
5
From Chemistry to Circuitry
In an uncanny way, the images of everyday speech can sometimes anticipate formal scientific knowledge. Who at one time or another has not compared an idea to an electrical spark or spoken of the “chemistry” of a particular mood? The brain does indeed convey its signals by means of electricity and chemical compounds; so much is well known. But the finer details of how it actually manages these transmissions, and at prodigious speeds—sometimes firing several hundred nerve impulses in a second —still make for fascinating inquiry.
The scientific understanding of electricity in the nervous system has come a long way since the 1700s, when Luigi Galvani of Bologna noticed that disembodied frog legs hanging on copper hooks from an iron balcony would occasionally twitch, as if still animated. On the basis of this observation and subsequent experiments, he concluded that the motive force in the nervous system was electricity rather than the traditionally conceived “animal spirits.” Galvani could scarcely have imagined the bacterial robots, electron diffraction microscopes, and positron emission tomography devices that would come to replace the accoutrements of an eighteenth-century Italian kitchen as the proper equipment for scientific observation. And he would be
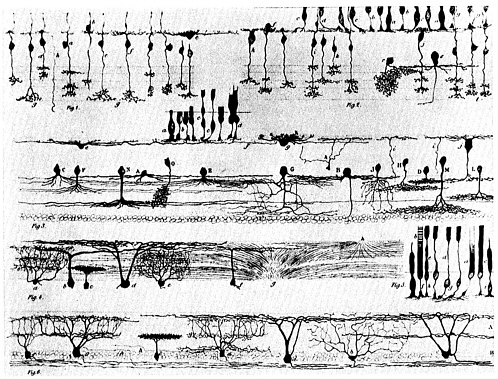
FIGURE 5.1. Pen-and-ink drawings were the first scientific illustrations to depict the actual structure of individual nerve cells. Several types of neurons are represented here; each has a recognizably thicker portion that is the cell body, and most have a single axon that projects like a thin wire. The number and the branching patterns of each cell 's dendrites show more variation. Source: D. Santiago Ramón y Cajal, 1889. Manuel de Histología Normal y Técnica Micrográphia. The National Library of Medicine.
equally astonished by today's cogent, nearly molecule-by-molecule explanation of just what was twitching in those frog legs.
CHEMICALS AS SIGNALS
Unlike electricity, the brain's chemical messengers, the neurotransmitters, are difficult to identify from first observations. The action of electricity could be confirmed or disproved by home-style tests; but the activity and nature of a chemical compound that may be involved in the brain's signaling system demand more rigorous examination. The compound must meet a half dozen specific criteria to be considered a neurotransmitter —as opposed to, say, a “second messenger” in the brain, which broadcasts signals within a cell rather than conveying a signal from one cell to another. (Distinctions such as this, which may seem overly fine at first, have a way of turning out later to be crucial for understanding new, otherwise inexplicable data.)
To be recognized as a neurotransmitter, a chemical compound must satisfy six conditions: It must be (1) synthesized in the neuron, (2) stored there, and (3) released in sufficient quantity to bring about some physical effect; (4) when administered experimentally, the compound must demonstrate the same effect that it brings about in living tissue; and (5) there must be receptor sites specific to this compound on the postsynaptic membrane, as well as (6) a means for shutting off its effect, either by causing its swift decomposition or by reuptake, absorbing it back into the cell. Of course, before any of these items on the checklist come into question, the compound must somehow be detectable in the human brain—not always an easy matter, in view of the minute quantities involved.
One of the first substances to pass all the tests was acetylcholine (ACh). Widespread throughout the central nervous system, ACh usually has an excitatory function, but it can also
be inhibitory, depending on prevailing conditions at the receptor site. Because ACh acts briskly and is subject to prompt breakdown in the synaptic cleft, it is well suited as the transmitter for motor neurons. Acetylcholine also acts in the autonomic nervous system, where it is responsible for such functions as contracting the pupil of the eye, slowing heartbeat, and stimulating salivation and digestion.
Two poisons that are well known to readers of mystery novels work their deadly effects by blocking the action of ACh. Curare, a plant extract used by South American Indians to treat their arrows for hunting, rapidly causes paralysis; botulin, a toxin produced by bacteria in improperly canned foods, paralyzes the muscles that control breathing and thereby causes suffocation. In clinical practice, drugs that block the action of ACh are useful in many ways. Short-lived ACh inhibitors are given in eye drops to dilate the pupil for ophthalmic examination. More enduring forms, such as atropine, reduce the secretion of saliva and bronchial fluid, which is helpful for anesthesia; hyoscine, or scopolamine, another related compound, is sometimes used as a sedative but does have the side effect of causing a very dry mouth. Conversely, drugs that inhibit the chemical breakdown of ACh and thus extend its action in the synaptic cleft are at least temporarily effective against myasthenia gravis; this is a crippling disease in which the body's own immune system attacks the receptor sites for ACh on the skeletal muscles. The muscles gradually weaken as they receive fewer synaptic transmissions, but drugs such as eserine can effectively increase the amount of ACh available to the remaining receptor sites.
In the cerebral cortex, ACh is thought to play a role in storing short-term memories. The hippocampus, for example, has dense areas of receptor sites for ACh, and their degeneration is one of the biological signs of Alzheimer's disease (see Chapter 4 ). Thus far, attempts to reverse symptoms by giving drugs that mimic or enhance the action of ACh have not been successful.
Along with ACh, another transmitter that is widely encountered throughout the nervous system is norepinephrine (also known as noradrenaline). In the central nervous system, the function of norepinephrine usually complements that of ACh; thus nor-
epinephrine acts in the general direction of arousal, and ACh tends toward restorative functions. Norepinephrine dilates the pupil of the eye, strengthens and speeds the heartbeat, and inhibits processes of digestion, all under the heading of what has been called the “fight or flight” response; it also stimulates the adrenal glands to release epinephrine and the liver to release large quantities of glucose, which make more energy available to the muscles for action. Several drugs that work by means of the norepinephrine system are useful in asthma; these are known as the beta-agonists, because they are targeted to a specific group of “beta” receptors in the bronchial muscles, where they relieve constriction.
In some instances, norepinephrine may function not as a transmitter but as a neuromodulator, promoting or blocking the action of some other transmitter at the synapse. Experimental evidence on this point is still coming in; if the hypothesis holds up, it would add a new detail to an already crowded picture and could open up other lines of investigation as well.
The neurons in the brain that contain norepinephrine cluster in a small region of the brainstem; their axons project to the hypothalamus, the cerebellum, and even the forebrain, a good 10 to 15 centimeters away. Norepinephrine is associated not only with alertness and arousal but also with the dreaming phase of sleep and, by way of the hypothalamus and the limbic system, with the regulation of mood. For instance, a number of studies point to depleted levels of norepinephrine at brain synapses, or a reduced ability of receptors to use it, as a factor in depression. Not that this amounts to a scientific formula that a normal brain minus some amount of norepinephrine equals a depressed mind; such formulas are far too coarse—particularly in an area like the basis of mood, where any number of elements may interact. Moreover, some of the factors that are undoubtedly important for mood are unquantifiable, invisible, and perhaps irreproducible for laboratory study. One point of wide consensus, however, is that depression can be helped by two classes of drugs: one class blocks an enzyme that would normally break down norepinephrine in the synaptic cleft, and the other slows the reuptake of norepinephrine into the presynaptic cell.
The neurotransmitter serotonin is a powerful constrictor of
blood flow and an inhibitor of some sensations of pain; in recent years, its intriguing variety of effects on our mental life have also come under study. Serotonin is of great importance in regulating sleep; the old folk remedy for sleeplessness, a glass of warm milk before bedtime, may work because of the presence in milk of tryptophan, an amino acid that the brain uses to make serotonin.
This transmitter can affect many parts of the brain at once through the long-reaching axons of serotonergic (serotonin-using) neurons, which underlie the transmitter's role in such global phenomena as sleep and mood. The drugs that raise levels of available norepinephrine to alleviate depression also work on serotonin, by the same mechanisms. (Interestingly, the axons that carry serotonin are not myelinated. Without the electrical insulation afforded by the myelin sheath, impulses travel at less than the lightning speed achieved by, say, signals to the motor neurons, but this seems appropriate to the more global and subjective areas of life regulated by serotonin.) The remarkable effects of lysergic acid diethylamide, or LSD, in even the tiniest quantities, are based on its strong chemical resemblance to serotonin; it is as if a full system of preexisting receptor sites lies ready for the drug's use.
One further aspect of this versatile transmitter is that serotonin is featured in biochemical accounts of “sensitization,” the enhanced response to a stimulus as a result of training. Scientists have studied sensitization in extraordinary detail in simple animals such as the marine snail as a model for more complex processes of learning in the human brain (see Chapter 7 ).
Dopamine is chemically similar to serotonin and norepinephrine, and it overlaps with them in several biological functions. Formed, like serotonin, from an amino acid, dopamine is actually a precursor to norepinephrine—the same compound except for one different chemical bond—and a wide-ranging neurotransmitter in its own right. In many systems, dopamine acts as an “off” switch: it halts the release of prolactin (which is responsible for the function of the mammary glands), inhibits some cells of the olfactory tract, and also shuts off some of the action of autonomic nerve cells (although this function is not well understood).
Elsewhere in the nervous system, dopamine is important
for the control of movement; the degeneration of dopamine-using neurons in a portion of the midbrain leads to Parkinson's disease. A patient with this condition finds it difficult to initiate movement and also to stop, and to manage associated actions such as swinging the arms while walking. A slow tremor of the hands and head, present when the patient is at rest but not during movement, is probably what gave the illness its original name of “the shaking palsy.” Although the progress of the disease cannot be halted, the symptoms of Parkinson's disease can be effectively controlled in most patients by treatment with L-dopa. (This is not the actual transmitter itself but a precursor, a molecule that has the ability to pass through the blood-brain barrier and from which the brain can form dopamine.)
Because dopaminergic neurons are also well distributed in the limbic system, we would expect some role for this neurotransmitter in the creation of mood—and, indeed, evidence to this effect is accumulating. Most striking are the signs that a relative increase in dopamine activity in the frontal cortex may provide the biochemical basis for schizophrenia.
Evidence for the role of dopamine has come from several directions. Chlorpromazine, a drug first used widely in mental hospitals in the 1950s and 1960s to reduce the overt symptoms of schizophrenia, was found in the 1970s to block the action of dopamine at receptor sites. Then, too, some of the patients who were given the medication to help control their hallucinations and thought disorders developed, over the long term, a tremor and other physical symptoms that resembled those of Parkinson's disease. A third clue may be the ability of amphetamines, when taken in sufficient quantity, to bring on disturbances of the mind much like schizophrenia; it is known that amphetamines, or “uppers,” increase the levels of dopamine available in the brain.
To be sure, an illness as complex as schizophrenia cannot be reduced to a simple chemical explanation such as “excess of dopamine.” The question remains open whether the schizophrenic brain suffers from too much dopamine, too many dopamine receptors, a standard quantity of receptors with abnormally high sensitivity, or some other dysfunction entirely—to say nothing of the important genetic, social, and psychological factors also under study. Science has a long way to go yet in
explaining schizophrenia, and it seems likely that a complete account of the functions of dopamine in the brain may also take some time to fall into place.
Another major neurotransmitter derived from an amino acid is gamma-aminobutyric acid, or GABA. Unlike dopamine or serotonin, which have diverse roles, GABA consistently acts as an “off” signal; the cerebellum, retina, and spinal cord all use this transmitter to inhibit signals, as do many other parts of the brain and nervous system. GABA's inhibitory effect comes about in the following way: the transmitter opens a channel in the membrane through which negatively charged chloride ions can enter the cell. This influx hyperpolarizes the cell and makes it less likely to be excited by incoming stimuli.
GABA receptor sites show some tendency to bind barbiturates and the “minor tranquilizers,” the benzodiazepines. Curiously, the presence of GABA in low concentrations enhances the binding of benzodiazepines to receptor sites. This pattern indicates that GABA and the benzodiazepines cannot be competing for exactly the same sites. Instead, an array of recent studies have yielded the view that the GABA receptor site is in fact a multifunctional set of proteins that contain the chloride ion channel and distinct subsites for binding of benzodiazepines, other tranquilizers such as barbiturates, and GABA itself.
RECEPTORS PLAY AN ACTIVE PART
The neurotransmitters discussed thus far are just a few of the ones that have been known for a relatively long time (at least a decade or two). But likely additions to the list number at least 40, and new candidates continue to appear. Even from this brief survey, however, a curious fact emerges: a single neurotransmitter can be responsible for many different effects.
How can a chemical compound, unchanging in various parts of the body or brain, produce diverse results? The answer lies on the other side of the synapse in the receptor sites, which do change their properties to a surprising degree and in the end determine what effect a neurotransmitter has on a cell.
For example, it is by means of different receptors that norepinephrine, which supplies blood vessels in both skeletal muscle and skin, can cause constriction in the vessels of the skin and
dilation in those of muscle. As another example, ACh's effect of constricting skeletal muscle cells can be blocked by curare but not by atropine, although the two alkaloids are in some ways similar; in smooth muscles of the intestine, however, the action of ACh can be blocked by atropine but is unaffected by curare. Clearly, the receptor molecules in the two locations have somewhat different shapes, despite their both being devised to respond to ACh.
Acetylcholine also serves to illustrate another level of complexity in the brain's signaling system. In muscle cells, it acts directly, moving positively charged sodium ions into the cell and depolarizing it. In the brain, however, this transmitter works in tandem with a “second messenger,” which conveys the signal inside the cell, sometimes amplifying it greatly in the process. (In this sense, the neurotransmitter that traverses the synapse is the “first messenger,” although this term is rarely used.)
To date, researchers have identified only two second-messenger systems —which is perhaps just as well for those who wish to understand neurophysiology, because the other kinds of messenger systems identified in the brain are proliferating so rapidly. One system uses the small molecule cyclic adenosine monophosphate (cyclic AMP) as its second messenger; the other system uses the even smaller calcium ion (Ca2+) and two compounds made up partly from the cell membrane itself: inositol triphosphate (IP3) and diacylglycerol (DG).
Both second-messenger systems have the same goal: to bring about a change in the shape of proteins inside the cell, which either enables or halts such activities as contraction (in a muscle cell, for example) or secretion (in a glandular cell). And both second-messenger systems begin in the same way: the receptor site at the cell membrane surface activates one of the so-called G-proteins (which require guanosine triphosphate to carry out their function). The G-protein in turn activates an enzyme within the membrane, causing a chemical reaction that assembles the second-messenger molecules from precursor molecules available inside the cell. Here the two systems diverge: in one, the enzyme adenylate cyclase removes two phosphate groups from adenosine triphosphate (incidentally releasing energy to the cell) and converts it into cyclic AMP. In the other
system, the enzyme phospholipase C cleaves a large lipid molecule into two parts, DG and IP3 (again, releasing energy in the process).
The second messenger, in the form of many thousands of newly created molecules, is thus in a good position to amplify the signal of a neurotransmitter and to broadcast its message speedily throughout the cell. The second messenger may act directly—by simply binding to a cellular protein and thus changing its structure—or indirectly, by activating another enzyme called a protein kinase, which changes a protein's structure and electrical charge by adding a phosphate group to it. In either case, the result is a change in the shape of the protein and consequently a shift in the activities of the cell. Because of the great potential for amplifying the signal at each step (from first messenger to enzyme to cyclic AMP to protein kinase to phosphorylated protein to switched-on function), just a few molecules of a neurotransmitter can cause target cells to produce several million molecules of a reaction substance in far less time than it takes to read about it.
Of course, signals are propagated even more rapidly when the neurotransmitter works without a second messenger. In contrast, the second-messenger routes of transmission generally lead to effects that are longer lasting.
THE BRAIN'S OWN PAINKILLERS
The identification and study of neurotransmitters is still a very young field that tends to grow in spurts—for example, in the 1970s, when researchers seeking to understand the action of opium-based drugs discovered the existence of an entire system of “natural painkillers ” produced by the body itself. These neurotransmitters, known as endorphins, for “endogenous morphine-like substances,” reduce the body's sensation of pain and, according to some studies, may also play a role in managing stress. Their release, along with epinephrine, in moments of emergency, shock, or injury could help to explain the apparently unnatural feelings of calm and obliviousness to discomfort that are often reported on such occasions.
For investigators on the trail of these natural painkillers, the first clue was the well-known effectiveness of opiates, which
are derived from the juice of the opium poppy. Morphine, in particular, has been used for centuries to relieve pain and bring about a sense of well-being. Heroin, a more concentrated form of poppy juice, produces more intense euphoria and is, in addition, quickly addictive—another clue that these drugs must be working by means of some direct pathway in the brain. Researchers devised a way to test this hypothesis by treating rats with opium derivatives that had been “labeled” with radioactive isotopes. The resultant images showed where the opiates had settled in the brain—in other words, they amounted to a map of the brain's opiate receptors. The pattern of these receptors, which are concentrated in the spinal cord, the brainstem, and the limbic system of the cerebrum, is found consistently in rats, humans, and other mammals—indeed, throughout the vertebrates. Clearly, a pattern so widely conserved across the evolutionary scale must have value for the animal's survival.
The next question was why the vertebrate brain should come equipped with the ability to “get high” on morphine or heroin. Or, to reframe the question, were the opiates gaining ready access by fitting into receptor sites that were there to serve some other built-in function? If so, the opiates must all be similar in structure—which they are—and they must also resemble the “original” endogenous transmitters well enough to mislead the receptor sites. By this point, armed with a rough sketch of the transmitters ' structure and with clues as to their likely function, researchers had a good idea of what they were looking for. In the mid-1970s a handful of natural opiates, collectively termed “endorphins,” were identified; they included a group called enkephalins and another group headed by beta-endorphin.
From the perspective of a couple of decades, the endorphin story reads like one of the more straightforward detective novels, in which every clue is found at its proper time and the inquiry moves steadily forward. What was most striking at the time, however, was that the mapping of the receptors, a task that might be thought purely mechanical and unimportant, was what gave this investigation its major impetus. This approach, which uses the mapping of receptor sites as a first step, has proved valuable in other investigations as well, such
as studies of the innate function of receptor sites in the brain that bind delta-9 THC (tetrahydrocannabinol), the major psychoactive ingredient in marijuana.
FOCUSING IN ON RECEPTOR SITES
The close study of receptor sites is a lively subfield in itself. Previously unknown techniques of imaging and magnification can go beyond displaying the location of sites in the brain as a whole, down to the molecule-by-molecule layout of a single receptor. And the structures disclosed with these techniques are fascinating. No longer regarded as the passive “lock” of a “lock-and-key” mechanism, the receptors appear to work from a few simple elements and to achieve a wide range of effects.
Robert Lefkowitz and his colleagues at the Howard Hughes Medical Institute at Duke University have been looking closely at epinephrine and norepinephrine receptor sites for several years—practically the lifetime of this field so far. The adrenergic (epinephrine-using) receptors make a good model for studying a more general class of receptors, those that work through the G-proteins and bring about the production of second messengers inside the cell. Adrenergic receptors are found in most mammalian tissues and appear to fall into four types, called alpha-1, alpha-2, beta-1, and beta-2. They are associated with the two second-messenger systems discussed earlier: cyclic AMP and DG–IP3. Specifically, the beta receptors apparently increase the rate of production of cyclic AMP, and the alpha-2 receptors decrease it.
This was roughly the extent of what was known until about 1986. Then Lefkowitz and others succeeded in reading the full genetic sequence of the beta-2 receptor, which allowed them to clone the gene—in effect, to create thousands of copies of the beta-2 receptor in their laboratory.
What they learned in the process was astonishing: a single receptor site spans the cell membrane, like a built-in tunnel, no fewer than seven times. The arrangement consists of seven recurring clusters of 20 to 25 amino acids, each crossing the membrane and all held together by loops of amino acids within the cell and just outside the membrane. This pattern appears to hold good for the other kinds of receptors as well. In
a comparison of any two receptors, 40 to 50 percent of the sequence is identical—a high degree of conservation and an indication of how effective this structure must be.
As well as elucidating the fine structure of the beta-2 receptor, the cloning work of Lefkowitz and others turned up at least five more adrenergic receptors, all different from one another (in ways that had been imperceptible before the techniques of molecular genetics) and each with its own functions in the cell. From the point of view of clinical applications, this was, and continues to be, an exciting time for drug research, because it becomes possible to distinguish more closely among receptors and to select medications with increasing specificity. In the future it may even be possible to develop drugs targeted to particular subtypes of receptors and reduce unwanted side effects to a bare minimum.
The arrangement of seven membrane-spanning domains occurs widely throughout nature, in numerous guises. Many hormones, as well as transmitters, have receptors of this type; proteins called the opsins, which are precursors for the visual pigments, also fall in this category; and even in species as distant from ourselves as the slime mold, cyclic AMP is regulated through such a receptor. Meanwhile, with what is already known, many researchers are tackling the question of just what it is about the receptor's structure that determines function.
For questions of this sort, one technique is particularly effective —with the added advantage (or disadvantage, depending on one's taste) of sounding like something from a science fiction script. This technique is the creation of chimeras, creatures or structures that are artificially assembled from diverse genetic origins. Chimeric receptors created with recombinant DNA are excellent tools for study, because they allow the investigator to alter the receptor's structure, one small piece at a time, and then to observe any associated change in functioning. For example, researchers explored the activity of a chimeric receptor made up mostly of the alpha-2 type, with a small portion of beta receptor inserted into it. The receptor bound the substances that would be expected for an alpha receptor, but then it stimulated second-messenger production, as would be expected of a beta receptor. Thus, like the original chimera in Greek mythology (part-lion, part-goat, part-serpent),
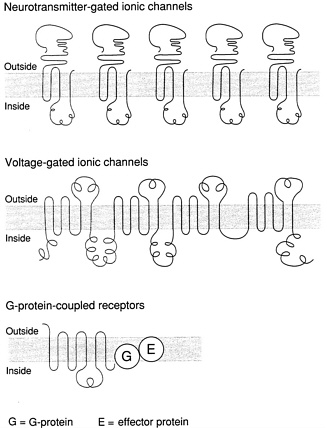
FIGURE 5.2. Receptor sites in the brain exhibit a variety of structures that each correspond with a particular function. Some (top row) receive the chemical signals of neurotransmitters and convert them into electrical signals, by allowing or halting the flow of charged ions into or out of the cell. Other receptors (middle) are triggered by electrical signals—changes in voltage—and respond by channeling ions into or out of the cell. The G-protein-coupled receptors (bottom row) react not to ions or neurotransmitters but to a “second messenger” (the G protein); the result is a chemical reaction that takes place inside the cell and, consequently, a change in the cell 's activities. What all receptor sites apparently have in common is a molecular structure that extends across the cell membrane (gray bar) and back again several times. The G-protein-coupled receptors span the membrane no fewer than seven times. Source: Marcus Raichle, Washington University School of Medicine, St. Louis, Mo.
this receptor is a fabrication, a blend of incongruous parts. With continued fine-tuning of the size and precise placement of chimeric insertions, researchers should be able to isolate the specific amino acids in the receptor that determine one or another biological function.
The dynamic nature as well as the versatility of the adrenergic receptors offers appealing avenues for further investigation. For instance, what takes place at the molecular level during the process of desensitization, in which the response drops in intensity over time even though the stimulus remains constant? All the evidence thus far suggests that there may be a new—previously unknown—protein kinase involved in desensitization, which essentially works the “off” switch for a receptor that has bound to a first messenger. Lefkowitz and his coworkers named such a substance, sight unseen, BARK—for beta adrenergic receptor kinase—and then proceeded to find it. BARK cooperated by revealing itself as a distant relative of the second messenger-sensitive protein kinases.
BARK works in cooperation with another newly discovered protein, barrestin (for “beta-arrestin”), to inactivate the receptor site temporarily. The mechanism works in two steps. When BARK is called into action by the binding of a neurotransmitter, it adds a phosphate group to the receptor site, thereby changing its shape. Barrestin can now bind easily to the receptor site and complete the action of the “off” switch.
The proving of a hunch can be its own best reward—particularly for the scientist, who is often pursuing something as intangible as a closer look at some aspect of nature. But the discovery of BARK and barrestin brings a practical bonus. In the development of pharmaceutical aids to reduce desensitization and thereby prolong the therapeutic effect of some drugs, BARK and barrestin offer two targets that were completely unknown only a few years ago.
THE INTRIGUING ROLE OF THE ION CHANNELS
While the discovery of neurotransmitters and elucidation of the structure of their receptor sites have been proceeding apace, another family of receptor sites is also becoming increasingly well known to neuroscience. This family comprises
all the voltage-gated channels, so named because they are activated by electricity—voltage differentials across the cell membrane—rather than by chemical factors such as the neurotransmitters. (Some receptors for neurotransmitters are coupled to voltage-gated channels in a sort of duplex arrangement.)
The main voltage-gated channels are of three types: potassium and sodium, by means of which the nerve cell fires an electrical impulse and transmits that impulse along the axon; and calcium, which reacts to the electrical charge at the end of the axon by releasing neurotransmitter into the synaptic cleft. The genetic sequences of these channels are all strikingly similar, even in animals as disparate as mammals and fruit flies. (Fruit flies, in particular, are a boon to the researcher, who can thus tackle many intriguing questions of neurophysiology in an animal model that produces a new generation every two weeks.)
The past few years have seen the emergence of new techniques for studying extremely precise sections of neuroanatomy, such as a particular kind of ion channel, isolated “in the dish,” in a laboratory setting. By these means, it may soon be possible to observe what goes on during the process of learning at the cellular level, where nerve cells store information in some physical form for later retrieval. (For an account of recent findings in this field, see Chapter 8 .) In addition, a better understanding of the genetic instructions for assembling various ion channels cannot fail to have clinical applications—for example, in the design of drugs for now-irreversible conditions, or in the pinpointing of genetic variations that may predispose an individual toward certain diseases.
Genetic studies have made it possible to clone several ion channels —the optimum method for a close study of channel structure. Cloning of the sodium channel gene has revealed that it includes four domains, each of which spans the membrane multiple times. The calcium channel, too, consistently contains four domains, each with multiple membrane-spanning sequences; the potassium channel shows more variation in form. The protein sequence for the potassium channel also is noticeably smaller, perhaps one-quarter that of the sodium channel.
Experiments in the past few years have shown that the diverse potassium channels tend to share a common core region
of their DNA sequence; the differences reside in the terminal regions. Potassium channels with different terminal regions are found in different parts of the brain. Diversity may also arise by another means: several of the relatively short sequences may coalesce in various ways to form a single potassium channel.
The research team of Lily Yeh Jan, at Howard Hughes Medical Institute and the Departments of Physiology and Biochemistry at the University of California, San Francisco, set out to test this possibility. They began by injecting an assortment of genetic material for potassium channels into developing egg cells of Xenopus, the African toad. The potassium channels thus created displayed an interesting combination of traits. For example, one set had lost most of the important “inactivation” function: not only did these channels remain open longer than usual but they were liable to open at any time rather than in precise response to a change in electrical polarization of the nerve cell. (Inactivation itself is thought to come about by a sort of molecule-sized tetherball, which swings into the mouth of the open potassium channel and thereby blocks it; negative charges at the mouth of the pore would temporarily hold the particle, which is presumably positively charged, in place. This model is being tested in several laboratories.) Other channels remained open for some time, but only in the first half of the depolarization phase. Clearly, while not all the combinations formed in this way are functional, a great many are. The net result is a great range of possible functions achieved with relative economy of means.
THE CASE OF THE 5-SECOND MESSENGER MOLECULE
In another trail of research, molecular biology and genetics are joining pharmacology to bring to light the workings of yet one more type of messenger molecule. The molecule nitric oxide fills a critical role in diverse tissues of the body, from the lining of blood vessels to the cerebellum, but its identity as a messenger of anything at all was completely unsuspected for a long time. For one thing, the compound—a single atom of nitrogen joined to one atom of oxygen—was very unstable, existing only for a matter of seconds. What could it be doing in the body?
It was the well-known effect of nitroglycerin on intense chest pains that first put investigators on the trail of nitric oxide as a messenger molecule. Nitroglycerin works in remarkably small doses to dilate the blood vessels and relieve chest pain. Pharmacologists already knew that nitric oxide was the active ingredient formed by the body from nitroglycerin. But what became clear only in the late 1980s was that nitric oxide was the very substance being sought independently in cardiovascular research as a “relaxing factor” that works in tandem with the neurotransmitter ACh in the lining of blood vessels. It soon emerged that nitric oxide does not bring about this effect alone; rather it stimulates the production of a second messenger, cyclic GMP (cyclic guanosine monophosphate).
As for its origin, nitric oxide is formed by the action of an enzyme from the amino acid arginine; another acid, citrulline, is given off as a by-product. One of the reasons nitric oxide has been so difficult to find in the body is that it is so shortlived (its half-life is five seconds). But the citrulline that is produced at the same time does remain in the system and it can be measured, providing a clue to the evanescent presence of nitric oxide.
At this point in the inquiry, brain researchers began to take an active interest. Solomon Snyder, director of the neuroscience department at Johns Hopkins Medical School, was intrigued by the actions of nitric oxide, and especially by their extraordinary rapidity. He felt sure a system as remarkable as that of nitric oxide could not have developed only for use in blood vessels—it must also be at work somewhere in the brain.
Snyder's research team used as their starting point the established fact that when the neurotransmitter glutamate binds to receptor sites, the calcium channels open and a great amount of cyclic GMP is produced. Once the investigators knew what to look for, they could follow two lines of evidence simultaneously: levels of citrulline and levels of cyclic GMP, both of which indicate the action of nitric oxide.
Sure enough, stimulating the glutamate receptors in the cerebellum tripled the levels of citrulline and increased the levels of GMP almost tenfold. (Conversely, when the cells were treated with a drug that inhibits the nitric oxide-forming enzyme, no cyclic GMP was produced, even when the glutamate receptors
were activated—a check on cause and effect that confirmed the researchers' hunch.) As another piece of evidence, these great increases in cyclic GMP production all took place within a few seconds. This was a remarkably swift reaction, well within the time frame of some of the more brisk neurotransmitters. It appears that the nitric oxide-forming enzyme is switched on as soon as the calcium channels open, and it begins at once to produce nitric oxide at full speed, with some help from calmodulin, a calcium-binding protein.
Thus, nitric oxide is indeed at work in the brain, and it is associated with one of the most important excitatory neurotransmitters—glutamate. Intriguingly, nitric oxide is neither a transmitter nor a second messenger but truly a different type of messenger between cells. By mapping the areas of the brain where the nitric oxide–forming enzyme tends to concentrate, investigators are beginning to understand more about the action of this substance.
Nitric oxide–forming enzyme is found in high concentrations in the cerebellum, which is largely involved in movement, and in the olfactory bulb; it also appears in the pituitary gland (the producer of many hormones), in sections of the eye, and in the intestine (where it may turn out to be the primary agent of muscle relaxation). One of the most exciting of recent results is the finding that certain arteries in the brain contain the enzyme not only in their interior lining but, more unusually, in the neurons that supply the outer layer—the very arteries and nerves recently shown to be involved in migraine headaches. These observations are already being applied in the rapid development of drugs to act at this site in the nitric oxide system, and may soon bring millions of migraine sufferers a new prospect of relief. This novel form of molecular signal, discovered so recently, suggests that there are still more intriguing trails to be explored within the messenger systems.
ACKNOWLEDGMENTS
Chapter 5 is based on presentations by Lily Yeh Jan, Robert Lefkowitz, and Solomon H. Snyder.



















