
THE SOCIAL BIOLOGY OF MICROBIAL COMMUNITIES
Beginning with the germ theory of disease in the 19th century and extending through most of the 20th century, microbes2 were believed to live their lives as solitary, unicellular, disease-causing organisms (Losick and Kaiser, 1997). This perception stemmed from the focus of most investigators on organisms that could be grown in the laboratory as cellular monocultures, often dispersed in liquid, and under ambient conditions of temperature, lighting, and humidity (Kolter and Greenberg, 2006). Most such inquiries were designed to identify microbial pathogens by satisfying Koch’s postulates.3 This pathogen-centric approach to the study of microorganisms produced a metaphorical “war” against these microbial invaders waged with antibiotic therapies, while simultaneously obscuring the
___________
1 The planning committee’s role was limited to planning the workshop, and the workshop summary has been prepared by the workshop rapporteurs (with the assistance of Pamela Bertelson, Rebekah Hutton, and Katherine McClure) as a factual summary of what occurred at the workshop. Statements, recommendations, and opinions expressed are those of individual presenters and participants, and are not necessarily endorsed or verified by the Institute of Medicine, and they should not be construed as reflecting any group consensus.
2 Microscopic organisms, including bacteria, archaea, fungi, protists, and viruses.
3 Koch’s postulates must be satisfied in order to state that a particular microbe causes a specific infectious disease. They include the following: (i) The parasite occurs in every case of the disease in question and under circumstances which can account for the pathological changes and clinical course of the disease. (ii) The parasite occurs in no other disease as a fortuitous and nonpathogenic parasite. (iii) After being fully isolated from the body and repeatedly grown in pure culture, the parasite can induce the disease anew (Fredricks and Relman, 1996; Koch, 1891; Rivers, 1937).
dynamic relationships that exist among and between host organisms and their associated microorganisms—only a tiny fraction of which act as pathogens.
A recent revolution in our collective understanding of microbes is that the vast majority of these organisms live in communities and lead intensely interactive lives, competing, cooperating, and forming associations with one another and with their living and nonliving host environments. As the earth’s first living inhabitants, communities4 of microorganisms have had several billion years to coevolve and adapt to one another and their environments, resulting in a world of spectacular diversity and interdependence. Indeed, microbial communities are intricately intertwined with all ecosystems on Earth—from the extreme environments of the human gut to deep-sea hydrothermal vents and the windswept plains of Antarctica.
This ecological view of microbial life has enormous potential for transforming our understanding of the world around us. Recent research on the communities of microorganisms that live in and on us (the human microbiome) suggests that many traits once assumed to be “human”—such as the digestion of certain foods or the ability to defend against disease—may result from human-microbe interactions (Dethlefsen et al., 2007; IOM, 2006). Such findings have dispelled the notion that “human beings are physiological islands, entirely capable of regulating [our] own internal workings” and replaced it with the notion of the human body as a complex ecosystem (Ackerman, 2012). This realization “promises to radically alter the principles and practices of medicine, public health, and basic science” (Relman, 2012).
Recognition of the ubiquity and importance of microbial communities not only advances an ecological view of microbial life but also raises intriguing questions about the formation of groups that behave collectively in ways that have consequences for their individual members. There is mounting evidence to suggest that molecular “conversations” take place among members of a broad spectrum of microbial communities, and also between a variety of microbes and host organisms. Having only recently become aware that such conversations exist at all, our ability to eavesdrop on them and to translate them into scientific knowledge can be described as rudimentary at best. Yet, there is the emerging sense that microbes interact in complex, diverse, and subtle ways that we have yet to fully appreciate, much less understand.
Despite their obvious importance, very little is actually known about the processes and factors that influence the assembly, function, and stability of microbial communities. Gaining this knowledge will require a seismic shift away from the study of individual microbes in isolation to inquiries into the nature of diverse and often complex microbial communities, the forces that shape them,
___________
4 For the purposes of this overview, and as suggested by speaker Joan Strassmann of Washington University at St. Louis, “microbial community” simply means “all the small forms of life occurring in the same place and time, where same implies a shared place, with some possibility they will encounter each other, or take resources the other might have used.”
and their relationships with other communities and organisms, including their multicellular hosts.
On March 6 and 7, 2012, the Institute of Medicine’s (IOM’s) Forum on Microbial Threats hosted a public workshop to explore the emerging science of the “social biology” of microbial communities. Workshop presentations and discussions embraced a wide spectrum of topics, experimental systems, and theoretical perspectives representative of the current, multifaceted exploration of the microbial frontier. Participants discussed ecological, evolutionary, and genetic factors contributing to the assembly, function, and stability of microbial communities; how microbial communities adapt and respond to environmental stimuli; theoretical and experimental approaches to advance this nascent field; and potential applications of knowledge gained from the study of microbial communities for the improvement of human, animal, plant, and ecosystem health and toward a deeper understanding of microbial diversity and evolution.
Organization of the Workshop Summary
This workshop summary was prepared by the rapporteurs for the Forum’s members and includes a collection of individually authored papers and commentary. Sections of the workshop summary not specifically attributed to an individual reflect the views of the rapporteurs and not those of the members of the Forum on Microbial Threats, its sponsors, or the IOM. The contents of the unattributed sections of this summary report provide a context for the reader to appreciate the presentations and discussions that occurred over the 2 days of this workshop.
The summary is organized into sections as a topic-by-topic description of the presentations and discussions that took place at the workshop. Its purpose is
___________
5 The original Statement of Task stated the following: An ad hoc committee will plan and conduct a public workshop that will feature invited presentations and discussions to explore the scientific and policy implications of the microbiome in health and disease. Topics to be discussed may include, but are not limited to, the social behavior of microorganisms to form and maintain stable communities; how the use of antibiotics and other drugs can influence the community composition of the microbiome; microbial evolution and co-adaptation; an exploration of the various microbiomes in mammalian/terrestrial/aquatic environments; and the impacts of globalization on the introduction, establishment and evolution of “novel” diseases in established microbial communities. In the course of planning this workshop, the planning committee decided to focus the workshop’s agenda on “the ecological, evolutionary, and genetic factors contributing to the assembly, function, and stability of microbial communities; how microbial communities adapt and respond to environmental stimuli; theoretical and experimental approaches to advance this nascent field; and potential applications of knowledge gained from the study of microbial communities for the improvement of human, animal, plant, and ecosystem health and toward a deeper understanding of microbial diversity and evolution.”
to present information from relevant experience, to delineate a range of pivotal issues and their respective challenges, and to offer differing perspectives on the topic as discussed and described by the workshop participants. Manuscripts and reprinted articles submitted by workshop participants may be found, in alphabetical order, in Appendix A.
Although this workshop summary provides a description of the individual presentations, it also reflects an important aspect of the Forum’s philosophy. The workshop functions as a dialogue among representatives from different sectors and allows them to present their views about which areas, in their opinion, merit further study. This report only summarizes the statements of participants at the workshop over the course of 2 consecutive days. This workshop summary is not intended to be an exhaustive exploration of the subject matter nor does it represent the findings, conclusions, or recommendations of a consensus committee process.
Glimpses of Microbial Community Dynamics
“We have to get away from this monolithic, one-dimensional perspective of a one bug–one-disease picture of health. The community is the unit of study.”
—David Relman (Buchen, 2010)
“One reason we may have a hard time remembering that all microbes exist in communities is due to an early focus of scientists on microbes that cause disease.”
—Joan Strassmann (2012a)
Observations of bacteria grown in the artificially simple environments of the Petri dish and the test tube have provided detailed knowledge of the physiology and cellular processes of organisms amenable to such culturing techniques (Little et al., 2008). With the recent development of “culture-independent” methods of microbial characterization,6 researchers have determined that such culturable species represent only a minuscule fraction of the microbial diversity around us. These techniques have further revealed the dynamic communities that the vast majority of microorganisms shape and inhabit—from simple communities composed of one to two species to complex, spatially diversified, host-associated communities comprising hundreds of species (Handelsman, 2004; Little et al., 2008; Nee, 2004).
This workshop’s focus on the community as the unit of study continues the Forum’s exploration of “a more realistic and detailed picture of the dynamic
___________
6 Various “culture-independent” techniques are discussed in the section “The Structure and Function of Microbial Communities (see page 25).”
interactions among and between host organisms and their diverse populations of microbes” (IOM, 2006, 2009). Newly recognized as social organisms, microbes also provide a fresh lens through which to view interactions both among and between species. Studies of such interactions among multicellular organisms inform the disciplines of social biology7 and ecology.8 While theoretical constructs derived from observations of the macroscopic world offer ways to interpret microbial interactions, it is also possible that these phenomena will require novel explanatory frameworks.
Microbial Communities in Biotic and Abiotic Environments
The following descriptions of microbial communities, adapted to several distinct habitats, provide glimpses of microbes interacting with each other and with their environments, and reveal collective functions that exceed the capabilities of individual members.
Biofilms The vast majority of microbes form and inhabit biofilms: complex, differentiated aggregations, typically of multiple species, that thrive on nearly every surface (Hall-Stoodley et al., 2004; Kolter and Greenberg, 2006; Parsek and Greenberg, 2005). Surrounded by a self-produced polymeric matrix,9 biofilms are characterized by structural heterogeneity, genetic diversity, and complex community interactions, as shown in Figure WO-1. For example, the microbial constituents of the biofilm known as dental plaque include hundreds of species and strains of bacteria, as well as various methanogens (archaea) whose collective metabolic activities are associated with tooth decay (Lepp et al., 2004; Relman, 2005). By analogy to human communities, biofilms are organized into divisions of labor, with individual cells taking on specific tasks (Kolter and Greenberg, 2006).
The structure of biofilms protects resident organisms from environmental extremes such as ultraviolet light, toxins (including antibiotics), pH, and dehydration—advantages that may have allowed the first microbes to populate Earth’s surface—as well as from host immune defenses (e.g., phagocytosis) and predation (Hall-Stoodley et al., 2004). The matrix polymer surrounding biofilms can store water and nutrients, and some biofilms have networks of channels that enable these resources to be distributed (IOM, 2011; Kolter and Greenberg, 2006; Stewart and Franklin, 2008).
In medical settings, biofilms contribute to hospital-acquired infections, most notably by colonizing in-dwelling medical devices such as catheters and
___________
7 The study of interactions within communities of single species.
8 The study of organisms’ interactions with each other and with their environment.
9 Cells in a biofilm secrete polymers of varying chemical composition that form an extracellular polymeric substance (EPS) or a slime matrix that gives the biofilm stability and helps it to adhere to a surface. Although generally assumed to be primarily composed of polysaccharides, the EPS can also contain proteins and nucleic acids (Hall-Stoodley et al., 2004).
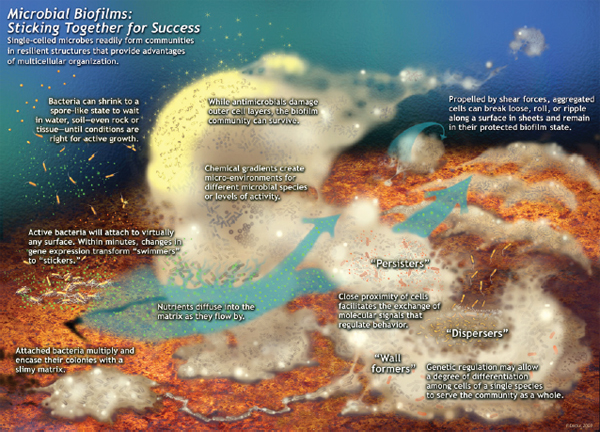
FIGURE WO-1 Microbial biofims: Sticking together for success. Single-celled microbes readily form communities in resilient structures that provide advantages of multicellular organization. This schematic was drawn by Peg Dirckx from the Center for Biofilm Engineering to incorporate various biofilm behaviors and concepts based largely on observations from confocal and time-lapse microscopy. An interactive version can be found at http://www.erc.montana.edu/MultiCellStrat/default.html.
SOURCE: MSU Center for Biofilm Engineering, P. Dirckx.
prostheses (Hall-Stoodley et al., 2004; Kolter and Greenberg, 2006). According to Freemont (IOM, 2011), bacteria within established biofilm communities have been shown to tolerate antimicrobial agents at concentrations as high as 1,000 times the dosage needed to kill genetically equivalent bacteria in liquid culture. Bacterial biofilms may also make certain infections, such as those found in chronic wounds and the respiratory tract of individuals with cystic fibrosis, very difficult to treat (Hall-Stoodley et al., 2004).
Multicellular structures for migration and dispersal The lifecycle of several types of microbes—including algae of the order Volvocales, social amoebae10 of the order Dictyosteliida, and more than 50 species of Myxobacteria11— contain stages in which these usually unicellular organisms aggregate to form multicellular structures (Brock et al., 2011; Kaiser, 2006; Strassmann and Queller, 2011). When the unicellular stage of the social amoeba Dictyostelium discoideum runs out of bacteria to prey upon, tens of thousands of amoebae aggregate into a multicellular migratory slug (Brock et al., 2011; Kuzdzal-Fick et al., 2011). It moves toward light and, once in a suitable location, the slug transforms into a fruiting body, a process during which one in five cells die in order to form the structure’s sterile stalk. The stalk aids in the dispersal of the remaining cells, which differentiate into spores. The social biology of D. discoideum is further discussed in Control of cheating in the social amoeba and Farming of bacteria.
Myxobacteria xanthus undergoes a similar transformation when nutrients are scarce, aggregating into groups of more than 100,000 cells that then form elaborate fruiting bodies for spore dispersal as illustrated in Figure WO-2. Chemical and cell-contact signals have been found to coordinate developmental gene expression with cellular movement, leading to the construction of fruiting bodies in this bacterium (Kaiser, 2006).
The bacterium and the squid The Hawaiian squid Euprymna scolopes forms a persistent association with the Gram-negative luminous bacterium Vibrio fischeri (Nyholm and McFall-Ngai, 2004). Incorporated into the squid’s light organ, the bacterium emits luminescence that resembles moonlight and starlight filtering through ocean waters, camouflaging the nocturnal squid from predators (Figure WO-3) (Nyholm and McFall-Ngai, 2004). The forces supporting the formation and stability of this association were discussed by several workshop speakers.
V. fischeri is the exclusive partner of the host squid in a special adaptation of the squid’s light organ. Colonization of the squid’s light organ by the bacterium
___________
10 Although they are amoeboid protists, not fungi, members of this order are commonly known as “cellular slime molds.”
11 Any of numerous Gram-negative, rod-shaped saprophytic bacteria (deriving nourishment from dead or decaying organic matter) of the phylum Myxobacteria, typically found embedded in slime in which they form complex colonies and noted for their ability to move by gliding along surfaces without any known organ of locomotion.

FIGURE WO-2 Myxobacteria build multicellular fruiting bodies. Each of the 50 species of myxobacteria inherits a plan to build a morphologically different fruiting body. Fruiting bodies are 100 to 400 microns high and contain about 100,000 terminally differentiated spores.
SOURCE: Kaiser (2006).
begins within an hour after hatching and appears to occur in stages, as shown in Figure WO-4, with each step enabling greater specificity between host and symbiont. Once established, V. fischeri drives the development of the tissues with which they associate, inducing the maturation of the squid’s light organ from a morphology that promotes colonization to one that promotes the maintenance of an exclusive association with V. fischeri through the life of the host (McFall-Ngai
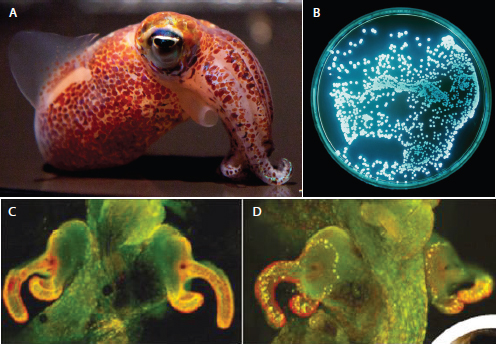
FIGURE WO-3 The bacterium and the squid. A persistent, symbiotic association between the squid Euprymna scolopes (A) and its luminous bacterial symbiont Vibrio fischeri (B) forms within the squid’s light organ (C and D). After colonization of the host’s light organ tissue, V. fischeri induces a series of irreversible developmental changes that transform these tissues into a mature, functional light organ (Nyholm and McFall-Ngai, 2004).
SOURCE: (A) Images taken by C. Frazee, provided by M. McFall-Ngai and E. G. Ruby; (B) Image provided courtesy of Marianne Engel; (C and D). Reprinted by permission from Macmillan Publishers Ltd: Nature, Dusheck (2002), copyright 2002.
et al., 2012). Up to 95 percent of the resident symbiont population is expelled each day at dawn, followed by daily regrowth of bacteria within the crypts (McFall-Ngai et al., 2012). This simple model of persistent colonization of animal epithelia by Gram-negative bacteria provides a “valuable complement to studies of both beneficial and pathogenic consortial interactions, such as in the mammalian intestine, and chronic disease that involve persistent colonization by Gram-negative bacteria, such as cystic fibrosis” (Nyholm and McFall-Ngai, 2004).
Plant roots and their partners Plants establish associations with several microorganisms in a relationship somewhat analogous to that of mammals with their gastrointestinal microbiota. The roots of most higher plant species form mycorrhizae, an association with specific fungal species that significantly improves the plant’s ability to acquire phosphorous, nitrogen, and water from the soil.12 A few plant families, including legumes, associate with nitrogen-fixing bacteria. They colonize the plant’s roots and form specialized nodules, where the bacteria
___________
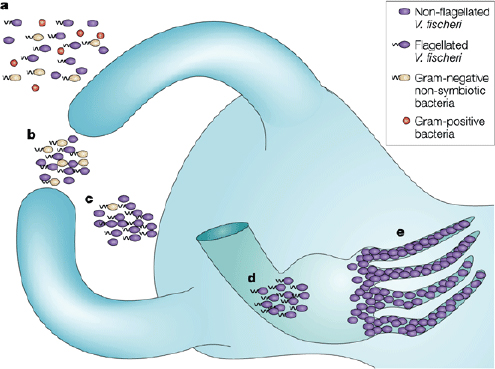
FIGURE WO-4 The winnowing. This model depicts the progression of light-organ colonization as a series of steps, each more specific for symbiosis-competent Vibrio fischeri. (a) In response to Gram-positive and Gram-negative bacteria (alive or dead) the bacterial peptidoglycan signal causes the cells of the ciliated surface epithelium to secrete mucus. (b) Only viable Gram-negative bacteria form dense aggregations. (c) Motile or nonmotile V. fischeri out-compete other Gram-negative bacteria for space and become dominant in the aggregations. (d) Viable and motile V. fischeri are the only bacteria that are able to migrate through the pores and into the ducts to colonize host tissue. (e) Following successful colonization, symbiotic bacterial cells become nonmotile and induce host epithelial cell swelling. Only bioluminescent V. fischeri will sustain long-term colonization of the crypt epithelium.
SOURCE: Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Microbiology, Nyholm and McFall-Ngai (2004), copyright 2004.
receive energy from the plant and convert atmospheric nitrogen to ammonia, which the plant can then assimilate into amino acids, nucleotides, and other cellular constituents (Desbrosses and Stougaard, 2011). This partnership furnishes much of Earth’s biologically available nitrogen,13 a key contributor to agricultural productivity that has long been achieved by growing legumes in rotation with nonlegume crops.
___________
13 Nitrogen is a critical nutrient for plants, but often it is not readily available in soil, hence the extensive use in agriculture of chemical fertilizers containing nitrogen.
Partnerships between plant roots and microbes are established through chemical and genetic “cross-talk.” During nodule formation, legume roots release flavonoid compounds that trigger nitrogen-fixing Rhizobium bacteria to express modified chitin oligomers known as nodulation (Nod) factors, which in turn facilitate infection of the root by the bacteria, as well as nodule development (Desbrosses and Stougaard, 2011; Ferguson et al., 2010; Long, 2001; Riely et al., 2006) (see Figure WO-5).
Other plants produce chemical signals called strigolactones that increase their contact with arbuscular mycorrhizal fungi; this triggers the fungi to release diffusible factors that, when recognized by the plant, activate genes collectively known as Myc factors (Parniske, 2008). Both Nod and Myc factors promote plant growth, which may benefit microbes by increasing the availability of infection sites (IOM, 2009).
Microbial inhabitants of the human gut Just as microbes colonize the bobtail squid’s light organ shortly after hatching, microbes colonize the human body internally and externally during its first weeks to years of life and establish themselves in relatively stable communities in various microhabitats (Costello et al., 2012; Dethlefsen et al., 2007). Research to date suggests that the site-specific microbial communities—known as microbiota or microbiomes14—that inhabit the skin, intestinal lumen, mouth, vagina, etc., contain characteristic microbial
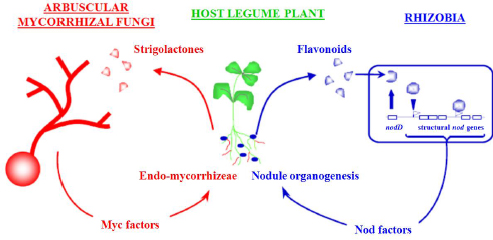
FIGURE WO-5 An example of nitrogen-fixing symbiosis between legumes and rhizobia bacteria.
SOURCE: Provided courtesy of Jean-Michel Ané, University of Wisconsin, Madison.
___________
14 The term microbiome is attributed to the late Joshua Lederberg, who suggested that a comprehensive genetic view of the human as an organism should include the genes of the human microbiome (Hooper and Gordon, 2001). Because most of the organisms that make up the microbiome are known only by their genomic sequences, the microbiota and the microbiome are from a practical standpoint largely one and the same (IOM, 2009).
families and genera (see Figure WO-6). The species and strains of microbes present on or in any given individual may be as unique as a fingerprint (Dethlefsen et al., 2007). The microbiotas of other terrestrial vertebrates are dominated by microbes that are related to, but distinct from, those found in humans. This suggests that host species have uniquely coevolved with and adapted to their microbial inhabitants.
The human gastrointestinal tract contains the highest cell densities of any known ecosystem (Ley et al., 2006a). A recent effort to catalog the genes of the adult human gut microbiome identified 3.3 million nonredundant microbial genes in fecal samples obtained from 124 individuals, suggesting the presence
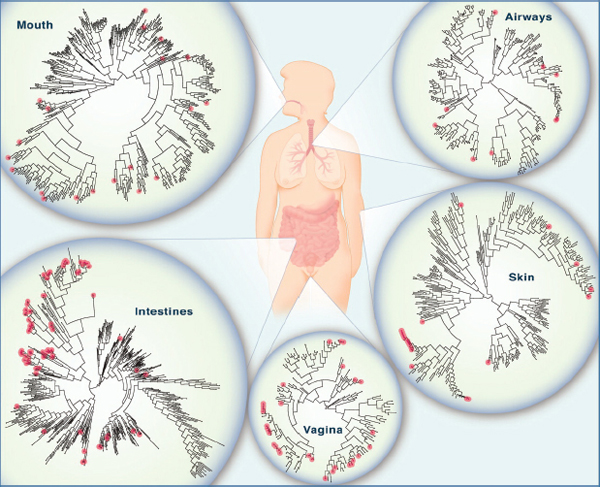
FIGURE WO-6 The microbiome of various anatomical locations of the human body. Numerous bacterial species colonize the mouth, upper airways, skin, vagina, and intestinal tract of humans. The phylogenetic trees show the speciation of bacterial clades from common ancestors at each anatomical site. Although the communities in different regions of the body share similarities, they each have a unique site-specific “fingerprint” made of many distinct microbes and genes. Each site has a very high level of diversity, as shown by the individual lines on the dendrograms. Data are from the National Institutes of Health (NIH)-funded Human Microbiome Project; circles represent bacterial species whose sequences are known.
SOURCE: Lee and Mazmanian (2010).
of 1,000 to 1,150 prevalent bacterial species, and found that each individual’s gut harbored at least 160 bacterial species (Qin et al., 2010). The vast majority of these microbes reside in the human distal gut, where they have been found to influence human health through a variety of functions, which include degrading and processing components of the human diet that would be otherwise indigestible (Gill et al., 2006); exchanging metabolites and cometabolizing substrates with its host (Nicholson et al., 2005, 2012); regulating drug metabolism and bioavailability and detoxifying ingested carcinogens (Gill et al., 2006; Turnbaugh et al., 2007); modulating responses to epithelial cell injury; and influencing the maturation of both the innate and adaptive immune systems (Gill et al., 2006; Lee and Mazmanian, 2010; Littman and Pamer, 2011).
While the relationship between humans and the microbial inhabitants of their gastrointestinal tracts tends to be mutually beneficial, shifts in microbial gene expression may result in immune responses that precipitate disease states such as inflammatory bowel disease, which is characterized by an unregulated immune response to gut microbes (Littman and Pamer, 2011). Evidence also suggests that increased susceptibility to enteropathogens in some individuals may stem from a failure of their gut microbiota to alter expression of host genes encoding antimicrobial compounds (Cash et al., 2006). The microbiota of the gastrointestinal tract has also been found to influence host predisposition to a number of diseases, including obesity (Ley et al., 2006b).
The mammalian gut microbiota also appears to be important in the development of the immune system (Hooper et al., 2010; Lee and Mazmanian, 2010; Littman and Pamer, 2011). Animals born and raised in sterile environments display developmental defects in tissue formation and compromised expression of receptors and molecules that are involved in pathogen sensing and antigen presentation. Intestinal and systemic immune responses are also influenced by the microbiota in ways that suggest that the host and its microbiota have coevolved to collaborate against infectious agents. Investigators recently discovered intriguing associations between the microbiota and the development of asthma in children born by Cesarean section—additional evidence that the “normal” balance of the composition of the microbiome may help guide the development of the immune system, and that when that balance is altered, disease may follow (Couzin-Frankel, 2010).
“When I started in biology, I was told that microbes were the proof that really small creatures didn’t need to engage in social activities. The smaller the creature, the less value there would be for social activity. That has given way to a new view, and now we talk about the social life of bacteria.”
—E. Peter Greenberg, Keynote Address
As the earlier examples suggest, microbes living in communities display coordinated, synchronized behaviors. They have also evolved specific interactions with each other and with their “host” environments. Like macroscopic organisms that cooperate to secure food, gain protection from predators, build shelters, and reproduce, microorganisms engage in activities with group-derived benefits (Bonner, 2010; Crespi, 2001). Social behaviors—broadly defined as actions with fitness consequences for both actor and recipient—enable microorganisms to build complex, interactive communities that often exhibit functions or social traits commonly associated with multicellular organisms, as shown in Table WO-1.
| Cooperative Behavior | Group-Derived Benefits | Microbe Examples | Higher Organism Comparisons | |
| Chemical communication (quorum sensing) | Coordinated population behavior | Vibrio fischeri, Pseudomonas aeruginosa, Staphylococcus aureus, etc. | Pheromone production in many social animals | |
| Biofilm formation | Protection from adverse environmental conditions | Many species of bacteria | Burrows, nests, hives, cities | |
| Nitrogen fixation: mutualistic behavior Foraging/hunting: nutrient acquisition | Nutrients and niche protection in nodules Enhanced growth and colonization sometimes in specialized niches | Rhizobium spp. with legume plants Siderophore production for iron acquisition in many bacteria | Yucca plant and yucca moth Wolves, lions, humans | |
| Autolysis (suicide) | Provides nutrients and DNA for biofilm development | P. aeruginosa | Apoptosis in eukaryotic cells | |
| Motility (swarming) | Coordinated motility to a nutrient source | Yersinia spp. Myxococcus xanthus, P. aeruginosa, | Ants, termites | |
| Antibiotic resistance | Production of extracellular enzymes (e.g., ß-lactamase) to break down antimicrobials | Escherichia coli, Klebsiella spp. | Group defense, antipredator vigilance | |
| Immune modulation | Modulation of immune response to facilitate survival within the host | P. aeruginosa, Porphyromonas gingivalis, Helicobacter pylori | Helminth parasites | |
SOURCE: Diggle et al., Evolutionary theory of bacterial quorum sensing: When is a signal not a signal?, Philosophical Transactions of the Royal Society B: Biological Sciences, 2007, 362(1483):1241-1249, reprinted by permission of the Royal Society.
Does the observation that microbes live in communities imply that all microbes are social? Not necessarily, according to workshop speaker Joan Strassmann of Washington University in St. Louis, who both raised and answered this question as follows (Dr. Strassman’s contribution to the workshop summary report can be found in Appendix A):
If there existed a microbe that moved through the soil as a single cell, propelled, perhaps, by a single flagellum, taking in nutrients as it encountered them, then dividing when it got big enough, I suppose you could call it solitary, not social. But if it bumped into another cell and exchanged some genetic material that would be the social act we could call a form of sex. If it sensed the presence of others through secretions that made it change its trajectory that would also be a social act. If it coordinated with others to move in swarms, that would be social. In short, I think it is safe to say, that all microbes are social. This does not mean they all do the same social things. (Strassmann, 2012a)
Social evolutionary theory interprets behavior in terms of fitness: the reproductive advantage actions confer on both actor and recipient. Evolutionary biologists have devoted considerable attention to the question of cooperation (Strassmann and Queller, 2011; West et al., 2007a), which for the purposes of this overview is defined as an action that an individual organism takes, at some “cost” to itself (that is, the action, in and of itself, diminishes its chance of reproducing), and which benefits the community as a whole.15 Social evolutionary theory explains cooperation in terms of enlightened self-interest: in return for their altruistic acts, individuals receive benefits that outweigh the cost of their actions either directly, or to related individuals. In the case of interactions among members of the same species, cooperation is expected to evolve when benefits of aiding another, weighted by relatedness between helper and altruist, outweigh the cost to the altruist in terms of reproduction. This is called Hamilton’s Rule, also called kin selection, and is the cooperative side of inclusive fitness (Hamilton, 1964). Cooperation among relatives can thus extend to altruism, in which an individual sacrifices its chance of reproducing to advance those of a relative.16
Applying social evolutionary theory to cooperative behaviors of microorganisms has provided insights into why these behaviors evolve and how they are maintained in microbial communities. Some have questioned, however, the appropriateness of the social evolution framework for interpreting the actions of microorganisms and, more generally, the notion that all microorganisms are inherently social (Zhang and Rainey, in preparation). This controversy was raised during the workshop and is addressed later in the Semantics section of this chapter.
___________
15 There can be positive acts that benefit self and community. For example, if a bacterium secretes something that facilitates movement, it could benefit itself and others.
16 Cooperation among relatives can also include spiteful behavior. Spiteful traits are harmful to both actor and recipient, but can be beneficial to a secondary recipient. According to West and Gardner (2010), spite can therefore be thought of as altruism toward the secondary recipients, with the actor sacrificing its chance of reproducing to advance those of a relative (West and Gardner, 2010).
Cooperation among microbes is also of interest to evolutionary biologists because it represents a likely stage in the evolution of multicellular organisms (Strassmann and Queller, 2011). Multicellularity has evolved multiple times. The process by which autonomous cells become an organism—and, thereby, subject to natural selection at this higher level of organization—often, but not always, involves the failure of dividing cells to separate and is an area of active research (Ratcliff et al., 2012).
Microbial Communities: The Ultimate Social Network
In his keynote address to the workshop, E. Peter Greenberg, of the University of Washington, defined the nascent field of sociomicrobiology as the study of the genetic basis of, and environmental influences on, the social activities of microbes (Dr. Greenberg’s contribution to the workshop summary report can be found in Appendix A) (Parsek and Greenberg, 2005). Among several reasons for pursuing such studies, microbes offer a novel perspective on the biology of sociality, and an efficient means to pursue basic evolutionary questions. “You could argue that what we understand of the microbes comes primarily from our understanding of social activities in higher organisms,” Greenberg said. “We’re just at the stage now where we can begin to jump ahead. We can use that knowledge, but we can also do things much more quickly and with a much less biased eye by studying bacteria.” As a result, he predicted, observations of microbial social biology will eventually produce hypotheses to be tested in macroscopic species.
Key to microbial interactions with other organisms and their surroundings are a range of microbial strategies for sensing and responding to environmental conditions (Bassler and Losick, 2006; IOM, 2009). The structure and function of microbial communities are influenced and modified by fluctuating biological, chemical, and physical factors (Maloy et al., 2011). In particular, microbial communities are awash in chemical information from the environments in which they reside and that the microorganisms themselves produce.
Research has associated specific chemical signals and communication mechanisms with a wide variety of behaviors and interactions between microorganisms (Bassler and Losick, 2006; Hughes and Sperandio, 2008); additional signals, cues, and systems of communication continue to be discovered (Han et al., 2011b; see also Bassler and Losick, 2006; Hayes et al., 2010; Shank and Kolter, 2009). Some pathogens appear to interfere with cell-to-cell signaling in order to subvert or evade host defenses, while others have been found to use signals to activate transcription of virulence genes coordinately, under circumstances favorable to infection (Diggle, 2010). Group behaviors need not arise from sophisticated interactions between
actors but may simply represent a collection of individual responses to a shared environment. For example, physical factors, such as chemical gradients, have been shown to produce heterogeneity in biofilm physiology and spatial structure without the active, coordinated behavior of individual cells (Nadell et al., 2009).
In the 1960s and 1970s, the discovery that bacteria produce diffusible signal molecules that trigger coordinated behavior among localized groups provided the first evidence that microbes communicate (Bassler and Losick, 2006; Sandoz et al., 2007). Since then, the general ability of cells to secrete small molecules and to sense their extracellular concentration—a reflection of population density and other features of the external environment—has been found in many organisms and even across taxonomic kingdoms (Xavier, 2011). In his keynote remarks, Greenberg provided a detailed description of the best-studied example of this form of microbial communication, known as quorum sensing. Additional workshop presentations summarized later in this overview described a range of mechanisms by which microbes interact with members of their own species and with other microbes, with host macroorganisms, and with their environment.
Quorum sensing17 Many microorganisms—as well as some cell types within multicellular organisms—secrete small signaling molecules and sense their concentration in the environment (Xavier, 2011). This signaling mechanism, called quorum sensing, is so named for the accumulation of signal molecules within a population of bacteria, which reaches a threshold when the population reaches a significant density, or quorum (Fuqua and Greenberg, 2002). The entire population responds when the signal threshold is reached, usually through the coordinated expression of specific target genes.
Quorum-sensing systems control the production of so-called public goods, Greenberg stated. Originating in the field of economics, this term is also used by social evolution theorists to describe metabolically “costly” products manufactured by individuals (in this case individual bacterial cells) that benefit other individuals (West et al., 2007a). Such products include exoenzymes, which are released by bacteria into their environment to perform such functions as breaking down food sources so that they can be transported within cells and defending against potential predators. “Secreted or excreted molecules are often toxins for human cells, for example, and often antibiotics for bacteria,” Greenberg observed.
In the best-studied quorum-sensing systems, according to Greenberg, an acyl-homoserine lactone (acyl-HSL) serves as the signaling molecule. This type of signaling system was first discovered in V. fischeri, the previously described bioluminescent bacterium that inhabits the light organ of the bobtail squid. In addition to this communal existence, individual V. fischeri also live in seawater,
___________
17 The term quorum sensing was first used by Fuqua et al. (1994). Since then, there have been many variations on the definition. According to Greenberg, quorum sensing and response is generally used to describe a cell-to-cell communication system that allows bacteria to monitor population density and control of specific genes in a density-dependent manner.
dispersed so widely that any light they might produce would be invisible to any known eye, he explained. Quorum sensing allows the bacterium to monitor which environment it inhabits, and to produce light only when there is a critical mass of bacteria in close proximity to induce bioluminescence.
Acyl-HSL quorum sensing occurs in more than 200 species of Proteobacteria, Greenberg noted; about 40 different acyl-HSL signals are known within this group, each with different specificities.18 The signaling molecule, which is produced by the LuxI family of signal synthases, binds to highly specific receptors in the LuxR family that act as transcription factors (see Figure WO-7). This coevolved pairing of signals and receptors marks quorum sensing as a formal communication system, he concluded.
Greenberg uses the bacterium Pseudomonas aeruginosa as a model to study the evolution of cooperative behavior and understand how quorum sensing is embedded in the complex regulatory networks of a cell. This microbe is “found wherever we look for it: in the soil, the water, on plants, invertebrates, and people,” he noted. It is also an opportunistic pathogen19 that can cause fatal infections in some humans and animals. P. aeruginosa’s acyl-HSL quorum-sensing system controls about 300 genes, including those involved in virulence and biofilm formation. This battery of genes “likely evolved and was selected for a purpose outside of an animal host,” Greenberg said. At high population densities, bacteria coordinate the expression of genes in the quorum regulon; this produces virulence factors in concentrations that can overwhelm host defenses, he explained.
The existence of quorum-sensing mutants of P. aeruginosa with reduced virulence suggests that quorum sensing may have promise as a therapeutic target, Greenberg reported. He further observed, however, that some of these same mutants are also commonly found in certain infected tissues, such as the lungs of patients with cystic fibrosis—an observation that calls into question whether quorum sensing is required to maintain virulence in P. aeruginosa (Sandoz et al., 2007). In the language of social evolution theory, these mutants are “cheaters”: they benefit from a public good (virulence factors released by “cooperators”), without paying the metabolic “cost” of production.
“One possible outcome of cheaters arising in a group is that there could be a tragedy of the commons,” Greenberg explained. “The cheaters have an advantage because they are not bearing the costs of making these things, so they should outgrow the cooperators, and if they overtake the cooperators, the ability to cooperate will be lost and the whole population will collapse.” Persistent infections
___________
18 There are numerous examples of quorum sensing in Gram-positive bacteria, Greenberg said, but instead of acyl-HSL signals, they use small peptides—a clear case of convergent evolution. Acknowledging what he called some “ill-defined reports” of quorum sensing in species of Archaea, as well as in halophilic and methanogenic bacteria, he observed that “it will be some time before we know whether those stand up to the test of time.”
19 Resulting from pathogen entry via wounds or weakened state of the host, or as a result of a disturbance of a normally benign host-microbe relationship.
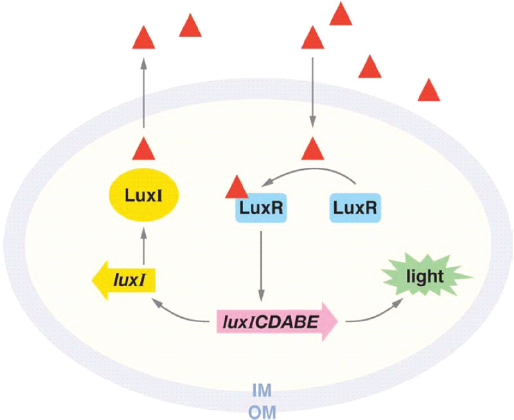
FIGURE WO-7 Mechanism of quorum sensing in the luminescent bacterium Vibrio fischeri. The luxI gene (yellow) encodes the LuxI signal synthase, which produces acyl-HSL (red triangle). Acyl-HSL is freely diffusible through the membrane of the cell, accumulating in the environment only when many cells, close together, are producing the signal molecule. When acyl-HSL concentrations reach nanomolar levels (both inside and outside the cells), acyl-HSL binds to the transcription factor LuxR (blue). LuxR interacts with genes that enable the bacterium to produce light, which are present on the same operon as luxI.
SOURCE: Republished with permission of Waters and Bassler, from “Quorum sensing: Cell-to-cell communication in bacteria,” Waters, C. M., and Bassler, B. L., Annual Review of Cell and Developmental Biology 21:319-346, copyright 2005; permission conveyed through Copyright Clearance Center, Inc.
include significant numbers of such cheaters—quorum-sensing mutants—suggesting that they are in equilibrium with the cooperators. Greenberg also noted a possible mechanism for maintaining this equilibrium, which he called “metabolic policing”: the quorum-sensing coregulation of certain “private goods” that benefit individual cells.
“We hypothesized that a relatively few cellular processes, these private goods, have been subsumed or retained in the quorum-sensing regulon to allow metabolic selection against quorum-sensing mutants as social cheats,” Greenberg said. Indeed, it turns out that quorum sensing also controls the ability of the bacterium to catabolize adenosine—and that the addition of adenosine to growth medium for P. aeruginosa suppresses the emergence of quorum-sensing mutants.
Workshop speaker Sam Brown, of the University of Edinburgh, suggested that the supply of adenosine might be greater under conditions that would favor the existence of a quorum, such as within a preferred host’s tissues (Dr. Brown’s contribution to the workshop summary report can be found in Appendix A). “That’s along the lines that we are thinking,” Greenberg agreed. He added that this policing mechanism may also reflect the possible original function of the quorum-sensing regulon: anticipation of the carrying capacity of the population. “The way to prepare for living in a world of maximum population is to activate all sorts of genes, most of which are private goods,” he said; and if that’s the case, he observed, other Proteobacteria quorum-sensing systems should have similar policing mechanisms.
Other environments may lend themselves to different types of policing systems, Greenberg noted. In biofilms and other structured environments, for example, cheaters multiply in place, eventually separating themselves from cooperators and the life-sustaining benefits they provide. Cooperation can happen without quorum sensing, he said, although the relative benefits and costs of quorum sensing as a means to control cooperation remain to be determined.
“This is a science in its infancy,” Greenberg observed. By learning about communication and control of cooperation and understanding the relationships between cooperation and conflict, it may eventually be possible to devise ways to induce a “tragedy of the commons” to resolve certain bacterial infections, or to encourage the growth of beneficial communities, Greenberg said.
Interactions with Other Species: Bioluminescence in Marine Microbial Communities
Underlying the complex dynamics of microbial communities—many of which include multiple species—are myriad interactions between community members and host organisms (Little et al., 2008). Interspecies relationships span the overlapping categories of mutualism, commensalism, and parasitism. In a mutualistic relationship, both (or all) members benefit. In a commensal20 relationship, two (or more) species coexist, one deriving benefit from the relationship without harm or obvious benefit to the other. A parasitic relationship results when one species inflicts harm upon the other (Little et al., 2008). A host-microbe interaction can evolve from one type of symbiosis21 to another over time, as circumstances change. Community-level behaviors may result from a network of direct and indirect effects of interactions between organisms.
___________
20 It has been suggested that purely commensal relationships may not exist. Rather, the benefit to the other partner simply may not yet have been discovered. For example, the microbial residents of the human gut were previously referred to as commensals, but are now known to contribute significantly to human well-being (Little et al., 2008).
21 Classically, the term symbiotic has been used to refer broadly to dissimilar organisms and/or species living in close association; however, it is often used interchangeably with mutualistic.
In a second keynote presentation Edith Widder, of the Ocean Research and Conservation Association, described her research into the dazzling world of bioluminescence (Dr. Widder’s contribution to the workshop summary report can be found in Appendix A). These light-producing chemical reactions occur in a wide variety of living organisms, the vast majority of which reside in the ocean (Widder, 2010). Witnessing these “light shows” in the ocean changed the course of Widder’s career as she “felt like bioluminescence had to be one of the most important processes in the ocean.” She described the basis of bioluminescence and its function of conveying information in the sea, as well as her work to develop tools that can detect and measure the emission of and response to bioluminescence.
The ubiquity and abundance of bioluminescence in the ocean suggests its importance in marine ecosystems: This trait spans marine phyla from bacteria to fish (Figure WO-8). In the upper 1,000 meters of the open ocean, 80-90 percent of animals are bioluminescent; below 1,000 meters it is estimated at about 50 percent. Even in the deep ocean, about 20 percent22 of the animals are bioluminescent. This trait appears to have evolved multiple times, she said. Bioluminescent species employ chemistries so diverse that the evolutionary origins of this phenomenon are uncertain (Widder, 2010). Bioluminescence most often results from chemical processes intrinsic to an organism, but it can also result from associations between bioluminescent bacteria23 and multicellular hosts (Widder, 2010).
Bioluminescence can appear as a “persistent glow of bioluminescent bacteria to brief flashes from lanternfish light organs” (Widder, 2010). Luminescent chemicals—called luciferins—may be released directly into the water or retained within cells called photocytes. The quality of the light can be adjusted by muscles or optical components within light organs that reflect, refract, or filter the light being produced. Patterns of light can also be created by motion or the placement of photocytes on the body surface. “All of these parameters carry information to the eyes of potential predators, prey, or members of the same species” (Widder, 2010).
Many researchers have suggested that bioluminescence “has no primary function”— an explanation often “given when you just don’t know what it does,” observed Widder. The many functions of bioluminescence reflect the unique ecology of the marine environment, particularly the nature of the visual environment (Widder, 2010). As the ocean filled up with ever swifter and nastier predators, she explained, potential prey species evolved the habit of staying in the dark depths during the day, then coming up to feed in food-rich surface waters under cover
___________
22 Based on limited sampling.
23 Four bacterial genera include bioluminescent species: Photobacterium, Vibrio, Shewanella, and Photorhabdus, Widder said; all of them make light through the oxidation of two substrates—a reduced flavin mononucleotide (FMNH2) and a long-chain aliphatic aldehyde (RCHO)—by molecular oxygen, catalyzed by luciferase. The blue light produced by this reaction enables DNA repair within cells (Czyz et al., 2003), but it is not visible to any known eye at the levels produced by individual bacteria, she noted.
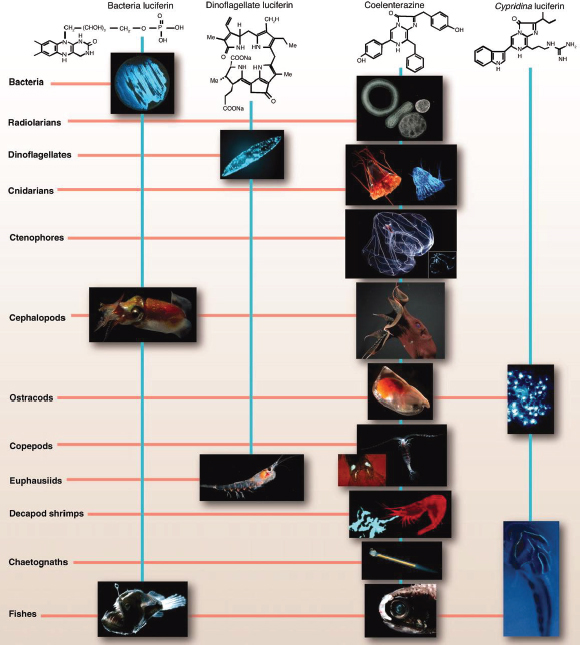
FIGURE WO-8 The chemical structures of the four best-known luciferins are as diverse as their phylogenetic distribution. Bacterial luciferin may occur in free-living or symbiont bacteria (e.g., in squid such as Heteroteuthis dispar) or in fish such as Melanocetous johnsoni. Dinoflagellate luciferin occurs not only in dinoflagellates (e.g., Pyrocystis fusiformis) but also in euphausiids (e.g., Meganyctiphanes norvegica). Some of those using coelenterazine as luciferin include radiolarians (e.g., unidentified polycystine radiolarians), cnidarians (e.g., scyphozoan Periphylla periphylla, as seen in the light and photographed by its own light), ctenophores (e.g., Bathocyroe fosteri, with bioluminescence display shown in inset), vampire squid (e.g., Vampyroteuthis infernalis), ostracods (e.g., Orthoconchoecia agassizi), copepods (e.g., Gaussia princeps releasing its bioluminescent chemicals from glands on its tail, shown in inset), decapods (e.g., Acanthephyra purpurea spewing luciferin and luciferase out of its mouth), chaetognaths (e.g., Caecosagitta macrocephala), and fish (e.g., the myctophid Diaphus sp. has a large preorbital light organ). Cypridina luciferin, which is an imidazopyrazinone like coelenterazine, is found in ostracods such as Vargula hilgendorfii and is the dietary source of luciferin for the midshipman fish Porichthys notatus.
SOURCE: Widder (2010). Photo credits: S. Haddock, radiolarians and chaetognath; K. Reisenbichler, V. infernalis; J. Case, copepod luminescent glands and midshipman fish photophores
of darkness24—a routine that favors animals with sensitive eyes and dark-defying signaling. “Bioluminescence has evolved many times because it serves three basic functions,” Widder stated. Animals use bioluminescence to survive by
- Finding food: Light organs aid in locating food either by means of built-in headlights or by the use of glowing lures that attract potential prey (Widder, 2010).
- Attracting mates: Species-specific spatial or temporal patterns of light emission can be used to attract a mate (Widder, 2010).
- Defending against predators: Bioluminescence emissions can be used to blind, distract, or serve as a warning to predators; when controlled to match ambient light color and intensity, bioluminescence provides counterillumination that camouflages organisms by obscuring their silhouette (Widder, 2010).
“In the case of luminous bacteria that form specific symbioses with certain marine fishes and squid, the adaptive value of the light emission is generally evident: The bacteria provide the host with light that can be used to attract prey, evade predators, or attract a mate, while the host provides the bacteria with an ideal growth environment” (Widder, 2010). For free-living bacteria where the adaptive value is less evident, she noted that organisms often form communities on the surface of fish fecal pellets and suggested that the collective glow they produce (on cue from quorum sensing) may lure other fish to consume the pellets, thereby introducing the bacteria to the nutrient-rich environment of the fishes’ digestive tract (Zarubin et al., 2012). This scenario favors not only bioluminescence, but also quorum sensing, she observed.
Widder described a fascinating—but unexplored—bioluminescent phenomenon: “marine snow” is a continuous shower of mostly organic detritus that falls through the water column and luminesces when stimulated with light. This energetically demanding and apparently widespread phenomenon represents a significant carbon flux in the ocean, she observed, “and nobody knows anything about it.” It looks like strings of glowing mucous-like material that respond to either photic or mechanical stimulation and likely contain bioluminescent bacteria. “This is clearly not any animal or organized thing,” she declared, but she has yet to identify what organisms or processes she is observing.
Explorations using submersibles and remotely operated vehicles are revealing new luminescent organisms. The use of far-red illumination and intensified imaging technologies have made it possible to develop unobtrusive methods to observe bioluminescence in natural settings. Widder developed an “electronic jellyfish”—essentially an optical lure that can imitate certain types of bioluminescent displays (Figure WO-9).
___________
24 This is known as vertical migration and is the most massive animal migration pattern on the planet.
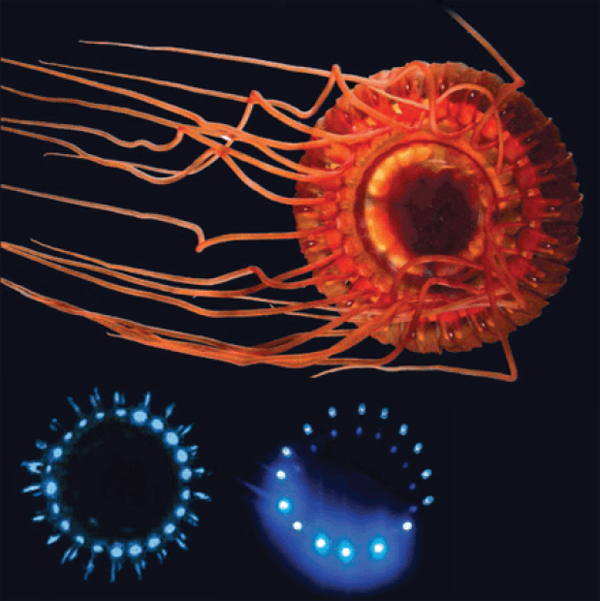
FIGURE WO-9 The burglar alarm jellyfish (top) lights up blue (lower left) to call in predators of its attackers; researchers copied this pattern with LEDs (lower right) to lure organisms to an underwater camera (Pennisi, 2012).
SOURCE: Edith Widder (published in Pennisi, 2012).
Using a camera system called “Eye-in-the-Sea” that uses far-red light that is invisible to most animals, she has been able to “eavesdrop” and communicate with different animals with the luminescence. For example, according to Widder a pinwheel of light display that imitates a certain type of jellyfish has been hugely attractive to squid. “We have gotten a lot of squid attacks on it. There is a rapid repetitive flash that we have used out in the Bahamas, and something talks back to us and leaves a lot of luminescence in the water. I think it’s a shrimp and I think we’re saying something sexy, but I’m not sure.”
STRUCTURE AND FUNCTION OF MICROBIAL COMMUNITIES
“Because of their ubiquity and centrality to life, microbial communities demand our attention. It will not be possible to understand fully the many services they perform without knowing which organisms are present and how each contributes to community function.”
—Jo Handelsman (2007)
Molecular technologies are revolutionizing our collective understanding of microbiology, expanding its purview from the study of individual organisms to considerations of microbial populations and communities, as illustrated in Figure WO-10 (Little et al., 2008). New technologies that catalog the broad diversity of microbial communities in their natural settings are allowing researchers to “see” microorganisms in a dramatically new way. But, as depicted in Figure WO-10, “more is different” (Anderson, 1972), and researchers are only just beginning to develop approaches to exploring the ecology of microbial communities. The ecological properties of microbial communities can be classified as structural (type and numbers of members) and functional (community behaviors resulting from interactions between community members and with external forces) (Little et al., 2008).
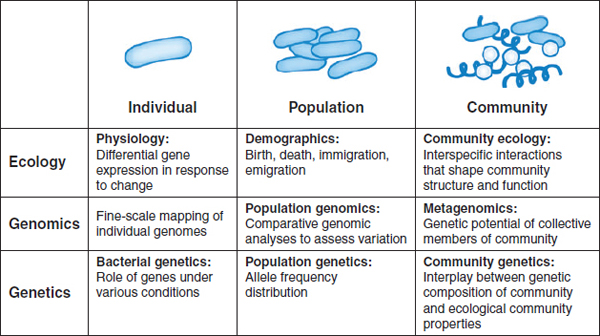
FIGURE WO-10 Progression from studies on the individual scale to studies on the community scale.
SOURCE: Little et al. (2008). Reprinted, with permission, from the Annual Review of Microbiology, Volume 62 © 2008 by Annual Reviews www.annualreviews.org.
The ability to survey “who is there” by analyzing 16S rRNA25 from environmental samples is complemented by the systems-level view provided by metagenomic analyses, which reveal the entire complement of genes present in communities of microbes and provide clues to their presumed functions (Little et al., 2008). Sequence-based metagenomics can be used to determine the complete genome of an individual microbial species or to analyze the genome of the community26 as a whole, which can offer insights about population ecology and evolution (NRC, 2007). Function-based metagenomic studies identify functions that are unknown in the limited number of microbes that can be grown in a laboratory, as well as novel proteins and metabolites (NRC, 2007).
The identification of core microbiomes—groups of organisms that are common to microbiomes in a particular habitat and which play an essential role in ecosystem function—is an area of active investigation. Understanding the communities of organisms present in states of “health” may also provide insights into dysfunction or disease that result from altered community composition, and how these microbial communities might be manipulated in order to achieve a particular outcome (Shade and Handelsman, 2011). The generation of metagenomic information from a variety of environments is supported by several active and proposed programs (Box WO-1).
Metagenomic analyses suggest that typical microbial communities contain a few “keystone” organisms, along with many members that are rare, but whose collective biomass and genome comprise a large fraction of the total. Deep sequencing of environmental samples is required to reveal such rare microbes, which have been collectively termed the“rare biosphere.” The consistent presence of a specific rare biosphere among microbial communities suggests its importance, and it has been speculated that its members may carry out critical physiological functions, respond to changes in the environment, or serve as a reservoir of novel genes (Reid and Buckley, 2011).
Several workshop presenters described the use of genomic information to determine the membership of microbial communities and to examine their ecology and evolution. The diversity of topics included among these presentations attests to the broad applicability of genomic analysis to the study of microbial communities and to the variety of inferences that may be drawn from such information. At the same time, several presenters and discussants noted the potential need to expand and refine existing methodologies in order to gain a more complete picture of the
___________
25 A component of the small subunit of prokaryotic ribosomes. The small subunit rRNA gene sequences contain hypervariable regions that can provide species-specific signature sequences. Polymerase chain reaction (PCR) amplification with primers that hybridize to highly conserved regions in bacterial or archaeal 16S rRNA genes (or eukaryotic microbial 18S rRNA genes) followed by cloning and sequencing yields an initial description of species present in a microbial community.
26 Metagenomic analyses have focused on identifying bacterial species; additional efforts are needed to characterize other microorganisms present within microbial communities—including Archaea, fungi, and viruses—because these organisms likely play important roles in shaping the properties of the community as a whole.
BOX WO-1
Microbiome Research Projects
NIH: The Human Microbiome Project (HMP) is a 5-year, US$157 million undertaking launched by NIH in 2007 (Buchen, 2010) to sequence the microbial communities of several hundred people in order to define commonalities and patterns, and a core microbiome if one exists. The first stage of the project was focused on the metagenomes of the human skin, nose, mouth, gut, and vagina of 300 healthy volunteers and has since expanded, sampling additional body sites. Beyond describing the human microbiota, the HMP seeks to understand aspects of communities such as function, including whether alterations to the microbiome can be correlated to changes in human health. Another project goal is to sequence 3,000 genomes from both cultured and uncultured bacteria, plus viral and small eukaryotic microbes isolated from human body sites.
Metagenomics of the Human Intestinal Tract (MetaHIT) is a project financed by the European Commission that seeks to establish associations between the genes of the human intestinal microbiota and health and disease. Launched in 2008, this 5-year project gathers 13 partners from academia and industry, from a total of 8 countries (China, Denmark, France, Germany, Italy, the Netherlands, Spain, and the United Kingdom). Focused on two disorders of increasing importance in Europe, inflammatory bowel disease (IBD) and obesity, MetaHIT has established an extensive reference catalog of microbial genes present in the human intestine and bioinformatics tools to store, organize, and interpret this information; developed tools to determine which genes of the reference catalog are present in different individuals and at what frequency; gathered cohorts of individuals, some sick and some healthy; determined for most which genes they carry; and developed methods to study the function of bacterial genes associated with disease aiming to understand the underlying mechanisms and host-microbe interactions.
The Earth Microbiome Project is a proposed effort to analyze microbial communities across the globe. Participants propose to “characterize the Earth by environmental parameter space into different biomes and then explore these using samples currently available from researchers across the globe.” This project seeks to “analyze 200,000 samples from these communities using metagenomics, metatranscriptomics, and amplicon sequencing to produce a global Gene Atlas describing protein space, environmental metabolic models for each biome, approximately 500,000 reconstructed microbial genomes, a global metabolic model, and a data-analysis portal for visualization of all information” (see http://earthmicrobiome.org).
_______________
SOURCES: http://commonfund.nih.gov/index.aspx; http://www.hmpdacc.org/reference_genomes/reference_genomes.php; The Human Microbiome Jumpstart Reference Strains Consortium (2010); http://www.metahit.eu; http://www.earthmicrobiome.org; Robinson et al. (2010).
membership and population dynamics of microbial communities and also to combine genomic analysis with experimental studies of microbial ecology and behavior.
Factors Influencing Community Formation and Function
Several presentations provided insights into some of the processes and factors that shape community formation and function. These presentations highlighted the contribution of a variety of factors to the development and dynamics of communities—including nutrient and resource availability, the development and maintenance of favorable ecological niches, and the adaptability of microorganisms to environmental change. The ecological context of these interactions was underscored by many speakers as a key driver for the nature and outcome of interactions between community members and their associated hosts.
The Role of Oxygen in the Structure and Function of Microbial Communities
“One of the spectacular features of microorganisms is their capacity to couple all kinds of chemical reactions,” observed Thomas Schmidt of Michigan State University (Dr. Schmidt’s contribution to the workshop summary report can be found in Appendix A). Many microbes use oxygen as a terminal electron acceptor for respiration, and “in any environment where microbes gather, there will be competition for oxygen and also an oxygen gradient,” he stated; thus, gradients and fluctuations of oxygen may influence both microbial gene expression and interactions within communities (Han et al., 2011a). In structured, oxygen-limited environments, such as biofilms or the largely anoxic environment of the human intestinal mucosa, fluxes in oxygen could have important implications for community structure and function.
Schmidt’s laboratory is particularly interested in low-oxygen (microoxic) environments such as the immediate exterior of the intestinal epithelium, where oxygen diffuses from capillary networks at the tips of villi (Marteyn et al., 2010). Organisms that require oxygen to survive but require its presence in lower concentrations than that found in the atmosphere are called microaerophiles.27Helicobacter thrive under such conditions, he observed, which also support Shigella and other organisms long classified as anaerobes28—organisms he has dubbed “microaerobes.” A recent study shows that even Escherichia coli grows at a slow rate, respiring aerobically, in a microoxic environment (Stolper et al., 2010), he noted. Microoxic conditions influence the expression of a number of Shigella genes involved in establishing an infection, including an effector secretion system (Marteyn et al., 2010). “Presumably Shigella, and perhaps other
___________
27 See http://medical-dictionary.thefreedictionary.com/microaerophile.
28 An organism that can or must live in the absence of oxygen (see: American Heritage Science Dictionary, 2011).
pathogens, sense low oxygen, sense that they are near the mucosa,29 and change their pattern of gene expression,” he speculated (Figure WO-11).
A defining feature of microaerobes is their requirement for high-affinity cytochrome oxidase. This form of a key respiratory enzyme captures oxygen at low concentrations and is essential to survival in microoxic conditions, Schmidt noted. His group performed searches of existing bacterial genome and metagenome sequences to determine the phylogenetic distribution of high-affinity cytochrome oxidases and found them to be present in nearly all phyla represented by genomic sequencing, and in nearly every shotgun metagenome30 they examined (Morris
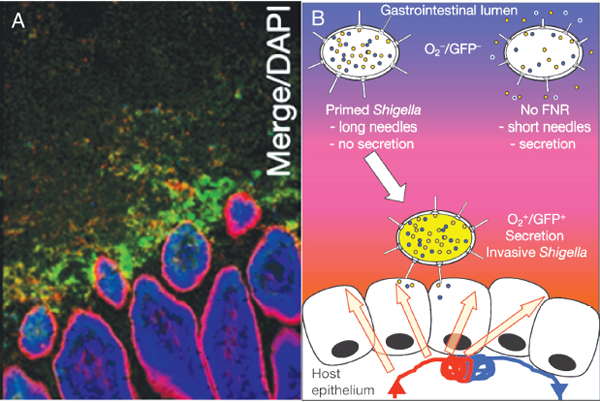
FIGURE WO-11 Oxygen gradients and microbial function. (A) Micrograph indicating the presence of oxygen adjacent to the intestinal tract mucosa. Here, Shigella expressing green fluorescent protein has been used as a marker of low oxygen, as well as of a change in gene expression. (B) Cartoon illustrating the hypothesis that in low-oxygen environments (such as those depicted in A) Shigella becomes invasive, expressing a number of genes involved in establishing an infection.
SOURCE: Reprinted by permission from Macmillan Publishers Ltd: Nature, 2012, Marteyn et al., “Modulation of Shigella virulence in response to available oxygen in vivo,” copyright 2012.
___________
29 Lining of cavities exposed to the external environment and internal organs that are covered in epithelium and are involved in absorption and secretion.
30 To gain insight into the metagenome—the genes and genomes present within a microbial community—researchers isolate DNA from these communities and sequence it in a “shotgun fashion”: the organisms’ genomes are fragemented into small pieces that can be sequenced. Fragment sequences are compared with known gene sequences to characterize the genes and genomes present.
and Schmidt, in preparation). In particular, many bacteria inhabiting the human gut possess high-affinity cytochrome oxidases, he observed. “Lots of the microbial world has the capacity to access these low concentrations of oxygen,” he concluded.
Based on these findings, Schmidt and coworkers are working to identify traits that increase fitness (as measured by both the rate and efficiency of growth relative to oxygen concentration) of bacteria in structured, oxygen-limited environments, such as the human intestinal mucosa. Such conditions are thought to favor slow-growing, oxygen-efficient organisms that produce adenosine triphosphate (ATP) at low rates, but at high yields (Pfeiffer et al., 2001)—a description that fits microaerobes in the intestinal mucosa, he noted.
These observations may also be relevant to the susceptibility of a community to invasion—in particular invasion by fast-growing microaerobes. By contrast, intestinal pathogens such as Vibrio and Shigella are capable of rapid growth under relatively high oxygen concentrations, which may affect their capacity to invade and establish infection in the human gut, Schmidt observed. “For that to happen in this model, there needs to be increased flux of oxygen, for instance, as a result of inflammation,” he continued. Those conditions, he explained, could decrease selection for “efficient” microaerobes and give fast-growing organisms the opportunity to establish themselves, which they could not have done if oxygen were scarce.
Source-Sink Dynamics: Host-Associated and Free-Living Chemosynthetic Symbionts
The waters surrounding deep-sea hydrothermal vents—fissures in the ocean floor through which geothermally heated water escapes—are home to abundant communities including tubeworms, bivalves, shrimp, and the chemosynthetic microbes upon which their survival depends. According to speaker Colleen Cavanaugh of Harvard University, the density of life in these communities vastly outstrips those of the surrounding ocean floor and rivals the biomass of rainforests (Dr. Cavanaugh’s contribution to the workshop summary report can be found in Appendix A). Chemosynthetic bacterial symbionts fuel these communities by converting energy in the form of reduced sulfur compounds or methane in the environment via oxygenation to provide their hosts with carbon and nutrients (Cavanaugh et al., 2006; Dubilier et al., 2008).
Researchers have found chemosynthetic symbionts in a variety of other environments—including hydrocarbon cold seeps, coastal sediments, mud volcanoes, and whale falls—in which oxic (O2) and anoxic (H2S) zones mix (Stewart et al., 2005). Cavanaugh noted that these symbionts have yet to be cultured in the laboratory, and molecular techniques of characterization have revolutionized researchers’ ability to gain insight into symbiont acquisition and transmission, population genetics, and ecology.
Vent-associated bacterial symbioses range from epibionts that colonize the external host surfaces, to symbionts that are extracellular but live inside specialized structures on the host, to endosymbionts that reside within host tissues. Host adaptations to their symbionts can be extreme, she said. Cavanaugh discussed her work on two host-microbe systems (Figure WO-12):
- The filamentous epsilon Proteobacterial epibionts31 of the shrimp Rimicaris exoculata, in which the shrimp are “characterized by unusual appendages and carapace shape,” which Cavanaugh believes act as a growth chamber to support the robust growth of the filamentous bacterial community.
- The obligate symbiosis that forms between gamma Proteobacteria and adult tubeworms (Vestimentifera spp.). The tubeworm, which lacks a mouth and gut, harbors its symbionts in a specially adapted organ in its
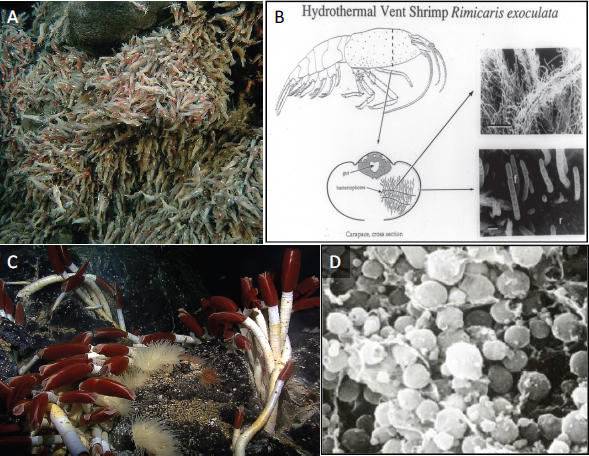
FIGURE WO-12 Hydrothermal vent organisms and their bacterial symbionts. Filamentous epsilon proteobacterial symbionts cover the exterior of the hydrothermal vent-dwelling shrimp, Rimicaris exoculata (A and B). Gamma Proteobacteria colonize the trophosome of adult vestimentiferan tubeworms (Riftia pachyptila) (C and D).
SOURCE: Figure A courtesy of NOAA Okeanos Explorer Program, MCR Expedition 2011, NOAA-OER; Figure C courtesy of NOAA Okeanos Explorer Program, Galapagos Rift Expedition 2011; Figures B and D courtesy of C. M. Cavanaugh.
___________
31 An epibiont is an organism that lives on the body surface of another organism.
trunk (troposome) and delivers oxygen and sulfide to the chemosynthetic bacteria with specially adapted hemoglobin.32
Host organisms acquire symbionts “vertically”—directly from their parents—or “horizontally” via free-living populations in the environment or from contemporary host individuals. The vent-associated shrimp frequently shed their carapace by molting and tubeworms appear to acquire symbionts early in life, suggesting that both symbionts are horizontally acquired, she noted. The mode of symbiont transmission impacts fundamental ecological and evolutionary processes, such as genome evolution and symbiont-host specificity. The heterogeneity and composition of the symbiont genome also suggested that these symbionts were free-living,33 because horizontal transmission permits genotypic variation whereas vertical transmission typically leads to symbiont populations that are genetically homogeneous,34 Cavanaugh noted.
To confirm the presence of free-living tubeworm endosymbionts, Cavanaugh and her colleagues collected environmental samples from two distinct habitats: seawater and biofilms attached to settlement devices deployed in hydrothermal-vent environments. Researchers tested these samples for the presence of the symbionts using polymerase chain reaction (PCR) amplification and DNA sequence analyses. Free-living tubeworm symbionts were “present among, adjacent to, and away from (within 10 meters) tubeworms and were also detected 100 meters outside the areas of hydrothermal activity” (Harmer et al., 2008). Noted Cavanaugh, “the question of what [these bacteria] are doing when they are not inside of a host is an open question.”
The presence of free-living symbiotic bacteria throughout a vent site suggests a potentially large environmental pool of symbionts (Harmer et al., 2008). As Cavanaugh and coworkers discovered, the presence of host-associated and free-living symbionts also influences microbial diversity in the surrounding ecosystem. “The symbiosis with microbial cells influences and impacts the freeliving microbial diversity of those environments. This can be on a local scale and potentially even on a very distant scale,” said Cavanaugh. Based on their study of bacterial symbionts of vent-dwelling shrimp and tubeworms, Cavanaugh’s group developed a “positive qualitative feedback model” by which the free-living population of symbiotic bacteria increases relative to nonsymbiotic microbes in the environment as a result of “inoculation” by the host-associated symbionts; at
___________
32 Cavanaugh noted that sulfide is able to be oxidized chemically in the presence of oxygen. The tubeworm hemoglobin binds the oxygen and sulfide separately (Flores et al., 2005), limiting this reaction until both elements are delivered to the chemosynthetic bacteria.
33Riftia pacyptila symbionts have a very large genome relative to vertically transmitted symbionts, a high GC content (typical of free-living bacteria), and sequence heterogeneity. Furthermore, in addition to sulfur metabolism and carbon fixation pathways, they also encode many of the enzymes and pathways that are used in a heterotrophic lifestyle, observed Cavanaugh.
34 Other features associated with vertical transmission include a reduced genome, a differential adenine thymine (AT) content, and loss of very specific genes.
the same time, free-living symbionts become increasingly available for recruitment into symbiosis by host organisms (Harmer et al., 2008; Polz et al., 2000).
Plant-Microbial Soil Communities
As part of her research on the vast, complex, and important soil microbiome, speaker Jo Handelsman, of Yale University, has examined interactions between one rare biosphere species, the bacterium Bacillus cereus, and other members of microbial communities associated with the interface between plant roots and the soil (Dr. Handelsman’s contribution to the workshop summary report can be found in Appendix A). As it turns out, she said, B. cereus is among several soil microbes that have been studied for many years, by virtue of their amenability to culture; as a result, much is known about its ecology, physiology, and relationships with other organisms. Culturing B. cereus allowed Handelsman and her colleagues to investigate molecular mechanisms underlying interactions between the bacterium, other microbes, and the plants that serve as their hosts. Dunn and Handelsman (2002) used the term communication networks to characterize the multiple molecular conversations that are carried on simultaneously between diverse organisms. Handelsman’s work on biocontrol of plant disease includes consideration of how signals are sent and received between soil microbiota, pathogens, and the plant host, providing a unique model for the study of communication among multiple organisms (Dunn and Handelsman, 2002).
Handelsman described multiple interactions involving B. cereus: its antagonistic interaction with a eukaryotic pathogen, its direct interactions with other bacterial genera, and its influence on the community as a whole (Figure WO-13).
Plant pathogens, such as the oomycete Phytophthora spp., “see” their host by detecting chemicals released from root exudates, explained Handelsman. By following these cues to find and invade a plant root, Phytophthora infects leguminous plants and starves them of nutrients and water. Infection ultimately rots the plant’s tissues. By culturing bacteria from the roots of healthy plants and the soil around them and testing each isolate, Handelsman’s group isolated an antagonist of Phytophthora spp. known as B. cereus. The molecular effector of this aggressive antagonism between the bacterium B. cereus and Phytophthora is an antibiotic they named zwittermicin (Emmert et al., 2004; He et al., 1994). The capacity to produce zwittermicin is common among soil strains of B. cereus, which occurs in most soils at ~105/g. This may make the genes for zwittermicin synthesis and resistance quite abundant on Earth, noted Handelsman.
Taking a different approach to understanding the role of B. cereus in disease suppression, a student in Handelsman’s laboratory examined the context of these interactions—the microbial community on the root. The rhizosphere—the dense microbial community that covers plant roots—teems with metabolic activity upon which it can be said that life on Earth depends, according to Handelsman. Its inhabitants, illustrated in Figure WO-14, provide plants with a range of services;
FIGURE WO-13 Molecular communication networks between organisms in the Bacillus cereus biocontrol system. One species of bacteria in the rhizosphere can have multiple interactions, often occurring through small molecules with the host plant, plant pathogens, and members of the microbial community.
SOURCE: Springer and Antonie van Leeuwenhoek, 81:565-574, “Toward an understanding of microbial communities through analysis of communication networks,” Dunn and Handelsman, Figure 2, copyright 2002, reprinted with kind permission from Springer Science and Business Media.
in return, the microbes gain access to a substantial portion of the plant’s carbon resources.
Microorganisms in the rhizosphere secrete numerous small molecules—root exudates that often help other microorganisms, including pathogens, find and colonize plant roots. Handelsman’s student hypothesized that B. cereus may suppress disease by changing the microbial community on the root and, consequently, the exudates from it. By comparing the physiological traits associated with communities of aerobic bacteria isolated from soy seedling roots treated with B. cereus with isolates from untreated roots, he demonstrated a substantial and significant difference between the two communities (Gilbert et al., 1993). This led him to propose that bacterial communities on disease-resistant roots more closely resemble soil communities than rhizosphere communities, providing“camouflage” to roots by reducing a pathogen’s ability to detect it as a potential host (Gilbert et al., 1994). “We can’t prove this hypothesis,” she said, “but we can say that there is a dramatic effect of B. cereus on the rhizosphere community.”35 Moreover, she added, this
___________
35 She noted that results from several studies from 1904 to the present are consistent with this hypothesis: plants bred for resistance, soil amendments, and bacterial biocontrol agents that were applied to suppress disease are all associated with a change in the rhizosphere community that makes it resemble the soil more than the typical rhizosphere (Gilbert et al., 1993, 1996; IOM, 2006).
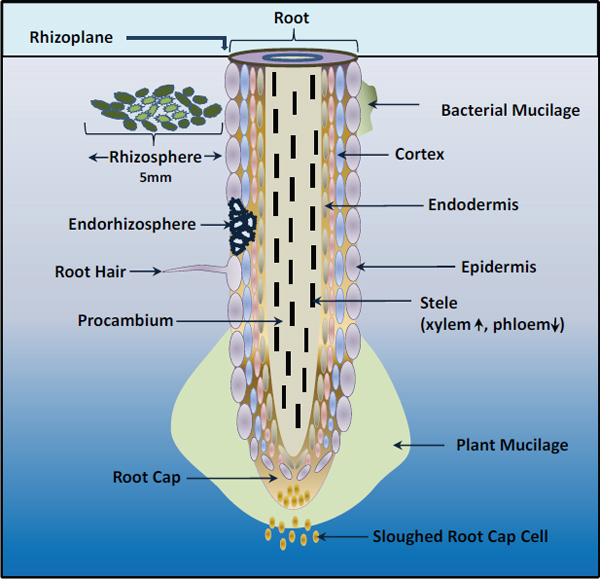
FIGURE WO-14 Parts of a root tip and areas of the rhizosphere.
SOURCE: Adapted from Maier et al. (2000).
global shift in community composition occurs after B. cereus is no longer present, so “a very small tweak in the community can have cascading effects down the line.”
One of the features of the B. cereus–associated shift in the rhizosphere community is a flush of growth of bacteria of the genera Flavobacterium and Cytophaga, Handelsman said. Upon further investigation, she and coworkers determined that Cytophaga and Flavobacterium—which are frequently co-isolated from field-grown roots along with B. cereus, earning them the nickname “hitchhikers”—needed the latter bacterium to grow. The relationship between B. cereus and its hitchhikers is specific, Handelsman noted. It involves some sort of physical association, and a nutritional relationship in which Cytophaga and Flavobacterium consume peptidoglycan produced by B. cereus to fuel a tremendous growth flush of these organisms on the root (Gilbert et al., 1993, 1994).
She observed that this finding raises a number of questions regarding the nature and ubiquity of the association between B. cereus and members of the Cytophaga/Flavobacterium group and the role played by these organisms or the peptidoglycan derived from B. cereus in the camouflaging shift in community. Such questions testify to both the complexity of the rhizosphere and the many relationships and interactions that occur among its inhabitants. As noted by Dunn and Handelsman, “the research challenge for microbiologists of the 21st century is to develop a portrait of the communication networks that link together organisms in communities” (Dunn and Handelsman, 2002).
Linking experimental studies with metagenomic information is an important challenge, Handelsman noted. At best, knowledge of “who is there” in a microbial community and their collective genetic potential raises hypotheses about the nature of interactions among community members and with their environment. These must be tested in single-variable experiments, she concluded—experiments that presently are performed readily on culturable microbes and are challenging to conduct on uncultured communities.
Phylogenetic and Phylogenomic Approaches to Studies of Microbial Communities
All cell-based organisms possess homologous genes for small-subunit ribosomal RNAs, but the sequences of these genes vary from species to species, in proportion to their relatedness. Comparisons of rRNA sequences offer a means to determine evolutionary relationships among organisms and to depict them as a “tree of life,” as illustrated in Figure WO-15; this process is known as phylogenetic analysis (Pace, 2009). One of the reasons that analysis of rRNA genes has been so powerful over the past 30 years is that it is relatively straightforward to determine the sequence of rRNA genes from organisms by making use of PCR. This is particularly important in studies of microbes where one can phylogenetically type (i.e., phylotype) organisms via analysis of their rRNA genes—even if one cannot grow the microbe in the lab. The ability to determine evolutionary relationships among organisms and to depict them as a tree of life has revolutionized our understanding of microbes in the world, said speaker Jonathan Eisen of the University of California, Davis (Dr. Eisen’s contribution to the workshop summary report can be found in Appendix A).
Despite the power of rRNA analysis there are some limitations (e.g., rRNA genes are not found in viruses; PCR can be biased). Metagenomics—where one reads the DNA sequence of random portions of the genomes of organisms from environmental samples—offers a more inclusive basis for phylotyping organisms via analysis of their DNA. Using metagenomics, researchers can phylotype using genes other than those for rRNAs—allowing an alternative perspective on cellular organisms and opening up new windows into studies of viruses. Referencing the pioneering work of Venter et al. (2004) to analyze microorganisms of the Sargasso Sea, Eisen described how a phylogenetic approach to interpreting genomic and
FIGURE WO-15 rRNA universal tree of life based on a comparison of nucleic acid sequences found in all cellular life (small subunit ribosomal RNA). The scale bar corresponds to 0.1 changes per nucleotide position. “A sobering aspect of large-scale phylogenetic trees, such as that shown in Fig. WO-15, is the graphical realization that most of our legacy in biological science, historically based on large organisms, has focused on a narrow slice of biological diversity. Thus, we see that animals (represented by Homo), plants (Zea), and fungi (Coprinus) [see BLUE arrows] constitute small and peripheral branches of even eukaryotic cellular diversity” (Pace, 1997).
SOURCE: From Pace, N. R. 1997. A Molecular View of Microbial Diversity and the Biosphere. Science 276:734-740. Reprinted with permission from AAAS.
metagenomic data provides important insights into microbial ecology and evolution. “The great thing about metagenomic data is we can build phylogenetic trees of other genes that are good phylogenetic markers,” Eisen observed. One can compare phylogenies based on protein-coding genes with those derived from rRNA. “The protein coding genes, even though they are not as richly sampled, are probably better markers for estimating relative abundance than ribosomal RNA sequences,” he noted.
Phylogenetic analysis not only tells us what we know about microbial diversity, but also helps reveal what we do not know, Eisen noted. For example, despite the discovery of dozens of major lineages of bacteria, archaea, or eukaryotes by phylogenetic analysis, most of the available genome sequences come from only a small number of those lineages. A project called the Genomic Encyclopedia of Bacteria and Archaea, which Eisen coordinates, is attempting to fill such critical gaps by selecting and sequencing genomes according to their phylogenetic novelty (Wu et al., 2009). The improved sampling of genomes from this project has been shown by Eisen and others to have many benefits, including improved ability to predict functions of uncharacterized genes as well as increased rate of discovery of genetic diversity.
Eisen also discussed another use of phylogenetic analysis in genomic and metagenomic studies: improving the accuracy of the prediction of gene function. For example, the function of uncharacterized genes can be predicted by analyzing a phylogenetic tree of the gene and examining its position in the tree relative to genes with known functions. This procedure, which Eisen developed and named phylogenomics, is now a widely used method to predict the functions of uncharacterized genes. In many cases it is not possible to use such phylogenomic analysis because none of the homologs36 of a gene of interest have been studied experimentally. In such cases, so-called“nonhomology” methods are powerful tools in functional prediction, and Eisen noted that these methods also can be improved by phylogenomic analyses. Such functional prediction methods can be used for sequences from cultured organisms as well as from metagenomic studies, although, Eisen conceded, exactly how you use nonhomology methods for metagenomic analyses “is still a work in progress.”
Eisen noted that phylogenetic analyses, which are primarily accomplished by hand, are overwhelming researchers. “I think as we get more and more sequence data, we can’t look at [phylogenetic] trees anymore,” he asserted. “We can’t look at sequence alignments. We can’t even handle all the data…. We certainly need to automate everything.” Eisen described several approaches he and coworkers have developed toward automating rRNA or metagenomic data. Tools designed for phylogenetic analysis of both rRNA and protein coding automatically build evolutionary trees of new sequences in relationship either to known sequences or exclusively to each other; another addresses the problem of nonoverlapping sequences
___________
36 One of two or more genes that are similar in sequence as a result of derivation from the same ancestral gene.
by anchoring them to reference sequences. “The final frontier in this is to try and build trees, even with different genes, when they don’t overlap with each other.” The combination of phylogeny and metagenomics provides a powerful tool for examining the evolutionary history of microbial communities and for discovering and characterizing novel genes and proteins, Eisen concluded.
Microbial Community Assembly and Dynamics: From Acidophilic Biofilms to the Premature Infant Gut
While metagenomic analyses can reveal the genetic and functional potential of microbial communities, their actual functional capacity is reflected in the type and number of their proteins. By analogy to the genome, every cell, organism, and community has a proteome: the complete complement of proteins encoded in its genome, individually expressed in response to a vast array of regulatory controls, and equally subject to modification and degradation. Metagenomic data offer clues to the contents of a microbial community’s proteome, which in turn suggests, in the words of speaker Jill Banfield of the University of California, Berkeley, “what are they doing and how are they doing it?” (Dr. Banfield’s contribution to the workshop summary report can be found in Appendix A).
For more than a decade, Banfield and her coworkers have characterized how acid mine drainage (AMD) biofilm communities “develop, how they first establish, and the process of ecological succession.”37 Her work has characterized the genetic and protein profiles of early and late stages of biofilm formation and illuminated how these communities organize, and how its members interact with each other and with the surrounding environment to form a relatively self-contained ecosystem (Denef et al., 2010b; IOM, 2009).
AMD streams and pools are home to a limited variety of microorganisms that can survive in this dark, very-low-pH, metal-rich, extreme subterranean environment (Denef et al., 2010b; IOM, 2009). AMD biofilms are “structured by oxygen and follow developmental stages through time,” noted Banfield. A nucleus—primarily a monospecies assemblage of Leptospirillium bacteria—begins at a subterranean stream’s margins and extends across the water’s surface toward its center, while simultaneously increasing in thickness. As the biofilms mature, their composition, structure, and function diversify across the community. “Layers of archaea, eukaryotes, and viruses are incorporated at various developmental stages, and chemical gradients become established across the biofilm” (Figure WO-16).
“There is organization in the vertical sense, and it is almost certainly structured by these gradients, but there is also functional variation—for example
___________
37Ecological succession is the phenomenon or process by which an ecological community undergoes more or less orderly and predictable changes following disturbance or initial colonization of new habitat.
FIGURE WO-16 Schematic illustrating important features that make the AMD system a good model for studying microbial communities (for example, relatively low species complexity, defined ecological succession patterns and trophic levels, tight biological-geochemical coupling, and high biological productivity).
SOURCE: Reprinted by permission from Macmillan Publishers Ltd: ISME Journal, Denef et al. AMD biofilms: Using model communities to study microbial evolution and ecological complexity in nature, 4(5):599-610, copyright 2010.
the activity of iron oxidation is higher toward the base of the biofilm,” reported Banfield, and this ecosystem can be reliably reproduced in the laboratory. “Despite living in what appears to be an extreme environment, these microbes have managed to find a niche, they are very well adapted to it, and they are growing very nicely.” These are conditions in which these microbes have “evolved in and probably existed in for billions of years.”
Banfield and coworkers used proteomic analyses (see Figure WO-17) to track changes in AMD community composition over time as reflected in shifts in protein expression and associated metabolic functions (Denef et al., 2010a; Wilmes et al., 2009). For example, the proteome of the dominant bacterium in the biofilm was found to shift between two distinct and stable states as the biofilm community became more species-diverse. This suggests that the main factor in how the bacterium “uses” its genome—that is, what proteins it expresses—is strongly influenced by the organisms around it, she reported (Mueller et al., 2010). Even when considering the metadata—the temperature, pH, flow rate, and other environmental factors—community composition affects and strongly influences the way the microorganism behaves, Banfield concluded.
The AMD system has “emerged as a really nice model system [with]in which to develop methods that can be then applied to other systems with the objective of trying to understand how microbial communities assemble and to understand and even predict their dynamics,” noted Banfield. She and her colleagues are
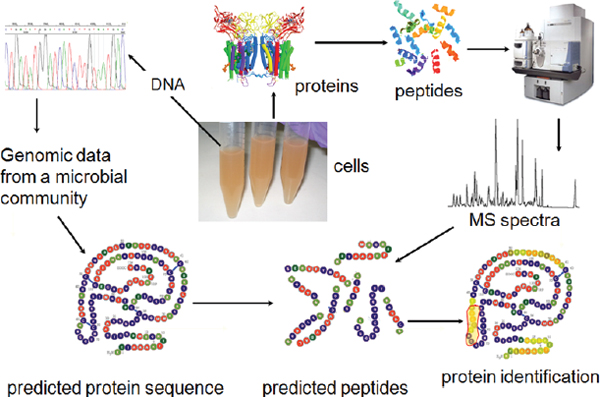
FIGURE WO-17 Microbial community proteomics: functional assays in situ.
SOURCE: Banfield (2012).
now applying similar methods to examine the establishment of the more complex—and yet relatively simple—microbial community of the human gut, as it is colonized during the first weeks of life (Buchen, 2010). Specifically, they are attempting to identify patterns in gut microbial succession associated with the development of neonatal necrotizing entercolitis (NEC) (Morowitz et al., 2010), a significant and potentially fatal disease in preterm infants.
As a first step toward this goal, researchers tracked colonization of a healthy preterm infant’s gut by analyzing daily fecal samples using both rRNA analysis (throughout the 3-week experiment) and sequence-based metagenomics (over the final week, the third of three colonization phases) (Morowitz et al., 2010). “By looking through the genome and comparing it to the other genomes, we can look at what is different, what is the same, what is novel about this organism, which genes are undergoing the most rapid evolution, and what do they encode,” Banfield said.
A key finding from this study concerned one of the dominant bacterial species, Citrobacter, among which two very closely related strains—with 99 percent sequence identity—behaved quite differently over the course of the time series, resulting in profound shifts in strain composition (Morowitz et al., 2010). Fine-grained comparative genomic analysis of the two strains revealed differences in traits associated with metabolism and pathogenicity—including virulence factors—as well as divergence in regulatory and intergenic regions. The difference between strains suggests that each strain plays a distinct ecological role in the nascent microbial community of the infant gut. Proteomic information supports the conclusion that the two strains are functionally different, Banfield reported.
Banfield and coworkers recently completed a second time-series study of another infant, using sequencing technology that allows them to identify subtle, strain-level differences such as those they detected between the Citrobacter strains (Sharon et al., 2012). Using this approach, she said, “I feel pretty confident that it is going to be possible to enable proteomic analyses of these colonization processes, and [also] to extend this to more complex systems.” Similar time series, including proteomic analysis, are planned to compare healthy preterm infants with those who develop NEC.
Ultimately, she added, they hope to test their findings from metagenomic analyses with experiments in the community context. Compounds could be designed to provoke changes in community function—whether those communities are AMD biofilms at the subsurface or the microbial flora in the human gut. “So long as we can maintain access to field sites where we can do those experiments, I think we will be able to really learn a great deal about this uncultivated diversity, and really how to harness microbial communities to do useful things, at least in terms of the environment,” she concluded.
The apparent significance of the 1 percent divergence in sequence between the two Citrobacter strains decribed by Morowitz and coworkers (2010) is but one illustration of the generally observed importance of fine-scale heterogeneity—depicted in Figure WO-18—to the ecology of microbial communities (Denef et al., 2010a, b; Wilmes et al., 2009). A major source of this heterogeneity is genetic exchanges that occur through transformation, phage transduction, and conjugation (IOM, 2006).
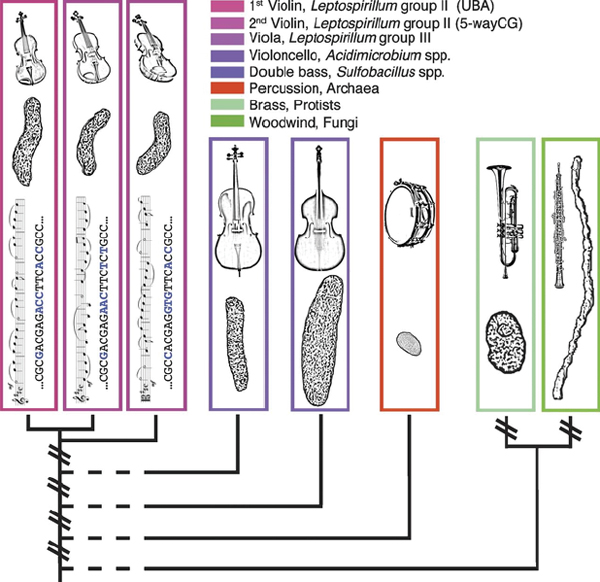
FIGURE WO-18 The dynamic genetic repertoire of microbial communities. The microbial orchestra analogy showing relatedness of individual community members in acid mine drainage biofilms with corresponding instrumental groups.
SOURCE: Wilmes et al., FEMS Microbiology Reviews, “The dynamic genetic repertoire of microbial communities,” 33(1):109-132, doi: 10.1111/j.1574-6976.2008.00144.x, reprinted with permission from Wiley Online Library.
Lateral gene transfer (also known as horizontal gene transfer) among members of some microbial communities appears to be so pervasive as to call into question whether the concept of speciation—founded on the existence of barriers to genetic exchange among multicellular organisms—applies to communal microbes (Eppley et al., 2007). For example, a comparison of 17 genomes from individual E. coli, which included both commensal and pathogenic strains, revealed that only about half the genes in each organism’s genome were conserved among the group; the rest were part of a vast reservoir of genes known as the pangenome (Rasko et al., 2008).
The apparent fluidity of gene exchange among microbes raises important topics for ongoing inquiry, such as how frequently and under what conditions it occurs, the extent to which lateral gene transfer contributes to the evolution of microbial communities and host-microbe relationships, and the potential of lateral gene transfer among microbes to influence microbial functions such as pathogenicity, virulence, antibiotic resistance, as well as host metabolism. These considerations were addressed in a workshop presentation by Sam Brown, which is discussed in Infectious Cooperation.
Clearly the species concept, as defined at the macroscopic level, fails to capture the complexity, interconnectedness, and diversity of genetic exchange among microbes. As Denef and colleagues (2010a) have observed, rather than investigate the ecological roles played by microbial species, “more widely accepted is the need to understand how differences in gene content and sequence lead to ecological divergence.”
DYNAMIC INTERACTIONS OF MICROBIAL COMMUNITIES
“Traditionally, microbiologists and evolutionary biologists have studied social behaviours from differing perspectives. Microbiologists are primarily interested in the genetic mechanisms controlling the behaviour (“how” questions) whereas the interest of the evolutionary biologist can be found in studying the fitness consequences of a particular behaviour which helps explain why these systems are found in nature (“why” questions). However, whilst these may be different approaches, they should be viewed as complementary and not contradictory. By combining both mechanistic and adaptive approaches we can begin to address questions such as, what factors influence cooperation and the evolution of virulence in microbes and how can we exploit these to develop new antimicrobial strategies?”
—Diggle et al. (2010)
“When we look at microbial communities and we think we know what’s going on in terms of interactions, what I would like to say is, ‘generally, we don’t.’”
—Joan Strassmann (2012b)
One way to gain an understanding of microbial communities is through characterizing their composition, genetic potential, and protein-associated functions. Another is to study how community members interact with each other and with their environment and how these interactions influence community structure and function as a whole. Studies of microbial communities have yet to define the ecological principles that can provide a foundation for predictive models of community dynamics (Little et al., 2008). General principles developed from observations of communities of macroscopic organisms, however, are informing efforts to interpret microbial community dynamics (Levin, 1998; Robinson et al., 2010; West et al., 2006).
Workshop speakers addressed the broad topic of microbial interactions from several perspectives: by exploring the variety of mechanisms by which microbes interact with each other, their hosts, and the environment; by describing specific microbial interactions across a range of ecological contexts; and by considering how ecology and natural selection have shaped microbial interactions and potentially led to the emergence of multicellular organisms.
Communication and Information Processing
For any organism to interact with any other organism or its environment, it must be capable of both sensing and responding to conditions beyond itself. That microbes possess such capacity has been recognized relatively recently, but this understanding underlies the discovery of several ways in which microbes interrogate their surroundings and react to the results of such interrogations (Bassler and Losick, 2006). However, what we currently know about mechanisms of microbial interaction may well represent but a few examples of a large, diverse, and complex repertoire.
The best characterized of these mechanisms is quorum sensing—described by Greenberg in his keynote presentation and discussed earlier in this overview—but it is just one among many. Two workshop presentations summarized below offer glimpses of additional sense-and-response systems that enable microbes to interact with other members of their species, with members of other kingdoms, and with their environment.
Intraspecies Interaction: Contact-Dependent Communication
Bacteria are known to use several distinct mechanisms to initiate a range of actions upon contact with neighboring cells (Hayes et al., 2010). These contact-dependent systems deliver effector molecules (e.g., enzymes, transcription regulators, toxins) that enhance the growth or survival of the “sender” cell at the expense of the “receiver.” They include the contact-dependent growth inhibition (CDI) system discussed by speaker David Low, of the University of California, Santa Barbara, as well as a virulence-associated secretion system, subsequently described by Vanessa Sperandio of the University of Texas Southwest Medical Center
(Dr. Low’s contribution to the workshop summary report can be found in Appendix A; Dr. Sperandio’s contribution to the workshop summary report can be found in Appendix A).
Low described the CDI system and noted that he and coworkers recently concluded that the CDI system’s function may not be limited to growth inhibition; rather, it appears to provide a more general mechanism of bacterial communication. Initial studies of the CDI system in E. coli produced the model shown in Figure WO-19, in which a stick-like protein displayed on the effector (CDI+) cell’s surface delivers its toxic tip to a receptor on a target cell. A tip-specific immunity protein, also produced by the CDI+ cell, protects it from the effects of this toxin (Aoki et al., 2010). Although several species of bacteria have functional CDI systems, each system operates almost exclusively within a single species, he said.
More than 60 different CDI-associated toxic tips have been identified in bacteria (Poole et al., 2011). Many are nucleases, according to Low, but they also include peptides that inhibit target cell growth by reducing energy-producing proton motive force across target cell membranes. In head-to-head competitions, wild-type cells out-compete mutants with nonfunctioning CDI systems up to 105 fold in a few hours, he observed. He also noted that while the toxic tip-immunity protein pairing is highly specific, evidence suggests that toxic tips from different pathogens can be displayed on other species’ “sticks” (Poole et al., 2011).
Study of a CDI system from a strain of uropathogenic38E. coli that uses a tRNase as its toxic tip led researchers to recognize the possibility that these systems play a more general role in cell-to-cell communication, Low recalled. When
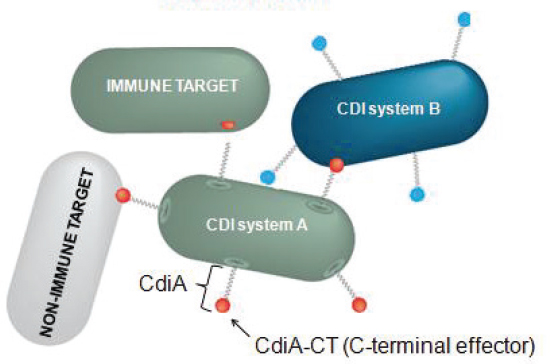
FIGURE WO-19 Contact-dependent growth inhibition (CDI).
SOURCE: Adapted from Aoki et al. (2011).
___________
38 Pathogenic organisms in the urinary tract.
Diner and Hayes purified the tRNase to demonstrate its function, they discovered the enzyme required another cell protein for function—CysK—a key enzyme in cysteine biosynthesis (Diner et al., 2012). Interaction between the tRNase and CysK in the target cell blocks the enzyme’s ability to synthesize cysteine; however, an isoenzyme,39 CysM, does not bind the tRNase and thus takes over cysteine production. These enzymes differ in the substrates they use to provide a sulfur moiety to cysteine: CysK uses sulfide, while CysM uses thiosulfate. This difference suggests that transition between them may be advantageous, depending on whether the bacterium finds itself in an aerobic or an anaerobic environment. As for the role of CDI in this shift, Low observed, “touch between cells could potentially affect their metabolism.”
Another observation of the same CDI system in action further supports its role in cell-to-cell communication. Bacteria with single point mutations that knocked out tRNase activity were found to be deficient in biofilm formation, Low reported. “This is likely communication, because we have an RNase activity that appears to be specifically required for biofilms,” he concluded. This phenomenon has been shown to occur in other organisms, he added, which suggests that CDI systems affect a broad range of physiological conditions. Taken in consideration with the fact that CDI systems orchestrate intraspecies interactions and the possibility that they could mediate metabolic processes, Low’s findings support the notion that CDI systems serve as communication links between“effector” and “target” cells.
Interkingdom Interactions: Symbiotic Microbes, Their Mammalian Host, and Invading Pathogens
Nowhere is interest in host-microbe interaction greater than in the complex context of the human gastrointestinal tract. Virulence traits are metabolically expensive, and, as previously discussed, their expression is tightly regulated. Underlying mechanisms of regulation may involve sensing of signals in the environment—ranging from specific molecules secreted by microorganisms to environmental cues, such as nutrient availability, temperature, and osmolarity. Signals may also be provided by the host, in a type of cell-to-cell communication known as interkingdom signaling (Hughes and Sperandio, 2008). Sperandio discussed her work on cell-to-cell signaling40 between humans, symbiotic microbes, and invading pathogens (Hughes and Sperandio, 2008). Her work builds on the well-established concept of quorum sensing as a means for bacteria to assess local population density and coordinate the expression of critical genes, including those that encode virulence factors (Njoroge and Sperandio, 2009).
___________
39 Isozymes (also known as isoenzymes or more generally as multiple forms of enzymes) are enzymes that differ in amino acid sequence but catalyze the same chemical reaction.
40 There are a variety of communication strategies—such as signals, cues, and coercion—used by hosts and their associated microorganisms. For more information on how different types of strategies may be explored and defined, see: Diggle et al. (2007a).
Sperandino described her work on cell signaling across kingdoms, which involves quorum-sensing signaling molecules (e.g., acyl-HSL) from bacteria and hormones produced by eukaryotes (Hughes and Sperandio, 2008). In the human gut, two-way communication occurs between epithelial cells and both commensal and pathogenic microbes, Sperandio noted; infection by enterohemorrhagic E. coli O157:H7 (EHEC) is a model of such interactions. EHEC is a highly infectious foodborne pathogen that typically causes bloody diarrhea, she said. However, in susceptible individuals (e.g., the very young and very old), it causes a more severe—and sometimes fatal—illness known as hemolytic uremic syndrome.
Sperandio observed that in healthy intestines the epithelium is shielded from contact with pathogens by a mucous layer that is densely populated with commensal bacteria. EHEC uses a suite of virulence traits to overcome these obstacles and gain access to epithelial cells, into which it injects Shiga toxin and other effector molecules through a syringe-like proteinaceous secretion system. The production of this toxin is metabolically costly for the bacterium, so it is advantageous for EHEC to produce the secretion system only if it is within range of “infectable” host cells. Sperandio noted that EHEC senses a quorum-sensing molecule called autoinducer-3 (AI-3) produced by commensal gut bacteria, and two hormones, epinephrine (adrenaline) and norepinephrine (noraderinaline), produced by intestinal cells (Hughes and Sperandio, 2008).
When a sensor kinase called QseC—a functional analog of an adrenergic receptor embedded in EHEC’s inner membrane—“receives” any of these chemical signals, it triggers a complex regulatory cascade that results in the transcription of virulence genes, Sperandio said. This system is present in dozens of other important human and plant pathogens, she noted, and it has been demonstrably associated with virulence in Salmonella typhimurium and Francisella tularensis (Curtis and Sperandio, 2011; Rasko and Sperandio, 2010). Researchers have pursued the therapeutic potential of interrupting the QseC-triggered virulence cascade by developing several animal models of this system, according to Sperandio. Her research group found that an EHEC-like pathogen, Citrobacter rodentium, caused lethal infections in mice unless the pathogen’s QseC gene was disabled by mutation.
The number and diversity of known microbial interactions suggests a universe of possible configurations and functions of microbial communities. Biofilms can form on nearly any surface, are composed of varied assemblies of bacterial species, and develop structural complexity and functional differentiation. Host-microbe interactions, such as highly specific mutualisms between bioluminescent bacteria and marine macrofauna, display a spectrum of coevolved features.
The workshop presentations summarized in this section further illustrate the intricacy and specificity of microbial interactions, and the mechanisms that make
these interactions possible. These interactions take place across a broad range of ecological and taxonomic contexts, yet all can be viewed as structure-function relationships shaped by evolution and natural selection.
Swarming Bacteria as Agents of Microbial Dispersal
An intriguing interspecies communication between bacteria and fungi was described by speaker Colin Ingham of Wageningen University, The Netherlands (Dr. Ingham’s contribution to the workshop summary report can be found in Appendix A). Instead of forming sessile biofilms, some surface-associated bacteria secrete surfactants41 that allow them to migrate collectively, powered by their flagella,42 in a process known as swarming (Kearns, 2010). Interest in swarming motility has benefited from the shift in the focus of microbiological research from planktonic to surface environments relevant to microbial communities in natural settings43 (Kearns, 2010). “You can ask quite a lot of interesting questions about these types of bacteria,” observed Ingham. For example, what happens when a mass of motile bacteria meets other microorganisms, something that can happen within a very short time frame (minutes)? How do they interact?
To pursue this line of inquiry, Ingham studies the Gram-positive bacterium Paenibacillus vortex, which he characterized as a curved rod-shaped microorganism, collectively a “versatile swarmer” that “forms complicated colonial patterns that are exceptionally sensitive to the environmental conditions” (Ingham and Ben Jacob, 2008). In what he called a “very simple experiment,” he and coworkers co-inoculated P. vortex with conidia (asexual spores) of the nonmotile fungus Aspergillus fumigatus on nutrient agar plates and found that the swarming bacterium could transport the spores over distances of up to 30 centimeters (Ingham et al., 2011). Both P. vortex and A. fumigatus are inhabitants of the rhizosphere, the soil immediately surrounding plant roots, so may plausibly interact in nature (Figures WO-20 and WO-21).
“For Aspergillus conidia this is a new method of dispersal and the first time a smaller microorganism has been shown to cooperatively transport a larger one,” Ingham pointed out. This mechanism may permit the fungus to target niches it could not reach by airborne spore dispersal, and it may also exploit a tendency
___________
41 Many swarming bacteria synthesize and secrete surfactants (short for “surface-active agent”). Surfactants are amphipathic molecules that reduce tension between the substrate and the bacterial cell and, in doing so, can permit spreading over surfaces.
42 A whiplike part extending from some single-celled organisms that moves back and forth to impart movement to the organism (American Heritage Science Dictionary, 2011).
43 According to Kearns (2010), swarming motility has been reported in species from three bacterial families, “almost certainly an underestimate, because swarming motility is often inhibited by standard laboratory media and genetically abolished during the domestication of commonly-used laboratory strains.” “The selection against swarming in these strains may be due to evolutionary forces that act when surface motility provides no advantage, for example in unstructured laboratory environments” (Kearns, 2010).
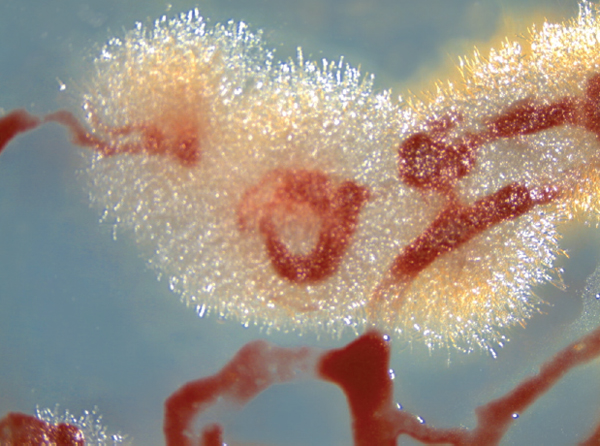
FIGURE WO-20 Photograph showing the result of transporting Aspergillus fumigatus conidia by swarming Paenibacillus vortex. The swarms of bacteria (stained red) carry fungal spores (conidia). This image (around 1 cm across) was taken a few days after transport of viable conidia over several centimeters was achieved; this is obvious because the conidia have germinated and are now growing out of the bacterial colonies, having being carried into a new niche.
SOURCE: King (2011); image provided courtesy of Colin Ingham and Eshel Ben-Jacob.
for Paenibacillus to seek (e.g., through chemotaxis) an environment that is also beneficial to the fungus. Not only was swarming by P. vortex found to permit efficient dispersal of the fungal conidia, but also it was shown capable of “rescuing” conidia by moving them from an antifungal-treated area on an agar plate to an untreated area, where they could grow. The researchers also discovered a potential benefit to the swarming bacteria in transporting fungal conidia: upon meeting an air gap (in experimental media, but presumably also in the soil) that the swarm cannot cross, they germinate to form mycelia that can serve as a bridge over which the bacteria can cross (Ingham et al., 2011).
“The image that we have taken from this is the possibility that a moving colony of this bacterium is kind of a mixed and highly dynamic ecosystem, with the potential to drop cargo cells off and have other microorganisms jump on and therefore the swarm may help redistribute organisms in the soil,” he concluded. However, he added, “we don’t know how important this is.” Ingham speculated that another possibility is that the extended colony can “effectively create a logistics system for
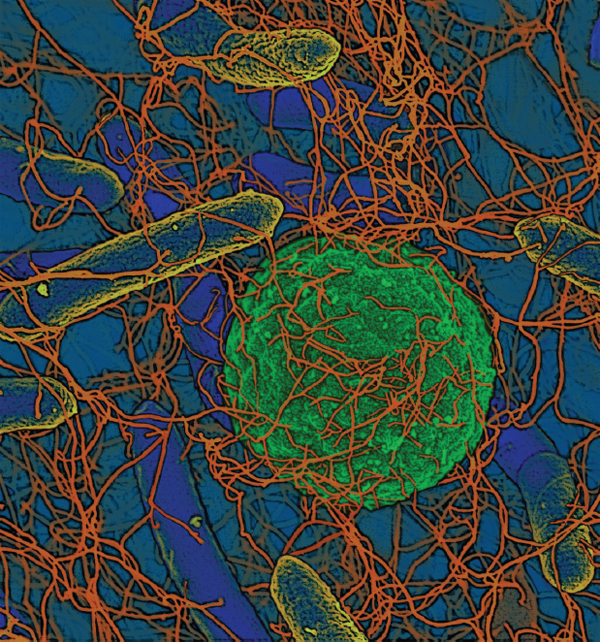
FIGURE WO-21 Colored scanning electron micrograph showing transport of a single conidium of Aspergillus fumigatus (central green sphere, 3 µm across) transported by swarming Paenibacillus vortex (blue rods with yellow highlights). There are many bacterial flagellar (orange filaments) apparently making contact with the conidium.
SOURCE: Image provided courtesy of Colin Ingham and Eshel Ben-Jacob.
resource optimization,” and he suggested that future studies could explore whether this form of colonial life is beneficial with regard to efficient foraging and distribution of nutrients from disparate sources throughout the colony.
The Fungal Gardens of the Leaf-Cutter Ants
The leaf-cutter ant occupies the center of an outstanding model of complex and dynamic interactions linking microbes, multicellular organisms, and the
environment. Among the most widespread and ecologically important insect species in the New World tropics, leaf-cutters are unique among ants because they cultivate a fungus44 that serves as their main source of food (Belt, 1874; Suen et al., 2011; Weber, 1966). The ants seek out and cut leaves upon which they grow the fungus in specialized “gardens,” engaging in behavior that in many ways parallels that of human agriculture. Colonies of leaf-cutter ants are so large—up to 600 cubic meters—as to be visible from satellite photos; the millions of workers present may harvest hundreds of kilograms of leaves per year.
This mutualistic relationship is thought to have originated 45 million years ago, according to speaker Cameron Currie of the University of Wisconsin-Madison, who noted that “the ants can’t survive without the fungus and the fungus can’t survive without the ants” (Dr. Currie’s contribution to the workshop summary report can be found in Appendix A). They are a “true superorganism” remarked Currie, with colonies often housing 5-10 million individuals that work together as a self-sustaining unit. These colonies can perform astonishing tasks he noted, including “defoliating a mature eucalyptus tree overnight.”
The mutualistic and parasitic relationships that sustain this system appear to be both ancient and specialized, said Currie. The actinomycete bacteria Pseudonocardia, which produces small molecules capable of inhibiting the growth of the specialized garden pathogen, is acquired horizontally from ant workers that tend the pupae, he said. A brief window of opportunity for acquisition occurs within 2 hours after pupae eclose to become an adult. Within 14 days of acquisition, the ants are fully covered with the actinobacteria, said Currie.
Currie described several “major” structural and physiological modifications exhibited by the ant that benefit its bacterial symbiont. Pseudonocardia resides in specialized crypts or cavities that cover the ants’ exoskeleton, he discovered (Figure WO-22). These crypts also appear to be attached to glands—suggesting a physiological modification in which the gland cells of the ants support the growth of the bacteria. “Presumably, these gland cells are producing the nutrient source for the growth of the actinobacteria.” Indeed, numerous animal and plant morphologies and developmental processes reflect ancient, coevolved relationships with host-associated microbiota. These specialized compartments and processes attest to the importance of symbiont function to host organisms, as well as to the adaptability of microorganisms.
Currie noted that “as with human agriculture, ant agriculture has major problems with disease.” The microfungi in the genus Escovopsis has been found only in gardens of fungus-growing ants, so it appears to be a specialized pathogen that has evolved to exploit this environment, he said; Escovopsis secretes proteolytic enzymes that break down the fungal cultivar’s tissue, allowing it to parasitize the cultivar. As previously described, the Pseudonocardia that reside in specialized
___________
44 Different species of leaf-cutter ants use different species of fungus, but all of the fungi the ants use are members of the Lepiotaceae family.
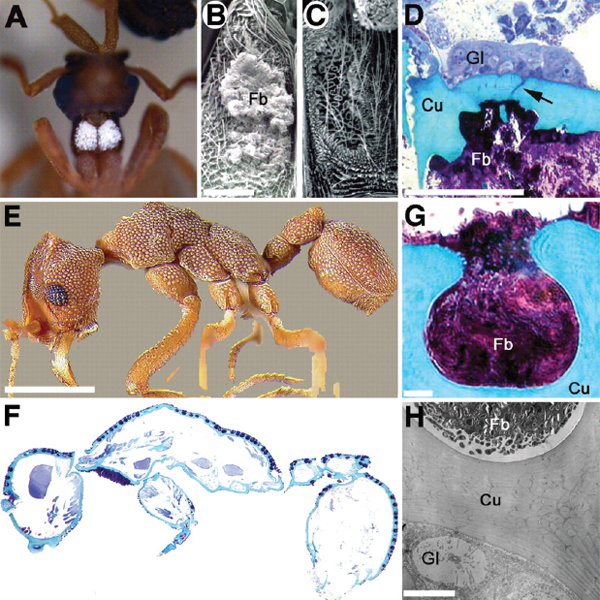
FIGURE WO-22 Coevolved crypts and exocrine glands support mutualistic bacteria in fungus-growing ants. (A) Photograph of Cyphomyrmex costatus showing the bacteria on the propleural plates. SEM of the plates in C. muelleri: The left plate is covered with bacteria (B), whereas they have been removed from the right plate, revealing the underlying fovea (C). (D) Light micrograph of a semithin cross-section through the propleural plate of C. longiscapus showing the gland (Gl) and duct cells (black arrow) associated with the fovea and the bacterium (Fb) on the plate (Cu for cuticle). (E) Photograph of C. longiscapus, illustrating foveae openings covering most of the cuticle. (F) Sagittal semithin section through a C. longiscapus worker, illustrating foveae outlining nearly the entire body of the ant. (G) Light micrograph of a single fovea within the cuticle (Cu) illustrating the abundance of mutualistic bacteria (Fb) within the crypt. (H) TEM of the lower section of a fovea showing a single glandular cell (Gl) and bacteria (Fb) within the crypt. Scale bars: 50 mm (A to C), 5 mm (E and G), and 0.5 mm (F).
SOURCE: Currie, C. R., M. Poulsen, J. Mendenhall, J. J. Boomsma, and J. Billen. 2006. Coevolved crypts and exocrine glands support mutualistic bacteria in fungus-growing ants. Science 311:81-83. Reprinted with permission from AAAS. Photograph in (A) by A. Little.
crypts that cover the ants’ exoskeleton secrete molecules that inhibit the growth of Escovopsis. But this is not the end of the chain of exploitation and mutualism, as Currie and coworkers identified yet another parasite: black yeasts (genus Phialophora; Ascomycota) that prey on Pseudonocardia (Little and Currie, 2008).
“To understand the dynamics of species interaction and complex communities, often you have these indirect effects, and you have to understand all the different components to start to even tease apart the dynamics,” Currie observed, noting that this phenomenon is known as ecological context dependence. In this case, he noted, focusing solely on the interaction between the ant and the “farmed” fungus would provide an incomplete view of the selective forces shaping their evolution, both as individual organisms and as partners in obligate mutualism. The concept of ecological context dependence resonated throughout the workshop, most notably in presentations and discussions concerning the evolutionary context of microbial interactions.
While the leaf-cutter ant system continues to generate questions—and, potentially, compounds for antibiotic development—it is worthwhile to investigate the possible existence of similarly complex, dynamic webs of interaction involving other organisms, Currie said.
Termites comprise an extraordinarily successful, globally distributed insect group and play an important role in the global carbon cycle because of their ability to derive energy from lignocellulose in wood (Warnecke et al., 2007). According to speaker Jared Leadbetter of the California Institute of Technology, many steps in the conversion of lignocellulose into host nutrients are accomplished by symbiotic gut microbial communities (Dr. Leadbetter’s contribution to the workshop summary report can be found in Appendix A). The termite hindgut is the residence of “a density and diversity of microbes that corresponds to all three domains of life,” he said, “diverse Archaea, diverse single-celled eukaryotes, and hundreds and hundreds of bacterial species that you find nowhere else on earth.” These polymicrobial, extracellular symbiont communities involve multiple microbial partnerships, thus providing an excellent system for probing the evolution of interactions between different symbiont species within a community.
As Leadbetter explained, the initial task of degrading polysaccharides in lignocellulose falls primarily to protozoa among one group, known as lower termites; in the less well understood higher termites, which lack protozoa in their hindguts, bacteria play a larger role. The main products of this fermentative process are acetate, carbon dioxide, and hydrogen, he said; acetate serves as the insect’s sole source of energy.45 The bulk of the produced hydrogen and carbon
___________
45 Thus, Leadbetter observed, “termites are not really eating wood—they are eating neutralized vinegar.”
dioxide are also converted into acetate by carbon dioxide–reducing homoacetogenic bacteria, Leadbetter noted.
The loss of symbiotic gut protozoa occurred early during the emergence of the lineage of higher termites, as did the development of their characteristic segmented gut architecture. “Since then, higher termites and their gut symbionts have found ways to access polysaccharides bound in forms other than wood (e.g., dry grass, leaf litter, organic compounds in soil)” (Zhang and Leadbetter, 2012). By comparing gene diversity in a wide range of termite host species, Leadbetter explored how environmental factors such as changes in termite biology, diet, and symbiont community composition may have impacted the evolution of the gut microbial communities of higher termites (Zhang and Leadbetter, 2012).
A key bacterial gene for the production of acetate, formate dehydrogenase (fdhF), has been shown to be widespread in the guts of lower termites and wood-roaches (Matson et al., 2010; Zhang et al., 2011). In contrast, phylogenetic analyses of fdhF sequences from eight species of higher termite revealed sweeping losses of fdhF diversity. These losses likely occurred during the evolution of the last common ancestor (LCA) of all higher termites over 50 Mya (Figure WO-23). Leadbetter hypothesized that the fdhF gene extinction events may be related to the extinction of hydrogen-producing gut protozoa and effects that “propagated down the microbial food chain to hydrogen-consuming symbionts that possess fdhF” (Zhang and Leadbetter, 2012). He noted that the lack of diversity of fdhF variants in higher termites is likely an example of “radiation from one of the fdhF genes and one of the organisms that was left standing after the loss of the protozoa and their associated microbes.”
Leadbetter also examined phylogenetic patterns of two functional variants of fdhF, which either have selenocysteine (Sec clade) or cysteine (Cys clade) as a key catalytic residue. The absence of Cys clade gene variants in higher termites suggests the relaxation of selective pressures related to selenium limitation in higher gut termite communities (Zhang and Leadbetter, 2012). Perhaps as higher termites changed their biology, diet, or habitat, they have been able to “alleviate that periodic limitation of selenium that lower termites face, and thus, they had a sweeping gene loss of every member of this clade,” he said.
Upon closer examination of variant sequences in one genus of termites from Costa Rica, Leadbetter and colleagues discovered that several sequences nested within the Sec clade actually encode for selenium-independent variants (Figure WO-23). These variants likely originated from “a duplication of a selenium-dependent gene, followed by mutational modification of the [catalytic residue] from selenocysteine into cysteine” (Zhang and Leadbetter, 2012). According to Leadbetter, possible factors driving this apparent “reinvention” or convergent evolution of selenium-independent function include changes in the termite’s foraging behavior or environment.
Sequence comparison of fdhF variants also revealed a “mystery clade of variants outside of the ones found in other termites” (AGR variants in Figure WO-23). Leadbetter speculated that this is likely an example of a “recent acquisition, by
FIGURE WO-23 Inferred evolutionary history for fdhF in the symbiotic gut microbial communities of lignocellulose-feeding insects. LCA, last common ancestor; FDH, formate dehydrogenase; Sec, selenocysteine functional variants of fdhF; Cys, cysteine functional variants of fdhF; and AGR, Amitermes-Gnathamitermes-Rhynchotermes.
SOURCE: Zhang and Leadbetter (in preparation).
these termites, of a gene or of an organism from a non-termite gut environment.” Although much is left to be discovered about these variants, Zhang and Leadbetter (2012) hypothesize they may be diagnostic markers for the subterranean and litter-feeding diets and behaviors specific to the termites in which they are found (Amitermes spp., Gnathamitermes spp., and Rhynchotermes spp.).
As illustrated by these examples, although coevolution between host and symbionts is an important force shaping mutualisms, other forces such as the loss of microbial community members or the relaxation and reemergence of nutritional pressures may also impact the evolution of a mutualism. As Zhang and Leadbetter (2012) note, “the metabolisms of community members form a network of dependent interactions: collapse of a functional population (or network node) within a symbiont community can have dramatic and long lasting effects on the genes encoded by symbionts occupying niches downstream in the chain of community metabolism” (Zhang and Leadbetter, 2012). Leadbetter concluded his presentation by suggesting that similar analyses of the gut communities of primates may reveal evidence of reverberations from past perturbations of these communities that occurred when hominids and humans moved into new biomes, migrated across different geographical regions, and adopted novel diets.
Evolution of Cooperation and Control of Cheating in the Social Amoeba
The social amoeba Dictyostelium discoideum uses a cooperative dispersal system when faced with starvation (Strassmann and Queller, 2011). Individual amoebas aggregate and form a migratory and multicellular slug that moves toward light, seeking a suitable location to transform into a fruiting body. About 80 percent of the cells differentiate into viable spores, while the remaining 20 percent commit the microbial equivalent of suicide to form a nonviable stalk that facilitates spore dispersal. The individual amoebae that aggregate to form the slug are largely but not always genetically identical—they may come from two or more clones—and are thus subject to selective pressure to cheat so as to be disproportionately included in the spore population and excluded from the nonpropagating stalk.
Cooperation among the individual amoeba in this system is maintained through a variety of selective mechanisms that limit cheating, according to Strassmann. These include kin selection, kin discrimination, and positive pleiotropy, in which an altruistic trait (e.g., ability to form the fruiting body stalk) is genetically linked with an essential function, so individuals must cooperate in order to survive (Strassmann and Queller, 2011). If kin selection were operative in maintaining multicellular behavior in D. discoideum, “that would mean that the stalk cells had to benefit enough to make it worthwhile for helping the spore,” she said.
To test this proposition, Strassmann and coworkers performed an experimental evolution study to determine what would happen if members of the multicellular community lost relatedness entirely, at least from the perspective of new
mutations (Kuzdzal-Fick et al., 2011). “If high relatedness is really important, and we get rid of it, we should be able to destroy this organism,” she explained. They allowed 24 lines of descendents of an ancestral strain to evolve through 30 generations of spore formation (and thereby drift by mutation from the ancestral genome), then mixed equal parts of each line with ancestral amoeba. Most of the resulting spores were dominated by the “evolved” lines, 18 of which cheated their ancestors, Strassmann concluded (Figure WO-24). “Some clones, even in this relatively short time, had actually completely lost the ability to form stalks on their own,” she noted.
Farming of bacteria Strassmann also discussed a recently discovered and intriguing interspecies interaction between bacteria and the amoebas. These bacteria appear to provide a selective advantage to the amoebas, facilitating survival in new environments. The discovery that D. discoideum contained bacteria not as contaminants, but within the amoebas’ fruiting bodies, led Strassmann’s group to
FIGURE WO-24 Cheating in low-relatedness studies. Eighteen (red numbers) out of 24 evolved lines showed significant evidence of cheating the ancestor by preferentially becoming spore, not stalk. In this study, Strassmann and colleagues artificially constructed low-relatedness conditions and allowed evolution to proceed for 31 generations of fruiting body formation. They assessed the cheating ability of the evolved lines in social competition with their ancestors, during fruiting body formation. They collected the resulting spores and calculated the percentage change of the ancestor in each experimental and control mix as 100 × [(% ancestor in spores) – (% ancestor in cells)]/(% ancestor in cells) (Kuzduzal-Fick et al., 2011).
SOURCE: Adapted from Kuzdzal-Fick et al. 2011. “High relatedness is necessary and sufficient to maintain multicellularity in Dictyostelium,” Volume 334, DOI: 10.1126/sci-ence.1213272 reprinted with permission from AAAS.
suspect that it engages in symbiotic behavior that resembles farming. Approximately one-third of D. discoideum clones they have tested to date carry bacteria and “seed” it into new environments, she reported. Similarities between this behavior and that of fungus-farming social insects such as leaf-cutter ants “makes sense because multigenerational benefits of farming go to already established kin groups,” the researchers observed (Brock et al., 2011). Some of the bacterial strains isolated from amoebas include those the organism consumes as food, according to Strassmann; however, others may serve as weapons against their nonfarming competitors by producing poisonous or defensive compounds. Such findings caused Strassmann to describe D. discoideum as a “selective sponge” capable of “telling us what bacteria are important to eukaryotes.”
Emergence and Robustness of Multicellular Behavior in Bacteria
Speaker Joao Xavier, of Memorial Sloan-Kettering Cancer Center, used swarming in P. aeruginosa as a model system for studying the selective pressures involved in this collective trait (Dr. Xavier’s contribution to the workshop summary report can be found in Appendix A). “Swarming is a multicellular trait, in the strictest definition,” Xavier noted, because individuals of this species are not capable of migrating across a surface, an important resource-seeking behavior. For that to happen, multiple cells in close proximity must together produce “massive amounts” of biosurfactants—up to 20 percent of their weight—on which the cells slide, Xavier explained.
Surprisingly, this trait appeared “uncheatable”; mixed cultures of wild-type bacteria and mutants incapable of producing the surfactant were not dominated (and then extinguished) by the mutant strain, but seemed to exist in equilibrium, Xavier reported. To investigate how the cooperators survived this competition, he and coworkers examined the genetic regulation of surfactant production, knowing that a gene associated with surfactant biosynthesis, rhlA, was expressed only when the bacteria had completed their initial period of rapid growth and were entering the stationary growth phase. If wild-type cells delay the costly expression of rhlA until most of their growth is complete, they would compete on equal footing with cheaters, he reasoned. He also knew that rhlA was under quorum-sensing regulation, yet simply supplying a quorum-sensing signal was not sufficient to trigger surfactant production during rapid growth, so there had to be an additional mechanism regulating surfactant biosynthesis.
That mechanism turned out to involve carbon and nitrogen availability, as shown in Figure WO-25, Xavier reported. In the medium in which the bacteria are grown, they have ample nitrogen and carbon for rapid growth, which depletes nitrogen supplies first. “At that point they start expressing rhlA,” he said; the bacteria then dedicate all of the carbon they take up to biosurfactant synthesis, which permits them to swarm toward more nitrogen-rich regions. Xavier’s group has named this strategy “metabolic prudence.”
FIGURE WO-25 rhlA regulation ensures metabolic prudence. As described by Xavier, P. auerginosa integrates multiple signals about environmental conditions—bacterial cell density and nutrient availability—in its regulation of the production of biosurfactants.
SOURCE: Adapted from Xavier et al. (2011).
“The cells, we believe, are integrating signals about the whole population—are we in enough numbers to start secreting biosurfactants, and do we have the right level of nutrients? Do we have an excess of carbon?—into this one molecular decision to start to express genes for a public good,” reported Xavier. As a first step to identify how such “molecular decisions” are made, they have shown that a mutant incapable of regulating expression rhlA (the gene has been engineered to include an inducible promoter) and induced to produce biosurfactants during the rapid growth phase while in competition with rhlA mutants, will be extinct within days.
Evolutionary Transitions to Multicellularity
Cooperation among microbes is also of interest to evolutionary biologists because it represents a likely stage in the evolution of multicellular organisms (Strassmann and Queller, 2011). Multicellularity has apparently evolved multiple times, but the process by which a group of previously autonomous cells become an organism—and thereby subject to natural selection at this higher level of organization—remains unknown. Speaker Paul Rainey, of the New Zealand Institute for Advanced Study and the Max Planck Institute for Evolutionary Biology, and his coworkers have examined the cyclical process of cooperation and cheating in bacteria as a possible route to multicellularity (Dr. Rainey’s contribution to the workshop summary report can be found in Appendix A) (Rainey, 2007; Rainey and Rainey, 2003).
When grown in unmixed liquid culture, the bacterium Pseudomonas fluorescens rapidly depletes oxygen, as illustrated in Figure WO-26, favoring
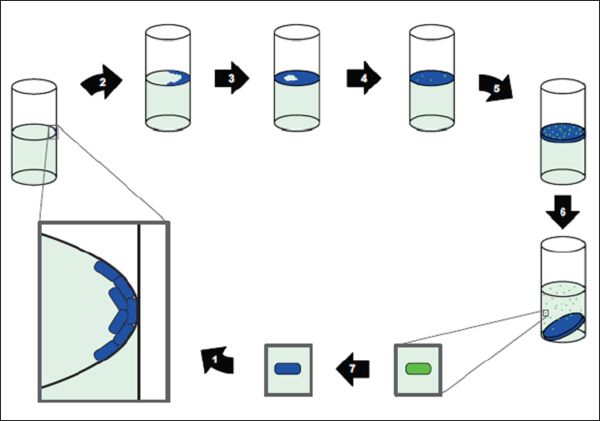
FIGURE WO-26 A putative life cycle for mat-forming bacteria. We start with a single bacterium (given in blue) capable of producing an extracellular adhesive. (1) It reproduces at the interface between liquid and air (in the case shown, starting at the inner surface of a glass tube). Daughter cells stick together because of the adhesive they produce. (2, 3) The resulting mat spreads over the liquid’s surface as a single-cell layer. (4) Because of prime access to oxygen, a robust mat forms. Mutation generates ‘‘cheats’’ (green cells that do not produce any adhesive polymer and grow faster as a consequence). (5) These cheats spread within the mat and contribute to (6) the collapse of the mat. Because the cheats do not produce the adhesive, they are liberated from the mat upon collapse. (7) Back mutation from one of these cheats to a mat-producing cell completes the life cycle. Of course, we do not imagine such a life cycle playing out in an environment where only a single mat can form (like a single tube). Rather, the back mutants from the liberated cheats could establish mats in different locations from their parent mat. Here the cell type leading to the death of the group also leads to its rebirth. The cheats amount to propagules (“germ line”) arising de novo from the mat-forming “soma” of an incipient multicellular individual.
SOURCE: Rainey and Kerr (2010).
mutants known as “wrinkly spreaders” that form mats at the air-liquid interface by secreting cellulose, which allows cells to stick to the walls of the container and to each other. Making cellulose is metabolically costly, and cheaters soon emerge within the mat. Ultimately, the increased proportion of cheaters within the mat leads to a classic tragedy of the commons, in which the drop in cellulose production leads to the collapse of the mat into the liquid medium. Back in the liquid phase, the cheaters easily free themselves from the mat; they continue
to live—and reproduce—as planktonic (free-swimming) organisms. Wrinkly spreader mutants arise again by random mutation, and the process repeats.
“Here we have the evolution of cooperating groups, and their demise,” Rainey said. “But there is the capacity for the group to evolve once again from the cheating types, to be destroyed, to re-evolve, and so forth.” He emphasized the critical role of ecology—in this case the liquid-air interface—in establishing this proto–life cycle. As Rainey and coworkers noted, the capacity to switch repeatedly between cooperating and cheating by mutation may not be widely possible, but clearly that capacity exists for this bacterium (Rainey, 2007). He added that “cooperating” bacteria could be viewed as behaving like somatic cells in multicellular organisms by providing support for cheaters—which resemble germ cells in that only they reproduce. This mechanism for collective reproduction—crude though it is—represents a critical step in the evolution of multicellularity, Rainey argued.
The emergence of higher-level individuality requires major evolutionary innovation: the capacity for one group of cells to beget another. This is a capacity that is difficult to reconcile with the central Darwinian concept of individuality, Rainey noted. “Any set of entities that have variation, reproduction, and heredity will evolve by natural selection,” he said. “What trick did nature play on individual cells that led them to give up their right to autonomous replication and come to reproduce solely as part of a corporate body?” he asked.
Returning to the proto–life cycle of P. fluorescens, the cycle is fueled by the niche constructing activities of each stage: the mat generates conditions that favor non-mat-formers (propagules) and the presence of non-mat-formers generates conditions that favor mat formers, and so the cycle proceeds. Rainey described an experiment in which his team observed the evolution of Darwinian individuality as a consequence of selection on the fecundity of mat-forming groups via the proto–life cycle. Not only did the selective regime result in the evolution of groups with enhanced fitness, but it also resulted in a decoupling of fitness: the fitness of derived mat-forming groups was no longer explicable on the basis of the fitness of the individual cells that made up the groups. “Enhanced fitness of derived mat-forming groups is attributable to a property selected at the collective level, namely, the capacity to transition through phases of a life cycle, and is not explained by improvement in cell fitness,” Rainey reported.
The various microbial communities that share our bodies contain characteristic and complex mixtures of microorganisms that have coevolved with humans (Costello et al., 2012; Dethlefsen et al., 2007). As previously noted, the gut microbiota is essential to human nutrition and immune system development. Ecological changes in the gut microbiota have been associated with such diseases as allergy, inflammatory bowel disease (IBD), and cancer, as well as two internationally
recognized epidemics, cardiovascular disease and obesity—findings which underscore the urgency for further research into the rules that govern the composition and stability of the human gut microbiome (Holmes et al., 2011; IOM, 2009; Littman and Pamer, 2011; Pennisi, 2011, Tremaroli and Bäckhed, 2012).
Several recent studies have analyzed the variability of human gut microbiota among individuals. An initial study of 39 individuals by Arumugam and coworkers (2011) indicated that each person’s gut microbiota belongs to one of three broad “enterotypes,” each of which contains relatively high levels of a single genus of bacteria: Bacteroides, Prevotella, or Ruminococcus. More recently, results of a similar study of 663 individuals identified archaea of genus Methanobrevibacter as a defining microbe in the Ruminococcus enterotype and failed to find clear separation between it and the Bacteroides enterotype (Yong, 2012). Other researchers have found more continuum than separation between the three enterotypes.
The distinction between the “type” and “continuum” models may have clinical significance given evidence linking the Bacteroides enterotype to diets high in fat or protein (Wu et al., 2011) and also to obesity and metabolic disorders (Yong, 2012); the Prevotella enterotype has also been associated with a high-carbohydrate diet (Wu et al., 2011). These findings suggest that enterotyping could be used to estimate a person’s risk of disease, or to predict his or her response to medications; however, such applications will demand not only a clearer picture of the nature of variation between individuals, but also an understanding of how individuals’ microbiota change over time and in response to perturbation (Yong, 2012).
Such knowledge can be gained through detailed longitudinal studies of the sort described by Banfield (Morowitz et al., 2010) and summarized in “Structure and Function of Microbial Communities.” Another time-series study of note monitored microbial taxa of the gut (as well as of the tongue and of the left and right palm) in two individuals for 6 months, in one case, and 15 months in the other, amassing nearly 400 time points (Caporaso et al., 2011). This analysis suggests that “there is pronounced variability in an individual’s microbiota across months, weeks and even days. Additionally, only a small fraction of the total taxa found within a single body site appear to be present across all time points” (Caporaso et al., 2011).
Communities of microbes and genes The human body provides a variety of habitats for its associated microbial communities, noted forum chair and speaker Relman of Stanford University (Dr. Relman’s contribution to the workshop summary report can be found in Appendix A). These environmental habitats and the associated ecological niches are shaped in part by the genetics of the host and in part by the history of all environmental exposures that have gone before, said Relman. Analyses of taxonomic and genetic diversity of the human microbiota suggest that body habitat is an important source for variation in community composition of the human microbiota. Within niche specialization also
appears to occur, with each body site “characterized by a small number of highly abundant ‘signature’ taxa,” and the relative representation of taxa and genes in each habitat varying considerably between individuals (Human Microbiome Project Consortium, 2012; Relman, 2012). Each person functions as a “unique and separate ecosystem” (Gonzales et al., 2011).
A recent survey of the gut microbiota produced a catalog of 3.3 million nonredundant, microbial genes,46 suggesting that these microbial communities have access to as many as 150 times as many genes as there are in the entire human genome (Figure WO-27) (Qin et al., 2010). The human genetic landscape may be better characterized as the sum of genes in the human genome and its microbiome (Turnbaugh et al., 2007). By providing traits that humans did not evolve on their

FIGURE WO-27 The number of genes distributed among the human-associated microbiota far outnumbers the number of genes humans inherit from their parents.
SOURCE: Reproduced with permission. Copyright © 2012, Scientific American, a division of Nature America, Inc. All rights reserved. Artwork by Bryan Christie Design (http://www.bryanchristiedesign.com).
___________
46 These microbial genes were detected in fecal samples obtained from 124 individuals, suggesting the presence of 1,000 to 1,150 prevalent bacterial species. Each individual’s gut harbored at least 160 bacterial species (Qin et al., 2010).
own, the gut microbiota may contribute to our ability to adapt to change, such as relocation to new environments, or adopting diverse diets (Balter, 2012).
The relative abundance of microbial genes associated with certain physiological pathways “varied less between samples from the same habitat than did the relative abundance of taxa,” suggesting that functional redundancy is likely between microbial communities despite variations at the species level (Relman, 2012). These studies have also revealed vast amounts of previously uncharacterized microbial and predicted functional diversity.
According to Relman, patterns of microbial community composition differ between healthy individuals and those with disorders such as obesity, IBD, chronic periodontitis, Crohn’s disease, and bacterial vaginosis (Dethlefsen et al., 2007; Ley et al., 2006b; Turnbaugh et al., 2006). He cautioned that the importance of these differences to the development of, or predisposition to, disease is not known. Disturbances in the microbiome could be a necessary initiating factor for disease, a factor in propagating disease pathology, or merely an effect of disease. Studies that monitor changes in the microbiota composition of individuals over time—before and after development of diseases—will be needed to assess the role of the microbiota in influencing states of health and disease, noted Relman.
Additional studies have tracked population shifts in the gut microbiota in response to a known disturbance: antibiotics (Dethlefsen and Relman, 2011; Dethlefsen et al., 2008; Jernberg et al., 2007). Relman described a study performed in his laboratory in which healthy subjects who had taken no antibiotics in the previous year were given two standard 5-day courses of the antibiotic ciprofloxacin (a fluoroquinolone) with a 6-month interval between courses (Dethlefsen and Relman, 2011). The researchers analyzed variability in fecal phylogenetic community composition by examining more than 1.7 million bacterial 16S rRNA hypervariable region sequences in more than 50 samples per individual, which they collected from 2 months prior to the first treatment through 2 months after the second treatment. In the pretreatment phase, they found some day-to-day variability within a relatively stable average community composition. These apparently routine fluctuations suggest that the gut community is stabilized not by resistance to change, but by its ability to function within a certain compositional range.
A profound and rapid loss of diversity and shift in the composition of the gut microbiota followed each course of ciprofloxacin, the researchers discovered; recovery began within a week after each treatment ended, but often failed fully to return the community to its pretreatment state (Dethlefsen and Relman, 2011). While the altered community apparently caused no gross alteration in the health of the human host, maintenance of ecosystem services, such as outcompeting pathogens, regulating host immunity, or co-metabolism, are not easily measured and may have been displaced by antibiotic treatment, the authors suggest. “Every course of antibiotics may represent another roll of the dice, potentially allowing displacement of a mutualist with a strain that may or may not provide the same benefit,” they observe.
Relman hypothesized that “external challenges such as antibiotic therapies can harm microbiota stability and make the host susceptible to pathogen invasion.” This point was reinforced by Xavier, who reported that mice given a single dose of the antibiotic clindamycin experienced a “severe loss of biodiversity” in the gut microbiota that took up to 28 days to resolve (Buffie et al., 2011). When treated mice were subsequently challenged with the pathogen Clostridium difficile, 40 percent died within 5 days, while all untreated mice that received the pathogen survived. In these control animals, he said, “Clostridium difficile wasn’t capable of colonizing the intestine and was, in fact, undetectable even from the sequencing data.”
Cooperative Survival Strategies
Unlike microbiologists, who tend to focus on the mechanisms by which microbes interact, evolutionary biologists attempt to determine the impact of interactions on the fitness of microbes as both individuals and populations, and thereby explain why particular behaviors have evolved (Diggle, 2010). Several workshop presentations offered this perspective through analyses of microbial conflict and cooperation observed both in the field and in model systems, and through the extension of one such model to demonstrate that cycles of cooperation and cheating among unicellular organisms provide a possible route to multicellularity.
Each of the evolutionary scenarios presented below depicts the forces of natural selection acting within a specific and influential ecological context, such as resource availability. The role of heterogeneous (“patchy”) environments in restricting gene flow, promoting genetic differentiation, and supporting local adaptation in microbial communities is particularly evident (Slatkin, 1987). This is the case in biofilms, where microscale gradients of chemicals and other environmental factors create a variety of ecological niches that are in turn reflected in the genetic and proteomic heterogeneity of their occupants (Denef et al., 2010b). Structural heterogeneity also drives diversification and coadaptation among community members in biofilms, where it has been associated with increasingly robust cooperative behavior (Brown, 2006). As speaker and Forum chair Relman observed, heterogeneity of all sorts causes microbial communities to behave differently than the homogeneous collections that are more typically studied.
A fundamental question regarding interactions within simple and complex communities of microorganisms concerns the coexistence of microorganisms within communities and in interactions with host organisms, and the stability of behaviors that support community-level function (Strassmann and Queller, 2011; West et al., 2007a). This overview uses the general term “cooperation” to describe actions that individual organisms take, at some reproductive cost, and which benefit their community as a whole. Microbial communities achieve a variety of benefits through cooperation, including shelter, nutrition, reproduction, defense, and dispersal.
Community Cooperation and the Expression of Virulence
The evolutionary theory of cooperation provides two basic answers to the question“why do individuals cooperate?,” according to Brown. The first reason is self-interest: in return for cooperation, individuals receive benefits that outweigh the cost of their actions; this description fits many mutualistic host-microbe interactions, he observed. The second reason applies only to certain cooperative interactions occurring within a species, in which cooperation occurs on the basis of relatedness, such that the cost of cooperation is outweighed by genetic representation in the population; this is known as kin selection, he stated. Cooperation among relatives may also extend to altruism, in which an individual sacrifices its chance of reproducing in order to increase those of a relative.
Host-associated microorganisms may have beneficial, neutral, or harmful effects on their host. According to Brown, “environmental” factors often influence the outcome of host-microbe interactions. Opportunistic47 pathogens, for example, are often commensal symbionts (e.g., Streptococcus pneumoniae) that become pathogenic following a perturbation to their host (e.g., wound, medication, immune deficiency) (Brown et al., 2012). Brown suggested that a focus on the diverse physical and social environments in which opportunistic pathogens grow may provide important insights into why these microorganisms cause disease in certain circumstances but not others.
Brown characterized virulence of a microorganism as the ability to cause damage to the host, and noted that a disproportionate number of genes encoding secreted, host-harming microbial products, known as virulence factors, are found on mobile genetic elements. “This supports the idea of virulence being intrinsically a cooperative trait,” he said, although the nature of the advantage provided by virulence seems paradoxical. “Why damage the source of your livelihood?” he asked.
The usual theoretical framework for thinking about the evolution of virulence is called the epidemiological trade-off model. In this model, virulence reflects an evolutionarily stable strategy that balances the exploitation of the host—which enhances microbial growth or transmission opportunities—with the cost of the shortened lifespan of the host. He noted that this framework was based upon the assumption of a “closed system” in which the pathogen is “always parasitic and always parasitic in the same host.” While this framework is “moderately successful for obligate, specialized pathogens,” Brown observed that disease-causing microorganisms only rarely satisfy these requirements. The trade-off model often fails to adequately describe the behavior of “opportunistic” pathogens, in part because of two key features of these microorganisms: generalism and phenotypic plasticity. They are generalists because these microorganisms can grow and thrive
___________
47 Resulting from pathogen entry via wounds or weakened state of the hosts, or as a result of a disturbance of a normally benign host-microbe relationship.
in a variety of environments; they can also modify phenotypic expression as a function of their changing environmental context (Brown et al., 2012).
Brown pointed to S. pneumoniae,48 which transitions from an asymptomatic and readily transmitted infection in the human nose to a highly virulent, non-transmissible infection of the blood. “As best as we can tell, this is a dead end for this bug,” he said. By taking an ecological approach to the question of “why pneumococcus shifts strategies?,” Brown and coworkers determined that another bacterial inhabitant of the human nose, Haemophilus influenzae, changes their shared environment in a way that harms pneumococcus (Lysenko et al., 2010). In the absence of H. influenzae, a less-virulent form of pneumococcus has a selective advantage. If H. influenza is present, however, a more virulent form of pneumococcus predominates. “The picture that is emerging is that virulence is an incidental byproduct of life in a distinct host compartment,” Brown said. “I believe this is a common theme for opportunistic pathogens,” he added. Virulence factors may provide advantages in a variety of nonparasitic contexts, noted Brown:“This could be a commensal compartment in the same host. It could be out there in the environment, in the soil, in the water. It could be in the same physical location, but in a different social state.”
The epidemiological trade-off model suggests that microbial virulence may be controlled by limiting transmission, Brown observed. The fact that improved hygiene is correlated with disease reduction appears to support this theory. Yet, in the pneumococcus scenario, the populations of the microbe that undergo transmission and those that cause disease are effectively separate—rendering hygiene ineffective as a means of disease control, he explained.
Brown also envisioned that opportunistic pathogens could pose a problem to proposed antivirulence drugs designed to disarm pathogens rather than kill them. Some have argued that such drugs are “evolution-proof,” or would at the very least slow the development of resistance (Clatworthy et al., 2007). Brown pointed out that resistance to an antivirulence drug would be selected for in the nasopharynx population of pneumococcus, as virulence confers fitness at that site, although it remains to be determined how rapidly such resistance would develop.
Responding to the suggestion that antivirulence drugs targeting cooperative or collective virulence traits should work because resistant pathogens would suffer the disadvantage of being the few cooperators in a sea of cheats, Brown replied that he would expect such a strategy would work in an unstructured environment. On the other hand, in a structured environment, he predicted, resistance to such a drug could spread. This is a hypothesis now being tested in his laboratory using Pseudomonas aeruginosa in what he described as an evolutionary screen for virulence traits. Such a screen could have general application in determining how easily microbes develop resistance to antivirulence drugs, he said.
___________
48S. pneumoniae may also be referred to as pneumococcus or pneumococci and may cause pneumococcal pneumonia and pneumococcal meningitis.
Cooperation, Cheating, and Coordinated Behaviors
In any scenario that involves cooperation, individuals can cheat by reaping the benefits of cooperation without contributing to it, for example, the quorum-sensing mutant bacteria described by Greenberg, which benefit from virulence factors excreted by their neighbors. Additional workshop presentations, summarized throughout this chapter, portray conflicts between individual- and population-level fitness, cheating, and the enforcement of cooperation by various mechanisms within microbial communities. They are representative of a range of studies demonstrating the combined influences of genes and environment on microbial social interactions (Brockhurst et al., 2006; Diggle et al., 2007b; Rainey and Rainey, 2003; Sandoz et al., 2007; Strassmann and Queller, 2011).
Social evolutionary theory provides a framework for analyzing the selective forces that shape microbial interactions such as cooperation, but it is important to recognize that microbial behaviors take place in ecological contexts that researchers are only beginning to define. For example, it may be unclear whether a microbial community such as a biofilm is behaving as a coordinated group (in response to active signaling) or as a collection of independent individuals (adjusting to chemical information). Such a distinction is critical to developing effective ways to stabilize or disrupt their activities. There are similar implications for conceptualizing behaviors that lead to pathogenesis and virulence in microbial communities and, therefore, for manipulating those behaviors for preventive or therapeutic purposes.
The semantics of cooperative behavior, the applicability of terms such as “cooperation” to microbial behavior, and indeed the broader question of the appropriateness of the social evolutionary framework for understanding microbial interactions are topics of ongoing debate (Nadell et al., 2009; West et al., 2007b; Zhang and Rainey, in preparation). These issues were reflected in workshop discussion and are explored in Semantics.
Multicellular behavior in bacteria Bacterial behaviors such as swarming and biofilm formation involve the secretion of metabolically costly substances that benefit their communities. As explained by Greenberg and other workshop speakers, such public goods are vulnerable to exploitation by cheaters, in the form of community members that do not produce the good, but are able to benefit from it (West et al., 2006). The cheater’s competitive advantage, if not otherwise constrained, eventually leads to a tragedy of the commons, as the community becomes overpopulated with cheaters and deficient in the associated public good.
Infectious cooperation As previously discussed, the fluidity of genetic exchange among bacteria via mobile genetic elements (e.g., plamids, phage) complicates the concept of speciation as it applies to macroorganisms. Genes for many secreted public goods, including exoenzymes and toxins, are frequently located on plasmids, leading some to speculate that this provides a mechanism for
populations to rescue themselves from loss-of-function cheats, by reinfecting them with cooperative genes (Nogueira et al., 2009; Smith, 2001). As Brown noted in his workshop presentation this scenario begs the question, “what about a plasmid that cheats?”
Brown and coworkers created a mathematical model of this situation, into which they introduced a second plasmid lacking the “public good” gene (McGinty et al., 2010). Their results suggest that cooperative genes on plasmids provide only short-term defense against cheaters in unstructured, well-mixed populations (e.g., liquid culture), after which the inevitable appearance of a cheater plasmid would lead to a tragedy of the commons. Brown explained that in a structured population (e.g., a biofilm), plasmids bearing cooperative genes are most likely to be passed between neighbors, thereby increasing both their relatedness and their productivity. This scenario favors the observed overrepresentation of public goods genes on mobile elements, he concluded.
Evolutionary Transitions Through Higher-Level Selection
Plausible on its face, the progression from multicellular microbial communities—with their previously noted capacities for intercellular communication, collective action, and division of labor—to multicellular organisms is difficult to reconcile with the central Darwinian concept of individuality, Rainey noted. “Any set of entities that have variation, reproduction, and heredity will evolve by natural selection,” he said. However, the transition from a state in which selection acts on a single-celled individual, to one in which selection acts on a multicellular one, he continued, means that cells that were capable of independent replication before the transition thereafter replicate solely as part of the whole.
The solution to this dilemma demands more than just cooperation, Rainey observed. As previously discussed, the emergence of higher-level individuality also requires major evolutionary innovation, such as the capacity for one group of cells to beget another. Such transitions are likely to show evidence of “fitness decoupling,” in which the fitness of collectives is no longer explicable based on the fitness of the individual cells that comprise the collectives. Life cycles can enable transitions to higher-level selection, he asserted, because they allow for individuality to shift from groups of single cells to a multicellular unit and also because they offer the potential to decouple fitness. For example, returning to the proto–life cycle of P. fluorescens, it is the oscillating shift between cooperating and cheating—and not cheating alone—that drives the cycle.
A series of painstaking experiments performed in Rainey’s laboratory demonstrated that fitness decoupling occurred over the course of several generations in his model, which the researchers demonstrated by comparing the number of cells making up mats with the reproductive success of cheater propagules (in preparation). As cell numbers in mats declined, the rate at which mats were replaced—the reproductive success of the cheater propagules—increased, he reported.
LOOKING AHEAD: MOVING TO THE
COMMUNITY AS THE UNIT OF STUDY
“The questions that biologists from diverse subdisciplines are asking have commonalities that make clear the continued existence of fundamental challenges that unify biology and that should form the core of much research in the decades to come. Some of these questions are as follows: What features convey robustness to systems? How different should we expect the robustness of different systems to be, depending on whether selection is operating primarily on the whole system or on its parts? How does robustness trade off against adaptability? How does natural selection deal with environmental noise and the consequent uncertainty at diverse scales? When does synchrony emerge, and what are its implications for robustness? When and how does cooperative behavior emerge, and can we derive lessons from evolutionary history to foster cooperation in a global commons?
These are among what we identify as fundamental questions in biology, cutting across subdisciplines and with the potential to reunify the subject.”
—Simon Levin (2006)
The “fundamental questions” identified above by Levin arose again and again in the workshop’s discussion. As Relman pointed out, these challenges urge researchers in the nascent field of social microbiology to reach beyond the immediate—and necessary—objective of understanding the mechanistic basis of microbial interactions, toward the yet-unrealized goal of predicting interactive and community behaviors.
The workshop presentations and discussion summarized in this section represent steps toward a predictive science of microbial sociality. They comprise four general areas: the development of theoretical and experimental frameworks to address questions such as those posed above; the adoption of analytical procedures to manage vast, multivariate data sets; the use of clear, well-defined description of phenomena; and the encouragement of transdisciplinary collaboration as essential to realizing a truly new field of scientific inquiry.
Developing Theoretical and Experimental Frameworks
Levin and Relman described theoretical and experimental frameworks for generating and testing hypotheses on structure-function relationships and interactions in microbial communities. Such investigations could take advantage of a wealth of analytical tools and mathematical models, as well as technologies such as genomics and proteomics for characterizing microbial community composition and function.
Microbial Communities as Complex Adaptive Systems
All ecosystems, including microbial communities, are examples of complex adaptive systems, according to speaker Simon Levin, of Princeton University (Dr. Levin’s contribution to the workshop summary report can be found in Appendix A). Such systems are characterized by the emergence of higher-level patterns from lower-level, localized interactions, and selection processes (Figure WO-28) (Levin, 1998). When individual microbes interact with their neighbors, their interactions are shaped by the forces of natural selection at the microscopic level, leading to macroscopic consequences, he observed. You have phenomena on multiple scales, he noted; “what’s going on at the macro scale is the result of lots of interactions at the micro scale, but is not easily deducible from them, and feedback to affect those behaviors,” he said. This phenomenon is “seen in a variety of systems including the biosphere, economic systems, social systems, and many physical systems,” he noted.
Anderson argued that a reductionistic approach to understanding the behavior of a complex adaptive system—for example, attempting to predict the behavior of an entire microbial community from knowledge of the behavior of individual community members—would prove inadequate (Anderson, 1972). Rather, he wrote, “the study of complex adaptive systems is a study of how complicated structures and patterns of interaction can arise from disorder through simple but powerful rules that guide change.”
In his workshop presentation, Levin focused on features of complex adaptive systems of particular relevance to interactions within microbial communities. First, he described attempts to develop descriptive laws for complex adaptive systems that relate emergent properties of such systems—macro-level characteristics such as patterns of biofilm growth, for example—to a set of rules that define behavior at the microscopic level (Bonachela et al., 2011). Taking a similar approach, he and coworkers determined that complex and apparently choreographed movements of animal groups such as schooling fish and migrating birds—as well as of swarming bacteria and motile slime molds—can be described with mathematical “rules” that determine (1) the speed and direction of each individual’s motion and (2) the criteria for any change in speed and direction (Couzin et al., 2005).
Analyses of complex adaptive systems also offer insights into the concept of robustness: a system’s ability to maintain function in response to perturbation. “We generally think about robustness as something we want to preserve,” Levin observed. “We don’t want our ecosystems to collapse. But in some cases robustness is something you want to overcome,” he added—for example, the robustness of a virulent biofilm infection. Complex adaptive systems collapse for a variety of reasons inherent to their structure, which makes them prone to sudden and hard-to-predict “regime” shifts. Shifts between states may also exhibit hysteresis, in which the “system goes from A to B and then getting back from B to A may not be so easy and may not occur via the same pathway.” Shifts may also exhibit
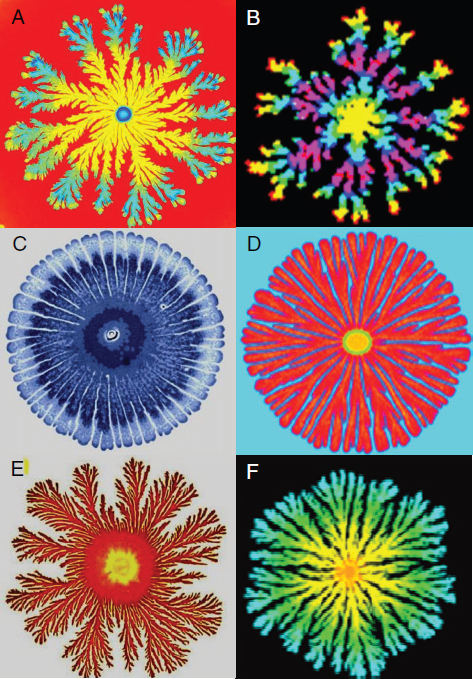
FIGURE WO-28 Patterns emerge from individual interactions in bacterial communities. Examples of different branching patterns formed during colonial development of the P. dendritiformis bacteria. To self-engineer their colonial structure these bacteria regulate the balance between attractive and repulsive chemotactic signaling as well as their food chemotaxis. Generic modeling of the growth can be used to test this idea by comparing the observed patterns shown on the left (A, C, and E) with the results of model simulations shown on the right (B, D, and F). The colors in the observed colonies are added according to the bacterial density. In the simulations the color code shows the time evolution of the growth. At the top (A, B) is shown the typical pattern when food chemotaxis dominates the growth at intermediate levels of food depletion. The middle (C, D) shows the typical pattern at higher food levels when attractive chemotactic signaling is activated, and the bottom (E, F) shows the very low level when repulsive chemotactic signaling is intensified. Comparing the patterns one should keep in mind that the real colonies have almost a million times more bacteria as well as additional mechanisms not included in the model.
SOURCE: Ben-Jacob et al. (2004), Bacteria harnessing complexity. Biofilms 1(4):239-263. Reprinted with permission.
contagious spread. Levin and colleagues grew concerned about“overconnectedness” in the global financial system that in 2008 resembled “a lot of food webs that collapse,” he said. The result was a publication published 6 months before the economic collapse titled “Complex Systems: Ecology for Bankers” (May et al., 2008). What makes systems prone to collapse, whether there are early warning indicators, and what might be done to avert collapse are areas of active investigation (Scheffer et al., 2009).
Complex adaptive systems can achieve robustness—often called resilience in ecology—through both rigidity (by resisting change from normal function) and flexibility (ability to recover from displacement from normal functioning). Both strategies may coexist at different levels in the same system, Levin noted, as illustrated by influenza A. “Depending on whether you think about influenza A at the level of the totality of all subtypes, or at the level of a single subtype or individual strain, it’s either very robust, because it has been around for a long time, or it’s not robust at all, because individual strains replace each other as surface proteins, like hemagglutinin and neuraminidase, replace each other,” he explained (Figure WO-29). “That makes it clear to us that if we are thinking about robustness, we really have to think about levels. Robustness at one level may depend upon the absence of it at lower levels.”
Influenza A also illustrates another important element of robustness: modularity. Nobel Laureate Herbert Simon described the advantages of modularity by comparing the work of two hypothetical watchmakers, Levin recalled: one made watches from beginning to end, and the other assembled several modules of multiple parts, which he set aside until he needed to combine them to make a complete watch. Both watchmakers“kept getting interrupted, and the first one never made a watch because he had to start over again each time, whereas the other one eventually assembled a watch because he saved his work along the way,” he explained.
Similar advantages are conferred on bacteria, which assemble biofilms in a modular fashion, Levin pointed out. Modules reduce risk to the entire system, and they provide building blocks for system complexity. Furthermore, modular bacterial assembly offers a pathway to multicellularity, as Rainey’s model suggests. Moreover, Levin added, by converting modules (of planktonic, free-living, individual bacteria) to elements of the next organizational level (a multicellular organism), evolutionary transitions reduce the potential for tragedies of the commons at lower organizational levels by shifting selection to a higher level. “If you build something modular, it allows you very easily to reconstitute the whole, to break it down and reconfigure things. If you are in an environment where you want to go back and forth between different forms, having a modular structure seems to me an extremely good strategy.”
Computer simulations of complex adaptive systems have provided insight into the dynamics of cooperation, competition, and the production and consumption of public goods in both human and microbial societies. Levin and
FIGURE WO-29 The persistence of influenza A illustrates how robustness can result from flexibility. Influenza A viruses are divided into subtypes based on the two surface proteins hemagglutinin (HA) and neuraminidase (NA), shown here as part of a 3D graphical representa - tion of a generic influenza virus (A). Robustness at the level of subtypes is mediated through high mutation rates of these surface proteins that allow the continual replacement of strains with novel ones as illustrated in (B), and at higher levels by reassortment events that create new subtypes. In B, HA1 sequence clusters are plotted as a function of calendar year of isolation. Each cluster is indicated by a different color, with the eight largest clusters shown in bold. The dashed line indicates the total number of isolates in the data set each year. The dominant sequence clusters tend to replace each other every 2-5 years. SOURCES: (A) CDC Public Health Image Library, image 11826; (B) From Plotkin et al., 2002, Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus, Volume 99, DOI: 10.1073/pnas.082110799, reprinted with permission from Proceedings of the National Academy of Sciences.
coworkers have used such techniques to compare the outcomes of simple competitive scenarios in heterogeneous and homogeneous environments, leading them to conclude that spatially restricted competition—which would occur in a patchy landscape of cooperators and cheaters—can enhance cooperation by providing some cooperators refuge from competition by allowing cooperators to self-organize into spatially contiguous ensembles.
Drawing comparisons with previously discussed policing mechanisms that preserve public goods in microbial communities by making cheating costly, Levin noted that similar strategies frequently arise in models of human behavior involving common-pool resources.49 In these situations, he noted, people will punish cheaters even at some cost to themselves, to the extent that punishment becomes a norm: an emergent property of that society. When participants questioned the extension of this analogy to microbial “societies,” Levin recognized their concerns, but said his comparison resulted from asking the question, “How much of human behavior can be explained with such simple assumptions? When simple models fail, only then must we develop more complicated ones.”
“Microbial communities are complex adaptive systems, made up of individual agents with their own selfish agendas,” he concluded. “The challenge is to understand them as emergent from individual interactions, with a complementarity of function that may arise.” Levin concluded that many analytic tools are available and are being developed to scale from individuals to collectives and to address problems of robustness and the particular evolutionary challenges due to the fact that microorganisms live in a commons.
Probing Resilience Through Perturbation
As discussed previously, the human gut is a highly complex series microbial communities in which “the fitness of a symbiont depends upon environmental features that can change, such as coexisting microbiota, the diet of the host, and which species and even particular individual is the host” (Dethlefsen et al., 2007). Relman described several experiments that examined the robustness of this ecosystem, as defined by Levin: the system’s ability to maintain function in response to perturbation. These studies tracked population shifts in the gut microbiota in response to a known perturbation, antibiotic treatment (Dethlefsen and Relman, 2011; Dethlefsen et al., 2008; Jernberg et al, 2007). These studies have revealed the human gut microbiota to be a “dynamic ecological system” with considerable resilience. However, repeated disturbances led to a persistent regime shift. Accoring to Dethlefsen and Relman (2011), “Although there are no immediate
___________
49 Levin explained that a public good, in economic terms, is something that all can use and to which all can contribute, such as a library, and that is nonrivalrous and nonexclusionary. A common-pool resource is something that can be depleted by one person, depriving another, such as a fishery.
signs of symptoms after antibiotic treatment, acute and chronic health problems are associated with antibiotic use.”50
Relman noted that robustness was achieved through resilience,51 as defined by ecologist C. S. Holling as “the capacity of a system to absorb disturbance and reorganize while undergoing change so as to still retain essentially the same function, structure, identity, and feedbacks” (Walker et al., 2004). He noted that ecologists often observe transitions between stable states in macroecosystems, and they have also correlated loss of resilience with the transition to diminished states of ecosystem “health,” as depicted in Figure WO-30 (Folke et al., 2004). Undergoing multiple severe perturbations within a relatively short time can increase the likelihood that an ecosystem will undergo a shift to a less-resilient state (Paine et al., 1998). Relman also noted that we have yet to grasp the full spectrum of attributes that contribute to resilience in the gut microbiota. A more complete understanding of these factors could lead to ways to measure resilience as a gauge of host health, and perhaps to the ability to increase the resilience of the gut community through manipulation.
Computer simulations and predictive mathematical models can be used to explore microbial community dynamics in complex communities and environmental settings. Xavier described the construction of a computational model of a simplified “microbiota” capable of shifting between alternative stable states of dominance by either antibiotic-tolerant or antibiotic-sensitive bacteria. This minimal ecological model of microbial interactions in the intestine can “explain how antibiotic mediated switches in the microbiota composition can result from simple social interactions between antibiotic-tolerant and antibiotic-sensitive bacterial groups.” Bucci et al. (2012) demonstrate how this model can analyze the temporal patterns of metagenomic data from the longitudinal study of Dethlefsen and Relman (2011).
New Tools and Approaches for an Emerging Field of Inquiry
New methods and analytical approaches are needed to evaluate the complex, high-dimensional data sets derived from studies of microbial ecosystems. Such data include not only gene sequences and population abundance distributions of cells, species, and phylotypes, but also sampling information and clinical or environmental covariates associated with each sample (e.g., physical, chemical,
___________
50 The hygiene hypothesis “asserts that increasing rates of autoimmune disorders in the developed world, such as asthma and inflammatory bowel disease, are related to the disruption of the normal interactions within and between the human microbiota and the host” (Dethlefsen and Relman, 2011).
51 As Levin pointed out, robustness and resilience are defined by the component that is being measured and the organizational level at which that measurement is made. For example, robustness in the intestinal microbiota as a whole might refer to its taxonomic composition, or to a particular function performed by the community. Either of these whole-system measures may include components (e.g., individual taxa or functions) that are not robust or resilient.
FIGURE WO-30 Alternative stable states, disturbance, and loss of resilience.
SOURCE: Adapted from Folke et al., 2004, and reprinted with permission from Annual Review of Ecology, Evolution, and Systematics.
biological, geographical, spatial, temporal information) (Fukuyama et al., 2012; Little et al., 2008). Progress will require collaborations between researchers in a variety of fields and disciplines (Levin, 2006).
Statistical Tools for Integrating Community Networks and Spatial and Clinical Data
Speaker Susan Holmes of Stanford University referred to this situation as a “perfect storm, in which we have data that are combining and coming in from every different possible level”; this includes metagenomic and phylogenomic data, as well as clinical data that describe host effects (Dr. Holmes’ contribution to the workshop summary report can be found in Appendix A). The solution, she asserted, is not to hire a statistician, but to take advantage of a free, user-friendly, open-source, flexible analytical tool called R52 (http://www.r-project.org). There are also multiple software packages to adapt the R platform to a variety of uses, including bioinformatics, phylogenetics, and
___________
52 Ecologists have long used R, which was developed in the mid-1990s, according to Holmes.
ecology. R offers a common statistical “home” for many of the scientific fields that contribute to the study of microbial communities, she observed.
The volume of metagenomic data has risen exponentially with the advent of high-throughput nucleic acid sequencing technologies; as sequencing becomes ever cheaper the volume of data is likely to continue to rise. According to Holmes, the volume of metagenomic data to be analyzed is readily matched by the speed and capacity of cloud computing to evaluate it. Holmes cautioned against “throwing away data” in attempts to streamline analysis. In about a third of the studies on which she has collaborated as a biostatistician, she said, “we realized that people thought that they were cleaning up, or standardizing, or making their data better in some way, and they threw out information.” She urged researchers to recognize that “raw data is the good data.” Moreover, she added, “most of us are funded by public agencies. It is our duty to then make the data publicly available so that people can combine studies.”
An important feature of statistical tools such as R is “to empower biologists to stay in contact with their data,” Holmes observed. To this end, she recommended that students be encouraged to enjoy data analysis. Luckily, she added, the current generation of computer-savvy biology students is far easier to interest in statistics than its predecessors.
Workshop participants embodied the broad spectrum of scientific disciplines currently contributing to the exploration of microbial communities. Many commented on the unprecedented, high level of collaboration among researchers in different disciplines. Benefits of this cross-fertilization will impact both the research side and the applied side, said Strassmann. Relman stressed the importance of moving beyond interdisciplinary research to transdisciplinarity, in which “you don’t simply bring together different ways of thinking, you try to adopt the ways of thinking of another in reformulating how you see the world, and what you end up with is something that is neither one nor the other.” Citing Levin’s presentation as an example, Relman encouraged researchers to “(view) the world through the lens of another kind of discipline, another set of rules and principles, or even language.”
Forum member Peter Daszak, of EcoHealth Alliance, agreed with this view and noted that the “transdisciplinary nexus” ultimately combines reductionist science, which has provided invaluable insights on biological mechanisms, “right down to the molecule,” with the holistic perspective of ecology. Noting that scientific institutions will need to be reorganized to support transdisciplinarity, forum member George Poste, of Arizona State University and Complex Adaptive Systems Initiative, Inc., decried“the anachronism of the contemporary training curriculum, and also the fragmentation of grant agencies to be able to deal with this type of very complex transdisciplinary research.”
Semantics Many participants suggested that, as this field of inquiry moves toward transdisciplinary work, the concepts and metaphors borrowed from other fields must be used with care. Metaphors are helpful tools, particularly when communicating complex ideas to diverse audiences, yet they can also serve as a barrier to understanding fundamental processes or perceiving novel mechanisms. As West and coauthors observe, progress in the field of social biology “is often hindered by poor communication between scientists, with different people using the same term to mean different things, or different terms to mean the same thing. This can obscure what is biologically important, and what is not.” In addition, “the potential for such semantic confusion is greatest with interdisciplinary research” (West et al., 2007b). As described in the section that follows, participants raised several examples of how the terms used to describe the constituents, traits, and activities of a microbial community may also limit the ways in which researchers think about and investigate these systems.
Many workshop participants debated the use of several key terms, beginning with “social.” Some participants observed that nearly every behavior is social, because everything an organism does affects another organism, in some direct or indirect way. It follows, therefore, that evolution proceeds in a social context. Rainey agreed, but noted that “social” is often misrepresented as “cooperative.” “The sense that the microbial world is inherently cooperative is not a rigorously tested idea at all,” he asserted. “That there are interactions governing the shape and structure of microbial communities” Rainey added, “is an absolute given.”
The language evolutionary biologists use to describe cooperative behavior was developed from observations of the macroscopic world, Rainey pointed out. As such, he said, the notion of cooperation is “well defined and understood,” but when applied to microbes, it may not be relevant, for we know little about the ecological conditions under which microbial behaviors evolved. Thus, he continued,
When we see an extracellular product, we should not leap [to] a conclusion that it is a public good. It could have a variety of explanations [such as]: copious production of extracellular products may reflect the fact that in the lab they are typically over-fed—in the wild, however, it is possible that they never excrete excessive amounts of any extracellular product. [Likewise,] (w)hen we see a non-producer, we shouldn’t leap to the conclusion that it’s a cheat. It may be that non-producing types evolve because production of the extracellular product under some conditions may be maladaptive. Alternatively, non-producing types might be cross feeders that take advantage of the resource partitioning activities of producing types. There are a range of alternate possibilities that need to be considered.
“The point you make is an extension of a debate that has gone on in evolutionary biology for a long time, which is that almost any trait of an organism people will interpret as being adaptive,” Levin responded. Although natural selection favors adaptation, “there are many, many things in individual organisms
and in communities…that are in fact not adaptive,” Eisen explained. “There are constraints upon selection, there are historical contingencies, there are nonequilibrium dynamics,” Eisen continued. Organisms may, therefore, display traits that are, in and of themselves, detrimental to themselves or their neighbors, but which were positively selected over the course of evolutionary history.
Several workshop participants also discussed whether the use of anthropomorphic terminology such as “cooperation” and “cheating” to describe nonhuman behavior encourages anthropocentric interpretations of nonhuman behaviors. Forum member Gerald Keusch, of Boston University, expressed concern that “you are shaping the phenomenon that you are looking at by the use of the terms rather than really understanding what those behaviors and interactions really are.” Several speakers who employ these terms agreed that they must be chosen with care and defined precisely whenever they are used. Even more subtle terminology is prone to anthropocentrism, Brown noted. The labeling of “virulence factors” as such, for example, implies that they were selected for causing virulence.
Strassmann reminded participants that although the words used in social biology are weighted by possible human interpretations, the field is “a really new and exciting field of understanding how selection operates.” Underlying the language of social biology is the important understanding that “natural selection operating on social behaviors is really powerful and takes into account competition among individuals in social contexts.”
Insights into Life on Earth and Other Worlds
The sea change that accompanies the shift to viewing microbial communities as the unit of study will extend to the obvious and subtle implications of this work. Microorganisms drive some of the largest-scale phenomena on the planet, from the conversion of energy from the sun to nitrogen fixation in plants. Research exploring the processes that drive microbial community formation and function will reveal the basis for these processes as well as the intricate interdependencies between the microbial communities and other forms of life on Earth. With this knowledge comes the opportunity to better harness the seemingly unlimited potential of these organisms to improve human, animal, plant, and ecosystem health and well-being.
Exploration of the intricate biology of life—in all of its varied environmental and ecological contexts—may reveal previously unknown activities that support life on Earth. The discovery of deep-sea hydrothermal vents in 1977 “revolutionized our understanding of the energy sources that fuel primary productivity on Earth” (Davis and Joyce, 2011). Hydrothermal vent-fluid chemistry fuels these ecosystems, which comprise communities of microorganisms and animals (Figure WO-31). Despite the absence of sunlight, and the extreme temperatures and pressures experienced in these environments, hydrothermal vents teem with life because of the extremophile bacteria that convert energy from the oxidation
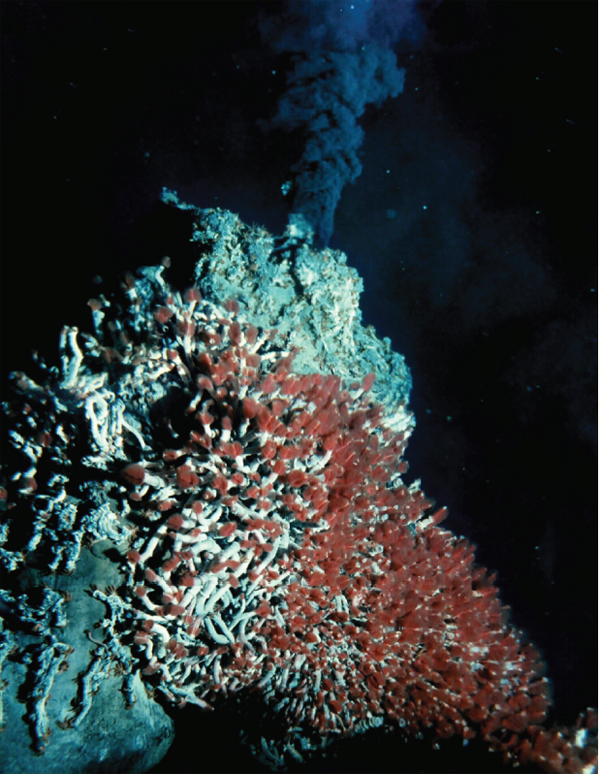
FIGURE WO-31 In 1977, the deep-ocean submersible Alvin led scientists to discover tubeworms living at the edges of hydrothermal vents in the deep sea.
SOURCE: University of Washington; NOAA/OAR/OER.
of reduced sulfur compounds and methane in the environment to provide their hosts with carbon and nutrients (Dubilier et al., 2008).
Cavanaugh and Widder emphasized the promise of yet unexplored phenomena in the world’s oceans. Widder noted that many of these phenomena are likely to stay that way because of the high cost of conducting deep-sea research and the low rate of funding for marine science in the United States. “I think it is really tragic that we don’t have better access to our oceans,” she said. “The U.S. is losing our capabilities. We sold the Johnson Sea Link submersibles to Brazil. China is now going to have the deepest diving submersible as of next year [, which will provide] access to 99 percent of the deep ocean.” What is needed, she said, is a NASA equivalent for the oceans.
Exploration of extreme environments may provide clues to the basis for life on other planets. Viable Pseudomonas bacterial species were recently isolated from a lava tube in the Cascade Mountains in Oregon. At an elevation of 5,000 feet, the bacteria were found deep inside the icy cave, an environment believed to be similar to some of the surface environments on Mars (Popa et al., 2012). The current NASA Mars Rover mission is gathering data to assess whether the Mars environment is, or previously was, capable of supporting microbial life (NASA, 2011).
Water samples obtained from the recently breached, prehistoric Lake Vostok in Antarctica will be examined for life forms that can withstand extreme conditions. The lake has been trapped deep beneath Antarctica for the past 14 million years and is an extreme habitat—with high pressure, constant cold, low nutrient input, high oxygen concentration, and an absence of sunlight. Life forms found to survive such conditions would strengthen the case for life in the outer solar system, because similar conditions are thought to exist on the moons of Jupiter (Europa) and Saturn (Enceladus) (Russian drill penetrates 14-million-year-old Antarctic lake, 2012).
Untapped Innovation and Functional Novelty
The characterization of microbial communities has informed and inspired a host of applications. Several presentations touched upon the ability of microorganisms to receive, integrate, and respond to multiple forms of inputs. One remarkable aspect of many of these systems is the sophistication of resulting behaviors in the absence of centralized control. The study of how organisms “solve” dynamic problems in nature, and how optimization processes are constructed and embedded into self-organized systems in general,53 may provide insights into new and efficient computing algorithms and network design (Box WO-2).
___________
53 See http://sydney.edu.au/science/biology/social_insects/people/madeleine_beekman/optimisation.shtml.
BOX WO-2
Biologically Inspired Computing Algorithms
The behavior of the multinucleate slime mold Physarum polycephalum is studied alongside that of colonies of honeybees and ants to inform “biologically inspired” algorithms for computing applications. Physarum are syncytial, multinucleate “plasmodia” that can transform into a network of highly dynamic, interconnected vein-like tubes when searching for food. Through tube extension and movement of the entire plasmodium, Physarum have been shown to seek out the shortest, most efficient path to a preferred food source (Dussutour et al., 2010; Marwan, 2010; Tero et al., 2010). Indeed, when baited with food sources, Physarum has navigated mazes and mimicked existing transportation networks (Figure WO-2-1) (Bonner, 2010; Tero et al., 2010).
Perhaps even more remarkable is this organism’s capacity to balance its own diet when presented with different foods—carbohydrates and proteins—at different sites. Physarum appears to choose the most nutritionally appropriate food source (Bonner, 2010). These adaptive, self-organizing and self-optimizing behaviors may provide insights important to the development of “scalable, multicomponent networks that need to function in the absence of central control mechanisms” (Marwan, 2010). Potential applications include the development of mobile communication networks, or networks for dynamically connected computational devices (Marwan, 2010).

FIGURE WO-2-1 Network formation in Physarum polycephalum. Clockwise from upper left panel: At t = 0, a small plasmodium of Physarum was placed at the location of Tokyo in an experimental arena bounded by the Pacific coastline (white border) and supplemented with additional food sources at each of the major cities in the region (white dots). The plasmodium grew out from the initial food source with a contiguous margin and progressively colonized each of the food sources. Behind the growing margin, the spreading mycelium resolved into a network of tubes interconnecting the food sources. The horizontal width of each panel is 17 cm.
SOURCE: Tero et al. (2010).
Genetic and Metabolic Diversity
The microbial biosphere is an important reservoir of genetic and metabolic diversity. Microorganisms have long been “mined” for small molecules that may be useful in the biomedical or agricultural contexts (Caetano et al., 2011). Investigations into how processes are carried out by communities of microorganisms may unlock even more potential benefits, as will continued investigation of the substantial proportion of putative genes identified in metagenomic studies that have no known functional analog. Several workshop participants described prospects for developments in a number of areas, amid a general sense of the unlimited potential of microbial function.
Widder’s research on bioluminescence led to her use of bioluminescent bacteria as a marker for pollution in waterways such as the Indian River Lagoon, a 156-mile estuary that scientists say is one of Florida’s most precious and threatened ecosystems (Olsen, 2011). Sediment samples are mixed with a bioluminescent bacterium (Vibrio fischeri) and the concentrations of toxic chemicals are determined by monitoring how quickly the bioluminescent glow fades (how quickly the sample contents kill the bacteria) (Olsen, 2011). The test is portable and has proved to be an effective and inexpensive way to detect sources of pollution and target interventions (Pennisi, 2012).
Understanding how microbial communities convert plant matter into less complex sugars and starches may promote the technological goal of converting cellulosic plant biomass into renewable biofuels. Metagenomic analysis of the gut community of a wood-feeding, higher termite species has revealed a rich reservoir of genes coding for enzymes relevant to wood degradation and the conversion of lignocelluloses into biofuels (Warnecke et al., 2007). Leaf-cutter ants harvest hundreds of kilograms of leaves each year to feed a mutualistic fungus that serves as the colony’s primary food source. Studies of the metagenome and metaproteome of the fungal gardens of leaf-cutter ants have revealed a previously unknown and diverse assembly of bacteria as well as many genes that are believed to encode enzymes for biomass degradation, and biosynthesis of amino acids and other nutrients, that could promote the growth of the fungal cultivar or even nourish the ants themselves (Aylward et al., 2012; Suen et al., 2010).
Most antibiotics currently in use were derived from bacteria that inhabit the soil, a rich source of microbial diversity that has barely been tapped (Handelsman, 2009). As previously described, the soil continues to yield new classes of antibiotic compounds with potential agricultural and medical benefits. More recently, insect symbionts have emerged as a promising source of antibiotic lead compounds.
Through close collaboration with Jon Clardy and his lab at Harvard, Currie has contributed to the isolation and characterization of several novel small-molecule drug leads from insect-associated Actinobacteria. Southern pine beetles are known to associate with both a beneficial fungus in the genus Entomocorticium and with the antagonistic fungi Ophiostoma minus. Based on
this information, he and colleagues searched for and found beetle-associated, antibiotic-producing Actinobacteria that mediate this fungal community, inhibiting O. minus without similarly affecting Entomocorticium (Scott et al., 2008). Another species of Actinobacteria they have isolated from honeybees produces a small-molecule antagonist to Paenibacillus larvae, the bees’ major bacterial pathogen. In total, Currie and coworkers have identified seven novel small molecules from Actinobacteria associated with insects; some of which are currently being tested as potential drug leads.
Insights into microbial interactions—and ways to disrupt them—could lead to new therapeutic approaches. Current approaches to infection, such as antibiotics and other antimicrobials, are nonspecific and create strong selective pressures for the development of resistance (Xavier, 2011). Targeting social strategies that underlie virulence, or the mechanisms by which microorganisms become pathogenic within certain environments may prove a more efficient and effective means to treat disease (Brown et al., 2009; Rasko and Sperandio, 2010). Indeed, a more ecologically-informed view of antibiotic production and resistance in bacteria may lead to new approaches to treat bacterial infections. While antibiotic resistance is generally thought to be driven by brief, cyclic invasions of populations by antibiotic-producing and antibiotic-resistant bacteria, recent research suggests that non-clonal communities of bacteria in structured, wild habitats use cooperation as a strategy in antibiotic-mediated competition with neighboring populations (Cordero et al., 2012). Reflecting the concept of “ecological context dependence,” noted by Currie and many others throughout the workshop, this research suggests that within a population, only a few members produce the antibiotic to which all others are resistant, creating interaction networks within and between populations that prevent invasion while also maintaining diversity (Cordero et al., 2012; Morlon, 2012).
As noted by Dethlefsen et al. (2007), it is “crucial to consider the role of microbial communities, and not just individual species, as pathogens and mutualists.” Recent investigations have revealed links between altered microbiota ecology (dysbiosis) and infectious and noninfectious diseases alike. These observations have prompted calls to transition clinical practice from “the body-as-a-battleground to the human-as-habitat perspective” and to consider system-level, adaptive management approaches to managing health. Adaptive management approaches are used to “manage biodiversity in a variety of habitats, including communities in highly disturbed environments affected by overfishing and by climate change” (Costello et al., 2012). This approach may better reflect health as “a product of ecosystem services provided by microbial communities” and would require the development of new diagnostic tools to inform health management decisions (Costello et al., 2012).
Relman observed that “there are all sorts of promises that are dangling out there in front of us in the way of diagnostics and predictive aspects of medicine. There is a lot of as yet unrealized potential and as yet unrealized promise.” Early investigations have revealed that there is a great deal left to discover about the patterns of microbial diversity in humans and the stability of these populations, particularly in the face of perturbations (i.e., resilience). This ecological perspective will likely provide new leads for the management of disease. Indeed, as noted by Lita Proctor, a program director of the Human Microbiome Project, “unlike the human genome, the microbiome is changeable; and it is this changeability that holds promise for prevention and treatment of disease” (Balter, 2012).
The increased recognition of the beneficial as well as benign host-microbe relationships will further drive the paradigm shift—in the way we collectively identify and think about the microbial world around us—first suggested by Joshua Lederberg more than two decades ago. The familiar “war metaphor” in which the only good bug is a dead bug will be replaced with a more ecologically informed view of the dynamic relationships within and between hosts, their microbiomes, and their environments (Lederberg, 2000). This perspective recognizes that microbes and their hosts ultimately depend upon one another for survival and encourages the exploration and exploitation of these ecological relationships in order to improve human, animal, plant, and environmental health and well-being (Lederberg, 2000).
Ackerman, J. 2012. The ultimate social network. Scientific American, June 27-43.
American Heritage Science Dictionary, 1st Edition. 2011. Boston: Houghton Mifflin Harcourt.
Anderson, P. W. 1972. More is different. Science 177(4047):393-396.
Aoki, S. K., E. J. Diner, C. T. de Roodenbeke, B. R. Burgess, S. J. Poole, B. A. Braaten, A. M. Jones, J. S. Webb, C. S. Hayes, P. A. Cotter, and D. A. Low. 2010. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468(7322):439-442.
Aoki, S. K., S. J. Poole, C. S. Hayes, and D. A. Low. 2011. Toxin on a stick: Modular CDI toxin delivery systems play roles in bacterial competition. Virulence 2(4):356-359.
Arumugam, M., J. Raes, E. Pelletier, D. Le Paslier, T. Yamada, D. R. Mende, G. R. Fernandes, J. Tap, T. Bruls, J. M. Batto, M. Bertalan, N. Borruel, F. Casellas, L. Fernandez, L. Gautier, T. Hansen, M. Hattori, T. Hayashi, M. Kleerebezem, K. Kurokawa, M. Leclerc, F. Levenez, C. Manichanh, H. B. Nielsen, T. Nielsen, N. Pons, J. Poulain, J. Qin, T. Sicheritz-Ponten, S. Tims, D. Torrents, E. Ugarte, E. G. Zoetendal, J. Wang, F. Guarner, O. Pedersen, W. M. de Vos, S. Brunak, J. Dore, M. Antolin, F. Artiguenave, H. M. Blottiere, M. Almeida, C. Brechot, C. Cara, C. Chervaux, A. Cultrone, C. Delorme, G. Denariaz, R. Dervyn, K. U. Foerstner, C. Friss, M. van de Guchte, E. Guedon, F. Haimet, W. Huber, J. van Hylckama-Vlieg, A. Jamet, C. Juste, G. Kaci, J. Knol, O. Lakhdari, S. Layec, K. Le Roux, E. Maguin, A. Merieux, R. Melo Minardi, C. M’Rini, J. Muller, R. Oozeer, J. Parkhill, P. Renault, M. Rescigno, N. Sanchez, S. Sunagawa, A. Torrejon, K. Turner, G. Vandemeulebrouck, E. Varela, Y. Winogradsky, G. Zeller, J. Weissenbach, S. D. Ehrlich, and P. Bork. 2011. Enterotypes of the human gut microbiome. Nature 473(7346):174-180.
Aylward, F. O., K. E. Burnum, J. J. Scott, G. Suen, S. G. Tringe, S. M. Adams, K. W. Barry, C. D. Nicora, P. D. Piehowski, S. O. Purvine, G. J. Starrett, L. A. Goodwin, R. D. Smith, M. S. Lipton, and C. R. Currie. 2012. Metagenomic and metaproteomic insights into bacterial communities in leaf-cutter ant fungus gardens. ISME Journal. Epub ahead of print.
Balter, M. 2012. Taking stock of the human microbiome and disease. Science 336:1246-1247.
Banfield, J. 2012. Session IV: Microbial community assembly and dynamics: From acidophilic biofilms to the premature infant gut. Paper presented at the Forum on Microbial Threats Workshop, The Social Biology of Microbial Communities, Washington, DC, March 7.
Bassler, B. L., and R. Losick. 2006. Bacterially speaking. Cell 125(2):237-246.
Belt, T. 1874. The naturalist in Nicaragua. London: E. Bumpus.
Ben-Jacob, E., Y. Aharonov, and Y. Shapira. 2004. Bacteria harnessing complexity. Biofilms 1:239-263.
Bonachela, J., C. Nadell, J. Xavier, and S. A. Levin. 2011. Universality in bacterial colonies. Journal of Statistical Physics 144:303-315.
Bonner, J. T. 2010. Brainless behavior: A myxomycete chooses a balanced diet. Proceedings of the National Academy of Sciences of the United States of America 107(12):5267-5268.
Brock, D. A., T. E. Douglas, D. C. Queller, and J. E. Strassmann. 2011. Primitive agriculture in a social amoeba. Nature 469(7330):393-396.
Brockhurst, M. A., M. E. Hochberg, T. Bell, and A. Buckling. 2006. Character displacement promotes cooperation in bacterial biofilms. Current Biology 16(20):2030-2034.
Brown, S. P. 2006. Cooperation: Integrating evolutionary and ecological perspectives. Current Biology 16(22):R960-R961.
Brown, S. P., S. A. West, S. P. Diggle, and A. S. Griffin. 2009. Social evolution in micro-organisms and a Trojan horse approach to medical intervention strategies. Philosophical Transactions of the Royal Society of London B: Biological Sciences 364(1533):3157-3168.
Brown, S. P., D. M. Cornforth, and N. Mideo. 2012. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends in Microbiology 20(7):336-342.
Bucci, V., S. Bradde, G. Biroli, and J. B. Xavier. 2012. Social interaction, noise and antibiotic-mediated switches in the intestinal microbiota. PLoS Computational Biology 8(4):e1002497.
Buchen, L. 2010. Microbiology: The new germ theory. Nature 468(7323):492-495.
Buffie, C. G., I. Jarchum, M. Equinda, L. Lipuma, A. Gobourne, A. Viale, C. Ubeda-Morant, J. Xavier, and E. G. Pamer. 2011. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to C. difficile–induced colitis. Infection and Immunity 80(1):63-73.
Caetano, L., M. Antunes, J. E. Davies, and B. B. Finlay. 2011. Mining bacterial small molecules. The Scientist, January 1, 26-30.
Caporaso, J. G., C. L. Lauber, E. K. Costello, D. Berg-Lyons, A. Gonzalez, J. Stombaugh, D. Knights, P. Gajer, J. Ravel, N. Fierer, J. I. Gordon, and R. Knight. 2011. Moving pictures of the human microbiome. Genome Biology 12(5):R50.
Cash, H. L., C. V. Whitham, C. L. Behrendt, and L. V. Hooper. 2006. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313(5790):1126-1130.
Cavanaugh, C. M., Z. P. McKiness, I. L. G. Newton, and F. J. Stewart. 2006. Marine chemosynthetic symbioses. Pp. 475-507 in The Prokaryotes, Vol. 1, edited by M. Dworkin et al. New York: Springer-Verlag. Available at http://link.springer-ny.com/link/service/books/10125.
Clatworthy, A. E., E. Pierson, and D. T. Hung. 2007. Targeting virulence: A new paradigm for antimicrobial therapy. Nature Chemical Biology 3:541-548.
Cordero, O. X. H. Wildschutte, B. Kirkup, S. Proehl, L. Ngo, F. Hussain, F. LeRoux, T. Mincer, and M. F. Polz. 2012. Ecological populations of bacteria act as socially cohesive units of antibiotic production and resistance. Science 337(6099):1228-1231.
Costello, E. K., K. Stagman, L. Dethlefsen, B. J. M. Bohannan, and D. A. Relman. 2012. The application of ecological theory toward and understanding of the human microbiome. Science 336:1255-1262.
Couzin, I. D., J. Krause, N. R. Franks, and S. A. Levin. 2005. Effective leadership and decision-making in animal groups on the move. Nature 433(7025):513-516.
Couzin-Frankel, J. 2010. Bacteria and asthma: Untangling the links. Science 330(6008):1168-1169.
Crespi, B. J. 2001. The evolution of social behavior in microorganisms. Trends in Ecology & Evolution 16(4):178-183.
Curtis, M. M., and V. Sperandio. 2011. A complex relationship: The interaction among symbiotic microbes, invading pathogens, and their mammalian host. Mucosal Immunology 4(2):133-138.
Czyz, A., K. Plata, and G. Wegrzyn. 2003. Stimulation of DNA repair as an evolutionary drive for bacterial luminescence. Luminescence 18(3):140-144.
Davis, R., and C. Joyce. 2011. The deep-sea finding that changed biology. NPR. http://www.npr.org/2011/12/05/142678239/the-deep-sea-find-that-changed-biology (accessed February 8, 2012).
Denef, V. J., L. H. Kalnejais, R. S. Mueller, P. Wilmes, B. J. Baker, B. C. Thomas, N. C. VerBerkmoes, R. L. Hettich, and J. F. Banfield. 2010a. Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proceedings of the National Academy of Sciences of the United States of America 107(6):2383-2390.
Denef, V. J., R. S. Mueller, and J. F. Banfield. 2010b. AMD biofilms: Using model communities to study microbial evolution and ecological complexity in nature. ISME Journal 4(5):599-610.
Desbrosses, G. J., and J. Stougaard. 2011. Root nodulation: A paradigm for how plant-microbe symbiosis influences host developmental pathways. Cell Host & Microbe 10(4):348-358.
Dethlefsen, L., and D. A. Relman. 2011. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences of the United States of America 108(Suppl 1):4554-4561.
Dethlefsen, L., M. McFall-Ngai, and D. A. Relman. 2007. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449(7164):811-818.
Dethlefsen, L., S. Huse, M. L. Sogin, and D. A. Relman. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biology 6(11):e280.
Diggle, S. P. 2010. Microbial communication and virulence: Lessons from evolutionary theory. Microbiology 156(Pt 12):3503-3512.
Diggle, S. P., A. Gardner, S. A. West, and A. S. Griffin. 2007a. Evolutionary theory of bacterial quorum sensing: When is a signal not a signal? Philosophical Transactions of the Royal Society B: Biological Sciences 362(1483):1241-1249.
Diggle, S. P., A. S. Griffin, G. S. Campbell, and S. A. West. 2007b. Cooperation and conflict in quorum-sensing bacterial populations. Nature 450(7168):411-414.
Diner, E. J., C. M. Beck, J. S. Webb, D. A. Low, and C. S. Hayes. 2012. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes & Development 26(5):515-525.
Dubilier, N., C. Bergin, and C. Lott. 2008. Symbiotic diversity in marine animals: The art of harnessing chemosynthesis. Nature Reviews Microbiology 6(10):725-740.
Dunn, A., and J. Handelsman. 2002. Toward an understanding of microbial communities through analysis of communication networks. Antonie van Leeuwenhoek 81:565-574.
Dusheck, J. 2002. It’s the ecology, stupid! Nature 418(6898):578-579.
Dussutour, A., T. Latty, M. Beekman, and S. J. Simpson. 2010. Amoeboid organism solves complex nutritional challenges. Proceedings of the National Academy of Sciences of the United States of America 107(10):4607-4611.
Emmert, E. A., A. K. Klimowicz, M. G. Thomas, and J. Handelsman. 2004. Genetics of zwittermicin A production by Bacillus cereus. Applied Environmental Microbiology 70(1):104-113.
Eppley, J. M., G. W. Tyson, W. M. Getz, and J. F. Banfield. 2007. Genetic exchange across a species boundary in the archaeal genus Ferroplasma. Genetics 177(1):407-416.
Ferguson, B. J., A. Indrasumunar, S. Hayashi, M. H. Lin, Y. H. Lin, D. E. Reid, and P. M. Gresshoff. 2010. Molecular analysis of legume nodule development and autoregulation. Journal of Integrative Plant Biology 52(1):61-76.
Flores, J. F., C. R. Fisher, S. L. Carney, B. N. Green, J. K. Freytag, S. W. Schaeffer, and W. E. Royer, Jr. 2005. Sulfide binding is mediated by zinc ions discovered in the crystal structure of a hydrothermal vent tubeworm hemoglobin. Proceedings of the National Academy of Sciences of the United States of America 102:2713-2718.
Folke, C., S. Carpenter, B. Walker, M. Scheffer, T. Elmqvist, L. Gunderson, and C. S. Holling. 2004. Regime shifts, resilience, and biodiversity in ecosystem management. Annual Review of Ecology, Evolution, and Systematics 335:557-581.
Fredricks, D. N., and D. A. Relman. 1996. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clinical Microbiology Reviews 9:18-33.
Fukuyama J., P. J. McMurdie, L. Dethlefsen, D. A. Relman, and S. Holmes. 2012. Comparisons of distance methods for combining covariates and abundances in microbiome studies. Pacific Symposium on Biocomputing 2012:213-224.
Fuqua, C., and E. P. Greenberg. 2002. Listening in on bacteria: Acyl-homoserine lactone signalling. Nature Reviews Molecular Cell Biology 3(9):685-695.
Fuqua, C., S. C. Winans, and E.P. Greenberg. 1994. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. Journal of Bacteriology 176(2):269-275.
Gilbert, G. S., J. L. Parke, M. K. Clayton, and J. Handelsman. 1993. Effects of an introduced bacterium on bacterial communities on roots. Ecology 74:840-854.
Gilbert, G. S., J. Handelsman, and J. L. Parke. 1994. Root camouflage and disease control. Phytopathology 84:222-225.
Gilbert, G. S., M. K. Clayton, J. Handelsman, and J. L. Parke. 1996. Use of cluster and discriminant analysese to compare, rhizosphere bacterial communities following biological perturbation. Microbial Ecology 32:123-147.
Gill, S. R., M. Pop, R. T. Deboy, P. B. Eckburg, P. J. Turnbaugh, B. S. Samuel, J. I. Gordon, D. A. Relman, C. M. Fraser-Liggett, and K. E. Nelson. 2006. Metagenomic analysis of the human distal gut microbiome. Science 312(5778):1355-1359.
Gonzalez, A., J. C. Clemente, A. Shade, J. L. Metcalf, S. Song, B. Prithiviraj, B.E. Palmer, and R. Knight. 2011. Our microbial selves: What ecology can teach us. EMBO Reports 12:775-784.
Hall-Stoodley, L., J. W. Costerton, and P. Stoodley. 2004. Bacterial biofilms: From the natural environment to infectious diseases. Nature Reviews Microbiology 2(2):95-108.
Hamilton, W. D. 1964. The genetical evolution of social behaviour. I-II. Journal of Theoretical Biology 7:1-52.
Han, H., J. Hemp, L. A. Pace, H. Ouyang, K. Ganesan, J. H. Roh, F. Daldal, S. R. Blanke, and R. B. Gennis. 2011a. Adaptation of aerobic respiration to low O2 environments. Proceedings of the National Academy of Sciences of the United States of America 108(34):14109-14114.
Han, S. W., M. Sriariyanun, S. W. Lee, M. Sharma, O. Bahar, Z. Bower, and P. C. Ronald. 2011b. Small protein-mediated quorum sensing in a gram-negative bacterium. PLoS One 6(12):e29192.
Handelsman, J. 2004. Metagenomics: Application of genomics to uncultured microorganisms. Microbiology and Molecular Biology Reviews 68(4):669-685.
———. 2007. Metagenomics and microbial communities. In Encyclopedia of life sciences. Chichester, UK: John Wiley & Sons.
———. 2009. Expanding the microbial universe: Metagenomics and microbial community dynamics. In Microbial evolution and co-adaptation. Washington, DC: The National Academies Press: A tribute to the life and scientific legacies of Joshua Lederburg: Workshop summary. Institute of Medicine.
Harmer, T. L., R. D. Rotjan, A. D. Nussbaumer, M. Bright, A. W. Ng, E. G. DeChaine, and C. M. Cavanaugh. 2008. Free-living tube worm endosymbionts found at deep-sea vents. Applied and Environmental Microbiology 74(12):3895-3898.
Hayes, C. S., S. K. Aoki, and D. A. Low. 2010. Bacterial contact-dependent delivery systems. Annual Review of Genetics 44:71-90.
He, H., L. A. Silo-Suh, J. Clardy, and J. Handelsman. 1994. Zwittermicin A, an antifungal and plant protection agent from Bacillus cereus. Tetrahedron Letters 35:2499-2502.
Holmes, E., J. V. Li, T. Athanasiou, H. Ashrafian, and J. K. Nicholson. 2011. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends in Microbiology 19(7):349-359.
Hooper, L. V., and J. I. Gordon. 2001. Commensal host-bacterial relationships in the gut. Science 292(5519):1115-1118.
Hooper, L. V., D. R. Littman, and A. J. Macpherson. 2010. Interactions between the microbiota and the immune system. Science 336(6086):1268-1273.
Hughes, D. T., and V. Sperandio. 2008. Inter-kingdom signalling: Communication between bacteria and their hosts. Nature Reviews Microbiology 6(2):111-120.
Human Microbiome Jumpstart Reference Strains Consortium. 2010. A catalog of reference genomes from the human microbiome. Science 328(5981):994-999.
Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207-214.
Ingham, C. J., and E. Ben Jacob. 2008. Swarming and complex pattern formation in Paenibacillus vortex studied by imaging and tracking cells. BMC Microbiology 8:36.
Ingham, C. J., O. Kalisman, A. Finkelshtein, and E. Ben-Jacob. 2011. Mutually facilitated dispersal between the nonmotile fungus Aspergillus fumigatus and the swarming bacterium Paenibacillus vortex. Proceedings of the National Academy of Sciences of the Untied States of America 108(49):19731-19736.
IOM (Institute of Medicine). 2006. Ending the war metaphor: The changing agenda for unraveling the host-microbe relationship. Washington DC: The National Academies Press.
———. 2009. Microbial adaptation and co-evolution: A tribute to the life and scientific legacies of Joshua Lederberg. Washington, DC: Workshop summary. The National Academies Press.
———. 2011. The science and applications of synthetic and systems biology: Workshop summary. Washington, DC: The National Academies Press.
Jernberg, C., S. Lofmark, C. Edlund, and J. K. Jansson. 2007. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME Journal 1:56-66.
Kaiser, D. 2006. A microbial genetic journey. Annual Review of Microbiology 60:1-25.
Kearns, D. B. 2010. A field guide to bacterial swarming motility. Nature Reviews Microbiology 8(9):634-644.
King, R. 2011. A bacterial platoon with fungal engineers. New York Times. http://www.nytimes.com/2011/11/29/science/a-bacterial-platoon-with-fungi-engineers.html (accessed July 25, 2012).
Koch, R. 1891. Uber bakteriologische Forschung Verhandlung des X Internationalen Medichinischen Congresses, Berlin, 1890, 1, 35. Berlin: August Hirschwald [in German]. Xth International Congress of Medicine, Berlin.
Kolter, R., and E. P. Greenberg. 2006. Microbial sciences: The superficial life of microbes. Nature 441(7091):300-302.
Kuzdzal-Fick, J. J., S. A. Fox, J. E. Strassmann, and D. C. Queller. 2011. High relatedness is necessary and sufficient to maintain multicellularity in Dictyostelium. Science 334(6062):1548-1551.
Lederberg, J. 2000. Infectious history. Science 288(5464):287-293.
Lee, Y. K., and S. K. Mazmanian. 2010. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330(6012):1768-1773.
Lepp, P. W., M. M. Brinig, C. C. Ouverney, K. Palm, G. C. Armitage, and D. A. Relman. 2004. Methanogenic archaea and human periodontal disease. Proceedings of the National Academy of Sciences of the United States of America 101(16):6176-6181.
Levin, S. 1998. Ecosystems and the biosphere as complex adaptive systems. Ecosystems 1:431-436. Levin, S. A. 2006. Fundamental questions in biology. PLoS Biology 4(9):e300.
Ley, R. E., D. A. Peterson, and J. I. Gordon. 2006a. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124(4):837-848.
Ley, R. E., P. J. Turnbaugh, S. Klein, and J. I. Gordon. 2006b. Microbial ecology: Human gut microbes associated with obesity. Nature 444(7122):1022-1023.
Little, A. E., and C. R. Currie. 2008. Black yeast symbionts compromise the efficiency of antibiotic defenses in fungus-growing ants. Ecology 89(5):1216-1222.
Little, A. E., C. J. Robinson, S. B. Peterson, K. F. Raffa, and J. Handelsman. 2008. Rules of engagement: Interspecies interactions that regulate microbial communities. Annual Review of Microbiology 62:375-401.
Littman, D. R., and E. G. Pamer. 2011. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host & Microbe 10(4):311-323.
Long, S. R. 2001. Genes and signals in the Rhizobium-legume symbiosis. Plant Physiology 125(1): 69-72.
Losick, R., and D. Kaiser. 1997. Why and how bacteria communicate. Scientific American 276(2): 68-73.
Lysenko, E. S., R. S. Lijek, S. P. Brown, and J. N. Weiser. 2010. Within-host competition drives selection for the capsule virulence determinant of streptococcus pneumoniae. Current Biology 20(13):1222-1226.
Maier, R. M., I. L. Pepper, and C. P. Gerba. 2000. Environmental microbiology. San Diego: Academic Press.
Maloy, S., J. Handelsman, and S. Singh. 2011. Dynamics of host-associated microbial communities. Microbe 6(1):21-25.
Marteyn, B., N. P. West, D. F. Browning, J. A. Cole, J. G. Shaw, F. Palm, J. Mounier, M. C. Prevost, P. Sansonetti, and C. M. Tang. 2010. Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465(7296):355-358.
Marwan, W. 2010. Amoeba-inspired network design. Science 327(5964):419-420.
Matson, E. G., X. Zhang, and J. R. Leadbetter. 2010. Selenium controls transcription of paralogous formate dehydrogenase genes in the termite gut acetogen, Treponema primitia. Environmental Microbiology 12:2245-2258.
May, R. M., S. A. Levin, and G. Sugihara. 2008. Complex systems: Ecology for bankers. Nature 451(7181):893-895.
McFall-Ngai, M., E. A. C. Heath-Heckman, A. A. Gillette, S. M. Peyer, and E. A. Harvie. 2012. The secret languages of coevolved symbioses: Insights from the Euprymna scolopes–Vibrio fischeri symbiosis. Seminars in Immunology 24:3-8.
McGinty, S. E., D. J. Rankin, and S. P. Brown. 2010. Horizontal gene transfer and the evolution of bacterial cooperation. Evolution 65(1):21-32.
Morlon, H. 2012. Microbial cooperative warfare. Science 337(6099):1184-1185.
Morowitz, M. J., V. J. Denef, E. K. Costello, B. C. Thomas, V. Poroyko, D. A. Relman, and J. F. Banfield. 2010. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proceedings of the National Academy of Sciences of the United States of America 108(3):1128-1133.
Mueller, R. S., V. J. Denef, L. H. Kalnejais, K. B. Suttle, B. C. Thomas, P. Wilmes, R. L. Smith, D. K. Nordstrom, R. B. McCleskey, M. B. Shah, N. C. Verberkmoes, R. L. Hettich, and J. F. Banfield. 2010. Ecological distribution and population physiology defined by proteomics in a natural microbial community. Molecular Systems Biology 6:374.
Nadell, C. D., J. B. Xavier, and K. R. Foster. 2009. The sociobiology of biofilms. FEMS Microbiology Review 33(1):206-224.
NASA (National Aeronautic and Atmospheric Administration). 2011. Mars science laboratory. http://www.jpl.nasa.gov/news/fact_sheets/mars-science-laboratory.pdf (accessed May 20, 2012).
Nee, S. 2004. More than meets the eye. Nature 429(6994):804-805.
Nicholson, J. K., E. Holmes, and I. D. Wilson. 2005. Gut microorganisms, mammalian metabolism and personalized health care. Nature Reviews Microbiology 3(5):431-438.
Nicholson, J. K., E. Holmens, J. Kinross, R. Burcelin, G.Gibson, W. Jia, and S. Pettersson. 2012. Host-gut microbiota metabolic interactions. Science 336:1262.
Njoroge, J., and V. Sperandio. 2009. Jamming bacterial communication: New approaches for the treatment of infectious diseases. EMBO Molecular Medicine 1(4):201-210.
Nogueira, T., D. J. Rankin, M. Touchon, F. Taddei, S. P. Brown, and E. P. Rocha. 2009. Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Current Biology 19(20):1683-1691.
NRC (National Research Council). 2007. Metagenomics: Revealing the secrets of our microbial planet (2007) by the committee on metagenomics: Challenges and functional applications. Washington, DC: The National Academies Press.
Nyholm, S. V., and M. J. McFall-Ngai. 2004. The winnowing: Establishing the squid-Vibrio symbiosis. Nature Reviews Microbiology 2(8):632-642.
Olsen, E. 2011. Illuminating the perils of pollution, nature’s way. New York Times. http://www.nytimes.com/2011/12/20/science/a-pollution-fight-powered-by-bioluminescent-sea-creatures.html (accessed June 14, 2012).
Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-740.
———. 2009. Mapping the tree of life: Progress and prospects. Microbiology and Molecular Biology Reviews 73(4):565-576.
Paine, R. T., M. J. Tegner, and E. A. Johnson. 1998. Compounded perturbations yield ecological surprises. Ecosystems 1:535-545.
Parniske, M. 2008. Arbuscular mycorrhiza: The mother of plant root endosymbioses. Nature Reviews Microbiology 6(10):763-775.
Parsek, M. R., and E. P. Greenberg. 2005. Sociomicrobiology: The connections between quorum sensing and biofilms. Trends in Microbiology 13(1):27-33.
Pennisi, E. 2011. Girth and the gut (bacteria). Science 332(6025):32-33.
———. 2012. Light in the deep. Science 335:1160-1163.
Pfeiffer, T., S. Schuster, and S. Bonhoeffer. 2001. Cooperation and competition in the evolution of ATP-producing pathways. Science 292(5516):504-507.
Plotkin, J. B., J. Dushoff, and S. A. Levin. 2002. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proceedings of the National Academy of Sciences of the Untied States of America 99(9):6263-6268.
Polz, M. F., J. A. Ott, M. Bright, and C. M. Cavanaugh. 2000. When bacteria hitch a ride: Associations between sufur-oxidizing bacteria and eukaryotes represent spectacular adaptations to environmental gradients. ASM News 66:531-539.
Poole, S. J., E. J. Diner, S. K. Aoki, B. A. Braaten, C. t’Kint de Roodenbeke, D. A. Low, and C. S. Hayes. 2011. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (RHS) systems. PLoS Genetics 7(8):e1002217.
Popa, R., A. R. Smith, J. Boone, and M. Fisk. 2012. Olivine-respiring bacteria isolated from the rock-ice interface in a lava-tube cave, a Mars analog environment. Astrobiology 12(1):9-18.
Qin, J., R. Li, J. Raes, M. Arumugam, K. S. Burgdorf, C. Manichanh, T. Nielsen, N. Pons, F. Levenez, T. Yamada, D. R. Mende, J. Li, J. Xu, S. Li, D. Li, J. Cao, B. Wang, H. Liang, H. Zheng, Y. Xie, J. Tap, P. Lepage, M. Bertalan, J. M. Batto, T. Hansen, D. Le Paslier, A. Linneberg, H. B. Nielsen, E. Pelletier, P. Renault, T. Sicheritz-Ponten, K. Turner, H. Zhu, C. Yu, M. Jian, Y. Zhou, Y. Li, X. Zhang, N. Qin, H. Yang, J. Wang, S. Brunak, J. Dore, F. Guarner, K. Kristiansen, O. Pedersen, J. Parkhill, J. Weissenbach, P. Bork, and S. D. Ehrlich. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59-65.
Rainey, P. B. 2007. Unity from conflict. Nature 446(7136):616.
Rainey, P. B., and B. Kerr. 2010. Cheats as first propagules: A new hypothesis for the evolution of individuality during the transition from single cells to multicellularity. Bioessays 32(10):872-880.
Rainey, P. B., and K. Rainey. 2003. Evolution of cooperation and conflict in experimental bacterial populations. Nature 425(6953):72-74.
Rasko, D. A., and V. Sperandio. 2010. Anti-virulence strategies to combat bacteria-mediated disease. Nature Reviews Drug Discovery 9(2):117-128.
Rasko, D. A., M. J. Rosovitz, G. S. Myers, E. F. Mongodin, W. F. Fricke, P. Gajer, J. Crabtree, M. Sebaihia, N. R. Thomson, R. Chaudhuri, I. R. Henderson, V. Sperandio, and J. Ravel. 2008. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. Journal of Bacteriology 190(20):6881-6893.
Ratcliff, W. C., R. F. Denison, M. Borrello, and M. Travisano. 2012. Experimental evolution of multicellularity. Proceedings of the National Academy of Sciences of the United States of America 109(5):1595-1600.
Reid, A., and M. Buckley. 2011. The rare biosphere. Washington, DC: American Academy of Microbiology.
Relman, D. A. 2005. Session II: Ecology of host-microbe interactions. Paper presented at the Forum on Microbial Threats Workshop, Ending the War Metaphor: The Changing Agenda for Unraveling the Host-Microbe Relationship, Washington, DC, Institute of Medicine, Forum on Microbial Threats, March 17.
———. 2012. Learning about who we are. Nature 486:194-195.
Riely, B. K., J. H. Mun, and J. M. Ane. 2006. Unravelling the molecular basis for symbiotic signal transduction in legumes. Molecular Plant Pathology 7(3):197-207.
Rivers, T. M. 1937. Viruses and Koch’s postulates. Journal of Bacteriology 33:1-12.
Robinson, C. J., B. J. Bohannan, and V. B. Young. 2010. From structure to function: The ecology of host-associated microbial communities. Microbiology and Molecular Biology Reviews 74(3):453-476.
Russian drill penetrates 14-million-year-old Antarctic lake. 2012. Wired. http://www.wired.com/wiredscience/2012/02/lake-vostok-drilled/ (accessed September 26, 2012).
Sandoz, K. M., S. M. Mitzimberg, and M. Schuster. 2007. Social cheating in Pseudomonas aeruginosa quorum sensing. Proceedings of the National Academy of Sciences of the United States of America 104(40):15876-15881.
Scheffer, M., J. Bascompte, W. A. Brock, V. Brovkin, S. R. Carpenter, V. Dakos, H. Held, E. H. van Nes, M. Rietkerk, and G. Sugihara. 2009. Early-warning signals for critical transitions. Nature 461(7260):53-59.
Scott, J. J., D. C. Oh, M. C. Yuceer, K. D. Klepzig, J. Clardy, and C. R. Currie. 2008. Bacterial protection of beetle-fungus mutualism. Science 322(5898):63.
Shade, A., and J. Handelsman. 2011. Beyond the Venn diagram: The hunt for a core microbiome. Environmental Microbiology 14(1):4-12.
Shank, E. A., and R. Kolter. 2009. New developments in microbial interspecies signaling. Current Opinion in Microbiology 12(2):205-214.
Sharon, I. M. J. Morowitz. B. C. Thomas, E. K. Costello, D. A. Relman, and J. F. Banfield. 2012. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Research [Epub ahead of print].
Slatkin, M. 1987. Gene flow and the geographic structure of natural populations. Science 236(4803): 787-792.
Smith, J. 2001. The social evolution of bacterial pathogenesis. Proceedings of the Royal Society of London B 268:61-69.
Stewart, F. J., I. L. G. Newton, and C. M. Cavanaugh. 2005. Chemosynthetic endosymbioses: Adaptations to oxic–anoxic interfaces. Trends in Microbiology 13(3):439-448.
Stewart, P. S., and M. J. Franklin. 2008. Physiological heterogeneity in biofilms. Nature Reviews Microbiology 6(3):199-210.
Stolper, D. A., N. P. Revsbech, and D. E. Canfield. 2010. Aerobic growth at nanomolar oxygen concentrations. Proceedings of the National Academy of Sciences of the United States of America 107(44):18755-18760.
Strassmann, J. 2012a. The language of sociomicrobiology: Report from a meeting for the Forum on Microbial Threats. http://sociobiology.wordpress.com/2012/03/08/273/ (accessed April 14, 2012).
———. 2012b. Session II: Evolution of cooperation and control of cheating in the social amoeba: Dictyostelium discoideum. Paper presented at the Forum on Microbial Threats Workshop, The Social Biology of Microbial Communities, Washington, DC, Institute of Medicine, Forum on Microbial Threats, March 6.
Suen, G., J. J. Scott, F. O. Aylward, S. M. Adams, S. G. Tringe, A. A. Pinto-Tomas, C. E. Foster, M. Pauly, P. J. Weimer, K. W. Barry, L. A. Goodwin, P. Bouffard, L. Li, J. Osterberger, T. T. Harkins, S. C. Slater, T. J. Donohue, and C. R. Currie. 2010. An insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genetics 6(9).
Suen, G., C. Teiling, L. Li, C. Holt, E. Abouheif, E. Bornberg-Bauer, P. Bouffard, E. J. Caldera, E. Cash, A. Cavanaugh, O. Denas, E. Elhaik, M. J. Fave, J. Gadau, J. D. Gibson, D. Graur, K. J. Grubbs, D. E. Hagen, T. T. Harkins, M. Helmkampf, H. Hu, B. R. Johnson, J. Kim, S. E. Marsh, J. A. Moeller, M. C. Munoz-Torres, M. C. Murphy, M. C. Naughton, S. Nigam, R. Overson, R. Rajakumar, J. T. Reese, J. J. Scott, C. R. Smith, S. Tao, N. D. Tsutsui, L. Viljakainen, L. Wissler, M. D. Yandell, F. Zimmer, J. Taylor, S. C. Slater, S. W. Clifton, W. C. Warren, C. G. Elsik, C. D. Smith, G. M. Weinstock, N. M. Gerardo, and C. R. Currie. 2011. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genetics 7(2):e1002007.
Tero, A., S. Takagi, T. Saigusa, K. Ito, D. P. Bebber, M. D. Fricker, K. Yumiki, R. Kobayashi, and T. Nakagaki. 2010. Rules for biologically inspired adaptive network design. Science 327 (5964):439-442.
Tremaroli V., and F. Bäckhed. 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489(7415):242-249.
Turnbaugh, P. J., R. E. Ley, M. Hamady, C. M. Fraser-Liggett, R. Knight, and J. I. Gordon. 2007. The Human Microbiome Project. Nature 449(7164):804-810.
Venter, J. C., K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A. Eisen, D. Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H. Knap, M. W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tillson, C. Pfannkoch, Y. H. Rogers, and H. O. Smith. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304(5667):66-74.
Walker, B., C. S. Holling, S. R. Carpenter, and A. Kinzig. 2004. Resilience, adaptability and transformability in social–ecological systems. Ecology and Society 9(2):1-9.
Warnecke, F., P. Luginbuhl, N. Ivanova, M. Ghassemian, T. H. Richardson, J. T. Stege, M. Cayouette, A. C. McHardy, G. Djordjevic, N. Aboushadi, R. Sorek, S. G. Tringe, M. Podar, H. G. Martin, V. Kunin, D. Dalevi, J. Madejska, E. Kirton, D. Platt, E. Szeto, A. Salamov, K. Barry, N. Mikhailova, N. C. Kyrpides, E. G. Matson, E. A. Ottesen, X. Zhang, M. Hernandez, C. Murillo, L. G. Acosta, I. Rigoutsos, G. Tamayo, B. D. Green, C. Chang, E. M. Rubin, E. J. Mathur, D. E. Robertson, P. Hugenholtz, and J. R. Leadbetter. 2007. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450(7169):560-565.
Waters, C. M., and B. L. Bassler. 2005. Quorum sensing: Cell-to-cell communication in bacteria. Annual Review of Cell and Developmental Biology 21:319-346.
Weber, N. A. 1966. Fungus-growing ants. Science 153:587-604.
West, S.A., and A. Gardner. 2010. Altruism, spite, and greenbeards. Science 327 (5971):1341-1344.
West, S. A., A. S. Griffin, A. Gardner, and S. P. Diggle. 2006. Social evolution theory for microorganisms. Nature Reviews Microbiology 4(8):597-607.
West, S. A., S. P. Diggle, A. Buckling, and A. L. Griffin. 2007a. The social lives of microbes. Annual Review of Ecology, Evolution, and Systematics 38:53-77.
West, S. A., A. S. Griffin, and A. Gardner. 2007b. Social semantics: Altruism, cooperation, mutualism, strong reciprocity and group selection. Journal of Evolutionary Biology 20(2):415-432.
Widder, E. A. 2010. Bioluminescence in the ocean: Origins of biological, chemical, and ecological diversity. Science 328(5979):704-708.
Wilmes, P., S. L. Simmons, V. J. Denef, and J. F. Banfield. 2009. The dynamic genetic repertoire of microbial communities. FEMS Microbiology Review 33(1):109-132.
Wu, D., P. Hugenholtz, K. Mavromatis, R. Pukall, E. Dalin, N. N. Ivanova, V. Kunin, L. Goodwin, M. Wu, B. J. Tindall, S. D. Hooper, A. Pati, A. Lykidis, S. Spring, I. J. Anderson, P. D’Haeseleer, A. Zemla, M. Singer, A. Lapidus, M. Nolan, A. Copeland, C. Han, F. Chen, J. F. Cheng, S. Lucas, C. Kerfeld, E. Lang, S. Gronow, P. Chain, D. Bruce, E. M. Rubin, N. C. Kyrpides, H. P. Klenk, and J. A. Eisen. 2009. A phylogeny-driven genomic encyclopaedia of bacteria and archaea. Nature 462(7276):1056-1060.
Wu, G. D., J. Chen, C. Hoffmann, K. Bittinger, Y. Y. Chen, S. A. Keilbaugh, M. Bewtra, D. Knights, W. A. Walters, R. Knight, R. Sinha, E. Gilroy, K. Gupta, R. Baldassano, L. Nessel, H. Li, F. D. Bushman, and J. D. Lewis. 2011. Linking long-term dietary patterns with gut microbial enterotypes. Science 334(6052):105-108.
Xavier, J. B. 2011. Social interaction in synthetic and natural microbial communities. Molecular & Systems Biology 7:483.
Xavier, J. B., W. Kim, and K. R. Foster. 2011. A molecular mechanism that stabilizes cooperative secretions in Pseudomonas aeruginosa. Molecular Microbiology 79(1):166-179.
Yong, E. 2012. Gut microbial “enterotypes” become less clear-cut. Nature. http://www.nature.com/news/gut-microbial-enterotypes-become-less-clear-cut-1.10276 (accessed September 26, 2012).
Zarubin, M., S. Belkin, M. Ionescu, and A. Genin. 2012. Bacterial bioluminescence as a lure for marine zooplankton and fish. Proceedings of the National Academy of Sciences of the United States of America 109(3):853-857.
Zhang, X., and J. R. Leadbetter. 2012. Evidence for cascades of perturbation and adaptation in the metabolic genes of higher termite gut symbionts. mBio (in press).
Zhang, X., E. G. Matson, and J. R. Leadbetter. 2011. Genes for selenium dependent and independent formate dehydrogenase in the gut microbial communities of three lower, wood-feeding termites and a wood-feeding roach. Environmental Microbiology 13:307-323.
Zhang, X.-X., and P. B. Rainey. In preparation. The asocial biology of pyoverdin-producing pseudomonas.
































































































