
4
The Role of the Brain in Mental Illness
The brain has not always been esteemed as the locus of that highly complex and contradictory entity, the human mind. In ancient Egypt, the soul and all mental functioning were thought to be located in the heart, although some scholars assigned this role to the liver. Hippocrates, in fourth-century (B.C.) Greece, identified the brain as “the interpreter of consciousness” and deduced that various forms of madness can arise from an unhealthy brain. He also held, though, that the brain was a gland and was most important as the site at which air, with its vital properties, was drawn into the body and entered the blood. Aristotle, for his part, considered the brain merely a cooling organ at the top of the body.
Early Christian thought located mental functions in the ventricles of the brain, along a progression from the front of the head toward the back: sensation and imagination in the anteri-or ventricle, reason and intellect in the third ventricle, and memory, as the most selective mental faculty, in the rearmost ventricle. In the eighteenth and nineteenth centuries, the popular science of phrenology sought to assign every conceivable trait of personality to its own specific location on the cerebral cortex, as if the mind were a physical entity like the brain. Most recently,
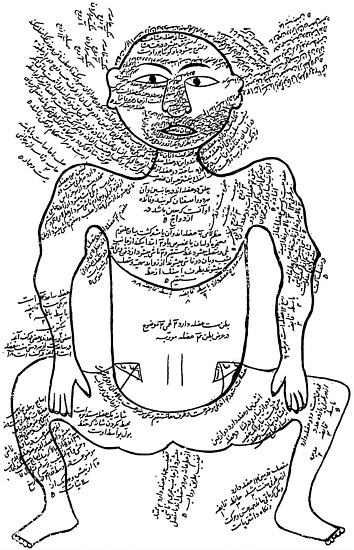
FIGURE 4.1. The medical basis of mental illness was under study as long ago as the fifteenth century, in Persia. This watercolor drawing from an anatomical textbook of 1488 reports beliefs about the correspondences between certain forms of mental illness and particular parts of the body. Source: Tashrih al-badan, 1488. The National Library of Medicine.
neuroscience has begun to uncover some of the intricate and less obvious relations between the mind and the brain, as investigative approaches from biochemistry, psychology, and genetics converge on an understanding of the physiology of mental illness.
In the 1980s, a new willingness to consider disorders of mental functioning as a proper concern of public health, and
thus an appropriate field for epidemiological study, led to the first attempt in this country to survey mental health on a national scale. The results, released in 1985 by the National Institute of Mental Health, were startling. Mental illnesses—syndromes and behavioral signs specific enough to be recognized, diagnosed, and, increasingly, treated effectively—are far from rare. On any given day, as extrapolated from the NIMH study population, 12 percent of adults in the United States—more than one in eight—suffer from a mental disorder. The toll is thus exceedingly high: in our collective lifetime, as many as 30 million Americans will at some point endure the disability, pain, and disruption of quality of life that come with a mental disorder. Many more will be touched, of course, through their concern for a family member, friend, or co-worker.
But the news is not all bad. With the efforts of numerous scientific disciplines—the combined approach that is a hallmark of neuroscience —the successes have begun to add up, at first slowly. Current work in the laboratory and in clinical practice builds on the basic research and clinical practice of the past, offering better prospects than before for alleviating a number of mental disorders. Multidisciplinary efforts have also made considerable progress toward untangling the factors at the root of some mental illnesses, bringing closer the goal of early detection or even of prevention to some degree, by interrupting a chain of factors.
MANIC-DEPRESSIVE ILLNESS
One of the better known success stories of this field has been the use of lithium to alleviate manic-depressive disorder. This bipolar illness, with its short phase of gregariousness, seemingly boundless energy, and ambition followed by deadening, slow-moving depression, tends to make its first appearance before the age of 35; the syndrome has been described (by the British psychoanalyst D. W. Winnicott) as “the mania being unreal and the depression being intolerable.” Little wonder that, according to public health studies, a manic-depressive patient without effective treatment could expect to spend almost half his or her adult life disabled, either in the hospital or at home grappling with or recovering from a cycle of illness. The use
of lithium in controlling this disorder was approved by the Food and Drug Administration in 1969; the benefits, in suffering averted and in recovered productivity, have been enormous. According to one estimate from NIMH, in a little more than 20 years, this use of lithium has saved the United States $39 billion.
Although the impact of lithium on manic-depressive illness is unmistakable, the biochemical means by which it works are not yet well understood. Family studies that examine inherited traits over several generations indicate at least some genetic basis for the illness: in sets of identical twins, if one twin suffers from the disorder, in almost 50 percent of cases the other twin does too. By contrast, the concordance rate in fraternal twins is only about 10 percent, the same as for other siblings. Adopted children have been found to suffer from the illness at a rate more in accord with that of their birth parents than of their adoptive parents, although environment, upbringing, and individual psychological makeup also clearly play a role in developing the illness (if this were not so, pairs of identical twins would tend to be affected in exactly the same way).
Lithium was first used for treating mania in 1948, in Australia, after psychiatrist John Cade observed that it had a calming effect on guinea pigs. The treatment for bipolar disorder nowadays is usually administered in the form of the common salt lithium carbonate; this compound appears to reduce the intensity of both the manic and the depressive phases, producing something of an evening out of mood. Just how the one compound could inhibit both extremes is unclear. One possibility is that as a salt, lithium may affect the passage of charged ions across cell membranes, which are controlled in part by sodium and calcium “gates” (as explained in Chapter 2 ).
OBSESSIVE-COMPULSIVE DISORDER
Obsessive-compulsive disorder, which had long challenged physicians with its almost limitless variety of forms, is a mental dysfunction that is only beginning to yield to biochemical therapy. Because the patient often is healthy and appears to function capably outside the area of the compulsion, the basis of obsessive-compulsive behavior was thought for some time
to be purely psychological, and the favored method of treatment was some form of psychotherapy. Physiological explanations seemed unable to account for the bewildering assortment of symptoms and behavioral tics brought about by this illness— from checking for the twentieth time that the door is locked before going out, to washing one's hands hundreds of times a day, to retracing a driving route yet again to make sure one had really not hit that pedestrian.
Certain drugs or injuries to the head have been known, however, to bring on compulsive behavior. Moreover, family studies show a significant rate of concordance for the illness in identical twins, regardless of whether they are brought up together or separately. Clearly, an explanation of obsessive-compulsive disorder must at least allow room for a physiological factor, traceable in part to the individual 's genetic makeup. And yet biology, even molecular biology, cannot give the full picture, because studies of twins have also shown that when both develop the illness, each experiences his or her own obsessions and enacts his or her own distinct compulsions. Most recently, investigators have been able to induce some of the symptoms of obsessive-compulsive disorder in experimental animals. With the identification of an animal model come strong leads for further study, such as selective mating of the animal to trace the genetic patterns in more detail and comparisons of different circumstances for raising the young. Following up these leads should help scientists gain a better idea of how and when a genetic predisposition toward the disorder may be amplified by the environment and to what degree.
On the clinical front, obsessive-compulsive disorder is yielding to treatment with a class of drugs that inhibits the uptake of the neurotransmitter serotonin. One of the most successful of these drugs has been clomipramine, which effectively blocks the uptake of serotonin at synapses; it also affects somewhat the uptake of another neurotransmitter, norepinephrine. In PET scans of patients given clomipramine, activity throughout the cortex and the basal ganglia (involved in the control of movement) is close to normal levels, rather than noticeably higher as is characteristic of obsessive-compulsive disorder. More tangibly, under clomipramine the patient's compulsive acts or rituals abate, sometimes by as much as half. However, while
medication can bring relief from the physical symptoms, it leaves untouched the obsessive state of mind that not only calls for the compulsive acts but also makes sense of them, attributing to them some rationale that lets them fit into the patient's world view. Thus both medication and some form of psychological counseling may be needed in the treatment of obsessive-compulsive disorder—truly a mind-brain disease.
PANIC DISORDER
Another malady to come under the scrutiny of neuroscience is panic disorder—a serious disability that has only recently been recognized as such, and whose physiological basis in the brain is just beginning to come to light. Estimated by some researchers to afflict as many as 1.5 million Americans, this disorder makes itself known by sporadic, inexplicable spells of panic: sudden intense feelings of fear and dread, choking or smothering, hyperventilation, irregular heartbeat, and extreme dizziness, together with a sense that one is about to die. Patients may suffer one panic attack a week or several attacks a day. Under this lash, some individuals grow afraid to leave their home; many are heavy users of emergency medical services, forming a large proportion of those mistakenly diagnosed with heart attacks, for example. In addition, the rate of attempted suicide among people with panic disorder is 20 times higher than among the population at large.
A first step toward understanding this illness was a clearcut definition that could be used consistently in studying the disorder from all angles. For example, if a particular compound or class of drugs was found to block panic anxiety specifically and consistently, this response could be used as a criterion for diagnosing the disorder. Certain antidepressant medications appeared indeed to work this way, as did several minor tranquilizers; this combination of effects set panic anxiety apart from less intense chronic anxiety and from transient feelings of panic, states that are unaffected by antidepressant drugs.
Also crucial to a definition, of course, was identifying what triggers panic attacks. The strong consensus of a number of research teams points not to external events or conditions but
to chemical factors in the blood. Panic attacks can be brought on by an excess of sodium lactate (like the lactate produced by muscles as a by-product of strenuous exercise), by substances that block the action of the “stress hormone” epinephrine, by at least one compound that facilitates the uptake of serotonin, and by inhalation of high concentrations of carbon dioxide.
At the cellular level, PET scans uncover a striking feature of panic disorder (see Plate 6 ). During an attack, the brain shows significant asymmetry between the left and right hemispheres, in a region that forms part of the limbic system (the “emotional brain”). In patients with panic disorder, the left side of this region shows lower blood volume and blood flow, and less use of oxygen—in short, less activity. This imbalance is not seen in normal patients, even in conditions of induced anxiety (for instance, when they are told by an experimenter to expect an electric shock), although the overall pattern for “normal” anxiety is similar. What distinguishes panic disorder is the asymmetry between the left and right hemispheres and the higher levels of activity overall.
With clear criteria for diagnosis in place, researchers can now pursue new lines of investigation into the origin of the disorder and its mechanism of action in the brain. As with obsessive-compulsive disorder, family studies show a pattern of inheritance—the sign of a genetic factor at work. For panic disorder, the rate of concordance among first-degree relatives is about 15 percent—not terribly high, but still many times the rate at which the illness is found in the general population. Among identical twins the concordance rate is 31 percent. The search is under way for more families touched by this disorder to provide a group of subjects for intensive genetic studies.
As with other important problems in biomedicine, investigating the nature of panic disorder has been fruitful for neuroscience in an unexpected way: it appears that the features of a panic attack can tell us something about the normal brain. In the late 1970s, several laboratory teams produced evidence of some interaction between the receptors for the neurotransmitter known as GABA (gamma-aminobutyric acid) and those for the benzodiazepines, a group of minor tranquilizers. The binding sites for the latter were found to lie close to the GABA receptor
sites, as did binding sites for the barbiturates, a class of sedative or hypnotic drugs. The discovery that makes sense of this arrangement is that both the benzodiazepines and the barbiturates work by means of the GABA receptors: they open the chloride ion channel so that negatively charged chloride ions enter the cell. The result is to hyperpolarize the cell membrane, thereby reducing the excitability of the neuron; this brings about a lower level of response to stimuli, which is experienced as a sedating or tranquilizing effect.
This mechanism in turn points back toward panic disorder, in which some patients apparently have a lower than usual sensitivity in their receptors to the benzodiazepines and an increased sensitivity to substances that block the action of those compounds. Animal models of panic disorder all show evidence of abnormality around the hippocampus, where the benzodiazepine-antagonist receptor sites are densely concentrated. Thus, from the perspective both of cell biology and of genetics, a general outline of panic disorder is being filled in with increasing detail.
So far we have considered several illnesses that interfere with the effective functioning of specific cells, usually at the level of chemical signal transmission. Not all mental disorders fall into this category, however. Another category, known as the dementias, arises from progressive degeneration of brain structures. Often, these illnesses pass through several stages of increasing severity.
ALZHEIMER'S DISEASE
Alzheimer's disease is the major form of dementia known in the United States today, affecting an estimated 4 million people. The progressive changes in mental functioning that it causes—disturbances of memory, a lessening ability to take in new information or to coordinate it with what is already known, and sometimes subtle changes in personality—may be slight at first but can soon become most distressing. The toll taken by Alzheimer's is clearly associated with advancing age: it afflicts less than 5 percent of the population below the age of 75, about 20 percent of those aged 75 to 84, and more than 40 percent of
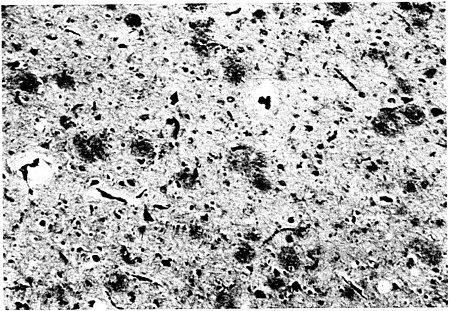
FIGURE 4.2. Tangles of neural fibers and “plaques” in the brain are two of the well-known signs of Alzheimer's disease. The plaques are lesions that contain a protein known as amyloid; their actual role in the course of the disease is still under debate. In this microscopic view of tissue from the cerebral cortex of a patient with Alzheimer's disease, the plaques appear as black knots against the gray background of the more healthy cells. Source: R. Cook-Deegan, taken from archives of the Department of Neurology, University of Colorado.
those aged 85 or older. This profile does not appear to be linked to an American life-style; study populations from various parts of the world have produced similar figures.
Although Alzheimer's disease is widely recognized, the formal criteria for diagnosing it are still under debate. In 1984, a working group of the National Institute of Neurological and Communicative Disorders and Stroke –Alzheimer's Disease and Related Disorders Association, headed by Guy McKhann, director of the Krieger Mind/Brain Institute at Johns Hopkins University, published a report concluding that to diagnose Alzheimer's disease with confidence, the physician should rely on specific signs in the brain. Chief among these were tangled knots of fine neural fibers and so-called “senile plaques,” actu-
ally microscopic lesions in the brain that contain a starchlike protein known as amyloid.
But McKhann is no longer satisfied with the conclusions of the report, which he now considers “rather myopic”—that is, too closely focused on the visible pathology of plaques and tangles. He believes instead that what is called Alzheimer's disease may not be a single entity at all and that using this simple name may in fact limit the thinking of some researchers, implying as it does that what is sought is the identity of a single disease-causing mechanism. An alternative explanation of the same clinical observations may be that several regions of the nervous system are involved, each one vulnerable to different factors. Combinations of these factors at work could give a clinical picture that is sufficiently similar, enough of the time, to hold together under the heading of Alzheimer's disease.
In the end stage of Alzheimer's, perhaps 5 to 10 years after the onset of disease, the brain shows some degeneration of the frontal and temporal lobes, particularly in the hippocampus, an area involved in short-term memory. Medical imaging reveals a significant loss of neurons, and an even greater loss of synapses—which may provide a better explanation for dementia than the tangles and plaques do. For that matter, the question of what causes dementia has any number of possible answers, most of them exceedingly difficult to disprove. There is even the possibility that such features as loss of short-term memory and reduced ability to learn are just a variant of normal aging. After all, the affected portion of the population forms a steady upward curve on epidemiological graphs. Perhaps everyone would eventually suffer dementia if they lived long enough. Since this theory is not provable, however, it is well to consider other possible causes of dementia. Among those suggested recently have been excitatory amino acids that exert a harmful effect, some form of infectious slow virus, the presence of toxic metals in the brain, vascular disease, timedelayed genetic programming, or some combination of these factors.
Excitatory amino acids can indeed cause severe deficits in the brain, including loss of memory. A dramatic illustration came recently from eastern Canada, where a number of people
became seriously ill after eating contaminated mussels. Several died; those who recovered sustained a major loss of memory. Brain scans from some of the victims showed an overwhelming loss of neurons from the hippocampus, similar to the pathology of the substantia nigra (a region involved in the control of movement) seen in Parkinson 's disease. The toxin from the mussels proved to be similar to glutamic acid, an excitatory neurotransmitter that occurs naturally in the central nervous system; it was also both highly potent and specific for those regions of the brain that process new memories. Such a substance is under study as a possible agent of Alzheimer's disease, as is the hypothesis that the disorder may be caused by a slow virus. Experimental testing of the virus theory would have to include a demonstrated ability to bring about recognizable “dementia” in laboratory animals solely by infection with the viral agent. Early claims of infection-induced dementia from one laboratory have not yet been replicated, despite attempts by the original group and a number of other research teams.
The theory that the presence of metal in the brain—specifically, aluminum—produces a toxic effect attracted lively interest when it was first advanced in the early 1970s. Evidence of aluminum in the neural tangles was plentiful and striking, and the theory circulated widely, to the point that the public began to wonder about the prudence of using aluminum cookware, for example. However, in the absence of evidence that further supports this theory or suggests how aluminum may work its harmful effects, the metal has lost some of its prominence as a possible cause of Alzheimer's. A current interpretation proposes instead that the aluminum in neural tangles may be a secondary effect, whose role (if any) in the development of this disease is unclear. Aluminum is, after all, an abundant element on earth, occurring naturally in water, soil, and much of our plant and animal food. Its presence in the brain may actually be the result of cells dying, rather than the cause; but this point is still under debate.
Vascular disease may figure in the onset of some dementias. The hippocampus is known to be an area of the brain that is highly susceptible to changes in blood flow; thus, some disruption of vascular function is being considered as at least a
factor in the deficits of memory and new learning that characterize Alzheimer's, if not the primary factor. The question that would remain, of course, is what gives rise to the vascular disease that causes these disabilities.
Genetic factors currently offer an exciting line of research into Alzheimer's disease. Recent success in locating the genetic region for another degenerative neurological disease, Huntington's chorea, has fostered the hope of a similar discovery for Alzheimer's. It appears that some families may have a gene on chromosome 21 that is associated with Alzheimer's disease. Another group of families among whom the disease develops later in life may have an affected gene on chromosome 19. Still other families in which Alzheimer's disease appears to be genetically based, however, show no evidence of an association with either chromosome. It seems likely that more than one gene can cause Alzheimer's disease, and the number of cases that are mainly genetic in origin remains uncertain.
A different sort of genetic lead comes from the observation that Down's syndrome, which is known to be caused by a mutation of chromosome 21, takes the form of mental retardation followed by dementia. The specific mutation is the existence of three chromosomes (or three copies of part of the chromosome) instead of the usual two, but the molecular biological mechanisms by which this surplus affects mental functioning are not yet understood.
An added reason to look at chromosome 21 is that the gene that codes for amyloid—the protein contained in senile plaques— is located there. Affected patients in a few families with Alzheimer's disease have shown a consistent defect in their amyloid gene. The normal aging human brain also contains amyloid, and the walls of blood vessels bear a similar material; some researchers have suggested therefore that the amyloid found in plaques is transported there from other, “healthy” sites. The mechanism for this process, which would involve a defect in the breakdown of amyloid from a larger precursor molecule, is under investigation by Dennis Selkoe, of the Harvard Medical School, and others.
Another explanation proposes that the low levels of acetylcholine observed in Alzheimer's disease could be an analogue to the low levels of another neurotransmitter, dopamine, ob-
served in Parkinson's disease. According to this rationale, methods of treatment that increase acetylcholine to normal levels should halt the degeneration and loss of neurons. (Acetylcholine can be increased by administering precursors of the neurotransmitter, which the patient's system then breaks down in a normal way, or by blocking the destruction of acetylcholine so that it is active longer.) In addition, several other neurotransmitters clearly suffer some deficit in Alzheimer's disease, so that multiple replacement of neurotransmitters, or the use of drugs that act like transmitters, are options for treatment that should be examined.
The administration of nerve growth factor has been a news-worthy step in laboratory attempts to prevent degeneration of the nerve fiber tracts known as the cholinergic pathways. In animals, nerve growth factor successfully protects the neurons of the hippocampus from dying, which should in turn preserve the workings of short-term memory. But applying such a therapy to humans raises several practical questions: how might a large molecule like nerve growth factor be made to cross the blood-brain barrier, and how would it then be directed to the specific region of the brain where it is needed? Two proposed solutions are a form of reservoir or pump implanted in the brain to give a continuous infusion of the compound, and the use of smaller molecules—analogues of nerve growth factor— that might pass more easily throughout the brain to the required sites. Preliminary reports of the use of nerve growth factor in a small trial, which have appeared recently, are exciting, but they have not yet been confirmed.
Meanwhile, stepping back to the perspective of epidemiology, researchers have posed a fruitful question by turning the usual query about dementia into its opposite. In other words, who does not get dementia? What are the factors that allow someone to reach an advanced age and continue to function successfully? For now, both this angle of investigation and the straightforward inquiry into agents of disease call for more and better understood study populations. In long-lived Americans it is especially difficult to sort out the factors that may have affected health over the course of, say, 80 years; however, studies in some regions of the world that have changed less rapidly than this country may be able to assemble populations
that show less variability—both among individuals and throughout the life-span.
The prospects for a thorough understanding of Alzheimer's disease are good. After an initial stage during which there appeared to be almost too much information, research from various branches of neuroscience should settle into a coherent picture that will be both satisfying in theory and applicable in clinical practice. Further work in this area will continue to be shaped by advances elsewhere in neuroscience and, at the same time, is well situated to help impel those advances.
Three areas of particular interest are the mechanisms at the cellular level that enable learning and memory (see Chapter 7 ), a clearer understanding of how Alzheimer's disease acts in the brain at each stage, and ways to protect specific regions of the nervous system from damage or perhaps even to reverse damage. Another advance in epidemiology would be to gather study populations whose health factors are thoroughly known and homogeneous, to address satisfactorily the two-sided question of what brings on dementia with increasing age and what permits increasing age without dementia. Many specialists in this area feel that the current position of research on Alzheimer's disease resembles that of work on Parkinson 's disease about 15 years ago, when the cause and some effective therapies were just about to come to light. Now, with the course of this disease broadly understood and the therapies continuing to improve, Parkinson 's is on its way to becoming a manageable disease. The mood is hopeful that Alzheimer's will follow suit.
AIDS-RELATED DEMENTIA
Not all dementias take a steady toll, like Alzheimer's, largely from among the elderly. One form that is spreading through a mixed-age population, on the basis of different risk factors altogether, is AIDS-related dementia (see Plate 2 ).
Human immunodeficiency virus (HIV) was established as the cause of AIDS in 1983. In 1985 it was recognized as a lentivirus—that is, a virus that primarily infects the macro-phages and leads to multisystem chronic disease, including encephalitis (inflammation of the brain). By 1986, it was clear that infection of the nervous system was to be a major feature
of AIDS. (Indeed, an HIV-positive but otherwise healthy individual who develops dementia can now be diagnosed, on the basis of that one symptom alone, as having AIDS.) What was not yet clear in 1986 was when the infections of the brain and nervous system were taking place, and whether they could be expected in most AIDS cases, given enough time. In other words, do infections of the brain and nervous system form an inherent stage in the course of AIDS in an individual, or should they be considered instead under the heading of the many opportunistic infections that attack the hobbled immune system in late-stage AIDS?
Questions of this degree of complexity call for the long-term cooperation of a consistent population of volunteers. In 1985 the Multicenter AIDS Cohort Study (MACS) recruited 480 homosexual and bisexual men into a cohort that included individuals who were HIV positive, some who seroconverted (that is, their blood began to test positive for HIV) at some point during the study, and others who remained HIV negative and served as control subjects. At the beginning, the study involved psychological testing and neurological examination; a smaller group underwent nuclear magnetic resonance imaging, electroencephalography, and a spinal tap at regular intervals, and they continue to volunteer for these painful and arduous tests as investigations proceed today with this same cohort.
The conclusions of this early component of the study were that infection of the nervous system occurs in most cases, and that it takes place quite early—perhaps even at the time of seroconversion, although the infected person may not suffer any symptoms for a considerable time. As to which infections are intrinsic to AIDS and which are opportunistic, the picture is still incomplete. Many diseases of the nervous system are known to be associated with the human immunodeficiency virus; some strike early in the course of infection and some later, when the patient's immune system is gravely suppressed. The diseases range from common to very rare, and they differ in their pathology: some destroy the myelin sheath that protects nerve fibers, others harm the axon or cause gradual loss of nerve cells. This broad variation suggests that the diseases work by means of different biological mechanisms and that a unifying explanation will not be easy to piece together.
A close look at AIDS-related dementia raises questions that apply to other diseases as well. Dementia is found at increasing rates throughout the course of AIDS infection: from about 3 percent at the time that AIDS is first diagnosed to 8–16 percent of HIV-positive outpatients at a hospital clinic, to more than 60 percent of AIDS patients at the time of death. In theory, the prevalence of AIDS-related dementia could be even higher; some specialists estimate that the pathology that underlies it is present in 90 percent of terminal AIDS cases. This observation appears to point toward some form of slow dementia, which incubates over a period of 5 to 10 years and eventually affects cognitive function. Most long-term studies so far suggest that even if the brain is infected early, neuropsychological processes continue unimpaired up to a late stage.
The physical signs of disease in the brain that can be gleaned from medical imaging are also quite subtle up to a late stage of the disease, as reported by Richard T. Johnson, director of the neurology department at Johns Hopkins Medical School. Close examination of the tissues of the brain reveals some frayed white matter, often containing macrophages, and occasionally an abnormally large cell that appears to contain viral antigen, but in general no striking appearance of disease. The pathology is also evident in the spinal cord; here, one finds many macrophages, some demyelination, and, curiously, at the same time some remyelination or resheathing of nerve fibers. The virus itself is thought to reside largely in the macrophages and in microglia, cells that develop from macrophages and whose function in the brain is unclear.
The features of AIDS-related dementia actually correlate best with a demyelinating disease, and, in fact, demyelination has been observed to some degree. The means by which it comes about, however, are still unclear. One promising line of study focuses on cytokines, which, like antibodies, are a product of the immune system. It appears that the infection may induce a particular cytokine to attack the myelin sheaths; this possibility is under examination. Another question is whether the viral proteins, those contained in the macrophages, for example, are toxic to cells or are harmful because they block the access of neurotransmitters to cells. Another possibility is transactivation, in which a gene from the AIDS virus acts as the “on”
switch for a genetic sequence outside itself—either in neural cells of the human host or in another virus infecting the patient at the same time. At present, opportunistic infections do not appear to be the agent of AIDS-related dementia; consequently, research is aimed toward deciphering the steps by which dementia could be produced by the AIDS virus itself. The major impact of the virus, according to scientific consensus, falls on T-4 cells, the “helper cells” of the immune system, and on monocytes, which later develop into macrophages. The monocyte is thought to be the essential cell that figures in the long incubation period between infection and the onset of disease; it may be that monocytes eventually enter the brain as infected macrophages, or they may infect macrophages already in the brain and cause them to release a substance injurious to the nerve cells.
The challenge of AIDS grows more pressing as the virus continues to spread and as more people infected several years ago reach later stages of disease. By current estimates, 1 to 2 million people in the United States are HIV positive; the figure worldwide is more than 5 million. One hopeful finding is that the neuropsychological damage, fairly early on, responds well to treatment with zidovudine, or AZT. That the dementia is reversible, even if only to a limited extent, bodes well for the future therapy of a debilitating disease. Meanwhile, it is crucial to widen the research perspective to include populations that have been neglected thus far and that bring their own factors to the epidemiological formula. For example, what are the special health risks associated with the AIDS virus in intravenous drug users or the ways the virus affects women differently from men or children differently from adults? Research in this field has taken shape quickly, moving in less than 10 years from the earliest observations of nervous system disease to focused molecular biological studies of the disease-causing virus.
DISORDERS OF MOOD AND MIND
To an observer of neuroscience, it is remarkable how permeable are the borders between the brain and the mind when both are considered as objects of study. There is a paradox
worth pointing out here, although it is well beyond the scope of this book to explore it: the more neuroscience devotes its attention to obtaining a thorough physiological account of what the brain is doing on a minute molecular level, the more the research tends to find itself in the realm of the psychological, offering information of an entirely different order than what had previously been understood about compulsive behavior, about panic, even about extremes of mood. Successful integration of the two forms of knowledge will provide the most help to patients of mental disorders, as well as, ultimately, yielding the most satisfying explanations. Such coordinated accounts of mental illness are being worked out for many disorders. Even chronic depression, which may be experienced entirely as a response of mood and mind to external events, has its biochemical side.
Observations from a variety of studies of depression have begun to fit together. For one thing, a handful of medications have proved to be effective in treating this disorder—so much so that they are now specifically called the antidepressants— and all of these seem to work by raising the brain's available levels of the same two neurotransmitters, serotonin and norepinephrine. For another thing, information about the drug reserpine has added to the base of knowledge. Once prescribed for high blood pressure, reserpine brought about depression as an unwanted side effect in about 15 percent of cases: in correcting hypertension, it reduced the levels of serotonin and norepinephrine available at the synaptic cleft. More recent studies seem to show that the receptors for at least one of these neurotransmitters, norepinephrine, grow less sensitive in depression; but whether this effect is instead of, or in addition to, a lower level of available neurotransmitter is not yet clear. Nor has it been established that the biochemical factors lead to or in any sense cause the drop in mood, energy, motivation, or enjoyment that characterizes depression. Several drugs that are effective in treating depression apparently act by influencing, in different ways, the levels of norepinephrine and serotonin available to receptors. The research into depression may be a good opportunity for obtaining some information on how a disorder that is experienced mainly in the mind can bring about its own chemical changes in the brain.
SCHIZOPHRENIA
The investigation of schizophrenia is a challenge of quite another kind. Here, there seem to be almost too many factors, and too many markers or manifestations. Schizophrenia is named for the abnormal division or split (“schizo”) between thought and emotion that characterizes the disorder in many cases. Striking about 1 percent of the population, schizophrenia can bring hallucinations, delusions, and profound withdrawal from normal social or family contact. An episode of schizophrenia can pass off spontaneously, but many individuals struggle for years in a snare of imagined scenes and voices, paranoia, and self-isolation, all of which reinforce one another. Patients suffering from schizophrenia number more than 1 million in the United States and occupy more than 100,000 hospital beds on any given day. As with so many mental disorders, the toll is unacceptably high; yet effective means of intervention or, ideally, of prediction and prevention have been largely out of reach while the illness itself is incompletely understood.
In the 1950s, a medication called chlorpromazine was found to reduce some of the more troubling symptoms, such as hallucinations and imagined voices. Although it did not address the delusional thinking that can come to shape the patient's whole world view in a case of chronic schizophrenia, its relief of overt symptoms promised to make life at least a little more manageable for many schizophrenic patients, and, in the 1960s, thousands were released from institutional care on the strength of that promise. But, although chlorpromazine offered relief from some symptoms, it could not cure the underlying disease.
It has remained for later decades to begin to unravel several lines of evidence about the nature of schizophrenia. Chlorpromazine, as well as several other drugs that relieve schizophrenic symptoms, works by blocking the brain's receptor sites for dopamine, particularly in the prefrontal cortex. More specifically, experiments at the National Institute of Mental Health and in Scandinavia in which schizophrenic patients were asked to perform cognitive tasks showed that these individuals have abnormally low activity in that area of the brain.
Such findings give rise to speculation about just what goes
on in the prefrontal cortex. It has been suggested that this area of the brain is concerned with the process of working memory, which underlies the ability to regulate behavior by ideas and concepts. For instance, Patricia Goldman-Rakic, professor of neuroscience at Yale University School of Medicine, has found that rhesus monkeys with lesions in the prefrontal cortex are unable to hold information “in mind” for even a few seconds; their behavior is erratic, distracted, and meaninglessly repetitive. They are also unable to follow a fast-moving target with their eyes, although their ability to follow a slow-moving target is unimpaired. Altogether, these deficits mimic some of the signs of schizophrenia. They also point to a defect in information processing: the loss of a capacity to predict where the target will be in the next fraction of a second, or the loss of ability to predict events in general.
It may appear unreasonable at first to single out one specific area of the brain to account for a disorder as global and wide ranging in its symptoms as schizophrenia. (Indeed, this area may well prove to be only one region out of many that play a role.) Still, its involvement seems plausible when we consider that the prefrontal cortex occupies one-quarter of the entire cerebral cortex and interconnects with many other regions and with the limbic system—including some areas that show evidence for related dysfunction in cases of schizophrenia. Disorders of the senses (hallucinations) and sudden urgent feelings of anger or fear can all be traced, at least in neurophysiological theory, to misfiring synapses in one area or another of the sensory association cortex or of the limbic system, which controls mood. Another point in favor of this line of thinking is that the frontal lobe contains the highest concentration of dopamine fibers in the cerebrum, and the specific dopamine receptor that is the primary site of action of antipsychotic drugs is known to reside in this area. Thus, a biological explanation for schizophrenia is beginning to evolve. It remains true, however, that the particulars of any case, the form of the hallucinations and delusions in an individual 's mind, can only be approached in psychological terms. Similarly, although dopamine blockers may relieve the brain somewhat of symptoms, psychosocial therapy can dramatically help the patient in day-to-
day living, by offering the chance to improve coping skills: how to remember to take medications reliably, how to manage in stressful situations, how to interact socially with others.
Research on schizophrenia has also brought neuroscientists a step closer to an understanding of how the prefrontal cortex functions in normal circumstances. Current scientific thinking is that the region guides behavior by its representations, or working memory, of stimuli rather than by direct perception of the stimuli themselves. (Hence the inability in schizophrenia to predict the location of a moving target or to foresee an expectable outcome in given circumstances, for instance, in social interactions.) In a general sense it can be said that this area of the brain—which is greatly enlarged in humans, relative to our near-cousins, the other primates—provides the physiological basis for abstract thought and the ability to plan.
Vigorous investigation into the many forms and factors of mental illness is thus a central task for neuroscience. Observations from clinical practice, laboratory study of the mechanisms of disease and the ability of chemical compounds to harm or help, and genetic analysis of family history all build on one another, offering the prospect of more efficacious treatments and of theoretical accounts that grow more solid as new details are filled in. While neuroscience looks more closely at the brain, it continues to enlarge our options for treating the mind.
ACKNOWLEDGMENTS
Chapter 4 is based on presentations by Patricia Goldman-Rakic, Richard Johnson, Lewis Judd, and Guy McKhann.





















