
at least some of the specializations that have been proposed are amenable to testing with noninvasive experimental techniques appropriate for the study of humans and apes.
The ability to sequence the whole genome of a species, along with other advances in molecular biology and in bioinformatics, has ushered in a remarkable new era of human evolutionary studies. We might reasonably expect that these developments have advanced our understanding of the evolution of the human brain and its functional capacities. Here, I will argue that this is the case, although the path connecting genes to phenotypes is not as straight as one might suppose.
COMPARATIVE GENETIC AND MOLECULAR BACKGROUND
To appreciate how far we have come in this field, and what we have yet to accomplish, it is useful to note where we were in the late 1990s, just before the comparative genomics revolution. What were scientists’ expectations about the kinds of molecular changes that occurred in human evolution? What was the nature of the phenotypic changes that they expected to explain or illuminate with comparative molecular studies?
It has long been understood that the evolution of biological features that do not fossilize, including molecules, can be reconstructed by comparing appropriately chosen species. Human specializations are, by definition, features of the human species that evolved in our lineage after it separated from the lineage leading to chimpanzees and bonobos, our closest relatives. A claim about human specializations requires comparing the human species to its sister taxa (chimpanzee and bonobos), to demonstrate that there are differences between these species, and then comparing the human–chimpanzee–bonobo group vs. other apes and monkeys, to estimate whether the common ancestor of humans, chimpanzees, and bonobos resembled humans or chimpanzees and bonobos (Fig. 14.1). The more species that can be studied, the more reliable the evaluation of evolutionary change. Unfortunately, many of the ape species—including bonobos, gorillas, orangutans, and gibbons—are not readily accessible even for noninvasive studies, so that comparative analysis often involves comparing humans, chimpanzees, and macaque monkeys (Fig. 14.1, Inset).
The beginning of comparative molecular biology (at least as regards human evolution) is usually traced to Nuttall (1904), who found that rabbit antisera raised to human blood reacted strongly with human, chimpanzee, and gorilla blood, but less strongly with orangutan or gibbon blood, indicating that the molecular differences between species are consistent with their evolutionary relationships as inferred from differences in anatomy
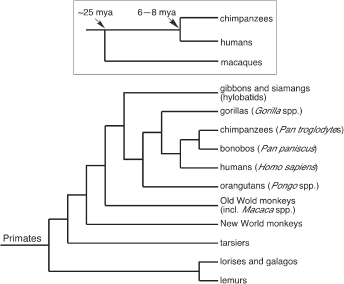
FIGURE 14.1 Identifying human specializations requires, first, determining that humans differ from the chimpanzee-bonobo group (the sister group of humans), and second, determining that the difference evolved in the human lineage. The latter judgement requires evidence about the character states of outgroup taxa, such as other apes and monkeys. For practical reasons, evaluations of human specializations often use a minimal set of comparisons, involving humans, chimpanzees, and macaque monkeys (Inset). Humans and chimpanzees diverged 6 to 8 Mya, and the lineage leading to macaque monkeys diverged from the human-ape lineage ~25 Mya. Modified from Preuss et al. (2004).
(Goodman and Sterner, 2010). The beginning of modern ideas about human molecular evolution, however, can be traced to a landmark paper published in Science by King and Wilson (1975). King and Wilson were struck by the great similarity between humans and chimpanzees in the amino acid sequences of proteins that had been sequenced to that point: the differences amounted to only 1%, which is less than that between anatomically indistinguishable sibling species in some animal groups. This great molecular similarity between humans and chimpanzees seemed paradoxical, given the profound differences in behavior, brain size, and gross anatomy between the two species, most of which, they assumed, evolved in the human lineage rather than the chimpanzee lineage. To resolve this paradox, they proposed that humans underwent a “relatively small number of genetic changes in systems controlling the expression of genes,” yielding a large phenotypic effect but only a small genetic signal (King and Wilson, 1975). Such changes could involve the sequences of
genes that code for regulatory proteins (including what we would today call “transcription factors”) or by DNA rearrangements (translocations, inversions, duplications, and deletions) that change the relationships of genes to regulatory elements. These ideas were subsequently championed by Stephen Jay Gould, in his book Ontogeny and Phylogeny (Gould, 1977), who emphasized that the phenotypic consequences of gene-expression changes would be magnified if they occurred early in development. These writings established the expectation that we should be able to discover a few key genetic changes that account for many, if not most, of the phenotypic differences between humans and chimpanzees.
NEUROBIOLOGICAL AND BEHAVIORAL BACKGROUND
In the classical process of “gene discovery,” geneticists start with a conspicuous phenotypic variant of a species, and then carry out mapping studies to identify the genetic locus that harbors the mutant allele. Thus, gene discovery begins with characterization of phenotypes. It seems reasonable to search for human genetic specializations related to characteristics of the brain, behavior, and cognition, as most people would regard humans as being highly specialized in these domains. Most people are not neurobiologists or behavioral scientists, however. Surprisingly, experts in these fields have often taken a very jaundiced view of supposed human evolutionary specializations, with the result that there is not at present a detailed, consensus view of human neurological or psychological specializations. In part, the reasons for this are pragmatic. Studying ape behavior and cognition is a daunting business, as the animals are large, powerful, and long-lived, and thus are difficult and expensive to maintain. Also, it has proven difficult to develop experimental procedures that convincingly tap the same psychological processes across multiple species. In neuroscience, the main difficulty has been the lack of suitable noninvasive methods for studying brain structure—and in particular, the axonal connections between brain regions that are such important components of the functional organization of the brain. Neuroscientists have powerful methods for studying connectivity, but until recently, these methods required invasive and terminal procedures, precluding their use in humans, as well as in apes. This situation led Crick and Jones (1993), to decry “the backwardness of human neuroanatomy.”
There are also ideological obstacles to identifying human specializations. Claims of human neural or psychological specializations have been seen by some as contrary to Darwin’s understanding of evolutionary continuity, and to represent special pleading on behalf of humans. In fact, Darwin (1871) insisted that there are no differences in kind between the minds of humans and related species; Huxley (1863a) made similar
claims with respect to the brain. However, Darwin and Huxley were not at all consistent about applying this principle to other animal groups or other biological systems, and modern evolutionary biology insists only that there be continuity across generations, not that every feature present in humans be present in some rudimentary form in other animals, a stance that would preclude the existence of any true evolutionary novelties (Preuss, 2009, 2012). Although some psychologists and neuroscientists now reject Darwin’s narrow construction of continuity (Povinelli, 1993; Preuss, 1995, 2012; Premack, 2007; Penn et al., 2008), it retains considerable currency in those fields, and informs the current public understanding of the meaning of evolution (Penn et al., 2008; Preuss, 1995).
There is an additional problem in the neurosciences. As new, powerful (and invasive) methods for studying brains became available in the 1970s, a doctrine developed that held that, apart from size, which is highly variable across species, the organization of the cerebral cortex (the largest part of the brain in most mammals) does not vary in important respects across species [reviewed in Preuss (2001)]. This concept of the “basic uniformity” of cortical organization, and kindred ideas, did little to encourage comparative studies of brain organization, nor did it cause many neuroscientists to be alarmed by the “backwardness of human neuroanatomy” (Crick and Jones, 1993) because there was no reason to think the human brain exceptional except with respect to size. Human brains are, indeed, for mammals of our body size, bizarrely large, approximately three times the volume of those of chimpanzees and other great apes, the result of an enormous evolutionary expansion of the cerebral cortex during the past 2 million years (Preuss, 2011).
Much has changed in recent years, as investigations of human psychological and neurological specializations have begun to flourish [reviewed in Tomasello and Call (1997), Subiaul et al. (2007), Gazzaniga (2008), Passingham (2008), Sherwood et al. (2008), Preuss (2009, 2011), Premack (2010)]. In the neurosciences, the development of noninvasive brain imaging techniques, in particular, has made human neuroanatomy much less backward today than in 1993. One new technique, diffusion-tensor imaging, makes it possible to study the long-range connections of the cerebral cortex. This means we can now compare the cortical connectivity of humans, chimpanzees, and other primates in considerable detail by using the same technique (Rilling, 2008; Preuss, 2010). The first comparative human–chimpanzee–macaque studies that used diffusion-tensor imaging have recently been published, demonstrating human specializations in the connections between regions of higher-order association cortex (Rilling et al., 2008, 2011). Studies of brain tissue acquired postmortem are also revealing human specializations of the cellular and histological organization of the cortex (Sherwood et al., 2008; Preuss, 2010).
Comparative psychological investigations, too, have highlighted human specializations, usually involving aspects of higher-order cognitive abilities, such as the ability to represent abstract or unobservable properties, like force, weight, and mental states (Povinelli and Eddy, 1996; Povinelli, 2000, 2012; Premack, 2007, 2010), and the capacity for symbolic or analogical reasoning (Deacon, 1997a; Penn et al., 2008; Premack, 2010). Another line of research focuses on adaptations of the human mind for culture and social cognition (Henrich and McElreath, 2003; Richerson and Boyd, 2006; Herrmann et al., 2007). Claims for human psychological specializations remain controversial, however, as reflected in the peer commentary on Penn et al. (2008).
There is, however, one human specialization that is widely, if not universally, acknowledged by scientists today: language, that remarkable instrument for organizing and sharing the contents of minds. The upshot of the ape-language projects of the 1960s and 1970s is that apes are understood to be able to learn elements of human language (e.g., manual signs), but they do not organize these elements into expressions possessing the formal characteristics of human language (Terrace et al., 1979; Wallman, 1992; Pinker, 1994; Rivas, 2005; Premack, 2010).
Although progress is being made in understanding human neurobiological and psychological specializations, this is relatively new science, and at present there is no real consensus among specialists about the precise nature of the human specializations. From the point of view of biologists looking to understand the genetic bases of human phenotypic specializations, the most salient ones today are, simply, big brains, advanced cognition, and language.
FOXP2: CASE STUDY
The advent of comparative genomics has been accompanied by a remarkable proliferation of research on human evolutionary biology. The resulting studies have compared humans with chimpanzees and other primates on nearly every imaginable dimension of genetics and molecular biology. These include studies that used high-throughput techniques to identify human specializations of gene expression, primarily in the brain, including whole-genome screens to identify genes that underwent human-specific sequence changes as a result of selection; studies of chromosome segment duplications; identification of differences in alternative splicing between humans and chimps; and more [reviewed in Preuss et al. (2004), Khaitovich et al. (2006), Sikela (2006), Varki and Nelson (2007), Varki et al. (2008), Johnson et al. (2009), Preuss (2010)]. The field continues to grow, but even at this early stage, it is apparent that the genetic and molecular differences between humans and chimpanzees are much greater than had
been supposed. For instance, the number of genes that show expression differences in adult cortex is on the order of hundreds (Preuss et al., 2004) at least, and more likely thousands (Konopka et al., 2009b). Similarly, the number of genes that underwent positive selection in humans is on the order of hundreds at least (Clark et al., 2003; Bustamante et al., 2005). Humans possess species-specific genes, as a result of the numerous tandem duplications of chromosome segments that occurred in human evolution, and also recombination events (Wu et al., 2011; Zhang et al., 2011). One consequence of the numerous duplications, insertions, and deletions is that the total DNA sequence similarity between humans and chimpanzees is not 98% to 99%, but instead closer to 95% to 96% (Britten, 2002; Wetterbom et al., 2006; Varki and Nelson, 2007), although the rearrangements are so extensive as to render one-dimensional comparisons overly simplistic.
There is, then, no shortage of human genetic specializations to work with: the problem is connecting the genes to phenotypes. There is at least one example, however, of a gene that underwent selection-driven sequence change in human evolution and is related to a human-specific cognitive and behavioral phenotype: FOXP2. The history of the discovery of this gene, its connection to speech and language, and the research inspired by the discovery, illustrate the range of approaches now available to scientists pursuing the genetic underpinnings of human nature, as well as the elusive character of the relationship between single genes and complex phenotypes.
FOXP2: Gene Discovery
The first part of the FOXP2 story is a straightforward narrative of gene discovery. Hurst et al. (1990), in England, identified an inherited defect of speech in three generations of a family, known as the KE family. The defect followed a pattern consistent with an autosomal dominant mutation, being present in approximately half the family members, both male and female. According to this initial report, the affected family members exhibited dysfluent, often simplified, speech, with difficultly constructing grammatical sentences. Hurst et al. attributed this to difficulties in making the rapid articulatory movements required in speech, although they did note problems with comprehension. They also reported that the affected members of the family had IQs in the normal range, although some were on the low side of normal. They characterized the disorder as “verbal apraxia” and “developmental verbal dyspraxia,” the suffix “-praxia” implying that the defect was in motor control (i.e., praxis) and was not a true linguistic defect (i.e., aphasia). Other researchers, however, favored a linguistic interpretation, either of grammar, mainly (Gopnik, 1990), or of language more broadly (Vargha-Khadem and Passingham, 1990).
The discovery of the KE family led to a search for the specific gene responsible for the speech defect. Linkage analysis with microsatellite markers suggested a locus on the long arm of chromosome 7, specifically a 5.6-cM region of 7q31, and the locus was dubbed SPCH1 (“speech and language disorder 1”) (Fisher et al., 1998). Subsequently, Lai et al. (2001) identified an unrelated individual (case CS) with a speech deficit similar to that of the affected KE members and a chromosomal translocation in 7q31. The breakpoint mapped onto a bacterial artificial chromosome that contained a known gene, FOXP2, a transcription factor with a forkhead-box DNA-binding domain. By screening the coding regions of FOXP2 with restriction-fragment mapping and direct sequencing, Lai et al. were able to identify the mutation shared by affected KE family members, which would result in an arginine-to-histidine substitution at position 553 (R553H). This falls within the DNA-binding forkhead domain of the FOXP2 protein, and the arginine residue in this part of the domain is highly conserved across members of the forkhead-box family of transcription factors. The implication is that R553H is a loss-of-function mutation.
At the same time geneticists were working to identify the mutation responsible for the deficit in affected KE family members, psychologists and neuroscientists were working to better characterize the phenotype. Vargha-Khadem and coworkers (Vargha-Khadem et al., 1995, 2005; Watkins et al., 2002a) ultimately argued for a primary deficit of orofacial apraxia, based in part on the fact that the mutation impairs the ability to repeat nonwords as well as words. While disputing the idea that FOXP2 is a grammar gene, they did acknowledge that grammatical and syntactic language deficits result from R553H, but left open the possibility that the language problems are a secondary consequence of the dyspraxia. They also confirmed that affected KE family members have lower IQs on average than nonaffected members (Vargha-Khadem et al., 1995; Watkins et al., 2002a), although, again, it is unclear whether this is a core deficit or a secondary consequence of language impairment.
The brain phenotype of affected KE family members has been assessed with structural and functional neuroimaging. These studies have highlighted abnormalities of the motor system. For example, affected family members show a 25% reduction of the volume of the caudate nucleus of the basal ganglia bilaterally, compared with unaffected family members and controls (Vargha-Khadem et al., 1998; Watkins et al., 2002b). Notably, caudate volume is significantly correlated with performance on tests of oral apraxia (Watkins et al., 2002b). Other motor-related structures also show reduced gray matter bilaterally, including the cerebellum and the cortex of the precentral gyrus, as does the inferior frontal gyrus (Broca’s area), whereas the superior temporal gyrus shows increased gray matter volume (Belton et al., 2003). Functional imaging studies using PET or functional
MRI reveal differences between affected KE family members compared with unaffected members and controls in tasks that required repeating words or nonwords, or in making orofacial movements (Vargha-Khadem et al., 1998; Watkins et al., 2002b; Liégeois et al., 2003, 2011). Although there are some inconsistencies in the functional imaging studies, they have also highlighted activation differences in motor structures—cortical motor areas, basal ganglia (striatum), and cerebellum—and Broca’s area. Vargha-Khadem et al. (2005), in their review of imaging studies, emphasize abnormalities in systems involving frontal lobe connections with the basal ganglia and cerebellum (i.e., frontostriatal and frontocerebellar systems) likely to be related to orofacial movements. Notably, however, in the structural and functional imaging studies, the differences between affected KE family members and unaffected members were not limited to those structures, but involved additional and extensive regions of cortex not usually associated with motor (or language) function, such as the postcentral (somatosensory) cortex and occipital (visual) cortex.
Examination of fetal human brain tissue also makes it clear that FOXP2 expression is not limited to brain regions usually associated with language (Ferland et al., 2003; Lai et al., 2003; Spiteri et al., 2007). For example, although it is expressed in the perisylvian cortical region (the cortex spanning the territory from Broca’s to Wernicke’s language areas), and is present in the striatum (caudate and putamen), as one might expect from neuroimaging studies of the KE family, it is expressed in the cortex of frontal pole and occipital pole, neither of which is critical for language. FOXP2 is also expressed in the thalamus, cerebellum, and brainstem, and moreover, is expressed in a wide variety of tissues other than the brain (National Center for Biotechnology Information, 2012).
FOXP2 in Human Evolution
If language is a human specialization, and FOXP2 plays an important role in the development of speech and language, it is natural to ask whether FOXP2 underwent evolutionary changes in its sequence or expression patterns in human evolution. Enard et al. (2002) addressed this question, and found that, although the FOXP2 protein sequence is very strongly conserved in mammalian evolution generally, human FOXP2 differs by two amino acids from that of chimpanzees, gorillas, and macaques, all of which have identical sequences. Both substitutions are in exon 7: a threonine-to-asparagine substitution at position 303 (T303N) and an asparagine-to-serine substitution at position 325 (N325S); the latter substitution creates a potential phosphorylation site. The occurrence of two amino acid fixations is highly unlikely to have occurred by chance, so it is reasonable to conclude that these changes were the result of positive selection,
a conclusion supported by analysis of variation in intronic regions of the gene. [Interestingly, at approximately the same time, two other groups independently reported that FOXP2 is among the genes likely to have undergone positive selection in human evolution, based on the ratio of nonsynonymous to synonymous nucleotide changes (Ka/Ks) in genes for which sequence information was available for humans, chimpanzees, and other species (Clark et al., 2003; Zhang et al., 2002a).] Enard et al. (2002) argued that the data are consistent with a selective sweep resulting in fixation of the two human-specific amino acid changes within the past 200,000 years, and speculated that this event occurred coincident with or subsequent to the appearance of modern Homo sapiens and is related to the ability to produce the orofacial movements required for speech.
With the publication of the FOXP2 evolutionary story, we now had a human-specific modification of a gene that seems to influence a human-specific phenotype. However, important questions remain. For one, were the sequence changes in human FOXP2 driven by selection for speech or language ability? This is not necessarily the case. The mutations in the KE family and in CS (Lai et al., 2001) do not recapitulate the evolutionary changes in the sequence of FOXP2: they are essentially knockout mutations, and the orthologs of FOXP2 present in other species are presumably functional. What does FOXP2 do in nonhuman species? If it is involved in speech or language in humans, are its actions narrowly tied to the development of vocalization systems in other animals, or does it also support the development of other systems?
FOXP2: Comparative Expression and Sequencing Studies
The expression of FOXP2 orthologs in the brains of nonhuman species has received considerable attention, and the anatomical patterns of expression have much in common with those documented in humans [reviewed in Scharff and Petri (2011)]. Fetal macaques express FOXP2 in the basal ganglia (specifically the striatum), thalamus, and extensive regions of cerebral cortex, especially in cortical layer 6 (Takahashi et al., 2008). In mice, the FOXP2 ortholog is expressed from embryonic day 12.5 through adulthood, and its expression is widespread, including the striatum (where it is very strongly expressed), layer 6 of the cerebral cortex, the thalamus, hypothalamus, the Purkinje cells of the cerebellum, the substantia nigra, superior and inferior colliculi, and the inferior olive (Ferland et al., 2003; Lai et al., 2003). Rats show similar patterns of expression, with especially strong expression in the striatum (Takahashi et al., 2003). The similarities in anatomical localization extend to other vertebrates as well, including birds and reptiles (Haesler et al., 2004; Teramitsu et al., 2004). FOXP2 orthologs are also expressed in the brains of amphibians and fish
(Bonkowsky and Chien, 2005; Shah et al., 2006; Schön et al., 2006; Itakura et al., 2008).
The presence of FOXP2 orthologs in birds has prompted investigations to determine whether song-learning birds underwent sequence changes in FOXP2 analogous to those documented in humans [reviewed in Scharff and Haesler (2005), Vargha-Khadem et al. (2005), Scharff and Petri (2011)]. Particular attention has been paid to zebra finches, a favorite species for students of bird song because the male develops its song at sexual maturity, the animal gradually shaping its song to match the memory of a tutor’s song (usually that of its father). In zebra finches, FOXP2 expression is especially strong in area X [Haesler et al. (2004), Teramitsu et al. (2004)], a critical element of the avian song-learning system that is composed of tissue homologous to the striatum of mammals. What is more, in zebra finches and in canaries, area X expresses more FOXP2 than the surrounding region of the striatum during the period of vocal learning (Haesler et al., 2004; Teramitsu et al., 2004). Zebra finches recruit new neurons during the period of song learning, and those neurons express FOXP2 (Rochefort et al., 2007). In addition, knockdown of FOXP2 in area X of zebra finches impairs song learning (Haesler et al., 2007). Thus, FOXP2 is probably involved in the development of the song system in zebra finches, and probably in other bird species as well. However, zebra finch FOXP2 lacks the specific nucleotide substitutions that evolved in the human terminal lineage (Haesler et al., 2004; Teramitsu et al., 2004).
A recent comparative sequencing study suggests that there is somewhat more evolutionary diversity in FOXP2 sequences than initially believed. Li et al. (2007) partly or completely sequenced FoxP2 in a large number of bats (42 species), including species that are echolocating and vocal learners, as well as other bats; in cetaceans (18 species of whales and dolphins, 15 of which are echolocating); in the African elephant (reputedly a vocal learner); and in other species from diverse mammalian orders. Although the FoxP2 sequence was confirmed to be highly conservative in most mammalian orders, bats proved to be markedly divergent, with nonsynonymous nucleotide changes concentrated in exons 7 and 17. Within exon 7, none of the bats or other mammalian species have a human-like asparagine substitution at position 303, but many echolocating bats, and also two carnivore species sequenced, do have human-like serine substitutions at position 325. In exon 17, no nonsynonymous substitutions were found in any of the mammals sequenced except bats, in which variation is considerable, with as many as eight substitutions in one species. Although they argued that FoxP2 was likely the target of positive selection in bats, however, Li et al. are skeptical that there is a relationship between vocal learning ability and specific FoxP2 substitutions, because they could identify no nonsynonymous substitution that is shared by all
the vocal learning species they examined or in other published studies of mammals or birds. Comparative studies of FOXP2, therefore, draw no clear connection between specific sequence changes and vocal learning capacities, although the increased levels of FOXP2 expression reported in birds during periods of song acquisition suggests that this transcription factor is involved in learning-related neural changes.
FOXP2: Mouse Model of R552H Substitution
One approach that has been adopted to understand the function of FOXP2 in humans is to make mouse models that express the mutant form of FOXP2 present in affected members of the KE family (R552H), as Groszer et al. (2008) have done. The homozygotes were severely developmentally delayed, and died by 3 to 4 wk after birth. The cerebellum was abnormally small, with decreased foliation, and behaviorally, the pups emitted fewer ultrasonic distress calls than heterozygotes or WT mice. The heterozygous pups, however, were fully viable and healthy, and were not developmentally delayed. These animals were screened for abnormalities of the motor system, particularly the striatum and cerebellum, brain regions that show structural and functional changes in affected KE family individuals. The brains of the R553H heterozygotes appeared to be grossly normal and the cerebellum was of normal size. Nevertheless, the animals showed impaired motor learning on a running wheel. Moreover, the physiology of the striatum and the cerebellum differed from WT mice. In the striatum, high-frequency electrical stimulation of glutamatergic synapses, which in WT animals produces long-term depression (LTD; a form of synaptic plasticity), failed to produce any significant LTD in R552H heterozygotes. Stimulation of the parallel fiber/Purkinje cell system in cerebellar tissue-slice preparations produced weaker effects on LTD, and in the opposite direction, with slightly stronger LTD in R552H heterozygous animals than in WT mice.
FOXP2: Mouse Model of Human Evolution?
A different approach to understanding the functions of FOXP2 in humans was adopted by Enard et al. (2009), who produced a transgenic mouse that expressed a humanized version of mouse FOXP2, with the characteristically human threonine-to-asparagine substitution at position 303 and asparagine-to-serine substitution at position 325. They also made Herculean efforts to screen the phenotype that involved behavioral, neurohistological, neurophysiological, and neurogenetic comparisons of the transgenic and normal mice. Unlike R552H mice, humanized FOXP2 homozygous mice are fully viable and fertile and show no gross behavioral
or anatomical abnormalities. Moreover, they expressed FOXP2 in the brain structures one would expect from previous studies: cerebral cortex (cells in layer 6), the striatum, thalamus, and cerebellar Purkinje cells. Behaviorally, the humanized mouse pups showed no difference with WT mice in the number of isolation calls made per unit time, although the spectral characteristics of their calls differed, with lower peak frequencies at the start of calls, and lower mean and maximum peak frequencies during calls. Interestingly, the humanized FOXP2 mice showed no evidence of motor impairment compared with WT mice, although they did have lower scores on several measures of exploratory behavior. Neurochemically, humanized FOXP2 mice showed lower dopamine concentrations in cortex, striatum, pallidum, and cerebellum compared with WT, but concentrations of other neurotransmitters (serotonin, glutamate, and GABA) did not differ. Humanized FOXP2 mice also showed differences in gene expression in the striatum compared with WT mice, with a number of genes downregulated in the humanized mice that are known to be expressed by medium spiny cells that express the dopamine D1 receptor, a class of cells that also express FOXP2 (Heiman et al., 2008). The electrophysiological characteristics of striatal medium spiny cells were examined in cells cultured from humanized FOXP2 mice, and found to support stronger LTD than WT cells, the opposite of the finding with R552H mice. Perhaps the most notable finding of this study (Enard et al., 2009) is that cultured striatal neuronal precursors from humanized FOXP2 mice had longer neurites (i.e., dendrites and axons) than those from the cells of WT mice. Subsequent examination of medium spiny neurons in brain sections from the striatum showed that neurons from humanized FOXP2 mice had longer dendrites that those from WT mice.
Regulation of Gene Expression by FOXP2
The study by Enard et al. (2009) suggests yet another, more mechanistic, approach to understanding the functions of FOXP2: identifying the genes regulated by FOXP2. Several studies have been carried out with this goal, using chromatin immunoprecipitation (CHiP) to identify DNA sequences bound by FOXP2 protein. DNA sequences bound with FOXP2 antibodies were then identified by hybridization to gene chips representing human promoters (i.e., ChIP-chip) or, more recently, by direct sequencing of the targeted sequences (i.e., ChIP-seq).
Spiteri et al. (2007) identified 285 gene targets in fetal human tissue from the inferior frontal gyrus and basal ganglia, and determined their representation in gene ontology (GO) and pathway categories. GO categories represented at greater than chance levels included morphogenesis, intracellular signaling, cation homeostasis, neuron outgrowth, and axonal
morphology. Overrepresented pathway categories included dendritic branching, calcium mobilization, calcium concentration, and learning. Notably, 14 genes that were targeted by FOXP2 (which underwent selective change in human evolution) also showed evidence of positive selection in human evolution. A comparison of published datasets identified 47 gene targets of FOXP2 that were differentially expressed in the cortex of humans and chimpanzees, including a number of genes involved in neural development, central nervous system patterning, and neural transmission. Vernes et al. obtained qualitatively similar results in GO and pathway analyses after examining expression in human neuron-like cells (SH-SY5Y cells) transfected with human FOXP2 (Vernes et al., 2007), and also in a ChIP study of FOXP2 promoter occupancy in embryonic mice (Vernes et al., 2011). The latter study also directly examined the effect of mouse Foxp2 expression on neurite outgrowth by transfecting mouse neuron-derived Neuro2a cells with Foxp2 or an empty vector (used as a control). They then treated the cells with retinoic acid, which causes Neuro2a cells to extend neurites as they adopt a more differentiated, neuron-like phenotype. Cells transfected with Foxp2 were found to have longer neurites than control cells.
To investigate the transcriptional consequences of the human-specific substitutions in FOXP2 (T303N and N325S) more directly, Konopka et al. (2009a) transfected SH-SY5Y cells with human FOXP2 or with a construct that restored the ancestral sequence found in chimpanzees, and used microarrays to assess gene expression. Although the two versions of FOXP2 regulated many of the same genes, there were also differences in transcriptional activity, with 61 genes being more strongly expressed with human FOXP2 than chimpanzee FOXP2 and 55 less strongly expressed with human FOXP2. They then examined expression of FOXP2-regulated genes in adult brain tissue from humans and chimpanzees and found that many of the same genes were expressed in tissue as in the SH-SY5Y cells. Moreover, the differences in expression between human and chimpanzee brain tissue mirrored the differences found in cells transfected with human FOXP2 or chimpanzee FOXP2. GO analysis of differentially expressed genes in the brain showed an enrichment of categories related to tissue and organ development and cell–cell signaling.
CONNECTING GENES AND PHENOTYPES
As a gene associated with a human-specific trait, FOXP2 would at first glance seem to be a dream come true for evolutionary geneticists. Moreover, it is hard not to be impressed by the depth and breadth of the research related to FOXP2. Nevertheless, there is still no clear or direct connection between the human-specific amino acid substitutions in FOXP2 and speech
or language—not from the comparative studies, or from the mouse-model studies, or from the gene expression studies. The fact that mutations of FOXP2 in humans result in speech impairments shows that it plays a role in speech development, but the nature of its role remains unclear. It might play a very specific role, for example, by orchestrating a whole set of genes that switch brain development from an ancestral program to a human program that causes cells and connections to differentiate into systems that sustain speech or language. It might even regulate the development of other parts of the anatomy, such as the lungs and larynx, involved in speech production. Alternatively, FOXP2 might have a permissive role, for example, by regulating some aspects of cell behavior required for the normal development of language systems, but also for the normal development of other structures and systems. Both options would be consistent with the action of a loss-of-function mutation in FOXP2, such as the R553H mutation in the KE family. In neither case, however, do we have a direct connection between language and the specific FOXP2 substitutions that took place in human evolution (T303N and N325S). There is not much question that these changes were the result of selection, and that they affect gene expression in the brain. However, given the widespread pattern of FOXP2 gene expression in the body, those substitutions are likely to affect gene expression in other organs, so it remains possible that the substitutions were driven by selection acting on non–speech-related parts of the brain or nonbrain tissues and organs. Humans are, after all, not just apes with unusually large, complex brains: other aspects of anatomy and physiology were extensively modified in human evolution as well. It could also be the case that FOXP2 has a speech- or language-specific function in the human brain, by virtue of the action of other transcription factors that bind to the same promoters in brain cells targeted by human FOXP2. However, then we would be talking about the interactions of genes involved in building a human organism, rather than a single gene, and it still would not be clear, without additional evidence, that the amino acid substitutions in FOXP2 were selected for their effects on developmental pathways specific to language.
It would seem that the crux of the problem with tying FOXP2 to language is that we are trying to relate a multifunctional gene to a complex, high-level phenotype, by which I mean a phenotype that encompasses a diverse collection of tissues and cell types. It is probably not realistic to think that the development of such systems has simple genetic triggers and they are the products of epigenetic programs acting in isolation from other epigenetic programs. This conclusion merely restates two of the important lessons of experimental population genetics: first, that most phenotypes arise through the interactions of multiple genes (the principle of epistasis), and, second, that most genes influence multiple phenotypes (the principle of pleiotropy) (Mayr, 1970; Dobzhansky et al., 1977).
These considerations, along with the great number and variety of human genetic specializations, make it unlikely that evolutionary changes in just a few regulatory genes, acting early in development, can explain most of the phenotypic differences between humans and chimpanzees, as King and Wilson (1975) and Gould (1977) suggested. There might, in fact, be a relatively small number of genes that do act early in development and do have profound effects, but as we have seen with FOXP2, the genes downstream of a regulatory gene themselves can undergo evolutionary changes (Spiteri et al., 2007). Without a deeper understanding of development and, especially, of the human-specific aspects of human development, we are not likely to be able to make many definite connections between high-level phenotypes and the role of early-acting regulatory genes.
This raises a matter of serious concern: our lack of direct information about the human organism. I have noted the lack of detailed accounts of the human neurobiological and psychological phenotypes, and I think it would be fair to say we are quite ignorant about many other human--specific features of human biology, including human-specific modifications of the developmental programs that generate human-specific phenotypes. In part, this lack of knowledge reflects technical limitations to our ability to study humans: just as we cannot do invasive neurobiological investigations, we cannot do invasive or terminal developmental investigations or transgenic experiments either. Given this, it is not surprising that we rely so much on studies of model animals to understand normal human biology and its development, with the hope that there is enough commonality in epigenetic systems across species that results in models will translate to humans. This approach seems particularly problematic when human-specific phenotypes are at issue. Presumably, no one expected that expressing human FOXP2 in a mouse would yield a mouse that talks. It is, after all, a human gene on a mouse background, and without understanding the similarities and differences between human and mouse developmental programs, predictions about the specific effects of a mutant human gene on the high-level phenotypes of mice are hazardous.
In offering this critique, I do not mean to say that mouse models are not useful or informative, but rather that they are not enough by themselves. Certainly, every aspect of our science would benefit from the better characterization of human-specific phenotypes, and the most direct way to obtain that information, especially for high-level phenotypes, is by studying humans directly. However, we do need other experimental paradigms to help us explore the phenotypic consequences of human-specific genetic changes. Investigations of gene functions in cell culture, as exemplified by a number of the studies cited earlier, provide another avenue for empirical research. This approach could be further empowered
by the emerging technology for generating differentiated cells of various types (including neurons) from induced pluripotent stem cells (Hansen et al., 2011; Shi et al., 2012).
There is another approach we can adopt, one that takes as its starting points not some known human phenotypic specializations (of which there are rather few), but instead starts with the genetic and molecular differences themselves and uses them as clues to previously unsuspected phenotypes: that is, “phenotype discovery” (Preuss et al., 2004; Varki et al., 2008; Preuss, 2010), in contrast to “gene discovery” (Fig. 14.2). How this works is illustrated by the studies reviewed in the previous section showing that FOXP2 expression affects neurite outgrowth and synaptic plasticity (Vernes et al., 2007, 2011), and that mice expressing human FOXP2 show more neurite outgrowth and synaptic plasticity than mice expressing mouse Foxp2 (Enard et al., 2009). There are additional lines of evidence suggesting that evolution targeted these features of human neuronal biology. Some of the first microarray studies comparing adult humans to other primates (Cáceres et al., 2003; Preuss et al., 2004; Uddin et al., 2004), and follow-up validation studies (Cáceres et al., 2007), noted expression changes in genes related to synaptic plasticity, as has a recent comparative study of metabolite concentrations (Fu et al., 2011). It is noteworthy, too, that there is genomic evidence for positive selection on genes
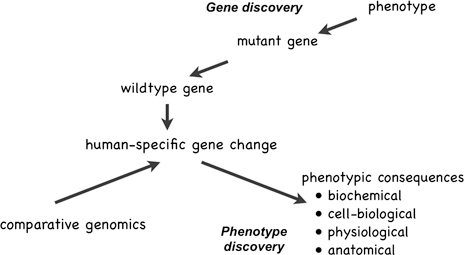
FIGURE 14.2 Gene discovery starts with an unusual, heritable phenotype, and then proceeds to determine the chromosomal locus of the mutation, and finally to identify the mutated gene itself. Phenotype discovery starts with species differences in genes or gene expression, identified through gene discovery, comparative genomics, or other comparative molecular methods, and proceeds to identify the biochemical, cell-biological, and other phenotypic consequences of the genetic differences.
involved in aerobic energy metabolism (Grossman et al., 2001; Goodman and Sterner, 2010), and evidence for upregulation of energy-metabolism genes in human evolution (Cáceres et al., 2003; Uddin et al., 2004). One study that examined patterns of gene coexpression in the adult cortex of humans and chimpanzees found evidence for differences both in genes related to synaptic plasticity and energy metabolism (Oldham et al., 2006). What is emerging is a picture of humans as having adult brains that are unusually active physiologically and unusually dynamic anatomically. This may reflect the extension in humans of patterns of gene expression normally restricted to early life stages well into adulthood (Somel et al., 2009, 2010; Fu et al., 2011), a phenomenon that has been termed “transcriptional neoteny” (Somel et al., 2009).
Although the idea that human brains are unusually active and malleable might make intuitive sense to many, there is, in fact, little in the primary scientific literature hinting at this. In fact, one of the most robust generalizations in physiology is that as biological entities—be they organisms, organs, or organelles—become bigger, they use less energy per unit mass of tissue (West and Brown, 2005). However, PET studies in awake individuals suggest that the human brain uses approximately the same amount of glucose per unit of tissue as do rhesus macaques, animals with brains less than one-tenth the volume of human brains (Bohnen et al., 1999; Bentourkia et al., 2000; Cross et al., 2000; Noda et al., 2002). This is consistent with the idea that human brains are “running hot” (Preuss, 2011).
What is notable about this example is not simply that the genomic evidence suggests phenotypic specializations of humans that were not previously suspected, but also that the claims can be tested empirically: the energetics claim by in vivo physiological techniques such as PET, and the claim of neural dynamism by studies in cell and tissue culture, and possibly even in postmortem tissue (Enard et al., 2009). Perhaps when we know enough about the cell-physiological consequences of enhanced neural dynamism, we can generate additional testable predictions at levels that are amenable to investigation by PET and other in vivo techniques.
I suggest that, in general, comparative molecular data are particularly well suited to phenotype discovery at the level of cells, and especially their biochemical and physiological characteristics, because the route from gene changes to cellular changes is more direct than that from genetic changes to high-level phenotypes [see also Varki et al. (2008)]. That does not mean we should abandon phenotype-driven gene discovery studies, but gene-driven phenotype discovery may represent an approach with a greater payoff, at least in the short term, and has the additional advantage of being able to reveal previously unsuspected aspects of human nature.
![]()
The integration of facial gestures and vocal signals is an essential process in human communication and relies on an interconnected circuit of brain regions, including language regions in the inferior frontal gyrus (IFG). Studies have determined that ventral prefrontal cortical regions in macaques [e.g., the ventrolateral prefrontal cortex (VLPFC)] share similar cytoarchitectonic features as cortical areas in the human IFG, suggesting structural homology. Anterograde and retrograde tracing studies show that macaque VLPFC receives afferents from the superior and inferior temporal gyrus, which provide complex auditory and visual information, respectively. Moreover, physiological studies have shown that single neurons in VLPFC integrate species-specific face and vocal stimuli. Although bimodal responses may be found across a wide region of prefrontal cortex, vocalization responsive cells, which also respond to faces, are mainly found in anterior VLPFC. This suggests that VLPFC may be specialized to process and integrate social communication information, just as the IFG is specialized to process and integrate speech and gestures in the human brain.
The area dedicated to language processing in the frontal lobe is located within the inferior frontal gyrus (IFG), which can be further subdivided into the pars opercularis (most posterior portion of
_______________
Department of Neurobiology & Anatomy, University of Rochester School of Medicine, Rochester, NY 14642. E-mail: liz_romanski@urmc.rochester.edu.




















