperitoneal injection or inhalation exposure in rats. Environ. Res. 40(2):411-420.
Mansuy, D., P. Beaune, T. Cresteil, M. Lange, and J.P. Leroux. 1977. Evidence for phosgene formation during liver microsomal oxidation of chloroform. Biochem. Biophys. Res. Commun. 79(2):513-517.
McDonald, M.N., and D.E. Vire. 1992. Chloroform in the endodontic operatory. J. Endod. 18(6):301-303.
Melnick, R.L., M.C. Kohn, J.K. Dunnick, and J.R. Leininger. 1998. Regenerative hyperplasia is not required for liver tumor induction in female B6C3F1 mice exposed to trihalomethanes. Toxicol. Appl. Pharmacol. 148(1):137-147.
Méry, S., J.L. Larson, B.E. Butterworth, D.C. Wolf, R. Harden, and K.T. Morgan. 1994. Nasal toxicity of chloroform in male F-344 rats and female B6C3F1 mice following a 1-week inhalation exposure. Toxicol. Appl. Pharmacol. 125(2):214-227.
MSZW (Ministerie van Sociale Zaken en Werkgelegenheid). 2004. Nationale MAC-lijst 2004: Chloroform. Den Haag: SDU Uitgevers [online]. Available: http://www.lasrook.net/lasrookNL/maclijst2004.htm [accessed Feb. 13, 2012].
Murray, F.J., B.A. Schwetz, J.G. McBride, and R.E. Staples. 1979. Toxicity of inhaled chloroform in pregnant mice and their offspring. Toxicol. Appl. Pharmacol. 50(3): 515-522.
NCI (National Cancer Institute). 1976. Report on the Carcinogenesis Bioassay of Chloroform. DHEW (NIH) 76-1279. U.S. Department of Health, Education, and Welfare, Public Health Service, National Institute of Health, National Cancer Institute, Bethesda, MD [online]. Available: http://ntp.niehs.nih.gov/ntp/htdocs/lt_rpts/trchloroform.pdf [accessed Feb. 13, 2012].
Newell, G.W., and J.V. Dilley. 1978. Teratology and Acute Toxicology of Selected Chemical Pesticides Administered by Inhalation. Report by Stanford Research Institute, Menlo Park, CA, for Health Effects Research Laboratory, Office of Research and Development, U.S. Environmental Protection Agency, Research Triangle Park, NC (as cited in ATSDR 1997).
NIOSH (National Institute for Occupational Safety and Health). 1994. Documentation for Immediately Dangerous to Life or Health Concentrations (IDLHs): Chloroform. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, Cincinnati, OH [online]. Available: http://www.cdc.gov/niosh/idlh/67663.html [accessed Feb. 13, 2012].
NIOSH (National Institute for Occupational Safety and Health). 2011. NIOSH Pocket Guide to Chemical Hazards: Chloroform. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, Cincinnati, OH [online]. Available: http://www.cdc.gov/niosh/npg/npgd0127.html [accessed Feb. 13, 2012].
NRC (National Research Council), 1984. Pp. 57-76 in Emergency and Continuous Exposure Guidance Levels for Selected Airborne Contaminants, Vol. 1. Washington, DC: National Academy Press.
NRC (National Research Council). 1993. Guidelines for Developing Community Emergency Exposure Levels for Hazardous Substances. Washington, DC: National Academy Press.
NRC (National Research Council). 2001. Standing Operating Procedures for Developing Acute Exposure Guideline Levels for Hazardous Chemicals. Washington, DC: National Academy Press.
Pohl, L.R., B. Bhooshan, N.F. Whittaker, and G. Krishna. 1977. Phosgene: A metabolite of chloroform. Biochem. Biophys. Res. Comm. 79(3):684-691.
Pohl, L.R., R.V. Branchflower, R.J. Highet, J.L. Martin, D.S. Nunn, T.J. Monks, J.W. George, and J.A. Hinson. 1981. The formation of diglutathionyl dithiocarbonate as a metabolite of chloroform, bromotrichloromethane, and carbon tetrachloride. Drug Metab. Dispos. 9(4):334-339.
Pohl, L.R., J.W. George, and H. Satoh. 1984. Strain and sex differences in chloroforminduced nephrotoxicity: Different rates of metabolism of chloroform to phosgene by the mouse kidney. Drug. Metab. Dispos. 12(3):304-308.
Puri, S.K., G.C. Fuller, and H. Lal. 1971. Effect of chloroform inhalation on barbiturate narcosis and metabolism in normal and phenobarbital pretreated rats. Pharmacol. Res. 3:247-254.
Schwetz, B.A., B.K. Leong, and P.J. Gehring. 1974. Embryo-and fetotoxicity of inhaled chloroform in rats. Toxicol. Appl. Pharmacol. 28(3):442-451.
Smith, A.A., P.P. Volpitto, Z.W. Gramling, M.B. DeVore, and A.B. Glassman. 1973. Chloroform, halothane, and regional anesthesia: A comparative study. Anesth. Analg. 52(1):1-11.
Smyth, H.F., C.P. Carpenter, C.S. Weil, U.C. Pozzani, and J.A. Striegel. 1962. Rangefinding toxicity data: List VI. Am. Ind. Hyg. Assoc. J. 23:95-107.
Snyder, R., and L.S. Andrews. 1996. Toxic effects of solvents and vapors. Pp. 737-772 in Casarett and Doull’s Toxicology: The Basic Science of Poisons, 5th Ed., C.D. Klaassen, M.O. Amdur, and J. Doull, eds. New York: McGraw Hill.
Templin, M.V., J.L. Larson, B. Butterworth, K.C. Jamison, J.R. Leininger, S. Méry, K.T.Morgan, B.A. Wong, and D.C. Wolf. 1996a. A 90-day chloroform inhalation study in F-344 rats: Profile of toxicity and relevance to cancer studies. Fundam. Appl. Toxicol. 32(1):109-125.
Templin, M.V., K.C. Jamison, C.S. Sprankle, D.C. Wolf, B.A. Wong, and B.E. Butterworth. 1996b. Chloroform-induced cytotoxicity and regenerative cell proliferation in the kidneys and liver of BDF1 mice. Cancer Lett. 108(2):225-231.
ten Berge, W.F., A. Zwart, and L.M. Appelman. 1986. Concentration-time mortality response relationship of irritant and systemically acting vapours and gases. J. Hazard. Mater. 13(3):301-309.
van Raaij, M.T.M., P.A.H. Janssen, and A.H. Piersma. 2003. The Relevance of Developmental Toxicity Endpoints for Acute Limit Setting. RIVM Report 601900004/2003. Rijksinstituut voor Volksgezondheid en Milieu [online]. Available: http://www.epa.gov/oppt/aegl/pubs/meetings/mtg35b.pdf [accessed Feb. 13, 2012].
Von Oettingen, W.F., C.C. Powell, N.E. Sharpless, W.C. Alford, and L.J. Pecora. 1949. Relation Between the Toxic Action of Chlorinated Methanes and Their Chemical and Physicochemical Properties. National Institutes of Health Bulletin No 191. Washington, DC: U.S. Government Printing Office.
Wang, P.Y., T. Kaneko, H. Tsukada, and A. Sato. 1994. Dose and route dependency of metabolism and toxicity of chloroform in ethanol-treated rats. Arch. Toxicol. 69(1):18-23.
Wang, P.Y., T. Kaneko, H. Tsukada, M. Nakano, and A. Sato. 1997. Dose-and routedependent alterations in metabolism and toxicity of chemical compounds in ethanol-treated rats: Difference between highly (chloroform) and poorly (carbon tetrachloride) metabolized hepatotoxic compounds. Toxicol. Appl. Pharmacol. 142(1):13-21.
Wennborg, H., L. Bodin, H. Vainio, and G. Axelsson. 2000. Pregnancy outcome of personnel in Swedish biomedical research laboratories. J. Occup. Environ. Med. 42(4):438-446.
Whipple, G.H., and J.A. Sperry. 1909. Chloroform poisoning-liver necrosis and repair.Bull. Johns Hopkins Hosp. 20:278-289 (as cited in NRC 1984).
Whitaker, A.M., and C.S. Jones. 1965. Report of 1500 chloroform anesthetics administered with a precision vaporizer. Anesth. Analg. 44:60-65.
Wolf, D.C., and B.E. Butterworth. 1997. Risk assessment of inhaled chloroform based on its mode of action. Toxicol. Pathol. 25(1):49-52.
Yamamoto, S., S. Aiso, N. Ikawa, and T. Matsushima. 1994. Carcinogenesis studies of chloroform in F344 rats and BDF1 mice [abstract]. Proceedings of the 53rd Annual Meeting of the Japanese Cancer Association (cited in Templin et al. 1996b).
APPENDIX A
DERIVATION SUMMARIES OF AEGL VALUES FOR CHLOROFORM
Derivation of AEGL-1 Values
AEGL-1 values were not recommended because it was not possible to identify a definitive effect consistent with the AEGL-1 definition. Concentrations of chloroform approaching those inducing narcosis or hepatic and renal effects are not accompanied by overt signs or symptoms. Furthermore, chloroform is not irritating and its odor is not unpleasant.
Derivation of AEGL-2 Values
| Key study: | Schwetz, B.A., B.K. Leong, and P.J. Gehring. 1974. Embryo-and fetotoxicity of inhaled chloroform in rats. Toxicol. Appl. Pharmacol. 28(3):442-451. |
| Toxicity end point: | No developmental effects in rats. |
| Time scaling: | Cn × t = k (default n = 3 for longer to shorter exposure durations; n = 1 for shorter to longer exposure durations) (100 ppm)1 × 7 h = 700 ppm-h (100 ppm) 3 × 7 h = 7000 ppm-h |
| Uncertainty factors: | An interspecies uncertainty factor was not applied because the available metabolism and kinetics data and PBPK models (Corley et al. 1990) indicate that humans may be less sensitive than laboratory animals to chloroform. Additional adjustments were considered unnecessary because a single 7-h exposure was assumed to produce effects rather than the full-exposure period specified in the study protocol (7 h/day on gestation days 6-15). 3 for intraspecies variability in metabolism and disposition of chloroform. Additional adjustment was not made because the point of departure and the assumption of a single-exposure effect were considered conservative. |
| Total uncertainty factor of 3 | |
| 10-min AEGL-2: | C3 × 0.1667 h = 7,000,000 ppm-h C = 348 ppm 348 ppm ÷ 3 = 120 ppm (rounded) |
| 30-min AEGL-2: | C3 × 0.5 h = 7,000,000 ppm-h C = 241 ppm 241 ppm ÷ 3 = 80 ppm (rounded) |
| 1-h AEGL-2: | C3 × 1 h = 7,000,000 ppm-h C = 191 ppm 191 ppm ÷ 3 = 64 ppm (rounded) |
| 4-h AEGL-2: | C3 × 4 h = 7,000,000 ppm-h C = 121 ppm 121 ppm ÷ 3 = 40 ppm (rounded) |
| 8-h AEGL-2: | C1 × 8 h = 700 ppm-h C = 87.5 ppm 87.5 ppm ÷ 3 = 29 ppm (rounded) |
| Derivation of AEGL-3 Values | |
| Key study: | Gehring, P.J. 1968. Hepatotoxic potency of various chlorinated hydrocarbon vapours relative to their narcotic and lethal potencies in mice. Toxicol. Appl. Pharmacol. 13(3):287-298. |
| Toxicity end point: | Lethality; 3-fold reduction in a 560-min LC50 of 4,500 ppm in mice was assumed to be a threshold for lethality (4,500 ppm ÷ 3 = 1,500 ppm). |
| Scaling: | Cn × t = k (default n = 3 for longer to shorter exposure durations; n = 1 for shorter to longer exposure durations) (1,500 ppm) 3 × 9.3 h = 3.1 × 1010 ppm3-h |
| Uncertainty factors: | An interspecies uncertainty factor was not applied because the available metabolism and kinetics data and PBPK models (Corley et al. 1990) indicate that humans may be less sensitive than laboratory animals to chloroform. |
| 3 for intraspecies variability in metabolism and disposition of chloroform (e.g., induction of P-450 enzymes and subsequent enhancement of toxicity). Comparison with available anesthesia data in humans precluded incorporation of additional uncertainty factor adjustment. Because results of PBPK models (Corley et al.1990; Delic et al. 2000) show that mice are considerably more sensitive (25-to 50-fold difference in rate of metabolism of chloroform) to the toxic effects of inhaled chloroform than are humans, an additional adjustment factor of 1/3 was applied and resulted in an overall net adjustment of 1. |
|
| 10-min AEGL-3: | Set equivalent to the 30-min value of 4,000 ppm to minimize uncertainty associated with extrapolating a 560-min exposure duration to 10 min. |
| 30-min AEGL-3: | C3 × 0.5 h = 3.1 × 1010 ppm3-h C = 4,000 ppm (rounded) |
| 1-h AEGL-3: | C3 × 1 h = 3.1 × 1010 ppm3-h C = 3,200 ppm (rounded) |
| 4-h AEGL-3: | C3 × 4 h = 3.1 × 1010 ppm3-h C = 2,000 (rounded) |
| 8-h AEGL-3: | C3 × 8 h = 3.1 × 1010 ppm3-h C = 1,600 (rounded) |
APPENDIX B
ACUTE EXPOSURE GUIDELINE LEVELS FOR CHLOROFORM
Derivation Summary for Chloroform
AEGL-1 VALUES
AEGL-1 values were not recommended because it was not possible to identify a definitive effect consistent with the AEGL-1 definition. Concentrations of chloroform approaching those inducing narcosis or hepatic and renal effects are not accompanied by overt signs or symptoms. Furthermore, chloroform is not irritating and its odor is not unpleasant.
| AEGL-2 VALUES | ||||
| 10 min | 30 min | l h | 4 h | 8 h |
| 120 ppm | 80 ppm | 64 ppm | 40 ppm | 29 ppm |
Reference: Schwetz, B.A., B.K. Leong, and P.J. Gehring. 1974. Embryo- and fetotoxicity of inhaled chloroform in rats. Toxicol. Appl. Pharmacol. 28(3):442-451.
Test species/Strain/Number: Sprague Dawley rats; 68, 8, 22, 23, and 3 dams for the control, pair-fed control, low-, mid-, and high-concentration groups, respectively.
Exposure route/Concentrations/Durations: Inhalation (whole body); 0, 30, 100, or 300 ppm, 7 h/day on gestation days 6-15.
Effects:
| Effect (litters affected litters examined) | Control | Pair-fed | 30 ppm | 100ppma | 300ppm |
|
|
|||||
| Total gross anomalies | 1/68 | 0/8 | 0/22 | 3/23b | 0/3 |
| Total skeletal anomalies | 46/68 | 3/8 | 20/22b | 17/23 | 2/3 |
| Total soft-tissue anomalies | 33/68 | 3/8 | 10/22 | 15/23 | 1/3 |
| Fetal body weight (g) | 5.69 | 5.19 | 5.51 | 5.59 | 3.42b |
| Fetal crown-rump length (mm) | 43.5 | 42.1 | 42.5b | 43.6 | 36.9b |
| aDeterminant for AEGL-2 (100 ppm); although the effects reported in the study were the result of 7-h exposures on gestation days 6-15, it was assumed that the effects were the result of a single 7-h exposure. | |||||
| bp < 0.05. | |||||
End point/Concentration/Rationale: Fetotoxicity (total gross anomalies), 7-h exposure at 100 ppm. It was assumed that a single 7-h exposure would produce the same effects as the 10-day exposure used in the study. Fetotoxicity was considered a sensitive indicator of potential serious and irreversible effects in a susceptible population.
|
|
||||
| 10 min | 30 min | 1 h | 4 h | 8 h |
| 120 ppm | 80 ppm | 64 ppm | 40 ppm | 29 ppm |
|
|
||||
Uncertainty factors/Rationale:
Total uncertainty factor: 3
Interspecies: None; metabolism and kinetics data and PBPK models (Corley et al. 1990) indicate that humans are less sensitive than rats to chloroform.
Intraspecies: 3 for individual variability in metabolism and disposition of chloroform and protection of individuals with altered metabolism and disposition (e.g., consumers of alcohol); the fetuses are a sensitive population but a larger uncertainty factor is unwarranted because the critical study involved effects on the fetus.
Modifying factor: None
Animal-to-human dosimetric adjustments: Insufficient data.
Time scaling: The concentration-time relationship for many irritant and systemically acting vapors and gases may be described by Cn × t = k, where the exponent n ranges from 0.8 to 3.5 (ten Berge et al. 1986). In the absence of chemical-specific data, temporal scaling was performed using n = 3 when extrapolating to shorter durations and n = 1 when extrapolating to longer durations.
Data adequacy: A conservative approach to select the point of departure was used by assuming that a single 7-h exposure would result in fetotoxicity. The values are considered to be protective of human health consistent with the AEGL-2 definition.
AEGL-3 VALUES |
||||
|
|
||||
| 10 min | 30 min | 1 h | 4 h | 8 h |
|
|
||||
| 4,000 ppm | 4,000 ppm | 3,200 ppm | 2,000 ppm | 1,600 ppm |
|
|
||||
Reference: Gehring, P.J. 1968. Hepatotoxic potency of various chlorinated hydrocarbon vapours relative to their narcotic and lethal potencies in mice. Toxicol. Appl. Pharmacol. 13(3):287-298.
Toxicol. Appl. Pharmacol. 13(3):287-298.
Test species/Strain/Number: Female Swiss-Webster mice (20/group)
Exposure route/Concentrations/Durations: Inhalation, various concentrations and durations
Effects: Lethality, 4,500-ppm LCt50 of 560 min (540-585 min, 95% CI)
End point/Concentration/ Rationale: Lethality threshold estimated by reducing the 560-min LC50 of 4,500 ppm by a factor of 3.
Uncertainty Factors/Rationale:
Total uncertainty factor: 1
Interspecies: None; laboratory animals metabolize chloroform more rapidly than humans and are, therefore, probably to be more susceptible to the toxic effects of the more rapidly formed toxic intermediates. PBPK models (Corley et al. 1990) also support not applying an uncertainty factor.
|
|
||||
| 10 min | 30 min | 1h | 4h | 8h |
|
|
||||
| 4,000 ppm | 4,000 ppm | 3,200 ppm | 2,000 ppm | 1,600 ppm |
|
|
||||
Intraspecies: 3 to account for individual variability in the sensitivity to chloroforminduced toxicity (e.g., alcohol-potentiated hepatotoxicity). An additional adjustment (weight-of-evidence factor of 1/3) was applied to account for the PBPK findings indicating that the mouse is more susceptible to chloroform.
Modifying factor: None applied.
Animal-to-human dosimetric adjustments: Insufficient data.
Time scaling: The concentration-time relationship for many irritant and systemically acting vapors and gases may be described by Cn × t = k (ten Berge et al. 1986), where the exponent n ranges from 0.8 to 3.5. In the absence of chemical-specific data, temporal scaling was performed using the default of n = 3 when extrapolating to shorter durations.
Data adequacy: Human lethality data are lacking and lethality data in laboratory animals have limitations. However, when compared with human anesthesia data, the AEGL-3 values appear to be sufficiently protective. PBPK models affirm that rodents, especially mice, are a considerably more sensitive species than humans to chloroform.
APPENDIX C
CATEGORY GRAPH FOR CHLOROFORM
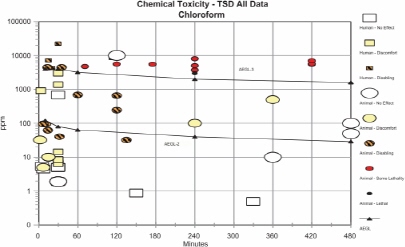
FIGURE C-1 Category graph of toxicity data and AEGLs values for chloroform.
APPENDIX D
CARCINOGENICITY ASSESSMENT FOR CHLOROFORM
Cancer Assessment of Chloroform
The cancer inhalation unit risk for chloroform is 2.3 × 10-5 per (μg/m3) (EPA 2001, 2012), and is based on a tumorigenic response (hepatocellular carcinomas) in B6C3F1 mice administered chloroform by gavage (NCI 1976). On the basis of this unit risk, the upper-bound unit risks of 10-4 to 10-7 are 4 × 10-3 to 4 × 10-6 mg/m3, assuming an inhalation rate of 20 m3/day for a 70 kg individual. At the 10-4 risk level, the virtually safe dose (d) is 4 μg/m3.
A 70-year exposure may be converted to a 24-h exposure by the following calculation:
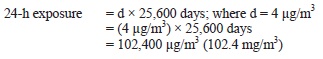
To account for uncertainty in the variability in the stage at which chloroform or its metabolites may act on the cancer process, a multistage factor of 6 is applied (Crump and Howe 1984):
![]()
Therefore, based on the potential carcinogenicity of chloroform, an acceptable 24-h exposure would be 17.07 mg/m3 (3.58 ppm). If the exposure is limited to a fraction (f) of a 24-h period, the fractional exposure becomes 1/f × 24 h (NRC 1984), resulting in the following values:
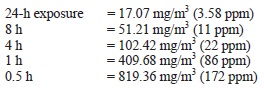
The AEGL-2 values based on acute toxicity were somewhat greater than the values derived based on potential carcinogenicity. However, the data are compelling that the carcinogenic response to chloroform has a threshold, such that repeated exposures are needed that result in tissue necrosis and regeneration.
A virtually safe dose of 0.01 ppm (48.7 μg/m3) was derived by Butterworth et al. (1995) and Wolf and Butterworth (1997) based on a no-observedadverse-effect level of 10 ppm in mice and the assumption that the tumorigenic response was secondary to necrosis and regenerative cell proliferation (a threshold response). Cancer risk based on this approach is 12-fold less than those derived from the 10-4 unit risk number.



















































